
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInverse potential scaling in co-electrocatalytic activity for CO2 reduction through redox mediator tuning and catalyst design†
Amelia G.
Reid
 ,
Juan J.
Moreno
,
Shelby L.
Hooe
,
Kira R.
Baugh
,
Isobel H.
Thomas
,
Diane A.
Dickie
and
Charles W.
Machan
*
,
Juan J.
Moreno
,
Shelby L.
Hooe
,
Kira R.
Baugh
,
Isobel H.
Thomas
,
Diane A.
Dickie
and
Charles W.
Machan
*
Department of Chemistry, University of Virginia, PO Box 400319, Charlottesville, VA 22904-4319, USA. E-mail: machan@virginia.edu
First published on 21st July 2022
Abstract
Electrocatalytic CO2 reduction is an attractive strategy to mitigate the continuous rise in atmospheric CO2 concentrations and generate value-added chemical products. A possible strategy to increase the activity of molecular systems for these reactions is the co-catalytic use of redox mediators (RMs), which direct reducing equivalents from the electrode surface to the active site. Recently, we demonstrated that a sulfone-based RM could trigger co-electrocatalytic CO2 reduction via an inner-sphere mechanism under aprotic conditions. Here, we provide support for inner-sphere cooperativity under protic conditions by synthetically modulating the mediator to increase activity at lower overpotentials (inverse potential scaling). Furthermore, we show that both the intrinsic and co-catalytic performance of the Cr-centered catalyst can be enhanced by ligand design. By tuning both the Cr-centered catalyst and RM appropriately, an optimized co-electrocatalytic system with quantitative selectivity for CO at an overpotential (η) of 280 mV and turnover frequency (TOF) of 194 s−1 is obtained, representing a three-fold increase in co-catalytic activity at 130 mV lower overpotential than our original report. Importantly, this work lays the foundation of a powerful tool for developing co-catalytic systems for homogeneous electrochemical reactions.
Introduction
The interest in electrochemical carbon dioxide (CO2) reduction is two-fold: the association of climate change with the global rise in atmospheric CO2 concentrations and the increasing need to transition from non-renewable fossil fuel-derived hydrocarbons as chemical feedstocks and energy sources.1 The reduction of CO2 to carbon monoxide (CO) could help to mitigate both of these issues through conversion of CO2 into useful building blocks for the synthesis of chemical feedstocks and fuels while using renewable energy sources, such as sunlight and wind, as the source of electricity.2,3 In recent years, the focus of molecular electrocatalyst development has shifted towards the use of abundant first-row transition metal centers to address the need for cost-effective and scalable systems.4–8Nature frequently leverages co-catalytic systems during reactions involving electron transfer. For example, during cellular respiration the electron transport chain uses several redox mediators (RMs) to shuttle protons and electrons between different redox-active cofactors. The final step of this cycle involves ubiquinone shuttling electrons and protons to cytochrome c oxidase as a part of the overall reduction of dioxygen to water.9 The use of RMs is necessary for enzymatic systems to transfer electron equivalents due to the limitations associated with relatively stationary active sites buried within protein superstructures that are spatially separated from redox centers in the electron transport chain.10 Co-catalytic systems which utilize RMs have been successfully developed for homogeneous O2 reduction, increasing the overall activity of the system and shifting the selectivity of the reaction.11–13 In these homogeneous systems, RMs deliver redox equivalents to the catalyst active sites with greater mobility than is possible in biological systems. Parallel developments have enabled electrocatalytic N2 reduction, where weak C–H bonds are generated in a metallocene-based RM to assist in the cleavage of inert bonds,14 and alcohol oxidation, where RMs are utilized to facilitate hydrogen atom transfer processes that work in conjunction with the catalyst.15–17 Additional work has focused on the use of small-molecule RMs to enhance the activation of photosensitizers,18–23 as catalysts in electrosynthesis,24 or to enhance the CO2 reduction reaction on heterogeneous surfaces.25,26 Smith et al. reported the first homogeneous co-electrocatalytic system for the reduction of CO2 using a NADH analogue as the RM and an Fe tetraarylporphyrin complex that shows enhanced catalytic activity as a combined system.27 The NADH analogue transfers protons and electrons during the reaction, although the exact reaction sequence is currently unknown.
Recently, we reported a co-electrocatalytic system comprised of a chromium-centered catalyst, Cr(tbudhbpy)Cl(H2O) (1), where the ligand precursor is (tbudhbpy(H)2) is 6,6′-di(3,5-di-tert-butyl-2-hydroxybenzene)-2,2′-bipyridine,8,28 and dibenzothiophene-5,5-dioxide (DBTD) as the RM.29 When both species are present under aprotic conditions, the co-catalytic reductive disproportionation of two equivalents of CO2 occurs to produce CO and carbonate (CO32−). Because neither the catalyst nor the mediator demonstrates intrinsic reactivity for CO2 reduction under these conditions, an outer-sphere mechanism could be excluded. For clarity, the following labeling scheme will be used for Cr complexes multiplicity# bound DMFCr(axial ligands)charge and for RM species multiplicityRMcharge; the ligand frameworks do not change the κ4 coordination mode during the reaction, so it is omitted in the notation where possible for clarity. These studies suggested that the assembly of 2DBTD−1 with an intermediate bis-CO2 adduct, 40Cr(CO2CO2)−1 through inner-sphere coordination produces a dianion, 30Cr(CO2CO2)(DBTD)−2 which mediates carbon–oxygen bond cleavage. Association between the catalyst and mediator monoanions under aprotic conditions is driven by contributions from dispersive interactions, Cr–O bond formation between the metal complex and [DBTD]−, and through-space electronic conjugation (TSEC, Fig. 1) between the ligand backbone of the catalyst and the RM.29 TSEC is a non-covalent interaction between cofacial aromatic rings based on the interaction of their spatially delocalized π electrons.30
 | ||
| Fig. 1 Overview of protic CO2 reduction co-electrocatalysis, through-space electronic conjugation, and pancake bonding interactions relevant to the results presented here. | ||
When phenol (PhOH) is added as a sacrificial proton source under co-catalytic conditions there is an increase in activity compared to when only 1 or DBTD is present.8,29 It was proposed that the association under protic conditions is driven in part by pancake bonding (PB, Fig. 1), which is a parallel π-stacking interaction between planar aromatic moieties with significant radical character.31–34 Under co-electrocatalytic conditions, PB favors an equilibrium solvent displacement from 31Cr(CO2H)−1 by 2(DBTD)−1 to form the dianion 40Cr(CO2H)(DBTD)−2, with contributions from chromium–sulfone coordination and dispersive interactions. In this bimolecular assembly under protic conditions, both the [RM]− and the bipyridine (bpy) fragment of the [Cr–CO2H]− complex are formally reduced by one electron, consistent with PB. However, since the Cr catalyst itself has intrinsic CO2 reduction activity when PhOH is present, we could not definitively discard the possibility that [DBTD]− enhanced reactivity by outer-sphere electron transfer (Fig. 1). We reasoned that the strength of the interaction between the RM and Cr complex could be increased by synthetic means, since it has been noted that the strength of PB generally scales with respect to the extent of π-electron delocalization in the participating radicals and their steric protection from sigma-bonding interactions.34
Here, we compare the activity and overpotential (η) of two Cr catalysts with four RM architectures, to better understand and optimize the co-electrocatalytic system under protic conditions. These results suggest that extended aromatic character in the RM structure has the dual benefit of lowering its standard reduction potential while also favoring its binding to the Cr complex, resulting in inverse reaction scaling where increased activity occurs at lower overpotentials. We propose that mediator designs which extend aromatic character and match redox potential with the metal complex induce stronger interactions that improve the stability and activity of co-electrocatalysis, without requiring changes in the coordinating strength of the axial sulfone ligand.
