
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGreen-/NIR-light-controlled rapid photochromism featuring reversible thermally activated delayed fluorescence and photoelectronic switching†
Ziyong
Li
 ,
Ji-Rui
Zhang
,
Xu-Ke
Tian
,
Shuren
Yang
,
Si
Chen
,
Hui
Zhou
and
Xiao-Gang
Yang
*
,
Ji-Rui
Zhang
,
Xu-Ke
Tian
,
Shuren
Yang
,
Si
Chen
,
Hui
Zhou
and
Xiao-Gang
Yang
*
College of Chemistry and Chemical Engineering, College of Food and Drug, Luoyang Normal University, Luoyang 471934, P. R. China. E-mail: yxg2233@126.com
First published on 21st July 2022
Abstract
Fluorescent dithienylethene-based photochromic materials have been attracting considerable attention owing to their wide applications in biological and materials sciences. However, the limitations of detrimental UV irradiation for photocyclization, short emission lifetime, and inefficient photoresponsive speed still need to be addressed. Herein, a novel dithienylethene photochromic molecule, BFBDTE, has been prepared by the incorporation of a difluoroboron β-diketonate (BF2bdk) unit. The strong electron acceptor BF2bdk not only reduces the energy gap of the open isomer, ensuring visible light-controlled fluorescence switching, but also promotes intersystem crossing for the generation of thermally activated delayed fluorescence (TADF). Upon alternating irradiation with green and NIR light, BFBDTE presents a rare example of photochromism, fluorescence and TADF switching in various polar solvents and a poly(methyl methacrylate) (PMMA) film. Meanwhile, it shows rapid and well repeatable cyclization (12 s) and cycloreversion reactions (20 s) in PMMA, accompanied by fast TADF switching within 11 s. Furthermore, photo-electrochemical measurements reveal a remarkable on-off photoelectronic response (photocurrent density ratio: Ilight/Idark = 684) between the open- and closed-form of BFBDTE. These remarkable merits make BFBDTE promising for photoswitchable molecular devices, optical memory storage systems, NIR detectors, and photoelectric switching.
Introduction
Photochromic fluorescence switches, whose emission can be reversibly switched via light irradiation, have recently aroused tremendous attention due to their potential in optoelectronic materials, bioimaging and super-resolution microscopy.1–6 As a core structural motif of a photochromic switch, the reversible cyclization/cycloreversion of dithienylethenes (DTEs) upon light irradiation has continued to attract substantial scientific interest.7 Recently, the combination of DTE photoswitching and a fluorescent unit has become a kind of appealing fluorescence switch arising from their thermo-stability, robust fatigue resistance and rapid response.8–11 A general scheme for designing such switches is a dyad system comprising one fluorophore and DTE unit in the same molecule, facilitating fluorescence quenching through intramolecular energy transfer (ET).12 As depicted in Scheme 1a, the ring-open state of DTE with high S1 energy restrains the energy transfer from the excited fluorophore to the open isomer, retaining the fluorescence emission of the fluorophore. Upon irradiation with UV light, the ring-closed state with low S1 energy enables energy transfer, and then quenches the fluorescence emission. Although great progress has been made in fluorescence switches in recent years,13–18 they still suffer from several drawbacks, e.g., the necessity of UV light to switch the fluorescence emission, inferior photoresponsive speed (several minutes), and short fluorescence lifetimes. Considering the high phototoxicity and low penetrability of UV light, numerous efforts have been devoted to fabricating visible light-activated fluorescence switches, of which simply extending the π-conjugation length by connecting aromatic fluorescent dyes on the peripheral thienyls of DTE is most appealing.19–25 However, the slow photoresponsive speed and short fluorescence lifetime with a time scale of nanoseconds (τ < 10 ns) still limit their further applications.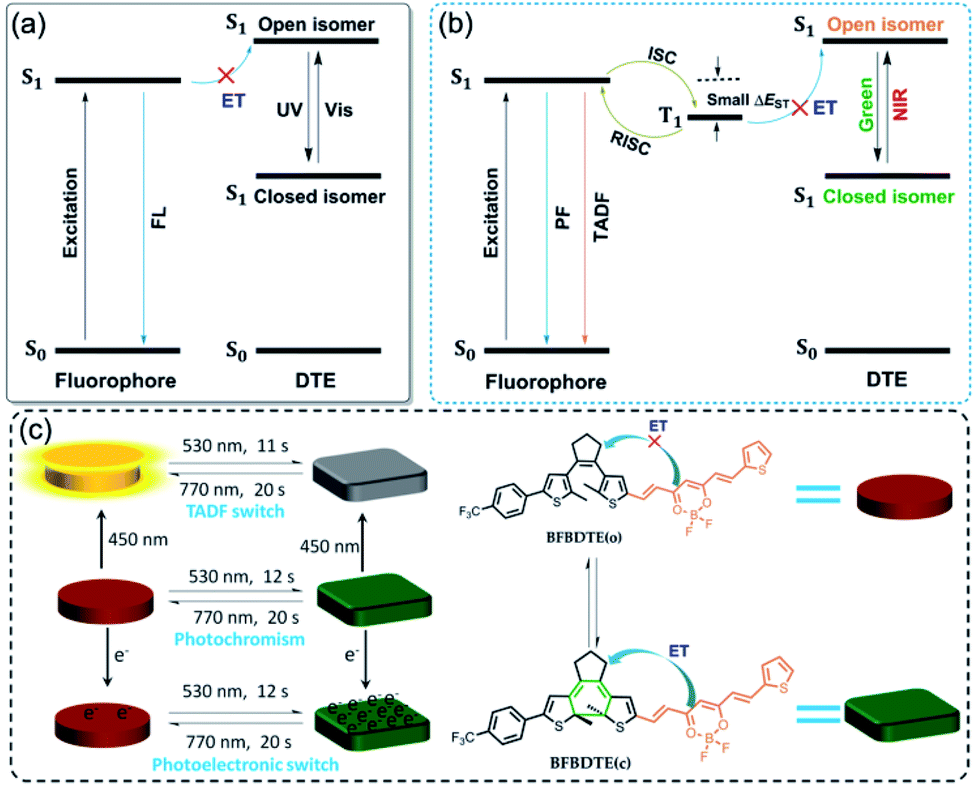 | ||
| Scheme 1 (a) Schematic illustrations of the energy transfer process upon UV and visible light irradiation in traditional photochromic fluorescence switching. (b) Proposed schematic illustrations of photochromic TADF switching. (c) Green-/NIR-light-controlled rapid photochromism featuring reversible TADF and photoelectronic switching. FL: fluorescence, PF: prompt fluorescence, TADF: thermally activated delayed fluorescence, S0: ground state, S1: excited singlet state, T1: excited triplet state, ET: energy transfer, ISC: intersystem crossing, RISC: reverse intersystem crossing, ΔEST: singlet–triplet energy gap. | ||
