
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceKinetic resolution of racemic tertiary allylic alcohols through SN2′ reaction using a chiral bisphosphoric acid/silver(I) salt co-catalyst system†
Satavisha
Kayal
,
Jun
Kikuchi‡
 ,
Naoya
Shinagawa
,
Shigenobu
Umemiya
and
Masahiro
Terada
*
,
Naoya
Shinagawa
,
Shigenobu
Umemiya
and
Masahiro
Terada
*
Department of Chemistry, Graduate School of Science, Tohoku University, Aramaki, Aoba-ku, Sendai 980-8578, Japan. E-mail: mterada@tohoku.ac.jp
First published on 20th July 2022
Abstract
A highly efficient kinetic resolution (KR) of racemic tertiary allylic alcohols was achieved through an intramolecular allylic substitution reaction using a co-catalyst system composed of chiral bisphosphoric acid and silver carbonate. This reaction afforded enantioenriched diene monoepoxides along with the recovery of tertiary allylic alcohols in a highly enantioselective manner, realizing an extremely high s-factor in most cases. The present method provides a new access to enantioenriched tertiary allylic alcohols, multifunctional compounds that are applicable for further synthetic manipulations.
Introduction
Tertiary alcohols and their derivatives are present in a wide range of natural products and biologically active molecules. Synthetic methods for their preparation in an enantioenriched form, however, are challenging.1 One of the most common approaches investigated thus far is the enantioselective nucleophilic addition to ketones using a chiral catalyst.2 Another potential method is the catalytic kinetic resolution (KR)3 of racemic tertiary alcohols. In contrast to the catalytic KR of racemic secondary alcohols,4 reliable methods for the non-enzymatic KR of tertiary alcohols remain few.5–9 Several groups have reported the use of chiral Lewis base (nucleophilic) catalysts6 and chiral transition metal catalysts.7 In the methodologies reported for the KR of racemic tertiary alcohols in recent years, chiral phosphoric acids (CPAs), which are one of the most powerful and privileged organocatalysts employed in a broad range of enantioselective transformations,10 have proven to be excellent catalysts.8 Several reaction systems, such as acetalization,8a–c transesterification,8d condensation,8e enamine/imine tautomerization,8f and SN1-type reaction,8g have been established in a highly resolved manner using CPAs as the efficient chiral Brønsted acid catalyst.With the aim of adding a new entry to catalytic KR using CPAs, we set out to develop a method for the KR of tertiary allylic alcohols,9d synthetically useful chiral building blocks. In order to establish a novel reaction system for the KR of racemic tertiary allylic alcohols, we envisioned a catalytic intramolecular SN2′ reaction using CPA,11,12i.e., KR through the formation of enantioenriched epoxides (Scheme 1).13 Considering the intramolecular SN2′ reaction, the relative position of the leaving group (LG) to the alcohol unit, namely, the oxygen nucleophile, is apparently defined by the geometry of the double bond. Hence, it was expected that the effective recognition of enantiomeric tertiary allylic alcohols would be realized by using a CPA catalyst despite the stereogenic centre having all non-hydrogen substituents.
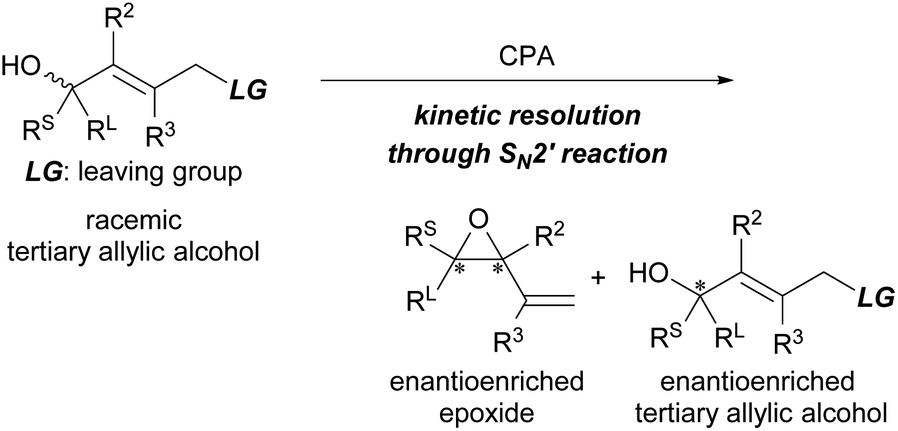 | ||
| Scheme 1 KR of enantiomeric tertiary allylic alcohols through an intramolecular SN2′ reaction. | ||
Recently, we have developed an enantioselective intramolecular SN2′ reaction using a co-catalyst system composed of chiral bisphosphoric acid catalyst (R)-1a12c,14 and arylboronic acid (or silver carbonate) as a weakly Lewis acidic additive (Scheme 2a).12c In the reported co-catalyst system, achiral tertiary allylic alcohols 2 having a normal ring structure (five- or six-membered ring) at the allylic position and trichloroacetimidate as the leaving group (LG) underwent an intramolecular SN2′ reaction smoothly, giving rise to enantioenriched diene monoepoxides (R)-3 as multifunctional products for further manipulation. In this enantioselective intramolecular SN2′ reaction, both arylboronic acid and silver carbonate were found to function as an efficient additive to suppress the catalyst deactivation process,§ and hence, to improve the yields of 3 markedly without compromising the enantioselectivities. We thus replaced the cyclic structure introduced at the allylic position with a tertiary stereogenic centre, resulting in racemic tertiary allylic alcohols (E)-4 (Scheme 2b). Here we report the development of a highly efficient KR of racemic tertiary allylic alcohols (E)-4 through the intramolecular SN2′ reaction using the co-catalyst system composed of chiral bisphosphoric acid (R)-1a and a weakly Lewis acidic additive.