Results
Electrochemistry of Cr catalysts
The synthesis of the (tbudhbpy(H)2) ligand and its subsequent metalation to generate Cr(tbudhbpy)Cl(H2O) (1) were carried out as previously reported (Fig. S1†).8 To improve the catalyst activity we synthesized a ligand framework with electron-donating tert-butyl groups on the bpy-backbone, 6,6′-di(3,5-di-tert-butyl-2-hydroxybenzene)-4,4′-di-tert-butyl-2,2′-bipyridine (tbudhtbubpy(H)2) which allows us to probe ligand electronic effects on the catalyst and co-catalytic system (see ESI and Fig. S2–S4†). The metalation of (tbudhtbubpy(H)2) to form Cr(tbudhtbubpy)Cl(H2O) (2) was carried out in a similar fashion to 1 (see ESI†) and 2 was characterized by UV-vis (Fig. S5†), NMR (Table S3†), electrospray ionization-mass spectrometry (ESI-MS) (see ESI†), microanalysis (see ESI†), and single-crystal X-ray diffraction (XRD) studies (Fig. 2).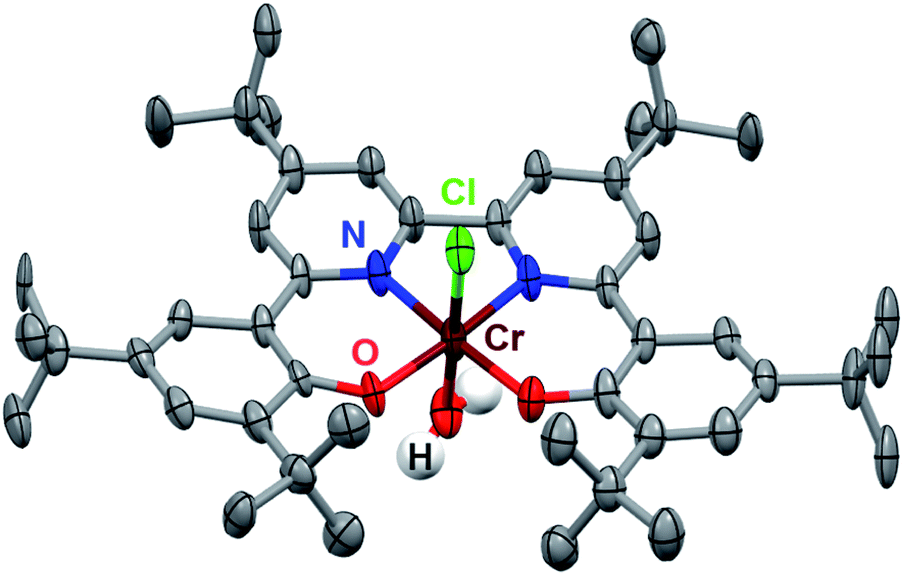 | ||
| Fig. 2 Molecular structure of Cr(tbudhtbubpy)Cl(H2O) (2) obtained from single-crystal X-ray diffraction studies. Blue = N, red = O, gray = C, green = Cl, maroon = Cr, white = H atoms of bound water molecule; thermal ellipsoids at 50%; ligand H atoms and occluded diethyl ether molecules omitted for clarity; hydrogen atoms of the Cr-bound water molecule were located in the diffraction map and refined isotropically. CCDC 2150930.† | ||
Cyclic voltammetry (CV) experiments were performed on 1 and 2 in N,N-dimethylformamide (DMF) with 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) as the supporting electrolyte. As reported previously, 1 exhibits three redox features under argon (Ar) saturation conditions, Ep = −1.66 and −1.78 V and E1/2 = −1.95 V versus the ferrocenium/ferrocene (Fc+/Fc) reduction potential.8 Complex 2 also exhibits three redox features, but due to the electron-donating quality of the tert-butyl groups on the bpy core of the ligand, all three are located at the more negative potentials of Ep = −1.76 and −1.87 V and E1/2 = −2.00 V vs. Fc+/Fc (Fig. S6†). The first two redox features of 1 were shown to be related to one another, previously assigned to the formation of a solvento species resulting from an equilibrium chloride displacement reaction.8,28 Due to the consistencies in redox potential, reversibility, and relative current intensities of these features in 2, the same can be concluded here. As was the case with 1, the first two reduction features coalesce at scan rates ≥2000 mV s−1, which is consistent with this assignment (Fig. S7†).
Upon the addition of PhOH under Ar saturation conditions, 1 and 2 demonstrate only a small change in the redox features, suggestive of either hydrogen bonding interactions between the reduced complex and PhOH or increased favorability of chloride anion solvation, but not formal protonation.35–37 Under CO2 saturation without PhOH, there are minimal changes to the redox features for both catalysts, meaning that aprotic CO2 reduction is not inherent to either 1 or 2. Variable scan rate analysis under both Ar and CO2 saturation shows that the electron transfer reaction is diffusion-controlled for 2 at the third reduction feature, indicating a homogenous electrochemical response (Fig. S7 and S8†) as was observed previously for 1.8
The similarity of the redox activity between complexes 1 and 2 extends to catalytic behavior: 2 also catalyzes CO2 reduction at the third redox feature (Ecat/2 = −2.00 V vs. Fc+/Fc) in the presence of PhOH. However, 2 more than doubles the amount of current density (1.38 mA cm−2, Fig. 3B, red) under the same conditions as 1 (0.554 mA cm−2, Fig. 3B, black). This significant increase in current density comes with a shift of only 50 mV to more negative potentials, increasing the η from 110 mV to 160 mV (see ESI†). Complex 1 was previously found to have first-order concentration dependences with respect to electrocatalytic current for PhOH, catalyst, and CO2.8 Performing the same variable concentration experiments with 2 (Fig. S9–S11†) established that in this system electrocatalytic current also has first-order concentration dependences with respect to PhOH (Fig. S9†), catalyst (Fig. S10†), and CO2 (Fig. S11†).