Fortunately, a novel class of fluorophores with long fluorescence lifetimes, i.e., thermally activated delayed fluorescence (TADF),26–28 may provide a possible solution to the above issues. As illustrated in Scheme 1b, the TADF process can undergo efficient ISC and then reverse intersystem crossing (RISC) for 100% internal quantum efficiency from T1 to S1 due to a remarkably small energy gap (ΔEST < 0.2 eV) between the lowest singlet excited states and the lowest triplet excited states, which makes them promising candidates for various applications in organic light-emitting diodes (OLEDs), triplet–triplet annihilation upconversion (TTA-UC), time-resolved fluorescence imaging (TRFI), photodynamic therapy (PDT) and organic photocatalytic synthesis.29–31 Inspired by the desired fluorescence efficiencies and lifetimes of TADF, we speculate that the incorporation of the TADF process into visible light-driven DTE systems could obviously improve the photoswitching efficiencies applied in photoswitchable molecular devices, optical memory storage systems and bioimaging with deeper light penetration. So far, only one example of TADF-based photochromic switching has been reported.32 Therefore, developing efficient visible light-controlled DTE systems with multi-mode TADF characteristics is highly desirable for photoelectric applications, but remains a severe challenge.
To prove our hypothesis, a novel DTE molecular switch modified by a difluoroboron β-diketonate (BF2bdk) moiety has been successfully prepared (Scheme 1c), in which the incorporation of active nonbonding p electrons on the BF2bdk group promotes the spin-orbital coupling (SOC) between the excited singlet and triplet states in favor of enhanced intersystem crossing (ISC).33 Moreover, the strong electron acceptor BF2bdk can bring about intramolecular charge transfer (ICT), which endows a typical TADF feature with small ΔEST for efficient ISC and RISC processes.34 On the other hand, merging BF2bdk and DTE can not only extend the π-conjugation, but also reduce the HOMO–LUMO energy gap of the open isomer, which will ensure excellent visible light-controlled fluorescence switching with high photoresponsive speed.35 As expected, the obtained fluorescent DTE (BFBDTE) shows rapid and long-term reversible tri-channels of photochromism, fluorescence and TADF switching in toluene and a poly(methyl methacrylate) (PMMA) film. It represents the first example of green-/NIR-light-controlled photoswitching with the combination of photochromism and TADF.
Results and discussion
As depicted in Scheme S1,†BFBDTE was prepared by the Knoevenagel condensation reaction between intermediates 4 and 5 under the catalysis of n-butylamine in anhydrous toluene in a yield of 69%. Its chemical structure was characterized by NMR spectroscopy, high-resolution mass spectrometry (Fig. S1–S3†) and single-crystal X-ray diffraction. From its 1H NMR spectrum, the coupling constants of the vinyl double bonds were 15.0 or 15.2 Hz, respectively, revealing the usual trans-configuration for both ethylene bonds in BFBDTE. An orange-red crystal was isolated by the slow volatilization of its chloroform solution. Single-crystal X-ray diffraction analysis reveals that BFBDTE crystallizes in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group (Table S1†). As depicted in Fig. 1a, BFBDTE presents an antiparallel conformation where the distance between the two reactive carbon atoms was as short as 3.57 Å (<4.2 Å), indicating that the photocyclization was easy to access.36 The BFBDTE molecules are linked together by C–H⋯F hydrogen bonding interactions giving rise to a 1D chain running along the a direction (Fig. 1b, S4 and S5†). Herein, the hydrogen bonding acceptors are all from the F atoms of difluoroboron. As shown in Fig. S6,† each chain is further extended to a 2D layer and then a 3D structure through C–H⋯F halogen bonding interactions with –CF3 as a hydrogen bonding acceptor. Viewed along the a direction, the packing structure exhibits an alternant arrangement of BF2bdk and DTE at the molecular level.
space group (Table S1†). As depicted in Fig. 1a, BFBDTE presents an antiparallel conformation where the distance between the two reactive carbon atoms was as short as 3.57 Å (<4.2 Å), indicating that the photocyclization was easy to access.36 The BFBDTE molecules are linked together by C–H⋯F hydrogen bonding interactions giving rise to a 1D chain running along the a direction (Fig. 1b, S4 and S5†). Herein, the hydrogen bonding acceptors are all from the F atoms of difluoroboron. As shown in Fig. S6,† each chain is further extended to a 2D layer and then a 3D structure through C–H⋯F halogen bonding interactions with –CF3 as a hydrogen bonding acceptor. Viewed along the a direction, the packing structure exhibits an alternant arrangement of BF2bdk and DTE at the molecular level.
