
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ modification of the d-band in the core–shell structure for efficient hydrogen storage via electrocatalytic N2 fixation†
Xiaohui
Yang‡
ab,
Jin
Wan‡
a,
Huijuan
Zhang
a and
Yu
Wang
 *ac
*ac
aSchool of Chemistry and Chemical Engineering, State Key Laboratory of Power Transmission Equipment & System Security and New Technology, Chongqing University, 174 Shazheng Street, Shapingba District, Chongqing City, 400044, P. R. China. E-mail: wangy@cqu.edu.cn
bKey Laboratory of Reservoir Aquatic Environment, Chongqing Institute of Green and Intelligent Technology, Chinese Academy of Sciences, No. 266, Fangzheng Avenue, Beibei District, Chongqing 400714, P. R. China
cSchool of Electrical Engineering, Chongqing University, 174 Shazheng Street, Shapingba District, Chongqing City, 400044, P. R. China
First published on 29th August 2022
Abstract
The electrochemical N2 reduction reaction (NRR) into NH3, especially powered by clean and renewable electricity, is a promising alternative to the capital- and energy-intensive Haber–Bosch process. However, the inert N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond and the frantic competition of the hydrogen evolution reaction lead to a poor NH3 yield rate and faradaic efficiency (FE). Here, we in situ construct a series of two-dimension core/shell V2O3/VN nanomeshes with a gradient nitride-layer thickness. Among them, V2O3/VN-2 exhibits the highest FE of 34.9%, an excellent NH3 yield rate of 59.7 μg h−1 mgcat.−1, and outstanding cycle stability, exceeding those of most of the NRR electrocatalysts reported to date. First-principles calculations reveal that the d-band center of VN shifts up in a nearly linear manner with the decrease of nitride-layer thickness, and V2O3/VN-2 with a d-band center closer to the Fermi level can strengthen the d–2π* coupling between the catalyst and N2 molecule, notably facilitating the N2-into-NH3 conversion.
N bond and the frantic competition of the hydrogen evolution reaction lead to a poor NH3 yield rate and faradaic efficiency (FE). Here, we in situ construct a series of two-dimension core/shell V2O3/VN nanomeshes with a gradient nitride-layer thickness. Among them, V2O3/VN-2 exhibits the highest FE of 34.9%, an excellent NH3 yield rate of 59.7 μg h−1 mgcat.−1, and outstanding cycle stability, exceeding those of most of the NRR electrocatalysts reported to date. First-principles calculations reveal that the d-band center of VN shifts up in a nearly linear manner with the decrease of nitride-layer thickness, and V2O3/VN-2 with a d-band center closer to the Fermi level can strengthen the d–2π* coupling between the catalyst and N2 molecule, notably facilitating the N2-into-NH3 conversion.
Introduction
With the climate change due to the excessive use of fossil fuels, a low-carbon energy transition is permeating many industrialized countries, which stresses the importance of hydrogen energy in optimizing their energy structures.1,2 Recently, ammonia (NH3), as a medium of clean hydrogen energy storage, has attracted considerable attention due to its high energy density (4.32 kW h L−1), low pressure storage, and convenient transport.3,4 Compared with the capital- and energy-intensive Haber–Bosch process, the electrochemical N2 reduction reaction (NRR) under ambient conditions is emerging as a promising alternative for the sustainable synthesis of NH3.5–8 Due to the high ionization energy (15.58 eV) of inert N2 and the sluggish reaction kinetics involving six proton-coupled electron transfer steps, an electrocatalyst is requisite in N2 adsorption and activation during the NRR process.9–11 Although tremendous efforts have been devoted to the screening and development of new electrocatalysts and optimizing the electronic structure, including noble-metal-based materials,12,13 metal-oxide materials,14,15 and metal-free materials,16,17 their NRR efficiency is still very poor and far from meeting the needs of practical industrial production.18 Thus, designing and fabricating potential electrocatalysts with high activity and selectivity is urgent and significant for the development of sustainable electrochemical NRR technology.Transition metal nitrides (TMNs) are considered to be one of the best promising NRR candidates because their inherent feature endows them with unique advantages in electrochemically reducing N2 into NH3 along with the more favorable Mars–van Krevelen (MvK) pathway.19–24 Several recent studies suggest that introducing dopants and/or defects into TMNs has also been proved to be an effective means to improve their electrocatalytic NRR performance compared with their counterparts.25–27 The interesting thing is that introducing an O atom onto the VN surface is beneficial for the activation of the surface N sites adjacent to the surface O, where the introduction of O can regulate the electronic structure of nitride and weaken the V–N bond energy, which lowers the energy barrier of NH3 release and thus facilitates the NRR progress.28 In this regard, it is possible to modify the TMN electron state using the relevant oxide to achieve high NRR performance.
Shifting the d-band position up or down in the active site of the electrocatalyst may be an effective strategy to tune the TMN electron state to achieve the desired catalytic performance.29,30 For instance, experimentally, Ling et al. reported that using the zeolitic-imidazole framework lowered the d-band position of Au/Pt to weaken H adsorption and concurrently constructed abundant electron-deficient sites to kinetically boost the NRR performance.31 Huang et al. developed a class of M–Te (M = Ru, Rh, Ir) glassy porous nanorods with a higher d-band center, showing better NRR activity.32 Unfortunately, regulating the d-band state close to the Fermi level to enhance the interaction of N2 with the substrate also leads to strong metal-H formation. Inspired by the above reports, how to develop a novel strategy by modifying the d-band center for improving NRR activity and simultaneously suppressing the competitive hydrogen evolution reaction (HER) will be an interesting topic.
In this work, we report the effect of d-band modification on NRR performance by engineering the core–shell nanostructure (CSN) consisting of an oxide core and a nitride shell, where d-band regulation is realized by controlling the thickness of the nitride shell in situ grown on the oxide core. Considering the influential role of vanadium (V) in nitrogenases,33 hypotoxicity of V2O3,34,35 and inactiveness of vanadium nitride (VN) for the HER,19,22 we take core/shell V2O3/VN as a proof-of-concept platform. Interestingly, the NRR catalytic results show that as the nitride-layer thickness increases, the NH3 yield of core/shell V2O3/VN first increases and then decreases, with the highest faradaic efficiency (FE) being 34.9% along with an excellent NH3 yield rate of 59.7 μg h−1 mgcat.−1, significantly better than most literature reports. Furthermore, density functional theory (DFT) calculations demonstrate a similar phenomenon that when the thickness of the nitride shell is 2 atomic-layer, the barrier of the potential-determining step (PDS) for the NRR is minimum, only 0.63 eV. These results suggest that the more appropriate the nitriding time, the better the NRR activity.