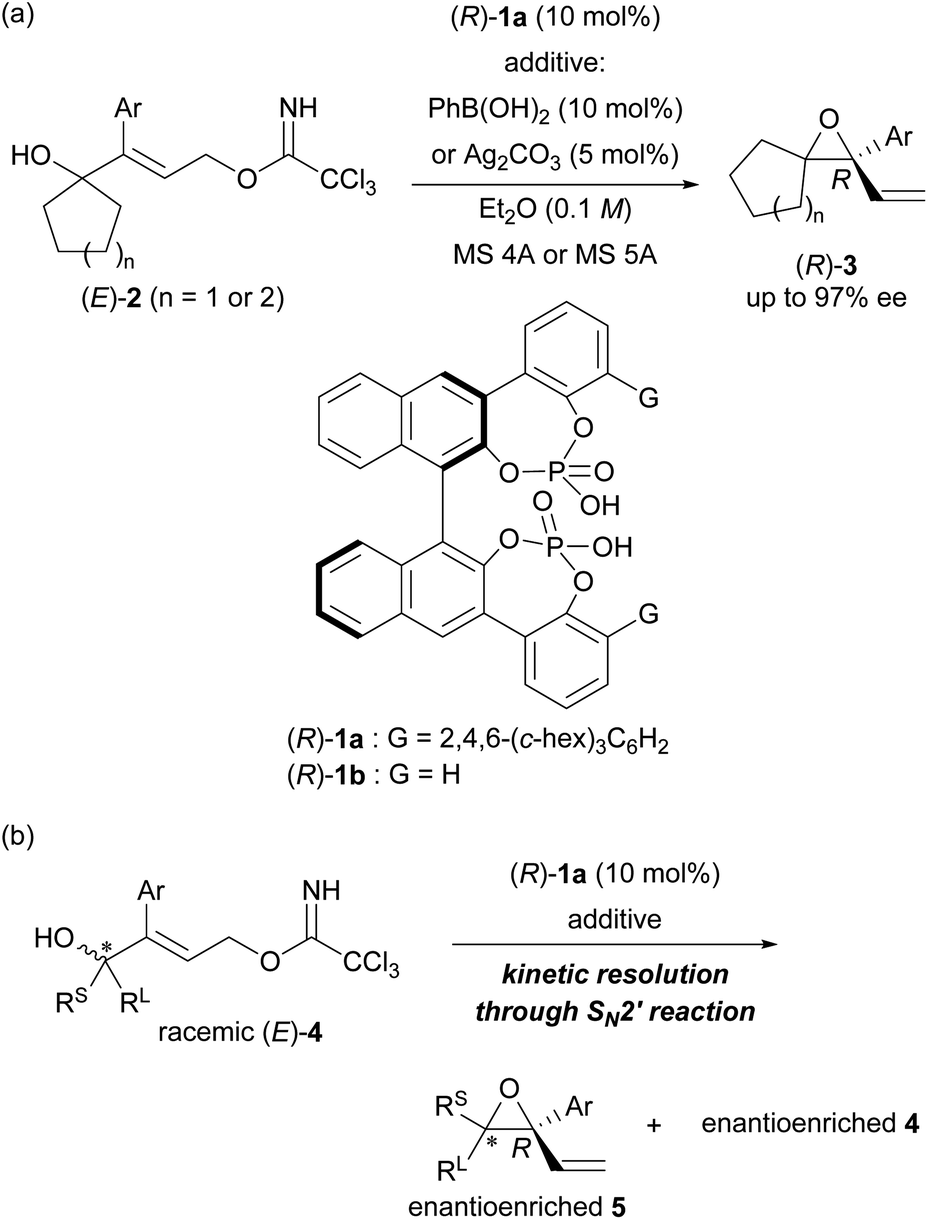 | ||
| Scheme 2 (a) Enantioselective intramolecular SN2′ reaction catalysed by (R)-1a (previous work). (b) Catalytic KR of racemic tertiary alcohols through the intramolecular SN2′ reaction developed in the present study. | ||
Results and discussion
At the outset of our studies, we predicted the stereochemical outcome of the present KR through the intramolecular SN2′ reaction (Fig. 1) by considering the previous results in the epoxide formation reaction of (E)-2 using the (R)-1a/additive co-catalyst system (Scheme 2a). We anticipated that the newly generated stereogenic centre at the allylic position of formed epoxide 5 should be (R)-stereochemistry in the present KR of (E)-4. Hence, the reaction of racemic (E)-4 under the influence of the (R)-1a/additive co-catalyst system would afford the diastereomers of trans-(R,R)-5 and cis-(S,R)-5 formed from (R,E)-4 and (S,E)-4, respectively (Fig. 1). Meanwhile, previous mechanistic studies on the enantioselective reaction of (E)-2 revealed that epoxide (R)-3 was formed in an anti-SN2′ fashion,12a,c¶ in which the leaving group and the oxygen nucleophile were oriented in an anti-relationship with respect to each other. On the basis of the anti-SN2′ pathway, the present intramolecular SN2′ reaction would proceed via transition states, TS-S and TS-R,|| depicted in Fig. 1. In these transition states, if chiral bisphosphoric acid (R)-1a would effectively distinguish the chirality at the allylic position, it was assumed that one of diastereomeric cis- and trans-epoxides 5 would form predominantly.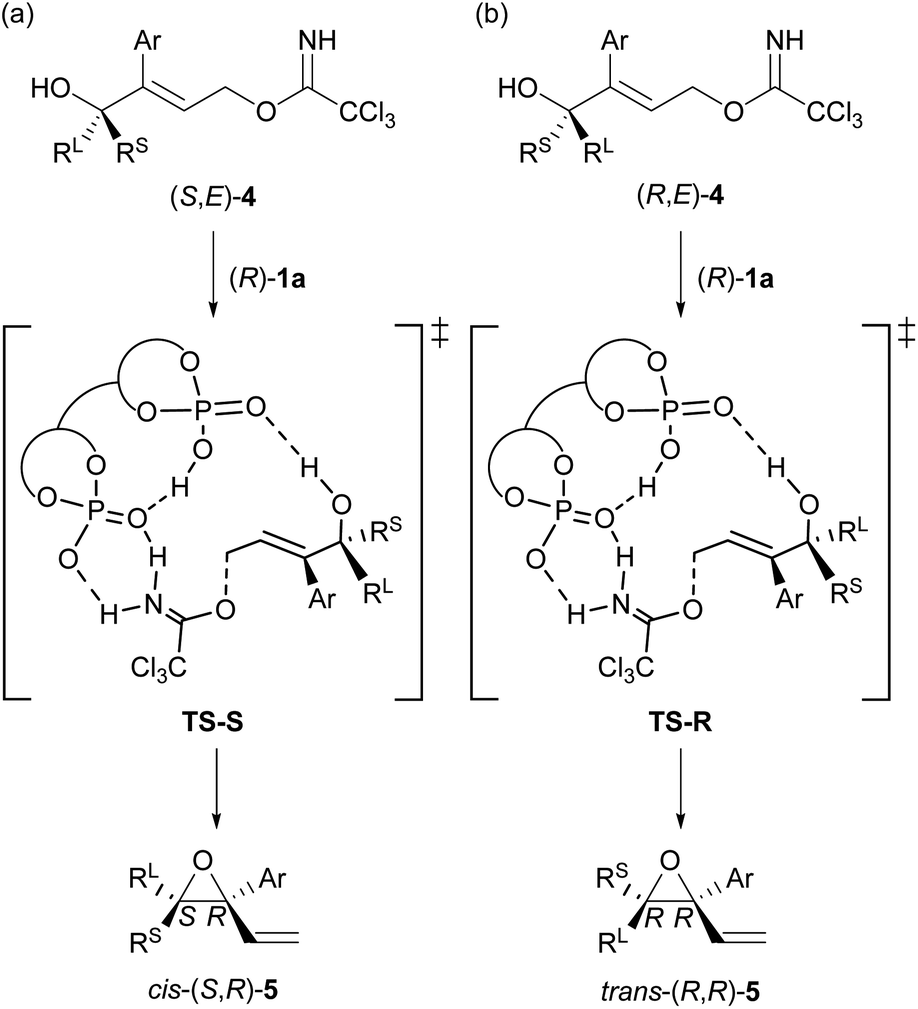 | ||
| Fig. 1 Prediction of stereochemical outcomes in the present KR of racemic (E)-4 through the intramolecular SN2′ reaction using the (R)-1a/additive co-catalyst system. The transition states in the absence of an additive are illustrated for clarity. (a) The reaction of (S,E)-4. (b) The reaction of (R,E)-4. | ||
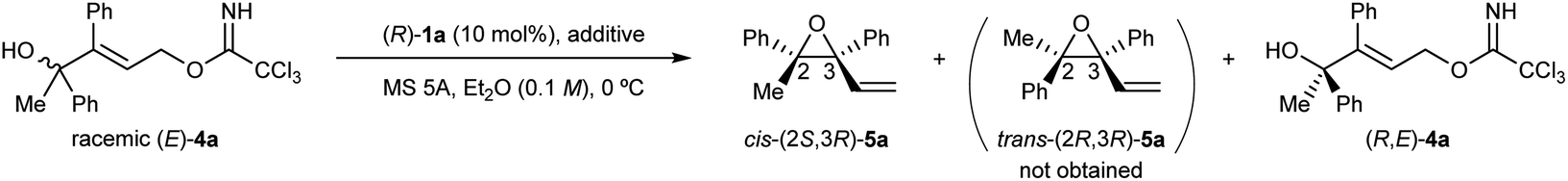
With the above prediction in mind, we commenced the intramolecular SN2′ reaction of racemic tertiary allylic alcohol (E)-4a using the (R)-1a/additive co-catalyst system for KR. We confirmed the validity of the previous reaction conditions12c using optimized chiral bisphosphoric acid (R)-1a and the additive, i.e., phenylboronic acid or silver carbonate, in the presence of molecular sieves (MS) 5A in diethyl ether at 0 °C. As shown in Table 1, the co-catalyst system was applicable to the intramolecular SN2′ reaction of tertiary allylic alcohol (E)-4a, and KR was achieved as intended with a high s-factor,15 regardless of the presence or absence of the additive (entries 1–3). Although phenylboronic acid functioned well as an additive in the previous intramolecular SN2′ reaction of (E)-2,12c the use of silver carbonate in the present KR resulted in a better material balance (entry 2 vs. 1), albeit with a slightly reduced s-factor. The absolute stereochemistry of recovered (E)-4a was determined to be R-isomer through derivatization into a stereochemically known compound.16 Since the efficient KR was achieved with the recovery of (R,E)-4a in a highly enantioselective manner, the substitution product, diene monoepoxide 5a, was formed as a single diastereomer with a cis-relative configuration and the 2S,3R absolute stereochemistry,** as predicted in Fig. 1. These results strongly suggest that the conformation at the stereogenic centre of the tertiary alcohol is strictly recognized by catalyst (R)-1a, in particular, with respect to substituent G introduced at the ortho-positions of the phenyl rings (vide infra). This is presumably because the transition states of the anti-SN2′ reaction pathway are well stabilized by catalyst (R)-1a through multiple interactions involving hydrogen bonds,¶ and hence, the relative position of the leaving group as well as the nucleophilic oxygen is firmly defined by the catalyst.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Additive | Time (h) | Yieldb of 5a/4a (%) | Calculated conversion cc (%) | drd of 5acis/trans | eee of 5a/4a (%) | s-Factorf |