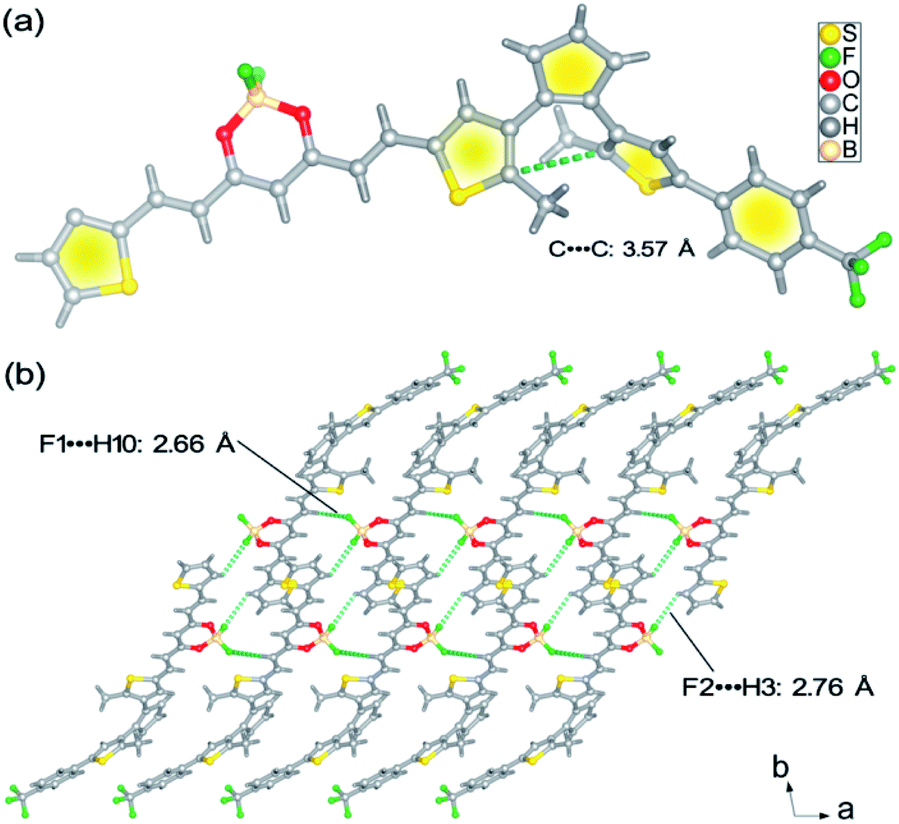 | ||
| Fig. 1 (a) Crystal structure of the BFBDTE molecule. (b) View of the 1D chain structure of BFBDTE extended by C–H⋯F hydrogen bonding. | ||
The photochromism of BFBDTE was then investigated in different polar solvents. As illustrated in Fig. 2a, it was clear that two absorption bands of the open isomer BFBDTE(o) centered at 478 nm (ε = 3.12 × 104 M−1 cm−1) and 509 nm (ε = 3.68 × 104 M−1 cm−1) were observed in toluene, which are derived from the intramolecular charge transfer (ICT) transition.37 Upon green-light irradiation (530 nm), a near-infrared absorption band centered at 727 nm (ε = 1.88 × 104 M−1 cm−1) gradually emerged along with the color changing from orange-yellow to green (Fig. 2a, insets), indicating the formation of the ring-closed isomer BFBDTE(c) (Scheme 1c). It is worth noting that the achievement of the photostationary state only takes 13 s of green-light irradiation. The response rate for BFBDTE is faster than that of the reported Pt-DTE (120 s)38a and POM-DTE complexes (65 min)38b detected in organic solvents. Moreover, an isosbestic point at 546 nm was observed, clearly indicating the photochromic transformation between BFBDTE(o) and BFBDTE(c). Irradiation with NIR light (λ = 770 nm, 80 s) triggered the cycloreversion reaction to regenerate the initial orange-yellow isomer (Fig. 2b). Further fatigue resistance results indicated good reversibility (without obvious degradation for ten cycles) between the open and closed forms under green/NIR light irradiation (Fig. 2c), which might be due to blocking of the pathway to photo-byproducts under the lower energy of light irradiation.39 As expected, a large cyclization quantum yield (φo–c) for BFBDTE in toluene was recorded as 0.56 and its cycloreversion quantum yield (φc–o) was 0.031. Subsequently, we further evaluated the photochromism of BFBDTE in CHCl3 and DMSO with larger polarity. Similar photochromic behavior was observed when these three solutions were exposed to the same light conditions (Fig. S7–S10†). In contrast, the absorption maximum of the open and closed isomers showed a distinct bathochromic shift in CHCl3 and DMSO (e.g.BFBDTE(o): λ = 523 nm in DMSO, λ = 516 nm in CHCl3; BFBDTE(c): λ = 758 nm in DMSO, λ = 756 nm in CHCl3) (Fig. 2d and Table S2†), indicating a unique solvent-dependent photochromic property. However, it took a longer time to reach the photostationary state in these two solvents, i.e., 28 s in CHCl3 and 70 s in DMSO, which implied that the higher the solvent polarity, the slower the cyclization. As expected, the corresponding cyclization quantum yields decreased as the solvent polarity increased (Table S2†). Therefore, BFBDTE presents exceptional photochromism triggered by green light in different solvents. Besides, the photoisomerization of BFBDTE was further analyzed via the 1H NMR spectral variations in toluene-d8 (Fig. S11†). The photocyclization conversion ratio in the photostationary state was determined to be 66.7% according to the 1H NMR results.