Results and discussion
It is widely accepted that a two-dimensional (2D) porous morphology of the catalyst could impart high catalytic activity due to the high active-site exposure and the favorable mass/charge transport. To this end, a 2D porous V2O3 nanomesh as the oxide core and an in situ grown nitride shell with controllable thickness are fabricated as shown in Fig. 1a (see ESI† for more details). Initially, the precursor of 2D Zn3(OH)2(V2O7)(H2O)2 nanosheets with a nanoscale thickness of ∼50 nm was prepared based on our previous studies (Fig. S1†).36 Then, this sheet-like precursor was annealed in an argon atmosphere to form 2D porous V2O3 nanomeshes (Fig. S2†). 2D porous core/shell V2O3/VN nanomeshes with a gradient thickness of the nitride shell were obtained via an appropriate nitriding treatment, where the target samples were defined as V2O3/VN-X (X = 0.5, 2, 10, representing the nitriding time). For comparison, 2D porous pure VN nanomeshes were also prepared by lengthening the time of nitriding treatment.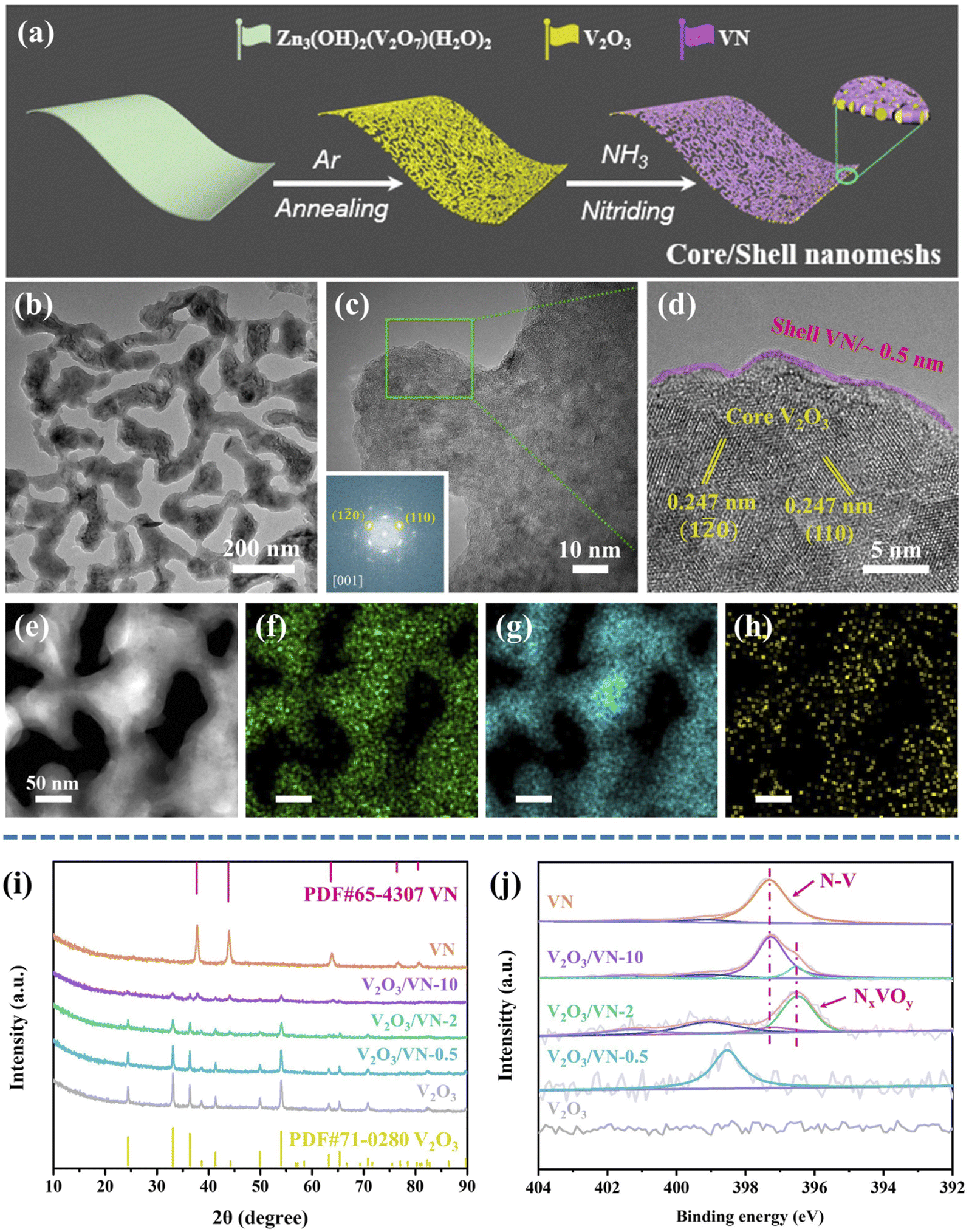 | ||
| Fig. 1 (a) Schematic illustration of the fabrication of 2D core/shell V2O3/VN nanomeshes. Morphology and structure characterization for 2D core/shell V2O3/VN-2. (b) TEM image, (c) magnified TEM image, and (d) the corresponding HR-TEM image in the edge. (e) HAADF-STEM image, and the corresponding EDS elemental mapping images of (f) V, (g) O, and (h) N based on image (d). (i) XRD patterns and (j) XPS spectra of N 1s for pure V2O3 nanomeshes, core/shell V2O3/VN-X and pure VN nanomeshes. Inset in (c) is the corresponding selected-area electron diffraction. | ||
Fig. 1b shows the transmission electron microscopy (TEM) image of 2D core/shell V2O3/VN-2 nanomeshes. Compared with the pure V2O3 core, no obvious change is observed in the morphology. The magnified TEM image shows that the surface of V2O3/VN-2 is rough, suggesting that a nitride layer has been formed (Fig. 1c). In addition, V2O3/VN-2 exhibits a crystalline structure, as evidenced by the selected-area electron diffraction image (inset in Fig. 1c). Notably, the corresponding high-resolution TEM (HRTEM) image of V2O3/VN-2 exhibits well-defined lattice fringes, and a lattice plane distance of 0.247 nm along different directions, which was indexed to the (110) or (1![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 0) facet of the rhombohedral V2O3 phase (PDF #71-0280). In contrast, the thickness of the VN shell is about 0.5 nm, and the crystallinity is low which may be attributed to the short time of nitriding. Furthermore, a high-angle annular dark-field scanning transmission electron microscope (HAADF-STEM) equipped with an energy-dispersive spectrometer (EDS) was employed to investigate the compositional distribution of this 2D core/shell V2O3/VN-2 nanomesh. The V element is uniformly distributed over the whole nanomesh skeleton, while the O element is distributed in the core area and the amount of N is comparatively higher in the boundary region (Fig. 1e–h). We further investigated other as-synthesized samples with different nitriding treatments. From the HAADF-STEM images and corresponding compositional distributions (Fig. S3†), it can be seen that the nitriding process is from outside to inside with the increase of nitriding time, indicating that the length of nitriding time determines the thickness of the VN shell in 2D core/shell V2O3/VN nanomeshes. Moreover, the structure of the 2D porous nanomeshes remains unchanged even if the oxide core is subjected to different degrees of nitridation or even converted to pure VN, which implies excellent structural stability.