| a Unless otherwise noted, all reactions were carried out using 10 mol% of (R)-1a, 0.1 mmol of racemic (E)-4a, and MS 5A (40 mg) in Et2O (1.0 mL) at 0 °C. b The yield of 5a is indicated, as determined by 1H NMR analysis of the crude reaction mixture using CH2Br2 as the internal standard. c Conversion c was calculated from eeproduct of cis-5a and eerecovered of recovered (E)-4a: c = eerecovered/(eeproduct + eerecovered). d The diastereomeric ratio (dr) of 5a was determined by 1H NMR analysis of the crude reaction mixture. e Enantiomeric excess (ee) was determined by HPLC analysis using a chiral stationary phase column. f s-Factor was calculated from the calculated conversion c and eeproduct of cis-5a: s = ln[(1 − c)(1 − eeproduct)]/ln[(1 − c)(1 + eeproduct)]. | |||||||
| 1 | PhB(OH)2 (10 mol%) | 96 | 35/30 | 48.9 | >98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :<2 :<2 |
98/94 | 351 |
| 2 | Ag2CO3 (5 mol%) | 80 | 46/52 | 47.3 | >98:<2 |
98/88 | 290 |
| 3 | None | 96 | 33/25 | 47.8 | >98:<2 |
98/90 | 305 |
With the suitable co-catalyst system composed of (R)-1a (10 mol%) and silver carbonate (5 mol%) in hand (Table 1, entry 2), we next investigated the generality of the present KR using a series of tertiary allylic alcohols (E)-4 having different electronic and steric properties. As shown in Table 2, high s-factors were achieved in most cases, whereas marked electronic and steric effects were observed in specific cases. A variety of RL substituents could be introduced at the stereogenic centre (entries 1–8), and efficient KR was achieved with (E)-4 having the aryl moiety as an RL substituent and methyl group as an RS substituent, except when the substrate had an electronically enriched aryl ring, i.e., the 4-methoxyphenyl group, as an RL substituent (entry 4). In this case, the vinylogous Wagner–Meerwein shift predominated over the desired intramolecular SN2′ reaction, affording 6e as a racemic mixture in 48% yield. The present KR was also applicable to substrate 4j, which had aliphatic substituents (RL = iPr, RS = Me) at the stereogenic centre (entry 9), leading to the formation of enantioenriched 5j with a relatively high s-factor. More interestingly, the high s-factor was substantially maintained even when the methyl group was replaced by the ethyl group as an RS substituent, which increased the steric demand (entry 10); efficient recognition was attained between the phenyl and ethyl groups. Further investigation was conducted by changing the Ar group at the carbon–carbon double bond (entries 11–19). Although high s-factors were achieved in most cases, some Ar substituents exerted marked electronic and steric effects (entries 14 and 17), presumably because having unfavourable Ar substituents destabilizes a positively charged transient species generated during the course of the intermolecular SN2′ reaction. In fact, an electron-withdrawing substituent, namely, the trifluoromethyl group, introduced at the para-position of the phenyl ring suppressed the SN2′ reaction completely, resulting in no product formation (entry 14). The sterically congested ortho-substituted aryl ring also impeded the intended reaction (entry 17). Meanwhile, the heteroaryl substituent could be introduced at the double bond, however the substrate having the thiophenyl group, (Z)-4t,†† underwent the SN2′ reaction to afford a diastereomeric mixture of cis- and trans-5t (80:20) with fairly good enantioselectivities for both diastereomers (entry 19). In addition, even though an almost half conversion of (Z)-4t (c = 48.5), moderate enantioselectivity was observed in recovered (Z)-4t. These results suggest that both enantiomers of (S,Z)- and (R,Z)-4t were consumed in parallel to some extent, affording cis-(2S,3S)-5t and trans-(2R,3S)-5t, respectively.‡‡ Consequently, the KR of (S,Z)- and (R,Z)-4t did not take place efficiently, although both diastereomers 5t were obtained in an enantioenriched form.