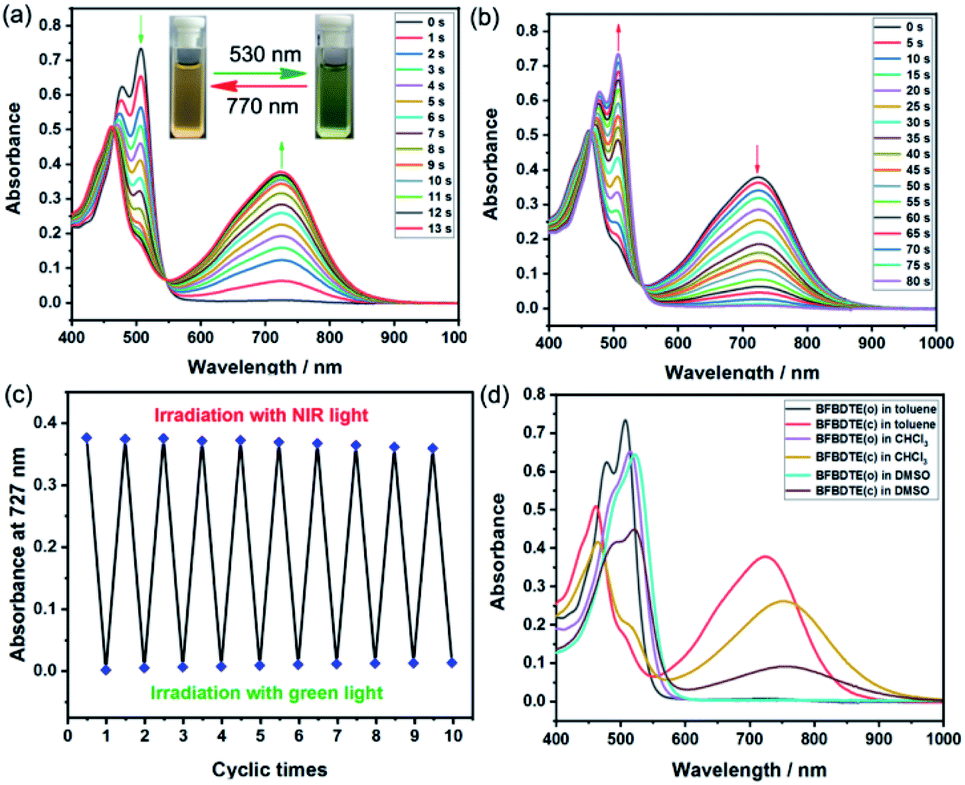 | ||
| Fig. 2 The absorption spectra changes of BFBDTE in toluene (2.0 × 10−5 mol L−1) upon alternating irradiation with green light at 530 nm (a) and NIR light at 770 nm. (b) The insets show the corresponding color changes upon photoirradiation. (c) The fatigue resistance of BFBDTE in toluene for ten cycles. (d) The absorption spectra of BFBDTE before and after irradiation with green light in various solvents (2.0 × 10−5 mol L−1). | ||
Given the excellent photophysical properties of BF2bdk-based complexes, the following exploration focused on the investigation of the photoluminescence (PL) performance of BFBDTE in solution under both prompt and delayed modes. Excited at 450 nm, BFBDTE exhibited bright yellow emission centered at ca. 556 nm in toluene. Alternatively, a new emission peak located at 572 nm was observed in the delayed mode in toluene (λex = 450 nm) (Fig. 3a), while very weak delayed fluorescence was detected in CHCl3 and DMSO (Fig. S12†). The delayed emission intensity increased with rising of the temperature from 77 to 300 K, indicating the TADF character of BFBDTE in toluene (Fig. 3b). Measured at 77 K, the prompt and delayed emission peaks afforded a small ΔEST of 0.09 eV between the excited S1 (559 nm, 2.22 eV) and T1 (582 nm, 2.13 eV) states. Upon irradiation with green light, the emission peak at 556 nm was gradually quenched (Fig. 3c) along with the fading of the yellow fluorescence in toluene (Fig. 3c insets), probably owing to efficient energy transfer between the excited BF2bdk and closed DTE core.40 After reaching the photostationary state in 16 s, the fluorescence intensity of BFBDTE(o) was quenched by ca. 88%. Its fluorescence returned to the original intensity after irradiation with 770 nm NIR light. Besides, this fluorescence switching process could be reversibly conducted for ten cycles with negligible degradation (Fig. S13†). Meanwhile, under the same green light irradiation, the time-resolved decay curves revealed that the prompt fluorescence lifetimes decreased from 1.73 to 1.04 ns at room temperature (Fig. 3d). Simultaneously, the TADF switching performance for BFBDTE was also observed in toluene. As depicted in Fig. 3e, the delayed fluorescence intensity at 572 nm was gradually decreased when exposed to green light irradiation, which in turn returned to the original intensity after irradiation with NIR light. This was accompanied by changes in the lifetimes of long-lived species from 28.57 to 13.03 μs at room temperature (Fig. 3f). Compared with that in toluene, the prompt emission peak of BFBDTE(o) displayed a significant red-shift in CHCl3 (577 nm) and DMSO (564 nm) (Fig. S14†). Moreover, similar prompt fluorescence switch behavior was observed in CHCl3 and DMSO under the same irradiation (Fig. S15 and S16, and Table S3†). To date, fluorescent photochromism materials controlled by visible light are highly desirable in both biological and materials sciences. As far as we know, the reported BFBDTE in our work is the first example of green-/NIR-light-controlled switching by the combination of photochromism, fluorescence and TADF with fast photoresponsive speed.