 | ||
| Fig. 3 (A) Structures of Cr(tbudhbpy)Cl(H2O) 1 and Cr(tbudhtbubpy)Cl(H2O) 2, where S is a solvent adduct of water or DMF. (B) Comparison of CVs for different Cr catalysts (1.0 mM) under CO2 saturation conditions and 0.1 M PhOH. Conditions: 0.1 M TBAPF6/DMF; glassy carbon disc working electrode, glassy carbon rod counter electrode, Ag/AgCl pseudoreference electrode; referenced to Fc+/Fc internal standard; 100 mV s−1 scan rate. | ||
To quantify the product selectivity and turnover frequency (TOF) of 2, controlled potential electrolysis (CPE) was performed with added PhOH under CO2 saturation conditions (Fig. S12†). Gaseous products were quantified by gas chromatography (GC, see ESI†). The results of this experiment (Table S4†) show that 2 is selectively reducing CO2 to CO under these conditions with 95 ± 8% FECO over 13.0 turnovers (turnover represents two electron equivalents of charge passed for each equivalent of 2 in solution) with a TOFCPE of 9.29 s−1 which is in good agreement with the TOFmax value of 14 ± 1 s−1 determined by CV (Fig. S13, S14 and Table S5†).38 Note that in all instances turnovers have been determined to show the catalytic nature of the process and do not represent a measurement to complete loss of activity.
Electrochemistry of redox mediators
To test the inner-sphere nature of the co-electrocatalytic mechanism we previously reported and further optimize the system,29 we prepared three new aromatic sulfone derivatives to establish structure–activity relationships with Cr catalysts 1 and 2. Triphenylothiophene-4,4-dioxide (TPTD, Fig. 4A) was synthesized by previously reported methods.39,40 Additionally, both 2,8-dimesityldibenzothiophene-5,5-dioxide (Mes2DBTD) and 2,8-diphenyldibenzothiophene-5,5-dioxide (Ph2DBTD) (Fig. 4A) were prepared using Pd-catalyzed cross-coupling and characterized by NMR (Fig. S15–S18†), ESI-MS (see ESI†), microanalysis (see ESI†), and XRD studies (Fig. 4A). CV experiments were carried out under the same conditions as 1 and 2, in 0.1 M TBAPF6 with DMF as the solvent. Under Ar saturation conditions, DBTD displays a reversible redox feature with an E1/2 = −2.25 V versus Fc+/Fc corresponding to a single-electron event.29 TPTD, Mes2DBTD, and Ph2DBTD also show single reversible one-electron redox features under Ar saturation conditions, but the E1/2 values for all three are shifted to more positive potentials compared to DBTD (Fig. 4B). Mes2DBTD has the most similar redox feature to DBTD with an E1/2 = −2.24 V versus Fc+/Fc, while TPTD and Ph2DBTD shift to more positive potentials with E1/2 = −2.19 V and −2.12 V versus Fc+/Fc, respectively. | ||
| Fig. 4 (A) Structures of redox mediators dibenzothiophene-5,5-dioxide (DBTD), triphenylothiophene-4,4-dioxide (TPTD), 2,8-dimesityldibenzothiophene-5,5-dioxide (Mes2DBTD), and 2,8-diphenyldibenzothiophene-5,5-dioxide (Ph2DBTD). TPTD, Mes2DBTD, and Ph2DBTD obtained from single-crystal X-ray diffraction studies. Yellow = S, red = O, gray = C; thermal ellipsoids at 50%; H atoms and occluded toluene molecules (Ph2DBTD only) omitted for clarity. CCDC 215497–215499.† (B) Comparison of CVs for different RMs (2.5 mM) under Ar saturation. Conditions: 0.1 M TBAPF6/DMF; glassy carbon disc working electrode, glassy carbon rod counter electrode, Ag/AgCl pseudoreference electrode; referenced to Fc+/Fc internal standard; 100 mV s−1 scan rate. | ||
Control experiments performed with TPTD, Mes2DBTD, and Ph2DBTD demonstrate little reactivity with CO2 or PhOH on the CV timescale, as was previously observed for analogous experiments with DBTD (Fig. S19–S21†). Variable scan rate experiments were performed for all four sulfone derivatives to calculate their diffusion coefficients (Fig. S22–S25, see ESI†).41 Unsurprisingly, the experimentally determined diffusion coefficients for these molecules scale with their molecular weight: DBTD (6.22 × 10−6 cm2 s−1) > TPTD (3.93 × 10−6 cm2 s−1) > Ph2DBTD (3.68 × 10−6 cm2 s−1) > Mes2DBTD (3.57 × 10−6 cm2 s−1).
Co-electrocatalysis under protic conditions
Since the addition of PhOH as a sacrificial proton donor led to a large increase in catalytic activity for 1 + DBTD in previous studies, we sought to see if a similar trend would be observed for 1 with the new RMs and 2 with all four RMs. Briefly, the observed trend in current density under CV conditions for all the RMs when added to a solution of 1 and PhOH can be summarized: DBTD exhibits the greatest amount of current followed by TPTD, and the use of Ph2DBTD produces more current than Mes2DBTD despite having a standard reduction potential which is 120 mV more positive (Fig. 5A). The addition of all four RMs causes a significant increase in current and completely irreversible waveforms. Variable concentration studies were completed for 1, all four RMs, PhOH, and CO2. These data show that the observed current is proportional to the concentration of 1 (Fig. S29–S31†), RM (Fig. S32–S34†), a fixed ratio of 1 and RM (Fig. S35–S37†), PhOH (Fig. S38–S40†), and CO2 (Fig. S41–S43†) where RM is DBTD,29 TPTD, Mes2DBTD, or Ph2DBTD. | ||
| Fig. 5 CVs of 1.0 mM Cr(tbudhbpy)Cl(H2O) 1 (A) or Cr(tbudhtbubpy)Cl(H2O) 2 (B) with 2.5 mM DBTD (black), TPTD (red), Mes2DBTD (green), and Ph2DBTD (blue) as the RM and 0.1 M PhOH under CO2 saturation conditions. Conditions: 0.1 M TBAPF6/DMF; glassy carbon disc working electrode, glassy carbon rod counter electrode, Ag/AgCl pseudoreference electrode; referenced to Fc+/Fc internal standard; 100 mV s−1 scan rate. | ||
When we compare the protic catalytic activity of 2 with all the RMs, we see a similar trend to the activity of 1 observed under comparable conditions. DBTD again shows the largest amount of current followed by TPTD under CV conditions (Fig. 5B). However, for complex 2 there is a significant difference in the activity of the co-catalytic system with Ph2DBTD relative to that with Mes2DBTD; Ph2DBTD again outperforms Mes2DBTD as a RM when paired with 2 despite its more positive standard reduction potential. Since 2 is an intrinsically better catalyst than 1 under CO2 saturation with a proton donor, we were delighted to find that co-electrocatalysis involving 2 and the four RMs under protic conditions also reached much larger current densities than any combination involving 1. Variable concentration experiments were also performed for 2, all four RMs, a fixed ratio of complex 2 and all RMs, PhOH, and CO2, demonstrating proportional increases in current with respect to each reaction component (see Fig. S48–S67†).