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | 4 | R L | R S | Ar | Yieldb of 5/4 (%) | Calculated conversion cc (%) | drd of 5 | eee of 5/4 (%) | s-Factorf |
| a Unless otherwise noted, all reactions were carried out using 10 mol% of (R)-1a, 5 mol% of Ag2CO3, 0.1 mmol of racemic (E)-4, and MS 5A (40 mg) in Et2O (1.0 mL) at 0 °C. b The yield of 5, as determined by 1H NMR analysis of the crude reaction mixture using CH2Br2 as the internal standard, is indicated. c Conversion c was calculated from eeproduct of cis-5 and eerecovered of recovered (E)-4: c = eerecovered/(eeproduct + eerecovered). d The diastereomeric ratio (dr) of 5 was determined by 1H NMR analysis of the crude reaction mixture. e Enantiomeric excess (ee) was determined by HPLC analysis using a chiral stationary phase column. f s-Factor was calculated from the calculated conversion c and the eeproduct of cis-5: s = ln[(1 − c)(1 − eeproduct)]/ln[(1 − c)(1 + eeproduct)]. g 6e was formed as a major product (48% yield) in a racemic form along with a small amount of desired 5e as a mixture of other unknown byproducts. h At −40 °C for 20 h. i (S,E)-4i was recovered due to the nomenclature of the substituent priority. j At −40 °C for 72 h. k ND: desired product 5 was not detected even at room temperature for 72 h. l At −40 °C for 120 h. m (R,Z)-4t was recovered due to the nomenclature of the substituent priority. n Cis-(2S,3S)-5t was formed as the major diastereomer with 86% ee along with trans-(2R,3S)-5t as a minor product with 90% ee. Therefore, conversion c was calculated from 51% ee for eeproduct averaged at the 2-position of cis- and trans-5t. See ESI for details. o s-Factor could not be calculated because trans-(2R,3S)-5t was formed from (R,Z)-4t as the minor diastereomer in this case. | |||||||||
| 1 | 4b | 4-CF3C6H4 | Me | Ph | 34/47 | 46.3 | >98:<2 |
94/81 | 81 |
| 2 | 4c | 4-ClC6H4 | Me | Ph | 43/45 | 49.5 | >98:<2 |
94/92 | 107 |
| 3 | 4d | 4-MeC6H4 | Me | Ph | 40/40 | 47.5 | >98:<2 |
94/85 | 88 |
| 4 | 4e | 4-MeOC6H4 | Me | Ph | —g/11 | — | >98:<2 |
—/67 | — |
| 5 | 4f | 3-ClC6H4 | Me | Ph | 40/45 | 47.2 | >98:<2 |
95/85 | 106 |
| 6 | 4g | 3-MeC6H4 | Me | Ph | 41/43 | 45.3 | >98:<2 |
94/78 | 76 |
| 7 | 4h | 3-MeOC6H4 | Me | Ph | 45/37 | 51.0 | >98:<2 |
90/94 | 67 |
| 8h | 4i | 2-ClC6H4 | Me | Ph | 37/49i | 37.3 | >98:<2 |
94/56 | 57 |
| 9 | 4j | iPr | Me | Ph | 32/52 | 35.3 | >98:<2 |
92/50 | 40 |
| 10 | 4k | Ph | Et | Ph | 49/39 | 50.7 | >98:<2 |
95/98 | 174 |
| 11j | 4l | Ph | Me | 4-MeOC6H4 | 44/36 | 49.4 | >98:<2 |
86/84 | 35 |
| 12 | 4m | Ph | Me | 4-ClC6H4 | 34/49 | 37.8 | >98:<2 |
92/56 | 42 |
| 13 | 4n | Ph | Me | 4-PhC6H4 | 26/71 | 27.5 | >98:<2 |
95/36 | 56 |
| 14 | 4o | Ph | Me | 4-CF3C6H4 | NDk | — | — | — | — |
| 15 | 4p | Ph | Me | 3-MeC6H4 | 30/63 | 36.8 | >98:<2 |
96/56 | 86 |
| 16 | 4q | Ph | Me | 3-ClC6H4 | 30/51 | 32.6 | >98:<2 |
97/47 | 105 |
| 17 | 4r | Ph | Me | 2-MeC6H4 | NDk | — | — | — | — |
| 18 | 4s | Ph | Me | 2-Naphthyl | 53/34 | 54.2 | >98:<2 |
84/99.5 | 65 |
| 19l | 4t | Ph | Me | 2-Thiophenyl | 40/49m | 48.5n | 80:20n |
86, 90n/48 | —o |
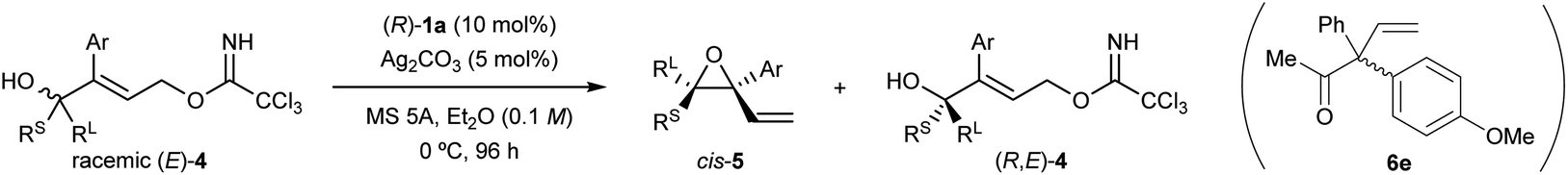
The efficiency of the developed KR was further demonstrated by scaling up the reaction (Scheme 3). Racemic (E)-4a (0.40 g, 1.0 mmol) underwent the intramolecular SN2′ reaction under the optimized reaction conditions while maintaining the high s-factor. Cis-(2S,3R)-5a was formed as the single diastereomer along with the recovery of (R,E)-4a in a highly enantioselective manner. As readily expected, formed epoxide 5a was easily separable from recovered tertiary alcohol 4a by silica-gel column chromatography.
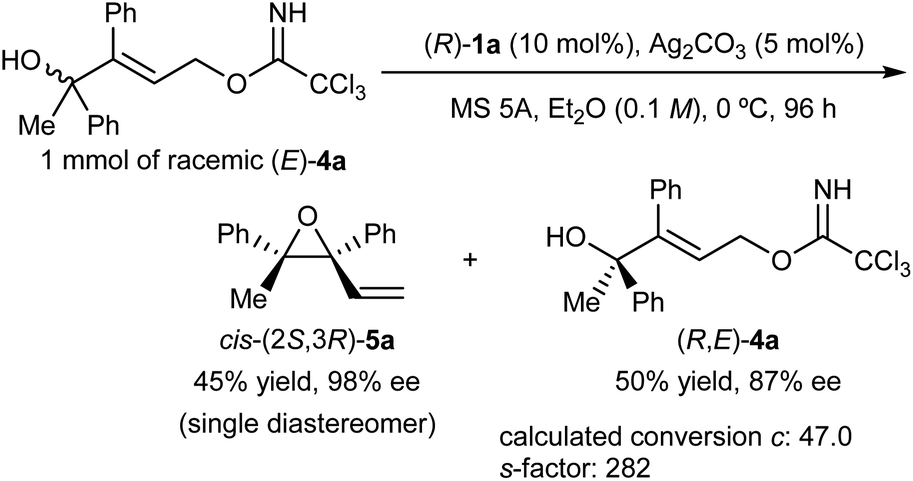 | ||
| Scheme 3 Large-scale experiment to demonstrate the utility of the present KR. | ||
In addition, simple derivatization of enantioenriched epoxide cis-(2S,3R)-5a and recovered tertiary alcohol (R,E)-4a was also carried out (Scheme 4). Treatment of the enantioenriched epoxide product cis-(2S,3R)-5a under acidic conditions using water as the nucleophile afforded corresponding allylic alcohol (S,E)-7 in acceptable yield (Scheme 4a). In the present SN2′ ring-opening reaction of the epoxide, the (E)-geometrical isomer of 7 was formed exclusively without marked loss of enantiomeric purity. Enantioenriched (R,E)-4a thus recovered was converted into (R,E)-7 in 72% yield without any loss of enantiomeric purity through the two-step transformation (Scheme 4b), i.e., the SN2 reaction of (R,E)-4a with trifluoroacetic acid, followed by the transesterification of the formed mono-trifluoroacetate of (R,E)-7 using methanol.