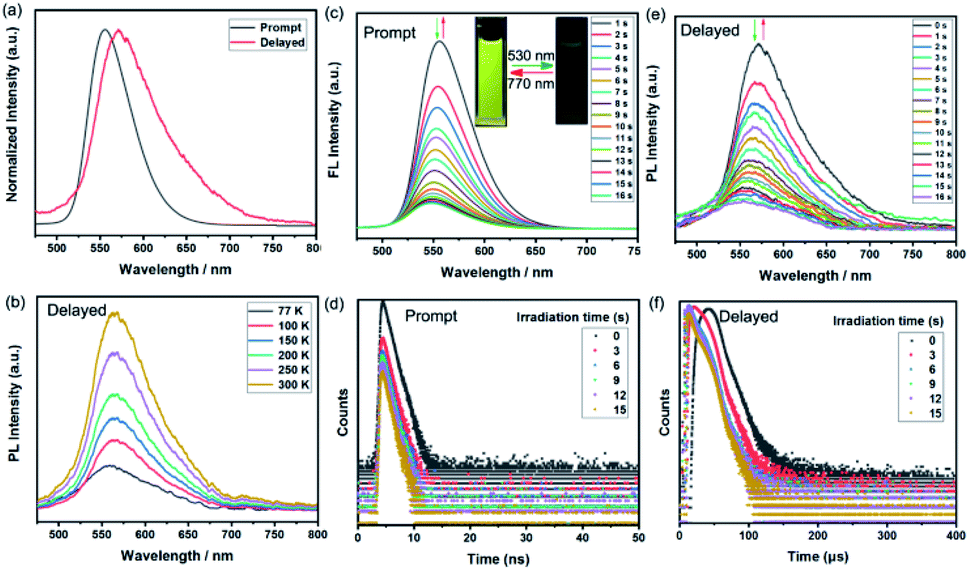 | ||
| Fig. 3 (a) PL spectra of BFBDTE in toluene measured under prompt and delayed mode at room temperature (λex = 450 nm). (b) Delayed fluorescence spectra of BFBDTE in toluene measured in the temperature range of 77 to 300 K. Prompt fluorescence spectra (c) and decay curves (d) of BFBDTE in toluene upon irradiation with green light (530 nm). The insets show the reversible fluorescence switching upon alternating irradiation with green and NIR (770 nm) light. Delayed fluorescence spectra (e) and decay curves (f) of BFBDTE in toluene upon irradiation with green light (530 nm). | ||
A possible photophysical process was speculated based on theoretical calculations for both BFBDTE(o) and BFBDTE(c). In the cycloreversion state (Fig. S17†), it shows complete separation of molecular orbitals owing to the twisty conformation of BFBDTE(o). The highest occupied molecular orbital (HOMO) mainly appears on the benzene ring and adjacent thiophene ring. The lowest unoccupied molecular orbital (LUMO) is exclusively located on the BF2bdk core, double bonds and thiophene rings. Therefore, the photophysical process for BFBDTE(o) can be assigned to charge transfer from the DTE moiety to the BF2bdk-based fluorophore. By contrast, the extended π-conjugated system of BFBDTE(c) possesses large overlap between the HOMO and LUMO (Fig. S18†). Delocalized molecular orbitals distribute on the entire cyclized molecule with a planar conformation. The extended π-conjugated system prevents the charge transfer process, leading to the quenching of the PL. Energy level (Fig. S19†) analysis shows a band gap of 2.28 eV (543 nm) for the first excited singlet state (S1) stemmed from excitation from the HOMO to the LUMO, corresponding to the prompt emission peak at 556 nm. The energy gap (ΔEST) between S1 and T1 was calculated as 0.14 eV, which is very close to the experimental one (0.09 eV). Combining the experiments and theoretical calculations, it was revealed that the presence of BF2bdk can largely reduce the energy level/gap and ΔEST, which enables the efficient energy transfer of S1 → T1, and then, the small energy gap between BF2bdk and DTE promotes energy transfer from T1 of BF2bdk to S1 of DTE, featuring rapid photochromism performance.
Encouraged by the excellent fluorescent photochromic performance of BFBDTE in toluene, its photochromism and dual-mode fluorescence switching performance were further evaluated in a poly(methyl methacrylate) film (PMMA). As shown in Fig. 4a, when exposed to green light irradiation (530 nm), a novel NIR absorption band at 744 nm gradually appeared, accompanied by the film color changing from orange-red to green (Fig. 4a, insets). During this process, it took only 12 s to reach the photostationary state and 20 s for the cycloreversion reaction to return to the initial orange-red film by irradiation with NIR (770 ns). The photoresponsive speed is faster than that in polar solvents mentioned in the preceding text. In addition, good fatigue resistance was also detected under green/NIR light irradiation (Fig. S20†). As expected, BFBDTE presents both prompt and delayed photoluminescence in the PMMA film under excitation at 450 nm (Fig. 4b). The prompt and delayed fluorescence lifetime was estimated as 1.85 ns and 48.75 μs, respectively (Fig. S21†). As illustrated in Fig. 4c, under constant green light irradiation for 11 s, the prompt yellow emission located at 569 nm sharply decreased along with the yellow fluorescence fading in the PMMA film. Irradiation of NIR light, in turn, reverted it back to the initial intensity. Furthermore, the BFBDTE PMMA film also displays fast delayed switching behavior (11 s) under the same irradiation conditions (Fig. 4d). Therefore, the NIR switching of the BFBDTE PMMA film with a fast response rate could serve as promising for infrared wavelength detectors applied in the fields of optical communications, anti-counterfeiting, bioimaging and optical memory storage systems.
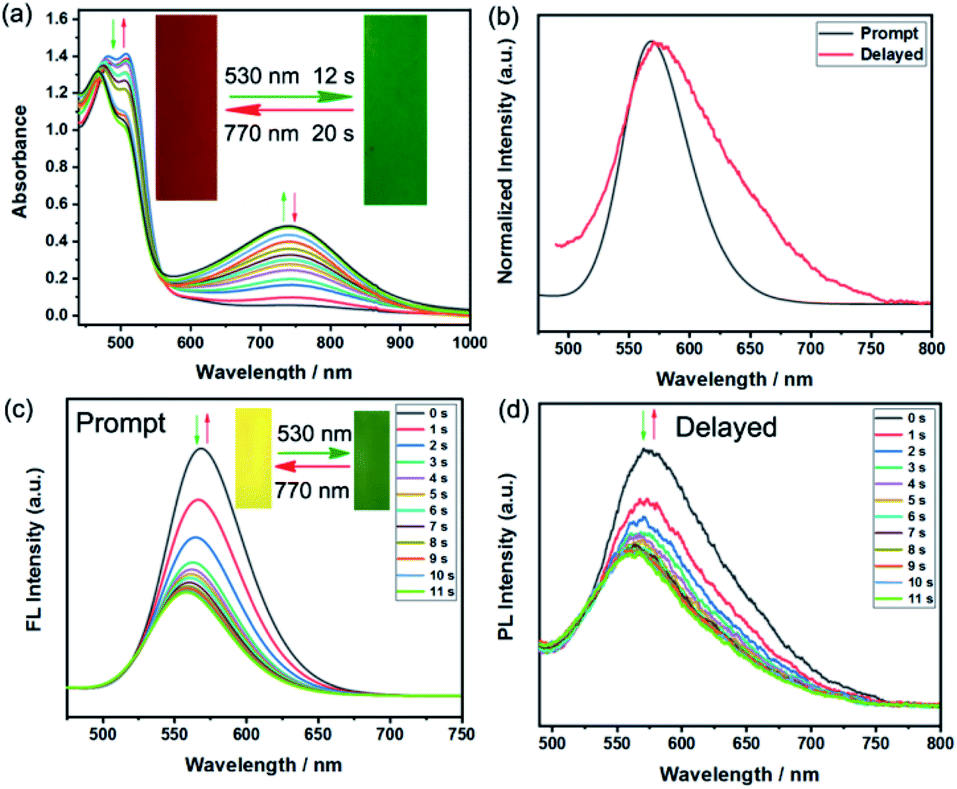 | ||
| Fig. 4 (a) The absorption spectra of BFBDTE in the PMMA film upon alternating irradiation with green and NIR light. Inset: corresponding color changes upon light irradiation. (b) Prompt and delayed fluorescence spectra of BFBDTE in the PMMA film. Prompt (c) and delayed (d) fluorescence spectra of BFBDTE doped in the PMMA film upon alternating irradiation with green and NIR light (λex = 450 nm). Insets: corresponding fluorescence changes upon light irradiation. | ||
By considering the different electron structures of the BFBDTE(o) and BFBDTE(c) isomers, photo-electrochemical measurements were carried out in 0.5 M Na2SO4 aqueous solution with a standard three-electrode system. The working electrodes were prepared by dropping chloroform solutions of BFBDTE(o) and BFBDTE(c) onto indium tin oxide (ITO) electrodes. Ag/AgCl and platinum wire were used as the reference and counter electrodes, respectively. The cyclic voltammetry (CV) curve (Fig. 5a) shows that BFBDTE(o) has little electrochemical activity, whereas BFBDTE(c) has large electrochemical activity in the negative voltage region. A small photocurrent density of only 12 nA cm−2 is generated for BFBDTE(o) under periodical on/off light irradiation without bias potential (Fig. 5b). In detail, it exhibits inverse photoelectronic response behavior: under the light-on state, the photocurrent decreases rapidly to about 2.8 nA cm−2 and then increases slowly to 8.3 nA cm−2, while it increases rapidly and then decreases slowly when the light is turned off. By sharp contrast, BFBDTE(c) generates a small photocurrent density of 46 nA cm−2 under dark conditions, and an instantaneous increase of the photocurrent density up to 958 nA cm−2 is observed when the light source is turned on (Fig. 5c). Thus, under light irradiation, the photocurrent density value of BFBDTE(c) is 684 times higher than that of BFBDTE(o). Electrochemical impedance spectroscopy (EIS) Nyquist plots (Fig. 5d) further suggest that the charge transfer resistance can be reduced under illumination for both BFBDTE(o) and BFBDTE(c), revealing that the additional light radiation can promote the charge transfer. By contrast, BFBDTE(c) exhibits higher charge transfer performance than that of BFBDTE(o).
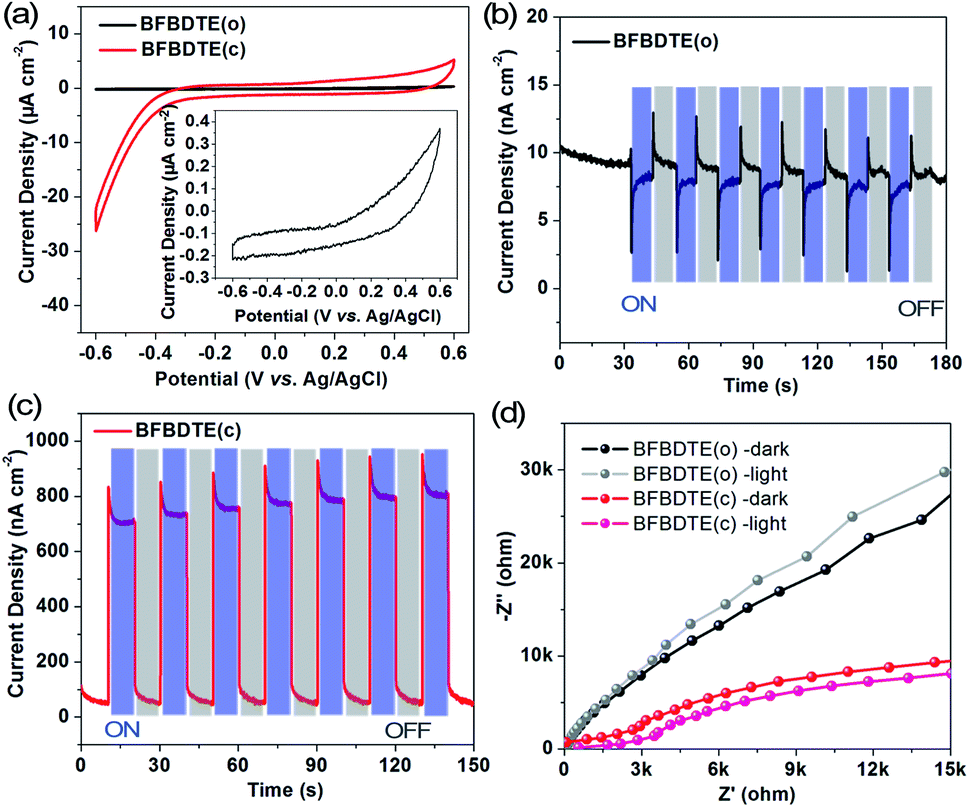 | ||
| Fig. 5 (a) Cyclic voltammetry curves of BFBDTE(o) and BFBDTE(c) measured in 0.5 M Na2SO4 aqueous solution. Transient current density–time curves of BFBDTE(o) (b) and BFBDTE(c) (c) without bias potentials. (d) Electrochemical impedance spectroscopy (EIS) Nyquist plots of BFBDTE(o) and BFBDTE(c) measured at 0 V bias potentials. | ||
The above results imply that the inefficient photoelectronic response performance of BFBDTE(o) can be assigned to the isolated π-conjugation localized in the two separated thiophene rings, which hinders the transport of carriers. In contrast, the extended π-conjugated system in BFBDTE(c) showing delocalized π-electrons can provide a charge transport channel, endowing highly efficient photoelectronic response performance. Scanning electron microscopy (SEM) images show a micrometer scale spherical morphology for both BFBDTE(o) and BFBDTE(c). Dense aggregation microspheres with a size of about 3 μm are observed for BFBDTE(c), whereas BFBDTE(o) exhibits a relatively sparse arrangement of microspheres with a size of about 10 μm (Fig. S22†). The reason for this morphology and arrangement diversity can be attributed to the smaller molecular size of BFBDTE(c) than BFBDTE(o) obtained from theoretical calculations (Fig. S23 and 24†). These results further demonstrate that the planar conformation of BFBDTE(c) with delocalized π-electrons tends to stack densely, resulting in high photoelectronic performance, which makes BFBDTE perfect for molecular switching controlled by NIR light.