0) facet of the rhombohedral V2O3 phase (PDF #71-0280). In contrast, the thickness of the VN shell is about 0.5 nm, and the crystallinity is low which may be attributed to the short time of nitriding. Furthermore, a high-angle annular dark-field scanning transmission electron microscope (HAADF-STEM) equipped with an energy-dispersive spectrometer (EDS) was employed to investigate the compositional distribution of this 2D core/shell V2O3/VN-2 nanomesh. The V element is uniformly distributed over the whole nanomesh skeleton, while the O element is distributed in the core area and the amount of N is comparatively higher in the boundary region (Fig. 1e–h). We further investigated other as-synthesized samples with different nitriding treatments. From the HAADF-STEM images and corresponding compositional distributions (Fig. S3†), it can be seen that the nitriding process is from outside to inside with the increase of nitriding time, indicating that the length of nitriding time determines the thickness of the VN shell in 2D core/shell V2O3/VN nanomeshes. Moreover, the structure of the 2D porous nanomeshes remains unchanged even if the oxide core is subjected to different degrees of nitridation or even converted to pure VN, which implies excellent structural stability.
To further study the crystal phases of all 2D porous nanomesh samples, X-ray powder diffraction (XRD) analysis was carried out. As shown in Fig. 1i, all the diffraction peaks of the core are well indexed to rhombohedral V2O3, in agreement with the TEM results. As nitriding time increases, the intensity of V2O3 diffraction peaks gradually decreases while new peaks assigned to the cubic VN (PDF #65-4307) are detected. It is noted that the diffraction peaks of nitride shells in V2O3/VN-0.5 and V2O3/VN-2 are not obvious, which is mainly caused by the inherent strong penetrability of XRD. Therefore, we further employ X-ray photoelectron spectroscopy (XPS) to research their surface chemical states. The XPS full spectra of all 2D porous nanomesh samples are shown in Fig. S4,† from which the existence of V, O, and N elements except for the pure V2O3 sample is verified. Fig. 1j shows the high-resolution N 1s spectrum of V2O3/VN-2 that can be deconvoluted into four subpeaks. The two subpeaks at a lower binding energy (BE) of 397.3 and 396.5 eV are attributed to the bond of N–V and the NxVOy species, respectively.37,38 The peak located at 401.4 eV is assigned to the residuals of ammonia/ammonium species adsorbed on the material surface during the nitriding treatment and the subpeak of 399.1 eV is considered a satellite peak.28 In contrast, V2O3/VN-0.5 has only a satellite peak, indicating that N atoms dope in V2O3. The N 1s spectrum of deeper nitrided V2O3/VN-10 can also be deconvoluted into four subpeaks, where the intensity of the N–V bond increases while the NxVOy species decreases. According to the above analysis, it is found that the thickness of the VN shell varies with nitriding time.
Based on the above-mentioned characterization, a series of 2D porous core/shell V2O3/VN nanomeshes with a gradient thickness of the nitride shell have been successfully prepared. Next, their NRR performance was measured by evaluating their electrochemical N2 reduction ability using a typical two-compartment cell separated by a proton exchange membrane. We first choose V2O3/VN-2 as the research object. Its linear sweep voltammetric (LSV) curves in Ar- and N2-saturated 0.05 M H2SO4 solution are shown in Fig. 2a, where an obviously increased reduction current density is detected in the N2-saturated acid solution, especially in the potential window between −0.1 V and −0.6 V vs. RHE, indicating an additional contribution from the conversion reaction of N2 to NH3 in this system. And then NH3 yield rates and corresponding FEs for V2O3/VN-2 were assessed by combining the chronoamperometry tests (Fig. S7†) and the corresponding UV-vis absorption spectra (Fig. S8†) at different applied potentials. As shown in Fig. 2b, the highest NH3 yield is 59.7 μg h−1 mgcat.−1, which is achieved at a driving potential of −0.4 V vs. RHE, while the highest FE value of 34.9% is reached at −0.2 V vs. RHE. To the best of our knowledge, such outstanding NRR performance is comparable with those of the most advanced NRR electrocatalysts recently reported in 0.1 M acid electrolyte under room temperature and atmosphere pressure (Fig. 2c and Table S1†). Meanwhile, V2O3/VN-2 also possesses excellent selectivity for NH3 formation, which is verified by no N2H4 being detected (Fig. S9†). To investigate the stability of V2O3/VN-2, consecutive cycling tests and long-term electrocatalytic tests were further carried out at −0.2 V vs. RHE. After eight consecutive cycling tests, there is no significant change in current densities, NH3 yields and FEs (Fig. 2d and e). After a long-term electrocatalytic test for 50 h, the activity of V2O3/VN-2 nanomeshes remains unchanged with an NH3 yield of 38.5 μg h−1 mgcat.−1 and FE of 35.6% (Fig. S10†). In addition, the SEM, TEM, and XRD results show that V2O3/VN-2 maintains its original morphology and crystal structure after electrocatalysis (Fig. S11 and S12†). All these results suggest that V2O3/VN-2 is a considerably robust NRR electrocatalyst.