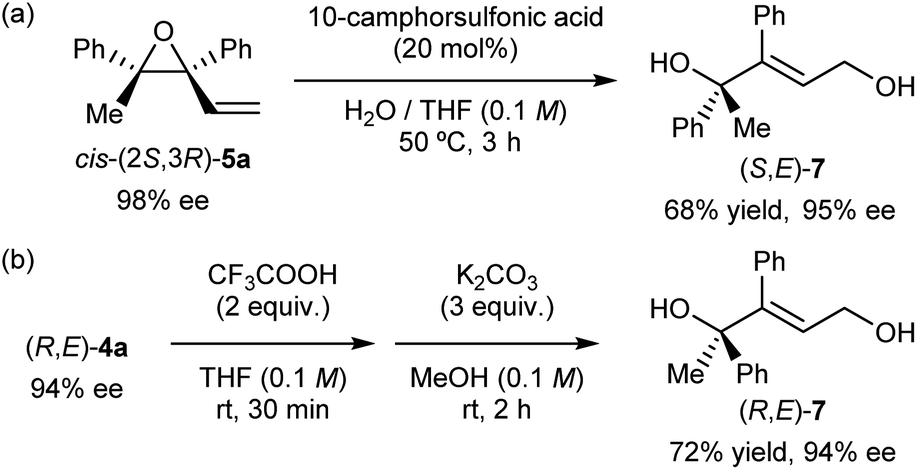 | ||
| Scheme 4 (a) Derivatization of enantioenriched epoxide cis-(2S,3R)-5a into allylic alcohol (S,E)-7. (b) Hydrolysis of enantioenriched (R,E)-4a into allylic alcohol (R,E)-7. | ||
As the developed KR through the intramolecular SN2′ reaction of racemic tertiary allylic alcohols (E)-4 was demonstrated to be highly efficient, our interest then turned to the origin of the high efficiency of the present KR and the stereochemical outcome. In order to gain a mechanistic insight of the present efficient KR, we carried out a reaction of racemic (E)-4a under the influence of (R)-1b (G = H) having no substituents at the ortho-positions of the phenyl rings (Scheme 5). The reaction was performed in chloroform using PhB(OH)2 as an additive given the low solubility of (R)-1b/silver carbonate co-catalyst in diethyl ether.§§ A mixture of cis- and trans-5a was formed with a slight excess of cis-5a. This result clearly suggests that the sterically bulky substituent (G = 2,4,6-(c-hex)3C6H2) of catalyst (R)-1a is key not only to controlling the newly generated stereogenic centre at the allylic position but also to recognizing the configuration of the racemic stereogenic centre.
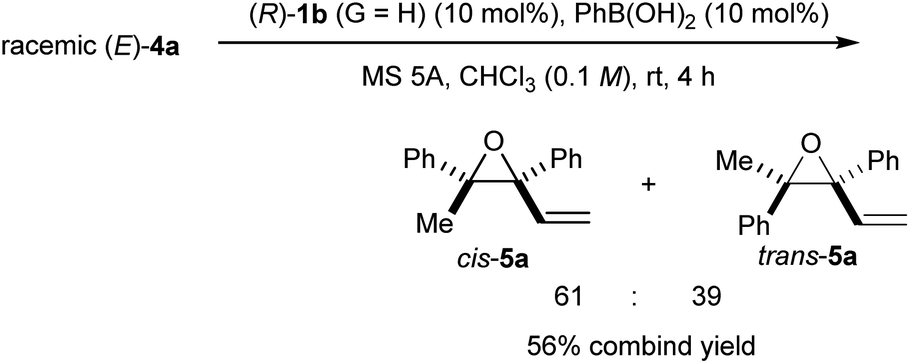 | ||
| Scheme 5 Intramolecular SN2′ reaction of racemic (E)-4a using the co-catalyst system of (R)-1b (G = H) and PhB(OH)2 in chloroform. | ||
As shown in Fig. 2, this remarkable substituent effect is readily anticipated from the 3D structures of the transition states, TS-Smodel and TS-Rmodel, using the model system composed of substrate (E)-4a and simplified catalyst (R)-1b (G = H). These transition states were optimized by DFT calculation,17–19|| in accordance with the anti-SN2′ reaction pathway.¶ Before considering the substituent effect of the catalyst, we summarize the intriguing features of these transition states, as follows: Two phosphoric acid units interact with each other through a hydrogen bond.14 These two phosphoric acid units have specific roles and their involvement in the bond recombination sequence is essential. One phosphoric acid forms a hydrogen bond between the phosphoryl oxygen P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and the hydroxy proton of the tertiary alcohol. This hydrogen bond is considered to promote the nucleophilic attack of the alcohol oxygen on the double bond, although the present intramolecular SN2′ reaction proceeds through a stepwise pathway and these transition states indicate the first C–O bond cleavage step of the leaving group. The nucleophilic attack of the alcohol oxygen follows the C–O bond cleavage and is involved in the subsequent epoxide formation step. In contrast, the acidic OH of the other phosphoric acid protonates the nitrogen of the trichloroacetimidate moiety to activate the leaving group. Furthermore, the phosphoryl oxygen interacts with the N–H proton of the imidate moiety and hence, the double hydrogen bonding interaction occurs between this phosphoric acid unit and the trichloroacetimidate moiety. These multiple hydrogen bonds stabilize the transition states and determine the relative position between catalyst (R)-1b and substrate (E)-4a. In other words, the unique features of these model transition states strongly suggest that chiral bisphosphoric acid catalyst plays a crucial role in the smooth acceleration of the present intramolecular SN2′ reaction. However, unlike the experimental result shown in Scheme 5 where the formation of cis-(2S,3R)-5a slightly dominated, TS-Rmodel (Fig. 2b), affording trans-(2R,3R)-5a, is energetically favorable, although the energy difference is not significant in the solution phase (ΔΔG≠ = 0.7 kcal mol−1) ¶¶.
O and the hydroxy proton of the tertiary alcohol. This hydrogen bond is considered to promote the nucleophilic attack of the alcohol oxygen on the double bond, although the present intramolecular SN2′ reaction proceeds through a stepwise pathway and these transition states indicate the first C–O bond cleavage step of the leaving group. The nucleophilic attack of the alcohol oxygen follows the C–O bond cleavage and is involved in the subsequent epoxide formation step. In contrast, the acidic OH of the other phosphoric acid protonates the nitrogen of the trichloroacetimidate moiety to activate the leaving group. Furthermore, the phosphoryl oxygen interacts with the N–H proton of the imidate moiety and hence, the double hydrogen bonding interaction occurs between this phosphoric acid unit and the trichloroacetimidate moiety. These multiple hydrogen bonds stabilize the transition states and determine the relative position between catalyst (R)-1b and substrate (E)-4a. In other words, the unique features of these model transition states strongly suggest that chiral bisphosphoric acid catalyst plays a crucial role in the smooth acceleration of the present intramolecular SN2′ reaction. However, unlike the experimental result shown in Scheme 5 where the formation of cis-(2S,3R)-5a slightly dominated, TS-Rmodel (Fig. 2b), affording trans-(2R,3R)-5a, is energetically favorable, although the energy difference is not significant in the solution phase (ΔΔG≠ = 0.7 kcal mol−1) ¶¶.