CPE experiments were performed in the presence of 0.12 M PhOH with 1 or 2 and across three concentrations of DBTD, TPTD, Mes2DBTD, and Ph2DBTD (Fig. S68–S74†). The current observed during the CPE experiments with 1 as the catalyst scaled with increasing RM concentration at the tested catalyst![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :RM ratios of 1:1, 1:3, and 1:5 (ref. 29 and Fig. S68–S70†). However, this did not hold true for all RM combinations with 2. When TPTD, Mes2DBTD, and Ph2DBTD were used as the RM, the observed current scaled with respect to their concentration. However, when DBTD was used as the RM with 2, saturation was observed at the 1:3 ratio with no further increase observed at the 1:5 ratio (Fig. S71†). Both catalysts with all mediators at all ratios tested were found to be quantitatively selective for CO (Tables S7–S27†). However, since the 1:5 (catalyst:RM) produced the largest amount of CO on average for all the systems, we will focus the remainder of our analysis on these experiments. All combinations of the protic co-electrocatalytic systems show high activity with TOF values ranging from 64.0 to 208 s−1 (Table 1), one to two orders of magnitude higher than the catalysts alone.
:RM ratios of 1:1, 1:3, and 1:5 (ref. 29 and Fig. S68–S70†). However, this did not hold true for all RM combinations with 2. When TPTD, Mes2DBTD, and Ph2DBTD were used as the RM, the observed current scaled with respect to their concentration. However, when DBTD was used as the RM with 2, saturation was observed at the 1:3 ratio with no further increase observed at the 1:5 ratio (Fig. S71†). Both catalysts with all mediators at all ratios tested were found to be quantitatively selective for CO (Tables S7–S27†). However, since the 1:5 (catalyst:RM) produced the largest amount of CO on average for all the systems, we will focus the remainder of our analysis on these experiments. All combinations of the protic co-electrocatalytic systems show high activity with TOF values ranging from 64.0 to 208 s−1 (Table 1), one to two orders of magnitude higher than the catalysts alone.
| Conditions | Potential (V vs. Fc+/Fc) | FECO (%) | TOFCPE (s−1) | η (V) | Turnovers of CO w.r.t [1 or 2] | Turnovers of CO w.r.t [RM] | i cat/ipe |
|---|---|---|---|---|---|---|---|
| a 0.5 mM catalyst and 0.6 M PhOH. b 0.5 mM catalyst, 2.5 mM RM, and 0.6 M PhOH. c 0.1 mM catalyst, 0.5 mM RM, and 0.12 M PhOH. d 0.1 mM catalyst and 0.12 M PhOH. e Scan rate = 100 mV s−1. | |||||||
| 1 + PhOH29a | −2.30 | 111 ± 14 | 7.12 | 0.11 | 11.4 | — | 3.4 |
| 1 + DBTD + PhOH29b | −2.30 | 102 ± 14 | 65.3 | 0.41 | 29 | 5.8 | 2.8 |
| 1 + TPTD + PhOHc | −2.25 | 98 ± 17 | 74.5 | 0.35 | 25 | 4.9 | 3.0 |
| 1 + Mes2DBTD + PhOHc | −2.30 | 98 ± 6 | 64.0 | 0.40 | 19 | 4.7 | 2.6 |
| 1 + Ph2DBTD + PhOHc | −2.20 | 100 ± 2 | 69.3 | 0.28 | 22 | 5.3 | 3.6 |
| 2 + PhOHd | −2.30 | 95 ± 8 | 9.29 | 0.16 | 13 | — | 8.4 |
| 2 + DBTD + PhOHc | −2.30 | 109 ± 9 | 163 | 0.41 | 28 | 6.5 | 5.8 |
| 2 + TPTD + PhOHc | −2.25 | 97 ± 6 | 208 | 0.35 | 31 | 8.0 | 6.6 |
| 2 + Mes2DBTD + PhOHc | −2.30 | 98 ± 4 | 149 | 0.40 | 27 | 5.9 | 5.7 |
| 2 + Ph2DBTD + PhOHc | −2.20 | 97 ± 5 | 194 | 0.28 | 35 | 8.8 | 7.1 |
As was the case for DBTD,29 control CPE experiments for TPTD, Mes2DBTD, and Ph2DBTD in the presence of PhOH without catalyst (Fig. S75–S77†) show non-quantifiable amounts of CO (Tables S28–S30†) and no H2 production, implicating the possibility of a degradation pathway in the absence of the Cr complex. To understand the stability of DBTD during electrolysis, a control CPE of DBTD and PhOH under N2 was performed (Fig. S78†). Characterization of the pre- and post-CPE solution by gas chromatography/mass spectrometry (GC/MS) with appropriate controls demonstrates a minor amount of DBTD is reduced to the corresponding sulfoxide, dibenzothiophene 5-oxide (DBTO; Fig. S78†). However, while comparable analysis of the CPE solution before and after the experiment under catalytic conditions (1, DBTD, and PhOH under CO2) also shows reduction of DBTD to DBTO, an additional product with a mass too high to be dibenzothiophene is detected, implying that reductive C–S bond cleavage is a possible degradation pathway (Fig. S79†). It is worth emphasizing that the high catalytic efficiencies observed under all measured conditions suggest that these pathways are minor contributors for all systems (Table 1). While our original report showed the appearance of aprotic CO2 reduction with 1 and DBTD, the new systems here showed passivation of the electrode during CPE experiments, suggestive of solubility issues under these conditions (see ESI†).
The systems with 1 and 2 as the catalyst followed the same trend in TOFCPE for CO formation when comparing across the RMs: Mes2DBTD < DBTD < Ph2DBTD < TPTD (Table 1). As introduced above, our proposed inner-sphere mechanism for this co-electrocatalytic reaction relies on the binding of the reduced RM to an intermediate [Cr–CO2H]− species before the rate-determining C–OH bond cleavage step.29 Although all RMs are reducing enough to transfer electron equivalents via an outer-sphere mechanism, the data presented in Table 1 are most consistent with the predominance of an inner-sphere RM pathway: the observed co-catalytic activity does not scale with the reduction potential of the RMs or their diffusion coefficients. For a purely outer-sphere mechanism, conformity to a Marcus theory-type model would be expected, where increased electrochemical driving force would equate to an increased rate of reaction (under the assumption that the inverted region or diffusion limit are not reached under experimental conditions).42 Instead, the two RMs with the most positive standard reduction potentials (TPTD and Ph2DBTD) demonstrate the greatest activity. Likewise, if outer-sphere electron transfer was the primary reaction pathway, the relatively slowly diffusing Ph2DBTD would be expected to show decreased activity compared to DBTD, however an opposite trend is observed experimentally. The relatively limited performance of the Mes2DBTD derivative despite a similar reduction potential to DBTD suggests that sterically encumbering the dibenzothiophene-5,5-dioxide core has a kinetic effect, which is also consistent with an inner-sphere pathway causing the greatest activity enhancement. The optimized combination of 2 + Ph2DBTD under protic conditions achieves a TOF of 194 s−1 at an η of 280 mV, which demonstrates a three-fold increase in activity at an η which is 130 mV lower in comparison to the previous system with 1 and DBTD.29
The trend in activity data derived from CPE is at first glance different than the overall current trend observed in the CV data (Fig. 5). However, homogeneous current density is dependent on the diffusion coefficients of all reaction components under experimental conditions. In an effort to account for the effect the diffusion coefficients have on the observed trend in the CV data, we have calculated the value of icat/ip (icat = catalytic plateau current and ip = Faradaic current) for each of the co-catalytic systems at the redox feature of the respective mediators (Table 1 and Fig. S80; see the ESI† for details). This experimental value can be used to calculate a TOFmax value from CV data,43 however, since the RMs and Cr complexes do not interact under Faradaic conditions, we are unable to use this method since diffusion coefficient and concentration are not properly normalized by the procedure. Therefore, the icat/ip ratio can only qualitatively represent relative catalytic activity when compared across systems. The trend observed for the icat/ip ratios is generally consistent with the TOFCPE: the co-catalytic Ph2DBTD and TPTD systems have larger activities than the other RMs.