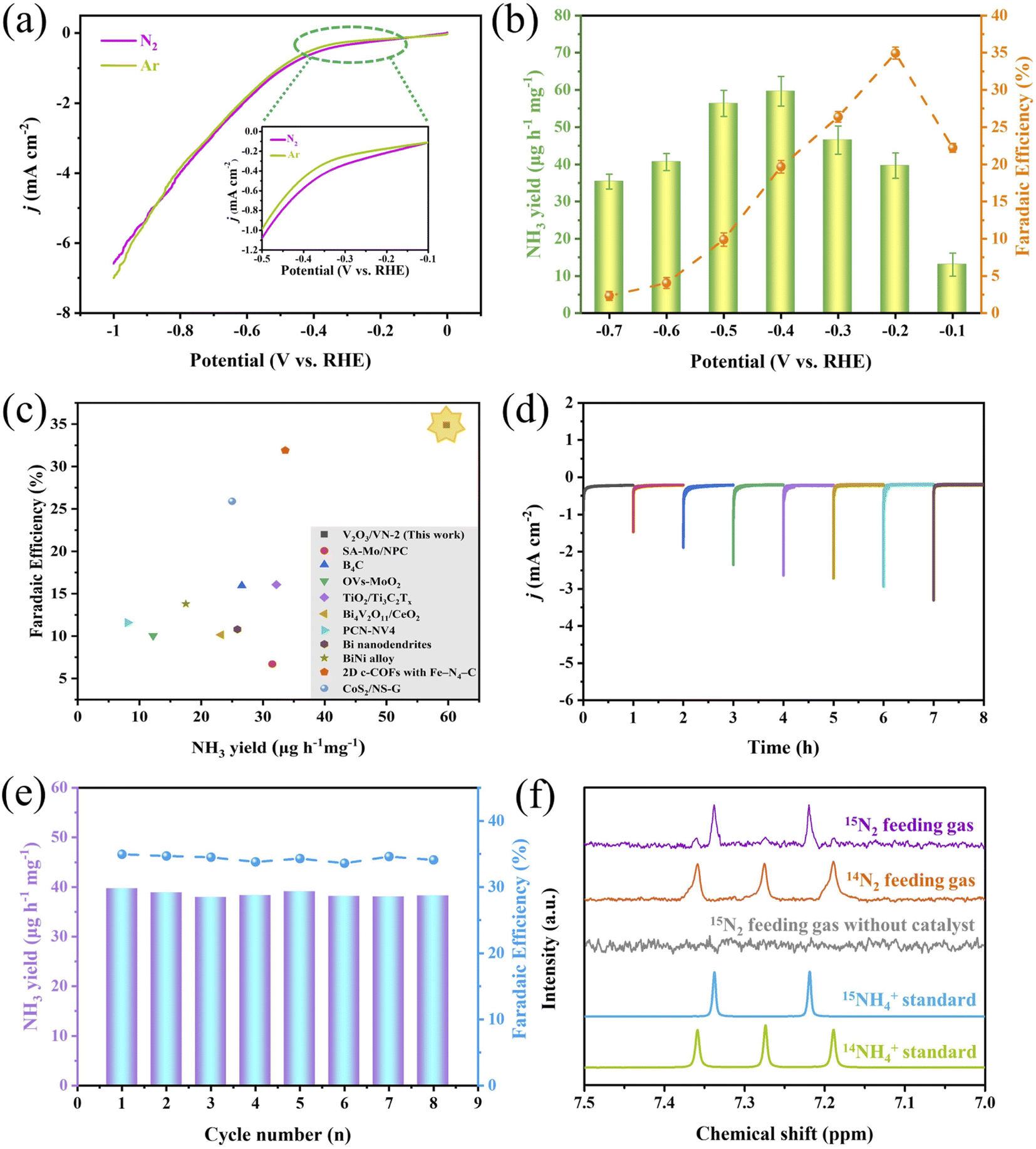 | ||
| Fig. 2 Electrocatalytic NRR performance of 2D core/shell V2O3/VN-2 nanomeshes. (a) Linear sweep voltammetric curves in Ar- and N2-saturated 0.05 M H2SO4 solution. Inset is the amplified details. (b) The average NH3 yields and faradaic efficiencies (FEs) at different applied potentials. (c) Comparison of NH3 yields and FEs with the recently well-developed NRR electrocatalysts in 0.1 M acid solution under room temperature and atmosphere pressure. (d) Cycling stability tests in N2-saturated 0.05 M H2SO4 solution at a potential of −0.2 V vs. RHE under consecutive recycling electrolysis. (e) The corresponding NH3 yields and FEs based on eight consecutive cycling tests. (f) 1H NMR spectra of the concentrated electrolyte fed with 15N2 and 14N2 after the electrolytic reaction. | ||
To confirm the safety of the test environment, we conducted blank experiments with the fresh 0.05 M H2SO4 solution and the cycled 0.05 M H2SO4 electrolyte treated by N2 gas bubbling for 1 h. No NH3 was detected in both of them, which is verified by UV-vis spectroscopy (Fig. S13†), indicating that the test environment is secure. And then, a series of control experiments using a bare carbon paper electrode (CPE) in N2-saturated electrolyte and a 2D core/shell V2O3/VN-2 nanomesh electrode in Ar/N2-saturated electrolyte were performed to examine the source of the produced NH3. The corresponding UV-vis absorption spectra are shown in Fig. S14.† Reasonably, a stronger signal for the core/shell V2O3/VN-2 electrode in N2-saturated electrolyte is observed compared with the other two control experiments, which indicates that the NH3 produced is derived from the electrochemical reduction of N2 to NH3 in the presence of the 2D core/shell V2O3/VN-2 catalyst. Furthermore, isotopic labeling experiments using 15N2 and 14N2 as feeding gases were carried out to identify the N source of the produced NH3 and to probe into the potential pathway of V2O3/VN-2 during the NRR process. The characteristic signal, a doublet coupling for 15NH4+ and a triplet coupling for 14NH4+ in the 1H nuclear magnetic resonance (1H NMR) spectra, is used as a criterion of judgment. As shown in Fig. 2f, when 14N2 as feeding gas is supplied, only triplet coupling from 14NH4+ was observed, and when 15N2 as feeding gas is supplied, primary doublet coupling from 15NH4+ along with weak triplet coupling from 14NH4+ is detected. Such results suggest that 2D core/shell V2O3/VN-2 nanomeshes as an active catalyst can effectively catalyze the conversion of N2 to NH3. Besides, it is reasonably inferred that the surface N atom in 2D core/shell V2O3/VN-2 nanomeshes is first reduced to NH3 and the rest of the N vacancies facilitate the continuous proceeding of the NRR, in line with the reported MvK pathway.