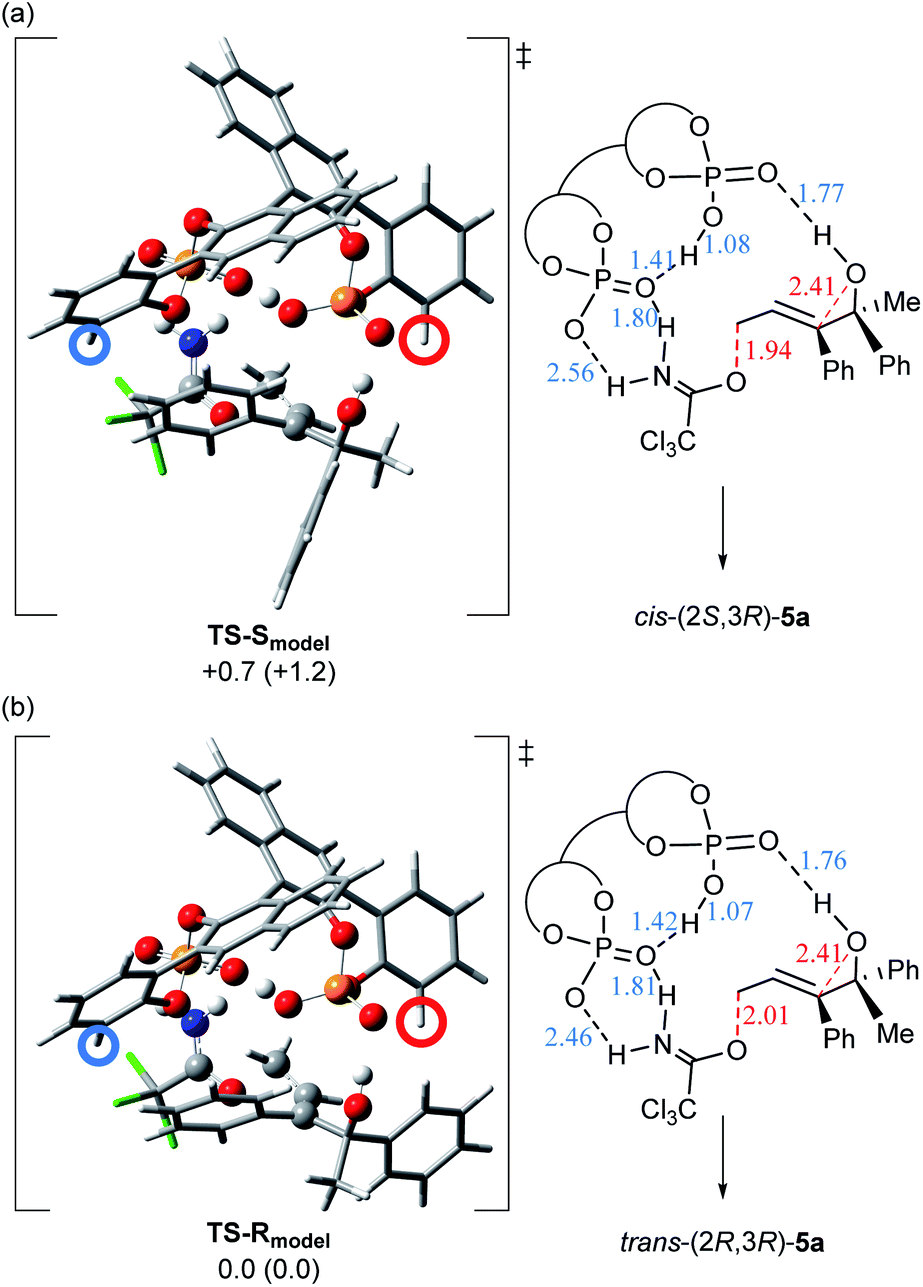 | ||
| Fig. 2 3D structures and schematic representation models of the most energetically favourable transition states for the C–O bond cleavage step, TS-Smodel and TS-Rmodel. The 3D structures of the fragments are represented as follows: phosphoric acid units and atoms involved in the bond recombination sequence and the hydrogen bonding interaction: “ball and bond type” model; and the other atoms, such as bisphosphoric acid backbone and substrate: “tube” model. The ortho-position of the phenyl ring is indicated by circles, where the substituent is introduced in the actual catalytic system. Relative free energies (kcal mol−1) of the optimized structures at the B97D/6-31G(d) level18 in the gas phase are shown in parentheses. Relative free energies (kcal mol−1) obtained by single-point energy calculations at the same level are shown for the optimized transition states in the solution phase according to the SCRF method based on CPCM (ether).19 Hydrogen bond lengths are indicated in blue (angstroms) and cleaved and formed C–O bond lengths are indicated in red (angstroms): (a) TS-Smodel generated from (R)-1b (G = H) and (S,E)-4a. (b) TS-Rmodel generated from (R)-1b (G = H) and (R,E)-4a. | ||
Further consideration of the real catalytic system was continued on the basis of these unique model transition states (Fig. 2). When a sterically bulky substituent (G = 2,4,6-(c-hex)3C6H2) is introduced at the ortho-position of the phenyl ring (indicated by circles in the 3D structures) of (R)-1b, giving (R)-1a, in TS-Rmodel generated from (R,E)-4a (Fig. 2b), it is readily assumed that a large steric repulsion occurs between the bulky substituent (the position indicated by the red circle) and the phenyl group attached to the stereogenic centre. Therefore, the formation of trans-(2R,3R)-5a from (R,E)-4a is efficiently suppressed. On the other hand, in the case of TS-Smodel generated from (S,E)-4a (Fig. 2a), the transition state structure is maintained without steric congestion because the sterically less hindered methyl group faces the substituent side (the position indicated by the red circle) of catalyst (R)-1a. Therefore, cis-(2S,3R)-5a is formed exclusively.