Computational studies
To gain insight into the assembly of RMs and catalysts, DFT calculations were done with the Gaussian 16 package at the B3LYP-D3(BJ)/def2-tzvp//B3LYP-D3(BJ)/def2-svp level of theory (see ESI† for details).44–52 As previously reported,29 including dispersion corrections at the optimization stage is of paramount importance due to the key role of non-covalent interactions. The free energies of formation of the dianionic 40Cr(CO2H)(RM)−2 assemblies for 1 and 2 summarized by eqn (1), where multiplicity# bound DMFCr(axial ligands)charge, are found in Table 2. | (1) |
| Cr Complex | RM | ΔGeqn (1) (kcal mol−1) | ΔG‡ C–OH (kcal mol−1) | RM0/−vs. Fc+/0 |
|---|---|---|---|---|
| [Cr(tbudhbpy)(CO2H)S]− | None | N/A | 13.5 | N/A |
| [Cr(tbudhbpy)(CO2H)S]− | [DBTD]− | −0.1 | 11.6 | −2.25 |
| [Cr(tbudhbpy)(CO2H)S]− | [Mes2DBTD]− | 0.5 | — | −2.24 |
| [Cr(tbudhbpy)(CO2H)S]− | [TPTD]− | −2.9 | 11.6 | −2.19 |
| [Cr(tbudhbpy)(CO2H)S]− | [Ph2DBTD]− | −3.1 | 11.6 | −2.12 |
| [Cr(tbudhtbubpy)(CO2H)S]− | None | N/A | 12.5 | N/A |
| [Cr(tbudhtbubpy)(CO2H)S]− | [DBTD]− | −2.3 | 10.7 | −2.25 |
| [Cr(tbudhtbubpy)(CO2H)S]− | [Mes2DBTD]− | −2.0 | — | −2.24 |
| [Cr(tbudhtbubpy)(CO2H)S]− | [TPTD]− | −6.6 | 10.9 | −2.19 |
| [Cr(tbudhtbubpy)(CO2H)S]− | [Ph2DBTD]− | −6.1 | 10.8 | −2.12 |
Despite Coulombic repulsion, the substitution of a DMF molecule for the reduced mediator is favorable in almost all cases, reaching large free energies of reaction of −6.1 and −6.6 kcal mol−1 for the combination of Ph2DBTD and TPTD with 2. These values show good correlation with the experimental TOF values for 2, while for 1 the trend is less pronounced (Fig. 6). Indeed, while transition state energies determined for the dianionic assemblies of DBTD, TPTD, and Ph2DBTD RMs with both complexes 1 and 2 show minimal variance with respect one another, they are uniformly lower than the comparable transition state energy of the monoanionic DMF adduct (Table 2). In other words, the solvento species for both complexes 1 and 2 lies on a higher-energy pathway than any of the co-catalytic combinations studied. Importantly, this result implies that the equilibrium represented in eqn (1) has a direct effect on catalyst speciation relevant to the observed activity.
 | ||
| Fig. 6 Correlation between computed DMF displacement energies and TOF. | ||
The apparent PB interaction can be visualized in the spin density plots and Kohn–Sham (KS) orbital representations of the singly occupied molecular orbitals of the 40Cr(CO2H)(Ph2DBTD)−2 adduct (Fig. 7). In addition to the favorable energetics of the association of the Ph2DBTD and the Cr complex, these plots show antiferromagnetic coupling between Ph2DBTD and the bpy fragment of the ligand in singly occupied orbitals of relevant symmetries. While a PB between [RM]− and [Cr–CO2H]− requires sufficient extended aromatic character, another component of fundamental importance for any bonding interaction is that in order to be maximized, orbitals need to be close both in terms of orbital shape and energy. In other words, we propose that the PB is maximized as the standard potential of the RM approaches that of the catalyst because the radical-containing orbitals of the reduced aromatic fragments become closer in energy. Nuclear Independent Chemical Shift (NICS(0)) calculations on all RMs in the neutral and radical anion states indicate that the five-membered sulfone-containing ring goes from antiaromatic to aromatic upon reduction (Tables S31–S34†). KS representations of all 40Cr(CO2H)(RM)−2 adducts show that orbitals from this five-membered ring are key to the proposed PB, interacting with the partially occupied π* orbitals of the bpy fragment in the Cr complex relevant to the co-catalytic pathway (Fig. S81–S87†).28,29
 | ||
| Fig. 7 Molecular geometry of the 40Cr(CO2H)(Ph2DBTD)−2 adduct (A) DFT-calculated spin density (B) Kohn–Sham orbital projection of SOMO (C) and SOMO−1 (D). | ||
It is worth stating again, however, that with the present data the observed correlation is weak for complex 1. This suggests that while RM redox potentials are to some extent an indirect measure of relevant orbital energies, there are additional kinetic components influenced by the steric bulk of the catalyst and the RM. This kinetic effect is seen most directly in the comparison of the experimental and computational data of DBTD and Mes2DBTD, which despite being nearly identical in terms of redox potential have significantly different activity.