To attest the superiority of 2D core/shell V2O3/VN-2 nanomeshes, the NRR activities of all comparative samples were tested at different applied potentials, including pure V2O3 nanomeshes, core/shell V2O3/VN-0.5 nanomeshes, core/shell V2O3/VN-10 nanomeshes, and pure VN nanomeshes. Based on the results of UV-vis absorption spectra (Fig. S15†), their respective NH3 yields are obtained and shown in Fig. 3a. It is obvious that as the thickness of the nitride shell in all 2D porous nanomesh samples increases, the corresponding NH3 yield first increases and then decreases at each given potential, where the sample of V2O3/VN-2 possesses the highest NRR activity. Furthermore, the electrochemically active surface area (ECSA) of each sample was obtained by evaluating their double-layer capacitance (Cdl) (Fig. S16 and S17†). To achieve the intrinsic activity (surface area normalized), here we introduce the concept of roughness factors (RFs) based on previous reports.39–41 The RF value of pure V2O3 nanomeshes is supposed to be 1, RFs of the other samples were calculated by dividing their capacitances by the Cdl value of pure V2O3 nanomeshes (Fig. 3b). As shown in Fig. 3c, the ECSA-normalized NH3 yields at different given potentials still first increase and then decrease with the growth of the nitride shell for all 2D porous nanomesh samples, indicating that only the sample with the appropriate thickness of the nitride shell has the best intrinsic NRR activity.
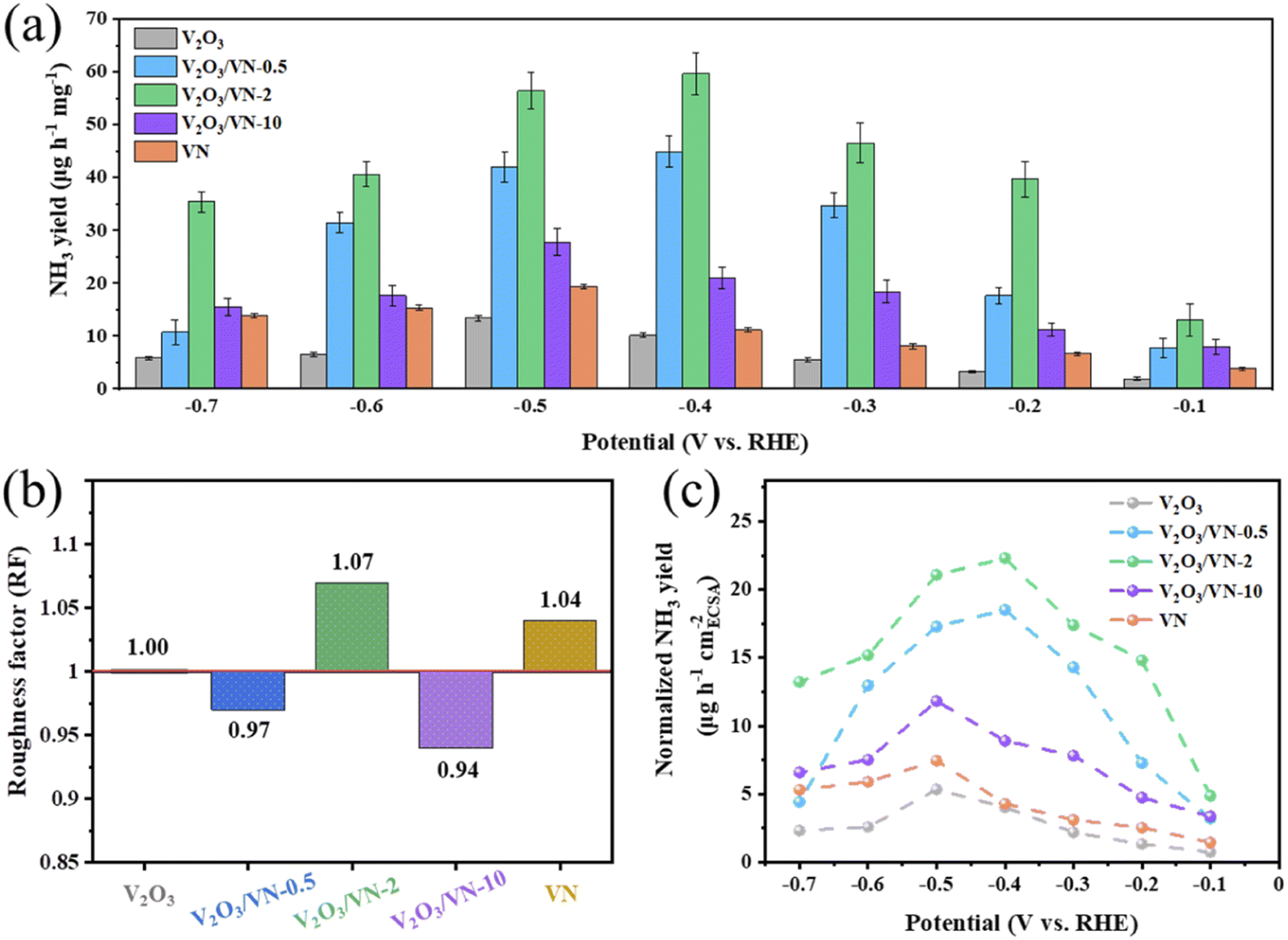 | ||
| Fig. 3 (a) Comparison of the NH3 yields for the pure V2O3 nanomeshes, core/shell V2O3/VN-X and pure VN nanomeshes in N2-saturated 0.05 M H2SO4 solution at different applied potentials. (b) The roughness factors of core/shell V2O3/VN-X and pure VN nanomeshes taken with pure V2O3 nanomeshes as a reference. (c) ECSA-normalized NH3 yields of all 2D porous nanomesh samples at each given potential. | ||
To further decode the origin of the improved NRR performance of 2D core/shell V2O3/VN nanomeshes, DFT calculations were performed to understand the response mechanism of reducing N2 into NH3 on these systems. The VN shell thickness observed in the HR-TEM image (∼0.5 nm, Fig. 1d) suggests that the VN covering the (001) surface of V2O3 is almost two atomic-layer thick. Furthermore, the amount of nitriding time will lead to different thicknesses of the VN shell. According to these characterization results, four systems were constructed, including V2O3/N-doped, V2O3/VN-2 layer, V2O3/VN-3 layer, and the (111) surface of pure VN, corresponding to V2O3/VN-X (X = 0.5, 2, 10) and pure VN (Fig. S18†).