Conclusion
A highly efficient KR of racemic tertiary allylic alcohols was developed through the intramolecular SN2′ reaction using the chiral bisphosphoric acid/silver carbonate co-catalyst system. In the established KR system, cis-epoxides were formed in a highly diastereo- and enantio-selective manner along with the recovery of tertiary allylic alcohols with high enantioselectivity, achieving a markedly high s-factor in most cases. Our protocol, namely, the intramolecular allylic substitution reaction catalysed by CPAs, provides a new entry to the catalytic KR of tertiary alcohols, which have been employed as synthetically useful chiral building blocks, in a highly enantioselective manner. Further elucidation of the mechanism of the present KR and application of the allylic substitution reaction to the enantioselective construction of the tetrasubstituted stereogenic centre is underway in our laboratory.Data availability
The exploratory investigation, experimental procedures, computational data, and characterization data are available.Author contributions
S. K.: conceptualization, data curation, formal analysis, and investigation (experimental studies). J. K.: data curation, formal analysis, and investigation (theoretical and experimental studies). N. S.: data curation, formal analysis, and investigation (mechanistic studies). S. U.: data curation, formal analysis, and investigation (mechanistic studies and derivatization). M. T.: conceptualization, project administration, writing – review & editing, supervision, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by a Grant-in-Aid for Scientific Research on Innovative Areas “Hybrid Catalysis for Enabling Molecular Synthesis on Demand” (JP17H06447) from MEXT (Japan).Notes and references
- (a) Quaternary Stereocenters, Challenges and Solutions for Organic Synthesis, ed. J. Christoffers and A. Baro, Wiley-VCH, Weinheim, 2005 Search PubMed; (b) Science of Synthesis: Stereoselective Synthesis Vol. 2: Stereoselective Reactions of Carbonyl and Imino Groups, ed. G. A. Molander, P. A. Evans and J. G. de Vries, Georg Thieme Verlag KG: New York, 2010 Search PubMed.
- (a) L. Pu and H.-B. Yu, Chem. Rev., 2001, 101, 757–824 CrossRef CAS PubMed; (b) M. Shibasaki and M. Kanai, Chem. Rev., 2008, 108, 2853–2873 CrossRef CAS PubMed; (c) Y.-L. Liu and X.-T. Lin, Adv. Synth. Catal., 2019, 361, 876–918 CrossRef CAS.
- (a) H. B. Kagan and J. C. Fiaud in Topics in Stereochemistry, ed. E. L. Eliel and S. H. Wilen, John Wiley & Sons, New York, 1988, vol. 18, pp. 249–330 Search PubMed; (b) J. M. Keith, J. F. Larrow and E. N. Jacobsen, Adv. Synth. Catal., 2001, 343, 5–26 CrossRef CAS; (c) E. Vedejs and M. Jure, Angew. Chem., Int. Ed., 2005, 44, 3974–4001 CrossRef CAS PubMed.
- (a) C. E. Müller and P. R. Schreiner, Angew. Chem., Int. Ed., 2011, 50, 6012–6042 CrossRef PubMed; (b) J. I. Murray, Z. Heckenast and A. C. Spivey in Lewis Base Catalysis in Organic Synthesis, ed. E. Vedejs and S. E. Denmark, Wiley-VCH, Weinheim, 2016, vol. 2, pp. 459–526 Search PubMed.
- For a recent review of non-enzymatic catalytic KR of tertiary alcohols, see: B. Ding, Q. Xue, S. Jiaa, H.-G. Cheng and Q. Zhou, Synthesis, 2022, 54, 1721–1732 CrossRef CAS.
- (a) E. R. Jarvo, C. A. Evans, G. T. Copeland and S. J. Miller, J. Org. Chem., 2001, 66, 5522–5527 CrossRef CAS PubMed; (b) M. C. Angione and S. J. Miller, Tetrahedron, 2006, 62, 5254–5261 CrossRef CAS; (c) S. Lu, S. B. Poh, W.-Y. Siau and Y. Zhao, Angew. Chem., Int. Ed., 2013, 52, 1731–1734 CrossRef CAS PubMed; (d) M. D. Greenhalgh, S. M. Smith, D. M. Walden, J. E. Taylor, Z. Brice, E. R. T. Robinson, C. Fallan, D. B. Cordes, A. M. Z. Slawin, H. C. Richardson, M. A. Grove, P. H.-Y. Cheong and A. D. Smith, Angew. Chem., Int. Ed., 2018, 57, 3200–3206 CrossRef CAS PubMed; (e) S. Qu, S. M. Smith, V. Laina-Martín, R. M. Neyyappadath, M. D. Greenhalgh and A. D. Smith, Angew. Chem., Int. Ed., 2020, 59, 16572–16578 CrossRef CAS PubMed; (f) T. Desrues, X. Liu, J.-M. Pons, V. Monnier, J.-A. Amalian, L. Charles, A. Quintard and C. Bressy, Org. Lett., 2021, 23, 4332–4336 CrossRef CAS PubMed; (g) S. Niu, H. Zhang, W. Xu, P. R. Bagdi, G. Zhang, J. Liu, S. Yang and X. Fang, Nat. Commun., 2021, 12, 3735 CrossRef CAS PubMed.
- (a) R. Shintani, K. Takatsu and T. Hayashi, Org. Lett., 2008, 10, 1191–1193 CrossRef CAS PubMed; (b) W. Zhang and S. Ma, Chem. Commun., 2018, 54, 6064–6067 RSC; (c) J. Seliger, X. Dong and M. Oestreich, Angew. Chem., Int. Ed., 2019, 58, 1970–1974 CrossRef CAS PubMed; (d) Y. Hua, Z.-S. Liu, P.-P. Xie, B. Ding, H.-G. Cheng, X. Hong and Q. Zhou, Angew. Chem., Int. Ed., 2021, 60, 12824–12828 CrossRef CAS PubMed; (e) R. Mao, Y. Zhao, X. Zhu, F. Wang, W.-Q. Deng and X. Li, Org. Lett., 2021, 23, 7038–7043 CrossRef CAS PubMed.
- (a) I. Čorić, S. Mgller and B. List, J. Am. Chem. Soc., 2010, 132, 17370–17373 CrossRef PubMed; (b) T. Yamanaka, A. Kondoh and M. Terada, J. Am. Chem. Soc., 2015, 137, 1048–1051 CrossRef CAS PubMed; (c) J. H. Kim, I. Čorić, C. Palumbo and B. List, J. Am. Chem. Soc., 2015, 137, 1778–1781 CrossRef CAS PubMed; (d) S. Rajkumar, S. He and X. Yang, Angew. Chem., Int. Ed., 2019, 58, 10315–10319 CrossRef CAS PubMed; (e) S. Rajkumar, M. Tang and X. Yang, Angew. Chem., Int. Ed., 2020, 59, 2333–2337 CrossRef CAS PubMed; (f) M. Tang, H. Gu, S. He, S. Rajkumar and X. Yang, Angew. Chem., Int. Ed., 2021, 60, 21334–21339 CrossRef CAS PubMed; (g) C.-H. Zhang, Q. Gao, M. Li, J.-F. Wang, C.-M. Yu and B. Mao, Org. Lett., 2021, 23, 3949–3954 CrossRef CAS PubMed.
- For other examples of non-enzymatic catalytic KR of tertiary alcohols, see: (a) S.-y. Tosaki, K. Hara, V. Gnanadesikan, H. Morimoto, S. Harada, M. Sugita, N. Yamagiwa, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2006, 128, 11776–11777 CrossRef CAS PubMed; (b) Y. Zhao, A. W. Mitra, A. H. Hoveyda and M. L. Snapper, Angew. Chem., Int. Ed., 2007, 46, 8471–8474 CrossRef CAS PubMed; (c) Z. Li, V. Boyarskikh, J. H. Hansen, J. Autschbach, D. G. Musaev and H. M. L. Davis, J. Am. Chem. Soc., 2012, 134, 15497–15504 CrossRef CAS PubMed; (d) J. L. Olivares-Romero, Z. Li and H. Yamamoto, J. Am. Chem. Soc., 2013, 135, 3411–3413 CrossRef CAS PubMed; (e) M. Pawliczek, T. Hashimoto and K. Maruoka, Chem. Sci., 2018, 9, 1231–1235 RSC; (f) J. Song and W.-H. Zheng, Org. Lett., 2022, 24, 2349–2353 CrossRef CAS PubMed.
- For seminal studies of chiral phosphoric acid catalysts, see: (a) T. Akiyama, J. Itoh, K. Yokota and K. Fuchibe, Angew. Chem., Int. Ed., 2004, 43, 1566–1568 CrossRef CAS PubMed; (b) D. Uraguchi and M. Terada, J. Am. Chem. Soc., 2004, 126, 5356–5357 CrossRef CAS PubMed . For selected reviews of chiral phosphoric acid, see:; (c) T. Akiyama, Chem. Rev., 2007, 107, 5744–5758 CrossRef CAS PubMed; (d) D. Kampen, C. M. Reisinger and B. List, Top. Curr. Chem., 2009, 291, 395–456 CrossRef; (e) M. Terada, Chem. Commun., 2008, 4097–4112 RSC; (f) M. Terada, Synthesis, 2010, 1929–1982 CrossRef CAS; (g) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS PubMed.
- For enantioselective SN2’ reactions catalysed by CPAs, see: (a) M. Rueping, U. Uria, M.-Y. Lin and I. Atodiresei, J. Am. Chem. Soc., 2011, 133, 3732–3735 CrossRef CAS PubMed; (b) P.-S. Wang, X.-L. Zhou and L.-Z. Gong, Org. Lett., 2014, 16, 976–979 CrossRef CAS PubMed; (c) M. Zhuang and H. Du, Org. Biomol. Chem., 2014, 12, 4590–4593 RSC; (d) Y. Kuroda, S. Harada, A. Oonishi, Y. Yamaoka, K. Yamada and K. Takasu, Angew. Chem., Int. Ed., 2015, 54, 8263–8266 CrossRef CAS PubMed; (e) J. Zhou and H. Xie, Org. Biomol. Chem., 2018, 16, 380–383 RSC.
- For our contributions to enantioselective SN2’ reactions catalysed by CPAs, see: (a) M. Shimizu, J. Kikuchi, A. Kondoh and M. Terada, Chem. Sci., 2018, 9, 5747–5757 RSC; (b) S. Kayal, J. Kikuchi, M. Shimizu and M. Terada, ACS Catal., 2019, 9, 6846–6850 CrossRef CAS . Also see, correction of this manuscript: ; (c) S. Kayal, J. Kikuchi, M. Shimizu, N. Shinagawa, S. Umemiya and M. Terada, ACS Catal., 2021, 11, 8956–8957 CrossRef CAS; (d) S. Kayal, J. Kikuchi, N. Shinagawa, S. Umemiya and M. Terada, Tetrahedron, 2021, 98, 132412 CrossRef CAS.
- For selected examples of kinetic resolution of alcohols through intramolecular cyclizations catalysed by CPAs, see: (a) I. Čorić, J. H. Kim, T. Vlaar, M. Patil, W. Thiel and B. List, Angew. Chem., Int. Ed., 2013, 52, 3490–3493 CrossRef PubMed; (b) G. Qabaja, J. E. Wilent, A. R. Benavides, G. E. Bullard and K. S. Petersen, Org. Lett., 2013, 15, 1266–1269 CrossRef CAS PubMed; (c) G. Qabaja, A. R. Benavides, S. Liu and K. S. Petersen, J. Org. Chem., 2015, 80, 133–140 CrossRef CAS PubMed . Also see ref. 8e and 8g..
- For chiral bisphosphoric acid catalysts, see: (a) N. Momiyama, T. Konno, Y. Furiya, T. Iwamoto and M. Terada, J. Am. Chem. Soc., 2011, 133, 19294–19297 CrossRef CAS PubMed; (b) N. Momiyama, K. Funayama, H. Noda, M. Yamanaka, N. Akasaka, S. Ishida, T. Iwamoto and M. Terada, ACS Catal., 2016, 6, 949–956 CrossRef CAS; (c) M. Terada, Y. Gupta and J. Kikuchi, Chem. Lett., 2019, 48, 260–263 CrossRef CAS; (d) J. Kikuchi, K. Takano, Y. Ota, S. Umemiya and M. Terada, Chem.–Eur. J., 2020, 26, 11124–11128 CrossRef CAS PubMed.
- M. D. Greenhalgh, J. E. Taylor and A. D. Smith, Tetrahedron, 2018, 74, 5554–5560 CrossRef CAS.
- S.-B. D. Sim, M. Wang and Y. Zhao, ACS Catal., 2015, 5, 3609–3612 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016. See ESI† for the full list of authors Search PubMed.
- For the B97D method, see: S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- For CPCM, see: V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, DFT calculations, stereochemical assignment, NMR spectra, and HPLC charts. See https://doi.org/10.1039/d2sc03052g |
| ‡ Current address: Graduate School of Pharmaceutical Sciences, Tohoku University, Aramaki, Aoba-ku, Sendai 980-8578, Japan. |
| § The formation of the corresponding phosphate ester through the SN2 reaction of substrate 2 with bisphosphoric acid 1a at the allylic position is responsible for the catalyst deactivation. These additives, arylboronic acid and silver carbonate, efficiently suppress the undesirable SN2 reaction to avoid the catalyst deactivation. |
| ¶ In the presence of the additive (either phenylboronic acid or silver carbonate), it has been confirmed that the reaction of 2 (Ar = Ph) proceeds in an anti-SN2′ fashion under the influence of catalyst (R)-1a. See ESI for details. |
| || As shown in Table 1, the presence or absence of additives has a notable influence on the yield but a minimal effect on both the stereochemical outcome and the efficiency of the KR. Hence, these schematic transition states were able to draw in the absence of additives, likewise transition states of the model systems were calculated without using additives. |
| ** On the basis of the absolute stereochemistry of recovered (R,E)-4a, the absolute stereochemistry at the 2-position of epoxide 5a was predicted to be 2S. On the other hand, the relative stereochemistry of epoxide 5 was assigned to be cis-isomer by the NOE experiment of 5l under the NMR measurement. Consequently, the combination of these stereochemical assignments resulted in the formation of cis-(2S,3R)-5a as the major product. See ESI for details. |
| †† Although the geometry of 4t is nomenclated to be Z due to the priority of the substituents, the relative location of the substituents attached to the CC double bond of (Z)-4t is the same as that of other substrates (E)-4. |
| ‡‡ The absolute stereochemistry of trans-5t was assigned as (2R,3S), which was derived from (R,Z)-4t, on the basis of the distribution of the stereochemical outcomes of cis- and trans-5t and recovered (Z)-4t. See ESI for details. |
| §§ The reaction of (E)-4a using the co-catalyst system of (R)-1a (G = 2,4,6-(c-hex)3C6H2) and PhB(OH)2 in chloroform at room temperature for 4 h afforded cis-(2S,3R)-5a (12% yield) as the single diastereomer along with the formation of a significant amount of the vinylogous Wagner–Meerwein shift product, ketone 6a (51% yield). See ESI for details. |
| ¶¶ A(1,2) strain, which causes steric congestion between the phenyl groups introduced at the stereogenic centre and the double bond in TS-Smodel, might be responsible for the calculated energy gap. |
| This journal is © The Royal Society of Chemistry 2022 |