Discussion
Here, both the experimental and computational data indicate that the RM is operating by an inner-sphere mechanism under protic conditions (Fig. 8). We propose that first, the four-coordinate neutral catalyst, i, is reduced to the monoanionic Cr species ii. Based on calculations previously performed on the co-electrocatalytic mechanism of 1, CO2 readily binds to ii to form [Cr–CO2]−iii.8,28,29 With PhOH present this species is protonated, binds DMF and is then reduced to form the monoanionic [Cr–CO2H]−iv. The one electron reduced [RM]− binds to this species to form v, which we proposed to be the key intermediate, 40Cr(CO2H)(RM)−2, stabilized by pancake bonding between the one-electron reduced bpy-backbone and the π-framework of the reduced RM. We propose that v is further stabilized when the π-framework of the RM is extended, due to closer energy levels of the orbitals involved in PB and greater dispersive interactions. Furthermore, extended aromatic character can protect the radical character of the RM in the reduced state, aligning with the improved performance of these mediators under electrolysis conditions. Following the addition of a second proton equivalent, the neutral RM is released along with water leaving [Cr–CO]−vi, which we have previously shown is not stable.8 This species then releases CO to form the monoanionic four-coordinate neutral Cr species ii, completing the cycle. | ||
| Fig. 8 Proposed catalytic mechanism for co-electrocatalytic CO2 reduction by Cr and RM under protic conditions where Cr is 1 or 2 and RM is DBTD, TPTD, Ph2DBTD, or Mes2DBTD. | ||
Computational results show that the barrier for C–OH bond cleavage in all computed 40Cr(CO2H)(RM)−2 adducts is roughly equivalent for all possibilities and lower than the corresponding solvento species 40Cr(CO2H)(DMF)−1. This means that the equilibrium displacement of the axial solvent ligand by [RM]−KRM dictates whether lower or higher energy catalytic pathways are accessible and will be directly reflected in the rate observed at co-catalytic potentials. Further, the minimal difference in barrier height across the 40Cr(CO2H)(RM)−2 adducts and the minimal changes in the bond between sulfone and Cr center are consistent with the proposal that changes in dispersion effects and PB are responsible for the differences in binding energy (Table S35†). Both TPTD and Ph2DBTD show increased activity compared to DBTD and Mes2DBTD as RMs when combined with 1 or 2, despite the more negative operating potentials of the latter pair, which is consistent with an inner-sphere process. Although the kinetic complexity of the system precludes straightforward analysis of variable concentration CV data, the proportional increase in current as the catalyst:RM ratio increased observed in CPE suggests that this equilibrium binding interaction is relevant to the catalytic rate expression. This leads to an inverse scaling effect between RM standard potential and the observed activity, with greater catalytic activity observed at lower reduction potentials.
Previously, the existence of inverse potential scaling relationships in molecular electrocatalysts for CO2 reduction has been primarily explained through the kinetic stabilization of key intermediates. Initial work from Savéant and co-workers in 2016 showed that the inclusion of trimethylammonium groups on the catalyst ligand framework resulted in significant kinetic enhancements to CO production under protic conditions at low overpotentials.6 This kinetic effect at more positive catalyst standard reduction potentials was initially proposed to be the result of coulombic stabilization of a key [Fe–CO2] intermediate, however, a later computational study showed that the relatively long distance (3.8 Å) resulted in effective charge screening by the implicit solvent used.53 Energy decomposition analysis showed that both through-space and through-structure effects contribute to the stabilization of the [Fe–CO2] intermediate, with through-structure being stronger, but both being necessary to kinetic enhancements at low applied potentials. Nichols et al. then reported in 2018 that when hydrogen-bond donors are properly positioned in the secondary coordination sphere, significant increases in activity occur with minor variance in the standard reduction potential of the catalyst.54 Mechanistic experiments again indicated that this observation resulted from stabilization of the [Fe–CO2] intermediate and not the inclusion of acidic amide protons near the catalytic active site. Subsequently, Gotico et al. demonstrated that using urea functional groups as multipoint hydrogen-bond donors in similar frameworks could further enhance this kinetic stabilization effect.55 A recent report from McCrory and co-workers on a non-porphyrinic Co-based catalyst revealed a previously unobserved effect where the combination of ligand reduction potential and electrostatic effects could be manipulated to facilitate the storing of additional charge equivalents, improving catalytic activity for CO2 reduction at lower catalyst standard potentials.56
Our proposed mechanism for inverse potential scaling is unique with respect to these previous examples. Unlike the use of electrostatic effects to stabilize bound CO2 intermediates or facilitate the storage of electron equivalents in the ligand framework, dispersive interactions and PB drive the association of RM and Cr complex, surmounting the Coulombic repulsion between the two fragments. This co-catalytic assembly presents a lower barrier for the rate-limiting C–OH bond cleavage step. Importantly, this barrier is equivalent across all RMs and instead it is the favorability of the pre-equilibrium comprising the formation of the co-catalytic assembly that dictates the extent the faster mechanism contributes to the observed activity. Mediators with extended aromaticity operate at lower redox potentials, but present stronger PB and dispersive interactions with the metal catalyst, favoring the formation of the co-catalytic assembly and thus yielding enhanced reaction rates at lower overpotentials.
Conclusion
These results show that by tuning properties of both catalyst and RM, an increase in activity for the co-electrocatalytic reduction of CO2 can be achieved at lower overpotential by means of inner-sphere electron transfer. Through the addition of electron-donating groups to the bpy backbone in complex 2, we have improved the activity of our Cr-centered catalyst for CO2 reduction. Consistent with the proposal in our initial report of co-catalytic systems with 1,29 these data provide a theoretical and experimental basis for an inner-sphere mechanism under protic conditions. By tuning the stereoelectronic properties of the catalyst and the RM, we have optimized our system to decrease the overpotential while increasing the activity, contrary to what would be predicted by a classical outer-sphere Marcus-theory type model.42 As a result, an equilibrium solvent displacement reaction can be used to access a lower energy catalytic pathway, with increased favorability driving the system to higher observed activity. Additionally, the selectivity of the parent Cr complexes is retained with all catalytically competent systems being quantitatively selective for CO. Importantly, implementing the strategies identified by this work will be a powerful tool for developing systems which improve the performance of molecular catalyst systems. We are exploring additional strategies to optimize the co-electrocatalytic response in ongoing studies.Note added in proof
During the final editorial process, a manuscript was published concerning the use of concerted proton–electron (CPET) mediators as part of a homogeneous co-electrocatalytic system for the reduction of CO2 to HCOOH.57Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
CWM conceptualized and supervised the project. AGR, JJM, SLH, KRB, IHT, DAD, and CWM designed and conducted experiments. The manuscript was written with analysis and contributions from all authors.Funding sources
This research was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Catalysis Science Program, under Award DE-SC0022219. Single crystal X-ray diffraction experiments were performed on a diffractometer at the University of Virginia funded by the NSF-MRI program (CHE-2018870). We acknowledge UVA for support of the Rivanna High-Performance Computing Center.Conflicts of interest
There are no conflicts to declare.References
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, What would it take for renewably powered electrosynthesis to displace petrochemical processes?, Science, 2019, 364(6438), eaav3506 CrossRef CAS PubMed.
- M. Aresta, A. Dibenedetto and A. Angelini, Catalysis for the Valorization of Exhaust Carbon: from CO2 to Chemicals, Materials, and Fuels. Technological Use of CO2, Chem. Rev., 2014, 114(3), 1709–1742 CrossRef CAS PubMed.
- T. P. Senftle and E. A. Carter, The Holy Grail: Chemistry Enabling an Economically Viable CO2 Capture, Utilization, and Storage Strategy, Acc. Chem. Res., 2017, 50(3), 472–475 CrossRef CAS PubMed.
- J. D. Froehlich and C. P. Kubiak, Homogeneous CO2 Reduction by Ni(cyclam) at a Glassy Carbon Electrode, Inorg. Chem., 2012, 51(7), 3932–3934 CrossRef CAS PubMed.
- C. Cometto, L. Chen, P.-K. Lo, Z. Guo, K.-C. Lau, E. Anxolabéhère-Mallart, C. Fave, T.-C. Lau and M. Robert, Highly Selective Molecular Catalysts for the CO2-to-CO Electrochemical Conversion at Very Low Overpotential. Contrasting Fe vs Co Quaterpyridine Complexes upon Mechanistic Studies, ACS Catal., 2018, 8(4), 3411–3417 CrossRef CAS.
- I. Azcarate, C. Costentin, M. Robert and J.-M. Savéant, Through-Space Charge Interaction Substituent Effects in Molecular Catalysis Leading to the Design of the Most Efficient Catalyst of CO2-to-CO Electrochemical Conversion, J. Am. Chem. Soc., 2016, 138(51), 16639–16644 CrossRef CAS PubMed.
- M. D. Sampson, A. D. Nguyen, K. A. Grice, C. E. Moore, A. L. Rheingold and C. P. Kubiak, Manganese Catalysts with Bulky Bipyridine Ligands for the Electrocatalytic Reduction of Carbon Dioxide: Eliminating Dimerization and Altering Catalysis, J. Am. Chem. Soc., 2014, 136(14), 5460–5471 CrossRef CAS PubMed.
- S. L. Hooe, J. M. Dressel, D. A. Dickie and C. W. Machan, Highly Efficient Electrocatalytic Reduction of CO2 to CO by a Molecular Chromium Complex, ACS Catal., 2020, 10(2), 1146–1151 CrossRef CAS.
- G. T. Babcock and M. Wikström, Oxygen activation and the conservation of energy in cell respiration, Nature, 1992, 356(6367), 301–309 CrossRef CAS PubMed.
- H. Chen, O. Simoska, K. Lim, M. Grattieri, M. Yuan, F. Dong, Y. S. Lee, K. Beaver, S. Weliwatte, E. M. Gaffney and S. D. Minteer, Fundamentals, Applications, and Future Directions of Bioelectrocatalysis, Chem. Rev., 2020, 120(23), 12903–12993 CrossRef CAS PubMed.
- C. W. Anson, S. Ghosh, S. Hammes-Schiffer and S. S. Stahl, Co(salophen)-Catalyzed Aerobic Oxidation of p-Hydroquinone: Mechanism and Implications for Aerobic Oxidation Catalysis, J. Am. Chem. Soc., 2016, 138(12), 4186–4193 CrossRef CAS PubMed.
- C. W. Anson and S. S. Stahl, Cooperative Electrocatalytic O2 Reduction Involving Co(salophen) with p-Hydroquinone as an Electron–Proton Transfer Mediator, J. Am. Chem. Soc., 2017, 139(51), 18472–18475 CrossRef CAS PubMed.
- S. L. Hooe, E. N. Cook, A. G. Reid and C. W. Machan, Non-covalent assembly of proton donors and p-benzoquinone anions for co-electrocatalytic reduction of dioxygen, Chem. Sci., 2021, 12(28), 9733–9741 RSC.
- M. J. Chalkley, T. J. Del Castillo, B. D. Matson and J. C. Peters, Fe-Mediated Nitrogen Fixation with a Metallocene Mediator: Exploring pKa Effects and Demonstrating Electrocatalysis, J. Am. Chem. Soc., 2018, 140(19), 6122–6129 CrossRef CAS PubMed.
- E. A. McLoughlin, K. C. Armstrong and R. M. Waymouth, Electrochemically Regenerable Hydrogen Atom Acceptors: Mediators in Electrocatalytic Alcohol Oxidation Reactions, ACS Catal., 2020, 10(19), 11654–11662 CrossRef CAS.
- C. M. Galvin and R. M. Waymouth, Electron-Rich Phenoxyl Mediators Improve Thermodynamic Performance of Electrocatalytic Alcohol Oxidation with an Iridium Pincer Complex, J. Am. Chem. Soc., 2020, 142(45), 19368–19378 CrossRef CAS PubMed.
- A. Badalyan and S. S. Stahl, Cooperative electrocatalytic alcohol oxidation with electron-proton-transfer mediators, Nature, 2016, 535(7612), 406–410 CrossRef CAS PubMed.
- B. Shan and R. Schmehl, Photochemical Generation of Strong One-Electron Reductants via Light-Induced Electron Transfer with Reversible Donors Followed by Cross Reaction with Sacrificial Donors, J. Phys. Chem. A, 2014, 118(45), 10400–10406 CrossRef CAS PubMed.
- E. L. Tyson, Z. L. Niemeyer and T. P. Yoon, Redox Mediators in Visible Light Photocatalysis: Photocatalytic Radical Thiol–Ene Additions, J. Org. Chem., 2014, 79(3), 1427–1436 CrossRef CAS PubMed.
- B. Happ, A. Winter, M. D. Hager and U. S. Schubert, Photogenerated avenues in macromolecules containing Re(I), Ru(II), Os(II), and Ir(III) metal complexes of pyridine-based ligands, Chem. Soc. Rev., 2012, 41(6), 2222–2255 RSC.
- E. S. Andreiadis, M. Chavarot-Kerlidou, M. Fontecave and V. Artero, Artificial Photosynthesis: From Molecular Catalysts for Light-driven Water Splitting to Photoelectrochemical Cells, Photochem. Photobiol., 2011, 87(5), 946–964 CrossRef CAS PubMed.
- T. S. Teets and D. G. Nocera, Photocatalytic hydrogen production, Chem. Commun., 2011, 47(33), 9268–9274 RSC.
- K. Sakai and H. Ozawa, Homogeneous catalysis of platinum(II) complexes in photochemical hydrogen production from water, Coord. Chem. Rev., 2007, 251(21), 2753–2766 CrossRef CAS.
- R. Francke and R. D. Little, Redox catalysis in organic electrosynthesis: basic principles and recent developments, Chem. Soc. Rev., 2014, 43(8), 2492–2521 RSC.
- Y. Oh and X. L. Hu, Organic molecules as mediators and catalysts for photocatalytic and electrocatalytic CO2 reduction, Chem. Soc. Rev., 2013, 42(6), 2253–2261 RSC.
- D. V. Vasilyev and P. J. Dyson, The Role of Organic Promoters in the Electroreduction of Carbon Dioxide, ACS Catal., 2021, 11(3), 1392–1405 CrossRef CAS.
- P. T. Smith, S. Weng and C. J. Chang, An NADH-Inspired Redox Mediator Strategy to Promote Second-Sphere Electron and Proton Transfer for Cooperative Electrochemical CO2 Reduction Catalyzed by Iron Porphyrin, Inorg. Chem., 2020, 59(13), 9270–9278 CrossRef CAS PubMed.
- J. J. Moreno, S. L. Hooe and C. W. Machan, DFT Study on the Electrocatalytic Reduction of CO2 to CO by a Molecular Chromium Complex, Inorg. Chem., 2021, 60(6), 3635–3650 CrossRef CAS PubMed.
- (a) S. L. Hooe, J. J. Moreno, A. G. Reid, E. N. Cook and C. W. Machan, Mediated Inner-Sphere Electron Transfer Induces Homogeneous Reduction of CO2 via Through-Space Electronic Conjugation**, Angew. Chem., Int. Ed., 2022, 61(1), e202109645 CrossRef CAS PubMed; (b) S. L. Hooe, J. J. Moreno, A. G. Reid, E. N. Cook and C. W. Machan, Angew. Chem., Int. Ed., 2022, 61, e202205139 CAS.
- J. Li, P. Shen, Z. Zhao and B. Z. Tang, Through-Space Conjugation: A Thriving Alternative for Optoelectronic Materials, CCS Chem., 2019, 1(2), 181–196 CAS.
- I. Garcia-Yoldi, J. S. Miller and J. J. Novoa, Structure and Stability of the [TCNE]22- Dimers in Dichloromethane Solution: A Computational Study, J. Phys. Chem. A, 2007, 111(32), 8020–8027 CrossRef CAS PubMed.
- I. Garcia-Yoldi, J. S. Miller and J. J. Novoa, [Cyanil]22− dimers possess long, two-electron ten-center (2e−/10c) multicenter bonding, Phys. Chem. Chem. Phys., 2008, 10(28), 4106–4109 RSC.
- F. Mota, J. S. Miller and J. J. Novoa, Comparative Analysis of the Multicenter, Long Bond in [TCNE]·− and Phenalenyl Radical Dimers: A Unified Description of Multicenter, Long Bonds, J. Am. Chem. Soc., 2009, 131(22), 7699–7707 CrossRef CAS PubMed.
- M. Kertesz, Pancake Bonding: An Unusual Pi-Stacking Interaction, Chem. – Eur. J., 2019, 25(2), 400–416 CrossRef CAS PubMed.
- S. L. Hooe, A. L. Rheingold and C. W. Machan, Electrocatalytic Reduction of Dioxygen to Hydrogen Peroxide by a Molecular Manganese Complex with a Bipyridine-Containing Schiff Base Ligand, J. Am. Chem. Soc., 2018, 140(9), 3232–3241 CrossRef CAS PubMed.
- A. W. Nichols, S. Chatterjee, M. Sabat and C. W. Machan, Electrocatalytic Reduction of CO2 to Formate by an Iron Schiff Base Complex, Inorg. Chem., 2018, 57(4), 2111–2121 CrossRef CAS PubMed.
- S. L. Hooe and C. W. Machan, Dioxygen Reduction to Hydrogen Peroxide by a Molecular Mn Complex: Mechanistic Divergence between Homogeneous and Heterogeneous Reductants, J. Am. Chem. Soc., 2019, 141(10), 4379–4387 CrossRef CAS PubMed.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, Turnover Numbers, Turnover Frequencies, and Overpotential in Molecular Catalysis of Electrochemical Reactions. Cyclic Voltammetry and Preparative-Scale Electrolysis, J. Am. Chem. Soc., 2012, 134(27), 11235–11242 CrossRef CAS PubMed.
- S. M. Chintala, J. T. Petroff II, A. Barnes and R. D. McCulla, Photodeoxygenation of phenanthro[4,5-bcd]thiophene S-oxide, triphenyleno[1,12-bcd]thiophene S-oxide and perylo[1,12-bcd]thiophene S-oxide, J. Sulphur Chem., 2019, 40(5), 503–515 CrossRef CAS.
- L. H. Klemm and R. F. Lawrence, The insertion and extrusion of heterosulfur bridges. X. Conversions in the triphenylene-triphenylo[4,5-bcd] thiophene system, J. Heterocycl. Chem., 1979, 16(3), 599–601 CrossRef CAS.
- J. E. Baur, Chapter 19 Diffusion Coefficients, in Handbook of Electrochemistry, ed. C. G. Zoski, Elsevier, 2007; pp. 829–848 Search PubMed.
- E. J. Piechota and G. J. Meyer, Introduction to Electron Transfer: Theoretical Foundations and Pedagogical Examples, J. Chem. Educ., 2019, 96(11), 2450–2466 CrossRef CAS.
- N. Elgrishi, B. D. McCarthy, E. S. Rountree and J. L. Dempsey, Reaction Pathways of Hydrogen-Evolving Electrocatalysts: Electrochemical and Spectroscopic Studies of Proton-Coupled Electron Transfer Processes, ACS Catal., 2016, 6(6), 3644–3659 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. B.01, Wallingford, CT, 2016 Search PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98(7), 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37(2), 785–789 CrossRef CAS PubMed.
- S. H. Vosko, L. Wilk and M. Nusair, Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis, Can. J. Phys., 1980, 58(8), 1200–1211 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields, J. Phys. Chem., 1994, 98(45), 11623–11627 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7(18), 3297–3305 RSC.
- F. Weigend, Accurate Coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8(9), 1057–1065 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132(15), 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32(7), 1456–1465 CrossRef CAS PubMed.
- Y. Mao, M. Loipersberger, K. J. Kron, J. S. Derrick, C. J. Chang, S. M. Sharada and M. Head-Gordon, Consistent inclusion of continuum solvation in energy decomposition analysis: theory and application to molecular CO2 reduction catalysts, Chem. Sci., 2021, 12(4), 1398–1414 RSC.
- E. M. Nichols, J. S. Derrick, S. K. Nistanaki, P. T. Smith and C. J. Chang, Positional effects of second-sphere amide pendants on electrochemical CO2 reduction catalyzed by iron porphyrins, Chem. Sci., 2018, 9(11), 2952–2960 RSC.
- P. Gotico, B. Boitrel, R. Guillot, M. Sircoglou, A. Quaranta, Z. Halime, W. Leibl and A. Aukauloo, Second-Sphere Biomimetic Multipoint Hydrogen-Bonding Patterns to Boost CO2 Reduction of Iron Porphyrins, Angew. Chem., Int. Ed., 2019, 58(14), 4504–4509 CrossRef CAS PubMed.
- W. Nie, D. E. Tarnopol and C. C. L. McCrory, Enhancing a Molecular Electrocatalyst's Activity for CO2 Reduction by Simultaneously Modulating Three Substituent Effects, J. Am. Chem. Soc., 2021, 143(10), 3764–3778 CrossRef CAS PubMed.
- S. Dey, F. Masero, E. Brack, M. Fontecave and V. Mougel, Electrocatalytic metal hydride generation using CPET mediators, Nature, 2022, 607, 499–506 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ESI includes synthetic summaries, NMR and UV-vis characterization, electrochemistry, and description of experimental details and methods, as well as a separate file containing computational coordinates. CCDC 1984949, 1984950, 2150929, 2150930, 2154597, 2154598, and 2154599. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc03258a |
| This journal is © The Royal Society of Chemistry 2022 |