As we discussed above, this reaction will follow the MvK mechanism (Fig. S19–S22†). In the reaction procedure, the pre-reacted *N can be hydrogenated into the *NH intermediate via the proton/electron (H+/e−) pair. Then, the adsorbed *NH captures the H+/e− pair, forming the *NH2. Subsequently, the *NH2 is further hydrogenated to form N defects, while releasing the NH3 molecule. Finally, the defect captures the N2 molecule which is stepwise hydrogenated into *NNH, *NNH2, and *NNH3 until the second NH3 molecule is released. Notably, the adsorption of N2 is the potential-limiting step (PDS) for the V2O3/VN-2 layer and V2O3/VN-3 layer with the maximum energy barrier of 0.63 and 0.70 eV, respectively. The V2O3/N-doped exhibits the largest reaction barrier of 0.88 eV when the first NH3 is released. In addition, the free energy change of the *N2 to *NNH step on V2O3/N-doped and pure VN surface is 0.83 and 0.80 eV, respectively (Fig. 4a). Thus, the calculated NRR performance on these systems follows the order of V2O3/VN-2 layer > V2O3/VN-3 layer > pure VN > V2O3/N-doped, which is highly consistent with the experimental results. In addition to the high NRR activity, an ideal catalyst should be able to suppress the HER to achieve high FE. Basically, if the adsorption free energy of the H adsorbate (ΔG*H) is more negative than that for *N2, *H can easily cover the active sites and block the NRR. As shown in Fig. S23,† V2O3/VN-2 layer presents the highest ΔG*H, which is much larger than the *N2 adsorption energy, indicating that d-band modification can effectively promote NRR actively while suppressing the competitive HER.
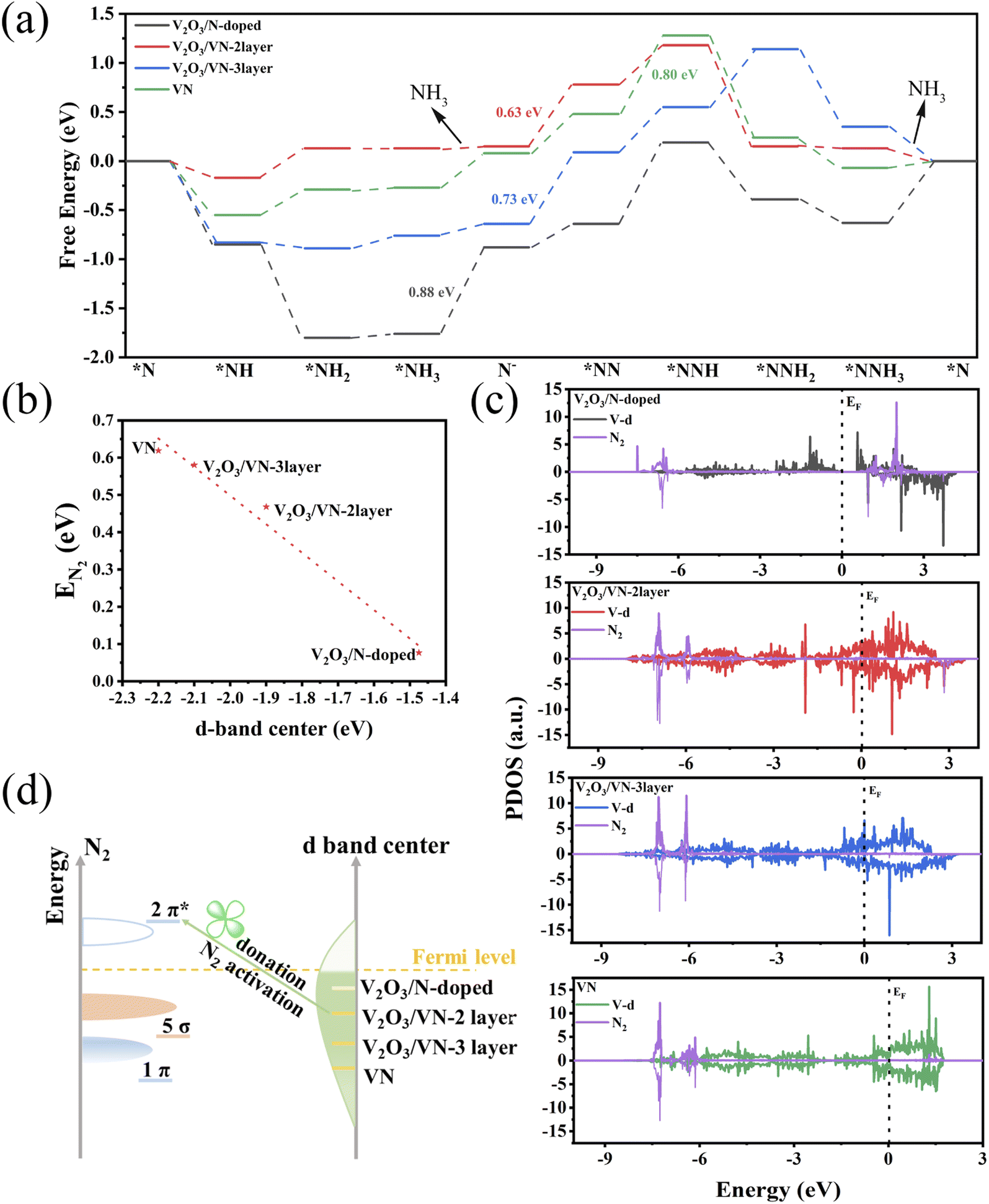 | ||
| Fig. 4 (a) Gibbs free energy diagrams for N2 electroreduction. (b) The linear relationship between the adsorption energy of N2 and the d-band center on these four systems. (c) The PDOS of N2 and the V d-band of N2@V2O3/N-doped, N2@V2O3/VN-2 layer, N2@V2O3/VN-3 layer, and pure N2@VN, respectively. (d) Schematic illustration explaining N2 activation by altering the d-band center. | ||
The electrocatalytic performance is closely related to the valence electron of the active sites. The d-band center is often used as a descriptor to predict and describe the interaction between the catalyst and the adsorbate. Inspired by previous work,42 the higher d-band center of the metal indicates a stronger interaction with the adsorbate state, which is beneficial for NRR performance. As shown in Fig. S24,† as the nitriding time increases, the d-band center of the active sites gradually shifted away from the Fermi level, indicating weakened binding strength. Except for the V2O3/N-doped, the d-band of the other three systems across the Fermi level exhibits a metallic character. Although the V2O3/N-doped exhibits the highest d-band center, there is a significant gap between the valence band and the conduction band, which limits charge transfer and electrocatalytic kinetics. Furthermore, the adsorption energy of N2 on these four systems decreases in a nearly linear manner with the downshifts of the d-band center (Fig. 4b).