Conclusions
In summary, a new photochromic fluorescence switching molecule, BFBDTE, was designed and synthesized by the conjugation of a difluoroboron β-diketonate (BF2bdk) fluorescent unit and dithienylethene. Reversible and repeatable cyclization–cycloreversion processes can be controlled by green- and NIR-light, accompanied by a remarkable color change as well as fluorescence and TADF switching. This reversible transformation tends to the photostationary state within only 20 s in a PMMA film. We can speculate that two reasons are responsible for the above lower energy induced rapid fluorescence photochromic performance. First, the highly electrophilic boron center in BF2bdk helps in reducing the HOMO and LUMO energy levels, as well as the energy gap, enabling the light absorption to red shift to the visible light region in the open-ring isomer. Additionally, the active nonbonding p electrons of the BF2bdk unit favor strong spin-orbital coupling and intersystem crossing, featuring typical TADF character with small ΔEST. Second, the extended π-conjugated system in the close-ring isomer with a planar configuration further broadens the absorption band to the NIR region. The presence of electron deficient BF2bdk, which possesses more ability to promote the homolytic bond, means that rapid cycloreversion can be realized by a lower energy of light irradiation. The delocalized π-electrons around the extended π-conjugated system in the closed-ring isomer promote charge mobility, exhibiting high electrochemical activity and photoelectronic response performance. In contrast, the open-ring isomer impedes the carrier transport, leading to turn-off of the photoelectronic response. Therefore, this study provides a new perspective into the future design of visible light-/NIR-controlled photochromic materials showing reversible TADF and photoelectronic switching.Data availability
All of data associated with this article are presented in the manuscript and ESI.†Author contributions
Z. Li, S. Yang, S. Chen and H. Zhou synthesized the complex. J. Zhang and X. Tian determined the single-crystal structure. S. Yang, S. Chen, H. Zhou, J. Zhang and X. Tian performed the characterization of the complex. Z. Li and X.-G. Yang designed and directed the studies and wrote the manuscript. All of the authors discussed the results and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21971100), the Natural Science Foundation of Henan Province (No. 222300420501), the Science and Technology Project of Henan Province (No. 212102210549), the Key Scientific Research Project of Higher Education of Henan Province (No. 22A430007), and the Innovation and Entrepreneurship Training Program for College students in China (No. 202110482010).Notes and references
- (a) K. Uno, A. Aktalay, M. L. Bossi, M. Iriec, V. N. Belov and S. W. Hell, Proc. Natl. Acad. Sci. U.S.A., 2021, 118, e2100165118 CrossRef CAS PubMed; (b) H.-B. Cheng, S. Zhang, E. Bai, X. Cao, J. Wang, J. Qi, J. Liu, J. Zhao, L. Zhang and J. Yoon, Adv. Mater., 2022, 34, 2108289 CrossRef CAS PubMed.
- N. M.-W. Wu, M. Ng and V. W.-W. Yam, Nat. Commun., 2022, 13, 33 CrossRef CAS PubMed.
- L. Kortekaas and W. R. Browne, Chem. Soc. Rev., 2019, 48, 3406–3424 RSC.
- D. Kim, K. Jeong, J. E. Kwon, H. Park, S. Lee, S. Kim and S. Y. Park, Nat. Commun., 2019, 10, 3089 CrossRef PubMed.
- R. Usui, K. Yamamoto, H. Okajima, K. Mutoh, A. Sakamoto, J. Abe and Y. Kobayashi, J. Am. Chem. Soc., 2020, 142, 10132–10142 CrossRef CAS PubMed.
- H. Yang, M. Li, C. Li, Q. Luo, M.-Q. Zhu, H. Tian and W.-H. Zhu, Angew. Chem., Int. Ed., 2020, 59, 8560–8570 CrossRef CAS PubMed.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- Y. Mu, Y. Liu, H. Tian, D. Ou, L. Gong, J. Zhao, Y. Zhang, Y. Huo, Z. Yang and Z. Chi, Angew. Chem., Int. Ed., 2021, 60, 6367–6371 CrossRef CAS PubMed.
- R. Haldar, L. Heinke and C. Wöll, Adv. Mater., 2019, 1905227 Search PubMed.
- E. C. Harvey, B. L. Feringa, J. G. Vos, W. R. Browne and M. T. Pryce, Coord. Chem. Rev., 2015, 282–283, 77–86 CrossRef CAS.
- (a) Y. Qin, Y.-T. Wang, H.-B. Yang and W. Zhu, Chem. Synth., 2021, 1, 2 Search PubMed; (b) G. Naren, W. Larsson, C. Benitez-Martin, S. Li, E. P. Inestrosa, B. Albinsson and J. Andreasson, Chem. Sci., 2021, 12, 7073–7078 RSC.
- K. E. Sapsford, L. Berti and I. L. Medintz, Angew. Chem., Int. Ed., 2006, 45, 4562–4588 CrossRef CAS PubMed.
- T. Fukaminato, T. Doi, N. Tamaoki, K. Okuno, Y. Ishibashi, H. Miyasaka and M. Irie, J. Am. Chem. Soc., 2011, 133, 4984–4990 CrossRef CAS PubMed.
- K. Uno, H. Niikura, M. Morimoto, Y. Ishibashi, H. Miyasaka and M. Irie, J. Am. Chem. Soc., 2011, 133, 13558–13564 CrossRef CAS PubMed.
- A. Nagai, R. Nishimura, Y. Hattori, E. Hatano, A. Fujimoto, M. Morimoto, N. Yasuda, K. Kamada, H. Sotome, H. Miyasaka, S. Yokojima, S. Nakamura and K. Uchida, Chem. Sci., 2021, 12, 11585–11592 RSC.
- C. Li, H. Yan, L.-X. Zhao, G.-F. Zhang, Z. Hu, Z.-L. Huang and M.-Q. Zhu, Nat. Commun., 2014, 5, 5709 CrossRef CAS PubMed.
- J.-X. Liu, B. Xin, C. Li, N.-H. Xie, W.-L. Gong, Z.-L. Huang and M.-Q. Zhu, ACS Appl. Mater. Interfaces, 2017, 9, 10338–10343 CrossRef CAS PubMed.
- A. Nagai, R. Nishimura, Y. Hattori, E. Hatano, A. Fujimoto, M. Morimoto, N. Yasuda, K. Kamada, H. Sotome, H. Miyasaka, S. Yokojima, S. Nakamura and K. Uchida, Chem. Sci., 2021, 12, 11585–11592 RSC.
- C. Li, K. Xiong, Y. Chen, C. Fan, Y.-L. Wang, H. Ye and M.-Q. Zhu, ACS Appl. Mater. Interfaces, 2020, 12, 27651–27662 CrossRef CAS PubMed.
- Z. Li, Y. Pei, Y. Wang, Z. Lu, Y. Dai, Y. Duan, Y. Ma and H. Guo, J. Org. Chem., 2019, 84, 13364–13373 CrossRef CAS PubMed.
- Z. Li, Y. Dai, Z. Lu, Y. Pei, H. Chen, L. Zhang, Y. Duan and H. Guo, Chem. Commun., 2019, 55, 13430–13433 RSC.
- T. Fukaminato, T. Hirose, T. Doi, M. Hazama, K. Matsuda and M. Irie, J. Am. Chem. Soc., 2014, 136, 17145–17154 CrossRef CAS PubMed.
- W. Tan, J. Zhou, F. Li, T. Yi and H. Tian, Chem. - Asian J., 2011, 6, 1263–1268 CrossRef CAS PubMed.
- T. Sumi, T. Kaburagi, M. Morimoto, K. Une, H. Sotome, S. Ito, H. Miyasaka and M. Irie, Org. Lett., 2015, 17, 4802–4805 CrossRef CAS PubMed.
- B. Seefeldt, K. Altenhöner, O. Tosic, T. Geisler, M. Sauer and J. Mattay, Photochem. Photobiol. Sci., 2011, 10, 1488–1495 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- V.-N. Nguyen, A. Kumar, M. H. Lee and J. Yoon, Coord. Chem. Rev., 2020, 425, 213545 CrossRef CAS.
- W. Chen and F. Song, Chin. Chem. Lett., 2019, 30, 1717–1730 CrossRef CAS.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- C. Wang and Y. Liu, Mater. Today Chem., 2022, 25, 100954 CrossRef CAS.
- Y. Xie, Y. Ge, Q. Peng, C. Li, Q. Li and Z. Li, Adv. Mater., 2017, 29, 1606829 CrossRef PubMed.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707–3714 CrossRef CAS PubMed.
- (a) C.-T. Poon, W. H. Lam, H.-L. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2010, 132, 13992–13993 CrossRef CAS PubMed; (b) C. C.-T. Poon, W. H. Lam and V. W.-W. Yam, Chem. - Eur. J., 2013, 19, 3467–3476 CrossRef CAS PubMed; (c) C.-L. Wong, C.-T. Poon and V. W.-W. Yam, Chem. - Eur. J., 2016, 22, 12931–12940 CrossRef CAS PubMed.
- (a) M. Morimoto, H. Miyasaka, M. Yamashita and M. Irie, J. Am. Chem. Soc., 2009, 131, 9823–9835 CrossRef CAS PubMed; (b) V. Ramamurthy and K. Venkatesan, Chem. Rev., 1987, 87, 433–481 CrossRef CAS.
- X. Ma, W. Chi, X. Han, C. Wang, S. H. Liu, X. Liu and J. Yin, Chin. Chem. Lett., 2021, 32, 1790–1794 CrossRef CAS.
- (a) Y. Qin, L.-J. Chen, Y. Zhang, Y.-X. Hu, W.-L. Jiang, G.-Q. Yin, H. Tan, X. Li, L. Xu and H.-B. Yang, Chem. Commun., 2019, 55, 11119–11122 RSC; (b) J. Xu, H. Volfova, R. J. Mulder, L. Goerigk, G. Bryant, E. Riedle and C. Ritchie, J. Am. Chem. Soc., 2018, 140, 10482–10487 CrossRef CAS PubMed.
- M. Herder, B. M. Schmidt, L. Grubert, M. Patzel, J. Schwarz and S. Hecht, J. Am. Chem. Soc., 2015, 137, 2738–2747 CrossRef CAS PubMed.
- T. Kawai, T. Sasaki and M. Irie, Chem. Commun., 2001, 711–712 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2171748. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc02662g |
| This journal is © The Royal Society of Chemistry 2022 |