The *N2 to *NNH step is widely considered to be the PDS due to the inert NN bond.43,44 To understand the underlying mechanism of the N2 activation, we investigated the partial density of states (PDOS) for free N2 and N2 adsorbed on these four systems with N vacancies. As shown in Fig. S25,† the occupied 1π and 5σ molecular orbitals of free N2 are located below the Fermi level, and can donate electrons to the unoccupied d orbitals of the systems, forming bonding states. Moreover, the unoccupied 2π* orbitals of N2 accept electrons from occupied d orbitals of the metal, leading to the partially occupied 2π* orbitals downshifting to the Fermi level. The “donation–back donation” between the metal and adsorbate can activate the adsorbed N2 (Fig. 4c). To be specific, for V2O3/VN-2 layer, V2O3/VN-3 layer, and pure VN, after adsorption of the free N2 on N defects, the antibonding 2π* orbitals of N2 strongly hybridize with the occupied d orbitals of active site V, especially for V2O3/VN-2 layer, indicating that more effective N2 activation occurs in V2O3/VN-2 layer. The results are in accord with the d-band center values for different core/shell V2O3/VN samples. Benefiting from the upper d-band of V2O3/VN-2 layer and thus closer energy level difference with unoccupied 2π* orbitals of N2, the π bond was constructed through side-to-side overlap of V-d orbitals and 2π* orbitals of N2 according to the molecular orbital theory. Thus, the effective electron conductive channel is built in V2O3/VN-2 layer through the V–N π bond; in other words, the electron transfer from partially occupied d-orbitals to the 2π* antibonding orbitals can easily occur in the catalytic process. In contrast, although the 1π and 5σ orbitals of N2 interact strongly with the d orbital of V2O3/N-doped, the antibonding orbitals 2π* of N2 are still located above the Fermi level and fail to activate the nitrogen. Furthermore, this strong interaction also leads to difficulties in the desorption of NH3. Consequently, suitable nitriding of V3O2 can balance the d-band center and electron conduction of the surface V, leading to outstanding NRR performance (Fig. 4d).
Conclusions
In summary, we have successfully constructed a stable NRR catalytic active intermediate of NxVOy species in 2D core/shell V2O3/VN nanomeshes and demonstrated that d-band modification is the key to enhance the NRR performance. Surprisingly, when employed in the electrochemical reduction of N2 to NH3, this 2D core/shell V2O3/VN-2 nanomesh achieves the highest FE of 34.9% among all recently reported advanced NRR electrocatalysts in an acid electrolyte under ambient conditions. Moreover, this catalyst also exhibits an excellent NH3 yield of 59.7 μg h−1 mgcat.−1 and outstanding stability without significant loss of activity after eight consecutive cycling tests and a long-term electrocatalytic test for 50 h. Isotopic labeling experiments combined with 1H NMR spectroscopy further confirm the reaction mechanism following the MvK pathway during the NRR process. DFT calculations reveal that the V2O3 core can tune the d-band structure of the VN shell, strengthen the interaction between N2 and the active site, and thus reduce the NRR potential barrier toward significantly enhanced electrochemical NRR performance. This study not only opens up a new avenue to develop high-efficiency NRR catalysts, but also guides to design other advanced catalysts for challenging electrocatalytic reactions for renewable energy conversion.Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.Author contributions
Y. W. and X. H. Y. developed the research concept, X. H. Y. prepared the materials, performed the electrochemical experiments, and analyzed the data. J. W. provided the theoretical calculations. X. H. Y. and J. W. contributed to the interpretation of the results and the writing of the manuscript, which was revised by H. J. Z. All authors contributed to the scientific discussions.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work is financially supported by the Fundamental Research Funds for the Central Universities (0301005202017, 2018CDQYFXCS0017, 106112017CDJXSYY0001), Thousand Young Talents Program of the Chinese Central Government (Grant No. 0220002102003), National Natural Science Foundation of China (NSFC, Grant No. U19A20100, 21971027, 21373280, 21403019), Beijing National Laboratory for Molecular Sciences (BNLMS) and Hundred Talents Program at Chongqing University (Grant No. 0903005203205), The State Key Laboratory of Mechanical Transmissions Project (SKLMT-ZZKT-2017M11), Natural Science Foundation of Chongqing (Grant No. cstc2019jcyj-msxmX0426), and Science and Technology Research Project of Education Agency in Chongqing (Grant No. KJZD-K201800102).References
- L. Pingkuo and H. Xue, Int. J. Hydrog. Energy, 2022, 47, 9485–9503 CrossRef
.
- X. Peng, H. X. Liu, Y. Zhang, Z. Q. Huang, L. Yang, Y. Jiang, X. Wang, L. Zheng, C. Chang, C. T. Au, L. Jiang and J. Li, Chem. Sci., 2021, 12, 7125–7137 RSC
- Y. Abghoui and E. Skulason, Catal. Today, 2017, 286, 69–77 CrossRef CAS
- L. Li, C. Tang, H. Jin, K. Davey and S.-Z. Qiao, Chem, 2021, 7, 3232–3255 CAS
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm, W. F. Schneider and R. R. Schrock, Science, 2018, 360, eaar6611 CrossRef PubMed
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. B. Chorkendorff, J. K. Norskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed
- G. Soloveichik, Nat. Catal., 2019, 2, 377–380 CrossRef CAS
- J. Wan, Y. Wang, W. Tian, H. Zhang and Y. Wang, Appl. Surf. Sci., 2021, 569, 151020 CrossRef CAS
- A. R. Singh, B. A. Rohr, J. A. Schwalbe, M. Cargnello, K. Chan, T. F. Jaramillo, I. Chorkendorff and J. K. Norskov, ACS Catal., 2017, 7, 706–709 CrossRef CAS
- X. Guo, J. Gu, S. Lin, S. Zhang, Z. Chen and S. Huang, J. Am. Chem. Soc., 2020, 142, 5709–5721 CrossRef CAS PubMed
- D. Yao, C. Tang, L. Li, B. Xia, A. Vasileff, H. Jin, Y. Zhang and S. Z. Qiao, Adv. Energy Mater., 2020, 10, 2001289 CrossRef CAS
- H. Huang, L. Xia, X. Shi, A. M. Asiri and X. Sun, Chem. Commun., 2018, 54, 11427–11430 RSC
- W. Cai, Y. Han, Y. Pan, X. Zhang, J. Xu, Y. Zhang, Y. Sun, S. Li, J. Lai and L. Wang, J. Mater. Chem. A, 2021, 9, 13483–13489 RSC
- T. Wu, H. Zhao, X. Zhu, Z. Xing, Q. Liu, T. Liu, S. Gao, S. Lu, G. Chen, A. M. Asiri, Y. Zhang and X. Sun, Adv. Mater., 2020, 32, e2000299 CrossRef PubMed
- K. Chu, Q. Q. Li, Y. H. Cheng and Y. P. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 11789–11796 CrossRef CAS PubMed
- S. Zhao, X. Lu, L. Wang, J. Gale and R. Amal, Adv. Mater., 2019, 31, e1805367 CrossRef PubMed
- Y. Yang, L. Zhang, Z. Hu, Y. Zheng, C. Tang, P. Chen, R. Wang, K. Qiu, J. Mao, T. Ling and S. Z. Qiao, Angew. Chem., Int. Ed., 2020, 59, 4525–4531 CrossRef CAS PubMed
- D. Yao, C. Tang, P. Wang, H. Cheng, H. Jin, L.-X. Ding and S.-Z. Qiao, Chem. Eng. Sci., 2022, 257, 117735 CrossRef CAS
- Y. Abghoui and E. Skúlason, Catal. Today, 2017, 286, 78–84 CrossRef CAS
- R. Zhang, Y. Zhang, X. Ren, G. Cui, A. M. Asiri, B. Zheng and X. Sun, ACS Sustain. Chem. Eng., 2018, 6, 9545–9549 CrossRef CAS
- L. Zhang, X. Ji, X. Ren, Y. Luo, X. Shi, A. M. Asiri, B. Zheng and X. Sun, ACS Sustain. Chem. Eng., 2018, 6, 9550–9554 CrossRef CAS
- Y. Abghoui, A. L. Garden, J. G. Howalt, T. Vegge and E. Skúlason, ACS Catal., 2016, 6, 635–646 CrossRef CAS
- X. Yang, S. Kattel, J. Nash, X. Chang, J. H. Lee, Y. Yan, J. G. Chen and B. Xu, Angew. Chem., Int. Ed., 2019, 58, 13768–13772 CrossRef CAS PubMed
- J. Nash, X. Yang, J. Anibal, M. Dunwell, S. Yao, K. Attenkofer, J. G. Chen, Y. Yan and B. Xu, J. Phys. Chem. C, 2019, 123, 23967–23975 CrossRef CAS
- X. Yang, F. Ling, J. Su, X. Zi, H. Zhang, H. Zhang, J. Li, M. Zhou and Y. Wang, Appl. Catal., B, 2020, 264, 118477 CrossRef CAS
- H. Jin, L. Li, X. Liu, C. Tang, W. Xu, S. Chen, L. Song, Y. Zheng and S. Z. Qiao, Adv. Mater., 2019, 31, 1902709 CrossRef PubMed
- Q. Li, L. He, C. Sun and X. Zhang, J. Phys. Chem. C, 2017, 121, 27563–27568 CrossRef CAS
- X. Yang, J. Nash, J. Anibal, M. Dunwell, S. Kattel, E. Stavitski, K. Attenkofer, J. G. Chen, Y. Yan and B. Xu, J. Am. Chem. Soc., 2018, 140, 13387–13391 CrossRef CAS PubMed
- H. Y. F. Sim, J. R. T. Chen, C. S. L. Koh, H. K. Lee, X. Han, G. C. Phan-Quang, J. Y. Pang, C. L. Lay, S. Pedireddy, I. Y. Phang, E. K. L. Yeow and X. Y. Ling, Angew. Chem., Int. Ed., 2020, 59, 16997–17003 CrossRef CAS
- J. Wang, B. Huang, Y. Ji, M. Sun, T. Wu, R. Yin, X. Zhu, Y. Li, Q. Shao and X. Huang, Adv. Mater., 2020, 32, e1907112 CrossRef PubMed
- H. Y. F. Sim, J. R. T. Chen, C. S. L. Koh, H. K. Lee, X. Han, G. C. Phan-Quang, J. Y. Pang, C. L. Lay, S. Pedireddy, I. Y. Phang, E. K. L. Yeow and X. Y. Ling, Angew. Chem., Int. Ed., 2020, 59, 16997–17003 CrossRef CAS PubMed
- J. Wang, B. Huang, Y. Ji, M. Sun, T. Wu, R. Yin, X. Zhu, Y. Li, Q. Shao and X. Huang, Adv. Mater., 2020, 32, 1907112 CrossRef CAS PubMed
- R. R. Eady, Chem. Rev., 1996, 96, 3013–3030 CrossRef CAS
- N. Rajendran, J. C. Seagrave, L. M. Plunkett and J. A. MacGregor, Inhalation Toxicol., 2016, 28, 618–628 CrossRef CAS
- R. Zhang, J. Han, B. Zheng, X. Shi, A. M. Asiri and X. Sun, Inorg. Chem. Front., 2019, 6, 391–395 RSC
- W. Fu, Y. Wang, H. Zhang, M. He, L. Fang, X. Yang, Z. Huang, J. Li, X. Gu and Y. Wang, J. Catal., 2019, 369, 47–53 CrossRef CAS
- N. C. Saha and H. G. Tompkins, J. Appl. Phys., 1992, 72, 3072–3079 CrossRef CAS
- A. Glaser, S. Surnev, F. P. Netzer, N. Fateh, G. A. Fontalvo and C. Mitterer, Surf. Sci., 2007, 601, 1153–1159 CrossRef CAS
- L. Hu, A. Khaniya, J. Wang, G. Chen, W. E. Kaden and X. Feng, ACS Catal., 2018, 8, 9312–9319 CrossRef CAS
- Y. Wang, M. M. Shi, D. Bao, F. L. Meng, Q. Zhang, Y. T. Zhou, K. H. Liu, Y. Zhang, J. Z. Wang, Z. W. Chen, D. P. Liu, Z. Jiang, M. Luo, L. Gu, Q. H. Zhang, X. Z. Cao, Y. Yao, M. H. Shao, Y. Zhang, X. B. Zhang, J. G. Chen, J. M. Yan and Q. Jiang, Angew. Chem., Int. Ed., 2019, 58, 9464–9469 CrossRef CAS PubMed
- X. Yang, F. Ling, X. Zi, Y. Wang, H. Zhang, H. Zhang, M. Zhou, Z. Guo and Y. Wang, Small, 2020, 16, 2000421 CrossRef CAS PubMed
- J. K. Norskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef CAS PubMed
- Y. Liao, J. Qian, G. Xie, Q. Han, W. Dang, Y. Wang, L. Lv, S. Zhao, L. Luo, W. Zhang, H.-Y. Jiang and J. Tang, Appl. Catal., B, 2020, 273, 119054 CrossRef CAS
- X. Yan, D. Liu, H. Cao, F. Hou, J. Liang and S. X. Dou, Small Methods, 2019, 3, 1800501 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc03975c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |