
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-electron bonds in copper–aluminum and copper–gallium complexes†
Brendan J.
Graziano
 a,
Thais R.
Scott
b,
Matthew V.
Vollmer
a,
Michael J.
Dorantes
a,
Victor G.
Young
Jr
a,
Eckhard
Bill
*c,
Laura
Gagliardi
*b and
Connie C.
Lu
*ad
a,
Thais R.
Scott
b,
Matthew V.
Vollmer
a,
Michael J.
Dorantes
a,
Victor G.
Young
Jr
a,
Eckhard
Bill
*c,
Laura
Gagliardi
*b and
Connie C.
Lu
*ad
aDepartment of Chemistry, University of Minnesota-Twin Cities, 207 Pleasant Street SE, Minneapolis, Minnesota 55455, USA. E-mail: clu@uni-bonn.de
bDepartment of Chemistry, The University of Chicago, Searle Chemistry Laboratory, 5735 South Ellis Avenue, Chicago, Illinois 60637, USA. E-mail: lgagliardi@uchicago.edu
cMax Planck Institut für Chemische Energiekonversion, Stiftstraβe 34–36, 45470, Mülheim an der Ruhr, Germany. E-mail: eckhard.bill@cec.mpg.de
dInstitut für Anorganische Chemie, Rheinische-Friedrich-Wilhelms Universität Bonn, Gerhard-Domagk-Straße 1, 53121, Bonn, Germany
First published on 5th May 2022
Abstract
Odd-electron bonds have unique electronic structures and are often encountered as transiently stable, homonuclear species. In this study, a pair of copper complexes supported by Group 13 metalloligands, M[N((o-C6H4)NCH2PiPr2)3] (M = Al or Ga), featuring two-center/one-electron (2c/1e) σ-bonds were synthesized by one-electron reduction of the corresponding Cu(I) ⇢ M(III) counterparts. The copper bimetallic complexes were investigated by X-ray diffraction, cyclic voltammetry, electron paramagnetic spectroscopy, and density functional theory calculations. The combined experimental and theoretical data corroborate that the unpaired spin is delocalized across Cu, M, and ancillary atoms, and the singly occupied molecular orbital (SOMO) corresponds to a σ-(Cu–M) bond involving the Cu 4pz and M ns/npz atomic orbitals. Collectively, the data suggest the covalent nature of these interactions, which represent the first examples of odd-electron σ-bonds for the heavier Group 13 elements Al and Ga.
Odd-electron σ-bonds, where the electrons are delocalized between two atoms, can occur as two-center/one-electron (2c/1e) or two-center/three-electron (2c/3e) interactions. Proposed by Pauling in 1931,1 odd-electron σ-bonds have garnered attention because of their fundamental importance to chemical bonding and their relationship to radical species generated during oxidative stress in biological systems.2–14 Examples of compounds exhibiting odd-electron bonding are typically homonuclear (like H2+, He2+, and alkali metal dimers) and transiently stable, limiting them to spectroscopic characterization.1,11,15–18
The first solid-state structure of a formally one-electron σ-bond was a tetraphosphabenzene species (Fig. 1a) which was formed by the coupling of two diphosphirenyl radicals.19 Following this discovery, the formation of discrete 2c/1e σ-bonds, where the odd-electron is delocalized between two homonuclear main group centers, was reported for B·B and then extended to P·P.8,17,20 Of note, the first solid-state structure of a B·B compound was reported in only 2014 (Fig. 1b).21 Examples of 2c/1e σ-bonds between the heavier Group 13 congeners are even more lacking because of the greater propensity for their unpaired spins to couple, forming larger more stable clusters.8 To our knowledge, there are only three structurally characterized examples of odd-electron bonds for the heavy Group 13 atoms,22 and these examples are all homonuclear π-radicals (Fig. 1c).23–26
 | ||
| Fig. 1 Select examples of structurally characterized molecules (a–d) featuring odd-electron bonds. | ||
Heteronuclear odd-electron σ-bonds are also rare. The Cu(TPB) complex, where TPB is a trisphosphinoborane, is the single structural example of a 2c/1e bond between heteroatoms (Fig. 1d).27 The authors described the bonding as Cu·B, where the unpaired electron is heavily polarized toward B. A theoretical study predicted that such a bond would also exist between Cu and Al, but no heavier analogues of Cu(TPB) have been synthesized to date.28 Furthermore, the heavier Group 13 elements by virtue of their lower electronegativity compared to B should facilitate greater covalent interactions with the Cu center.
Hence, we sought to target formally zerovalent Cu complexes supported by Al(III) or Ga(III) as an extension of the previously reported isoelectronic nickelate species and Cu(TPB).29 Herein, we describe the synthesis, structure, spectroscopic characterization, and DFT calculations of cationic [CuML]+ complexes (L = [N((o-C6H4)NCH2PiPr2)3]3−; M = Al and Ga) as well as their one-electron reduced metalloradical counterparts that feature discrete 2c/1e bonds.
Results and discussion
Synthesis and cyclic voltammetry
Initial synthetic attempts to produce cationic [CuML]+ complexes via metalation of ML with a copper halide followed by halide abstraction produced intractable mixtures. Hence, we sought an alternative Cu(I) precursor that eschews the halide and has labile ligands, such as [Cu(COD)2]A (COD = 1,5-cyclooctadiene and A = counteranion). This cation has received little attention as a precursor to low-coordinate copper complexes, which may stem from the known salts (A = ClO4−, NO3−, SbF6−, etc.) lacking sufficient solubility in weakly coordinating solvents.30–32 Because the BArF4 counteranion (BArF4 = tetrakis[3,5-bis(trifluoromethyl)-phenyl]borate) confers high solubility in low dielectric solvents, we targeted and isolated [Cu(COD)2]BArF4 by protonating the pentameric (CuMes)5 (ref. 33 and 34) (Mes = 2,4,6-trimethylphenyl) with [(H(OEt2)2]BArF4 (ref. 35) in the presence of excess COD (see ESI for details, Fig. S1†). Gratifyingly, [Cu(COD)2]BArF4 reacted with the metalloligands AlL or GaL in PhF to provide [1]BArF4 or [2]BArF4 in good yields as yellow or orange powders, respectively (Scheme 1, Fig. S3–S22†).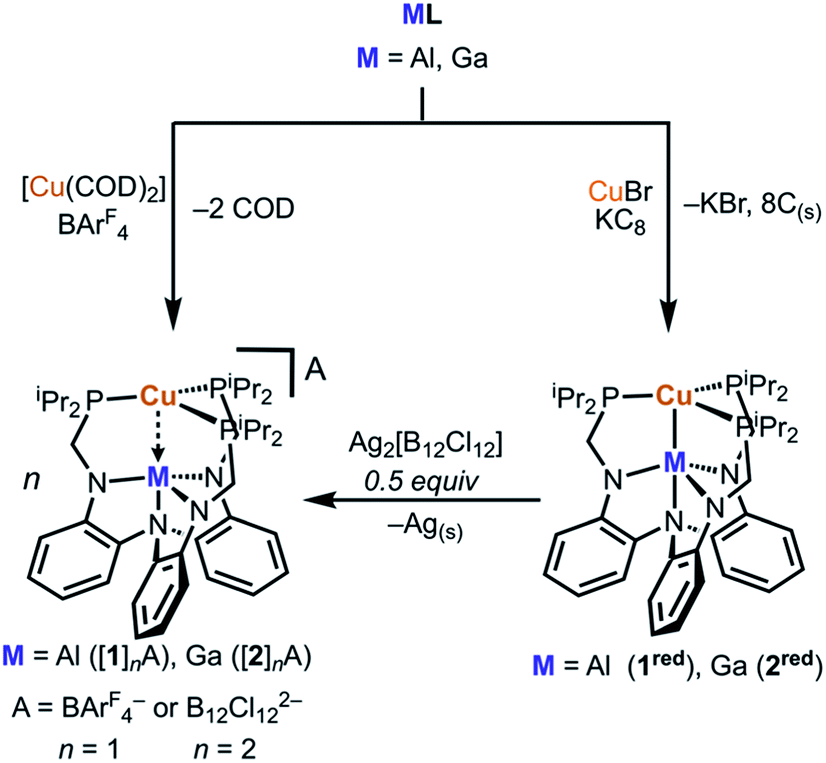 | ||
| Scheme 1 Synthesis of heterobimetallic copper-Group 13 complexes. | ||
Cyclic voltammetry (CV) studies of both [1]BArF4 and [2]BArF4 suggested that {CuM}11 complexes are accessible at low potentials (Fig. S31–36†). The CV of [1]BArF4 reveals a single reduction event at −2.03 V vs. [FeCp2]0/+, which corresponds to the [CuAlL]+/0 redox couple. The CV of [2]BArF4 shows two single-electron reductions at −1.79 V and −2.42 V vs. [FeCp2]0/+, which correspond to the [CuGaL]+/0 and [CuGaL]0/– redox couples, respectively. Of note, Cu(TBP) was described to exhibit a similar CV profile to 2red.27 Unfortunately, the second redox event for [1]BArF4 was not observed.36 Scan rate studies indicate that the [CuML]+/0 redox couple is reversible for both bimetallics species, while the [CuGaL]0/– redox process observed for [2]BArF4 is quasi-reversible (Fig. S32 and S34–S35†). For comparison, the Group 13 metalloligands and the monocopper complex, [Cu(LH3)]BArF4, exhibit no reversible redox processes in a similar potential window (Fig. S23–S30 and S37†), which suggests that the Cu–M unit may be necessary for accessing the reduced Cu states. Furthermore, the formal substitution of Al with Ga shifts the [CuML]+/0 redox couple by more than 200 mV. With this electrochemical insight, we sought to prepare the reduced {CuM}11 species. Chemical reduction of [1]BArF4 or [2]BArF4 was accomplished using KC8 to generate CuAlL (1red) and CuGaL (2red), respectively. However, high-yielding syntheses of 1red and 2red were achieved by first metalating ML with CuBr to generate an in situ Cu(I) halide complex followed by addition of KC8 in THF at −78 °C (Scheme 1). The resultant reductions produced deeply colored maroon (1red) and dark red (2red) solutions, from which the products were purified by removing all volatiles and then extracting into benzene.
X-ray crystallography
While [1]BArF4 and [2]BArF4 could be isolated, diffraction-quality crystals of these species were not readily obtained. To this end, we sought to incorporate the highly crystalline dodecachlorododecaborate dianion ([B12Cl12]2–). X-ray quality crystals of [1]2[B12Cl12] and [2]2[B12Cl12] were achieved by the oxidation of 1red and 2red, respectively, with Ag2[B12Cl12].37–39 Crystals of the neutral copper species were grown from saturated Et2O at −30 °C (1red) or toluene/pentane vapor diffusion at room temperature (2red). The solid-state structures of the four copper-Group 13 complexes are shown in Fig. 2, and the structural metrics are summarized in Table 1. | ||
| Fig. 2 Solid-state structures of [1]2[B12Cl12], [2]2[B12Cl12], 1red, and 2red. Thermal ellipsoids are depicted at 50% probability. Counteranions, co-crystallized solvent molecules, and hydrogen atoms are omitted for clarity. | ||
| Parameter | [1]2[B12Cl12] | 1 red | [2]2[B12Cl12] | 2 red |
|---|---|---|---|---|
| a Ratio of the Cu–Al/Ga bond length to the sum the Cu and Al/Ga Alvarez covalent radii.41 b Trigonal space groups only display one value by symmetry. c Average of three unique values. | ||||
| M–Cu | 2.6239(8) | 2.5298(4) | 2.5737(5) | 2.4541(6) |
| r | 1.04 | 1.00 | 1.01 | 0.97 |
| Cu–P | 2.2925(4)b | 2.2487(7)c | 2.2994(5)b | 2.2698(9)c |
| M–Neq | 1.85469(12)b | 1.8915(1)c | 1.9059(14)b | 1.9574(9)c |
| M–Nap | 2.000(2) | 2.0836(1) | 2.069(3) | 2.1948(9) |
| Cu-to-P3-plane | 0.188 | 0.118 | 0.180 | 0.136 |
| M-to-N3-plane | 0.07 | 0.246 | 0.142 | 0.355 |
| Σ(∡P–Cu–P) | 358.02(1) | 359.18(2) | 358.19(2) | 358.93(4) |
| Σ(∡Neq–M–Neq) | 359.47(1) | 355.01(1) | 358.36(2) | 350.36(4) |
The structures of [1]2[B12Cl12] and [2]2[B12Cl12] reveal weak to nonexistent Cu(I)–M interactions as their Cu–M distances of 2.6239(8) and 2.5737(5) Å, respectively, are longer than the sum of the metals' covalent radii (c.f. 2.53 and 2.54 Å, respectively).40,41 The formal shortness ratio (r),42 which is defined as the ratio of the metal–metal bond length to the sum of the single-bond covalent radii41 was also calculated: 1.04 for [1]2[B12Cl12] and 1.01 for [2]2[B12Cl12]. The strength of the copper-Group 13 donor–acceptor interaction in [1]2[B12Cl12] and [2]2[B12Cl12] is much weaker compared to the corresponding isoelectronic neutral Ni(0)43,44 and anionic Co(–I)45 bimetallics, which have r values <1, because the localized cationic change on the Cu(I) center should render it a weaker donor. The Cu center is nearly trigonal planar with minimal perturbation due to the Group 13 ion as both ∑(∡P–Cu–P) and ∑(∡Neq−M−Neq) approach 360°. For comparison, mononuclear [Cu(LH3)]BArF4 exhibits nearly perfect trigonal symmetry in the solid state (∑(∡P–Cu–P) = 359.96(2)° and Cu-to-P3-plane = 0.027 Å, Table S2 and Fig. S2†). The average Cu–P bond length in [Cu(LH3)]BArF4 of 2.3001(4) Å is similar to that of [1]2[B12Cl12] and [2]2[B12Cl12], which indicates that all three complexes possess a Cu(I) d10 center.
After reduction to 1red and 2red, the Cu–M bond contracts significantly by 0.094 Å (r = 1.00) and 0.120 Å (r = 0.97), respectively, indicating an increase in the metal–metal bonding. Of relevance, bonding between copper and a Group 13 centers have been structurally characterized within clusters46 and unsupported complexes,47–49 although these prior examples do not feature odd-electron bonds. In comparison to 1red and 2red, the known Cu–Al49 and Cu–Ga47–49 complexes have lower r values of 0.90–0.91. The greater r values for 1red and 2red are expected considering that their bonding involves only one electron. Another indication of a metal–metal interaction is the pyramidalization of the Group 13 center as indicated by the ∑(∠Neq–M–Neq) values of 355.01(1)° and 350.36(4)° for 1red and 2red, respectively, as both the M-to-(N3-plane) and M–Nap distances increase as the M center moves closer to Cu.50 The local coordination environment around the Cu center also changes, with the slight lowering of the Cu center into the P3-plane and the contraction of the Cu–P bonds upon reduction to the {Cu–M}11 core. The Cu–P bond contraction is consistent with an increased Cu electron density that more strongly π-backbonds into the phosphine ligands. Moreover, the significant perturbation of the Group 13 centers suggests their direct participation in stabilizing the reduced {Cu–M}11 core, presumably via bonding interactions to the Cu center.
EPR spectroscopy
To gain insight into the radical nature of the one-electron bonds, room-temperature EPR spectra were collected for solutions of 1red and 2red in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solvent mixture of 2-MeTHF and toluene (Fig. 3). Complex 1red gave rise to a complex signal centered at g ∼ 2.0, which corresponds to a highly overlapped manifold of hyperfine lines that can be decomposed into a leading 1:1:1:1:1:1 sextet due to hyperfine coupling (hfc) of the electronic spin S = 1/2 with 27Al nuclei (I = 5/2, 100% natural abundance) and additional splitting of each of these hyperfine transitions into two 1:1:1:1 quartets due to additional coupling to the isotopes 63Cu and 65Cu on the copper site (I = 3/2 both, with 69.2% and 30.8% natural abundance, respectively). The corresponding combined hyperfine transitions are assigned by stick spectrum shown on top of Fig. 3. The experimental spectrum was well modeled using an isotropic g value of 2.007 and three isotropic hyperfine values (A, in MHz): 269.7 (27Al), 171.4 (63Cu), and 182.6 (65Cu).
:1 solvent mixture of 2-MeTHF and toluene (Fig. 3). Complex 1red gave rise to a complex signal centered at g ∼ 2.0, which corresponds to a highly overlapped manifold of hyperfine lines that can be decomposed into a leading 1:1:1:1:1:1 sextet due to hyperfine coupling (hfc) of the electronic spin S = 1/2 with 27Al nuclei (I = 5/2, 100% natural abundance) and additional splitting of each of these hyperfine transitions into two 1:1:1:1 quartets due to additional coupling to the isotopes 63Cu and 65Cu on the copper site (I = 3/2 both, with 69.2% and 30.8% natural abundance, respectively). The corresponding combined hyperfine transitions are assigned by stick spectrum shown on top of Fig. 3. The experimental spectrum was well modeled using an isotropic g value of 2.007 and three isotropic hyperfine values (A, in MHz): 269.7 (27Al), 171.4 (63Cu), and 182.6 (65Cu).
 | ||
| Fig. 3 X-band (9.64 GHz) EPR spectra of 2.5 mM solutions of (a) 1red and (b) 2red in a 1:1 mixture of 2-MeTHF/toluene at 298 K (time constant, 22 ms). The plot shows the simulated (red) and experimental (black) spectra, as well as a simulation with DFT-derived parameters using the PBE0 functional and def2-TZVP/def2/J basis sets (blue). Inset is a zoom of the 63/65Cu hyperfine pattern at the superimposed mI = −1/2 69Ga/71Ga-transitions of 2red. Microwave power and modulation amplitudes used for 1red and 2red are 0.7 and 21.3 mW, and 0.98 and 0.50 mT, respectively. See ESI† for additional EPR spectra and details. | ||
Complex 2red likewise exhibited a complex signal with an isotropic g value of 2.002, which was well modeled as a combined hyperfine splitting of 1:1:1:1 quartets into 1:1:1:1 quartets due to dominating hfc with 69/71Ga (I = 3/2) and weaker coupling with 63/65Cu (also I = 3/2). The two larger quartets arise from the distinct couplings to the two naturally abundant Ga isotopes with A(69Ga) = 1199.3 MHz (60.1%) and A(71Ga) = 1524.3 MHz (39.9%), while the smaller quartets result from the similar coupling to the two Cu isotopes: A(63Cu) = 128.9 MHz and A(65Cu) = 137.3 MHz. Furthermore, superhyperfine coupling was observed at the superimposed mI = −1/2 transition for both the 69Ga and 71Ga quartets (inset of Fig. 3). A reasonable fitting was obtained by including the hyperfine interaction of three equivalent 31P nuclei (I = 1/2, A = 29.4 MHz), three equivalent 14N (I = 1, A = 20.1 MHz), and one unique 14N (A = 3.5 MHz). The simulation is consistent with the ligand's three equivalent amido donors and one apical amine donor.29
The distribution of the unpaired spin density across the Cu and Group 13 valence orbitals in the {Cu–M}11 core can be evaluated by comparing the corresponding experimental Aiso values with tabulated reference values (aiso0) for each specific elemental orbital.51 Using the reference values for a localized valence s-based spin for Al (3911 MHz) and Ga (12,210 MHz), the 27Al 3s and the 69Ga 4s character in 1red and 2red, respectively, were found to be small at 6.9 and 9.8%, respectively.51 A similar calculation was performed for the 63Cu 4s orbital (aiso0 = 5995 MHz) resulting in even smaller contributions of 2.9 and 2.2% in 1red and 2red, respectively. The frozen EPR spectra of 1red and 2red were also collected (Fig. S43 and S46†). However, only in the former case were we able to model the axial signal and thus extract the 27Al hyperfine tensor, A = [229.6, 229.6, 325.4] MHz. The anisotropic component of the hfc tensor, defined as b = [(Ax − Aiso), (Ay − Aiso), (Az − Aiso)] = [−32, −32, 64] MHz, was then compared to that of a fully localized spin in an Al 3pz orbital, where b0 = [−83, −83, 166] MHz. We found that the 27Al 3pz character is much greater at 38%. Similarly, the 63Cu 4pz contribution was found to be 19.9%.52 The total estimated spin density between Cu and Al is ca. 67%, wherein the majority of the unpaired spin likely resides in the bonding axis between the two metals.
Only a handful of formal Cu(0) complexes are known in the literature, and they exhibit a range of isotropic A(63Cu) values. The complexes Cu(PMe3)3 and Cu(TPB) have A(63Cu) values of 120 and 191 MHz, respectively, which compare well to those measured for 1red and 2red.27,53,54 For other literature complexes, the A(63Cu) values are significantly smaller (<30 MHz) because the unpaired spin is strongly delocalized over the ligand. For example, Cu(Me2-cAAC)2 (cAAC = cyclic (alkyl)(amino)carbene) displayed a small A(63Cu) coupling of 22.2 MHz, and the DFT-calculated spin density revealed that the majority of the spin density resides in the Ccarbene–N π-bond.55 Similarly, the unpaired spin in Cu(B2P2) (B2P2 = diphosphine–diboraanthracene) was proposed to reside on the redox-active ligand core as no Cu hyperfine was discerned.56
Further comparison can be made with hfc to the Group 13 supporting ions with 27Al and 69Ga hyperfine values for 1red and 2red of 269.7 and 1199.3 MHz, respectively. Due to their highly unstable nature, examples of mononuclear Al(II) and Ga(II) radicals are sparse. Both alane and gallane radical anions display isotropic hfc with their respective Group 13 centers with values of 432.2 MHz for [AlH3]˙− and 1179.5 MHz for [GaH3]˙−.57,58 In contrast, the more bulky [Al(SiMetBu2)3]˙− and [Ga(SiMetBu2)3]˙− complexes exhibit much smaller isotropic hfc of 174.0 and 346.9 MHz, respectively.59 Larger hfc in main group radicals has been shown to be directly related to increased s-character via pyramidalization.57,58,60,61 The hyperfine values of 1red and 2red are closer to those of [MH3]˙− because the Group 13 ions are similarly pyramidalized57,58 and correspondingly are different from [Al(SiMetBu2)3]˙−, which is more planar (Σ(∡Si–M–Si): 358.4°).59 Lastly, direct comparison to isoelectronic [NiML]−˙ analogues show similar hfc to 27Al (219.7 MHz) and 69Ga (1050 MHz).29
Theoretical computations
To further elucidate the electronic structure of these species, theoretical calculations were performed using Kohn–Sham density functional theory (KS-DFT). Geometry optimizations were performed on the structures without truncation using the M06-L functional62 with def2-series basis sets63 and strong agreement was found between the optimized geometries and the X-ray structures (Table S3 and S4†). Based on the in depth study by Hedegård et al.,64 the PBE0 (ref. 65 and 66) functional in conjunction with def2-TZVP and def2/J basis sets67 were used to determine the EPR parameters with spin–orbit coupling included. The DFT-calculated EPR parameters also matched well to the experimental g and isotropic A values, as well as the anisotropic A tensors, when available (Fig. 3, Tables S12 and S13†). The good agreement further supports the validity of the calculated electronic structures as models for the experimental species.Examination of the molecular orbitals (MOs) of cationic species [Cu(LH3)]+, [1]+, and [2]+ revealed five doubly filled d-orbitals as expected for trigonal Cu(I) species. In both bimetallics, the lowest unoccupied molecular orbital (LUMO, Fig. 4) was found to be a hybrid MO containing large contributions from the empty Cu 4pz and Group 13 ns and npz-orbitals.68 The neutral radical species 1red and 2red each contain a singly occupied molecular orbital (SOMO) that is similar to the LUMO of the cations, suggesting that one-electron reduction of the cations results in occupancy of the LUMO. The SOMO of 1red contains major contributions from Cu (8.4%), Al (33.8%), and P (35.2%). Similarly, the SOMO for 2red possesses contributions from Cu (12.7%), Ga (32.0%), and P (35.2%).
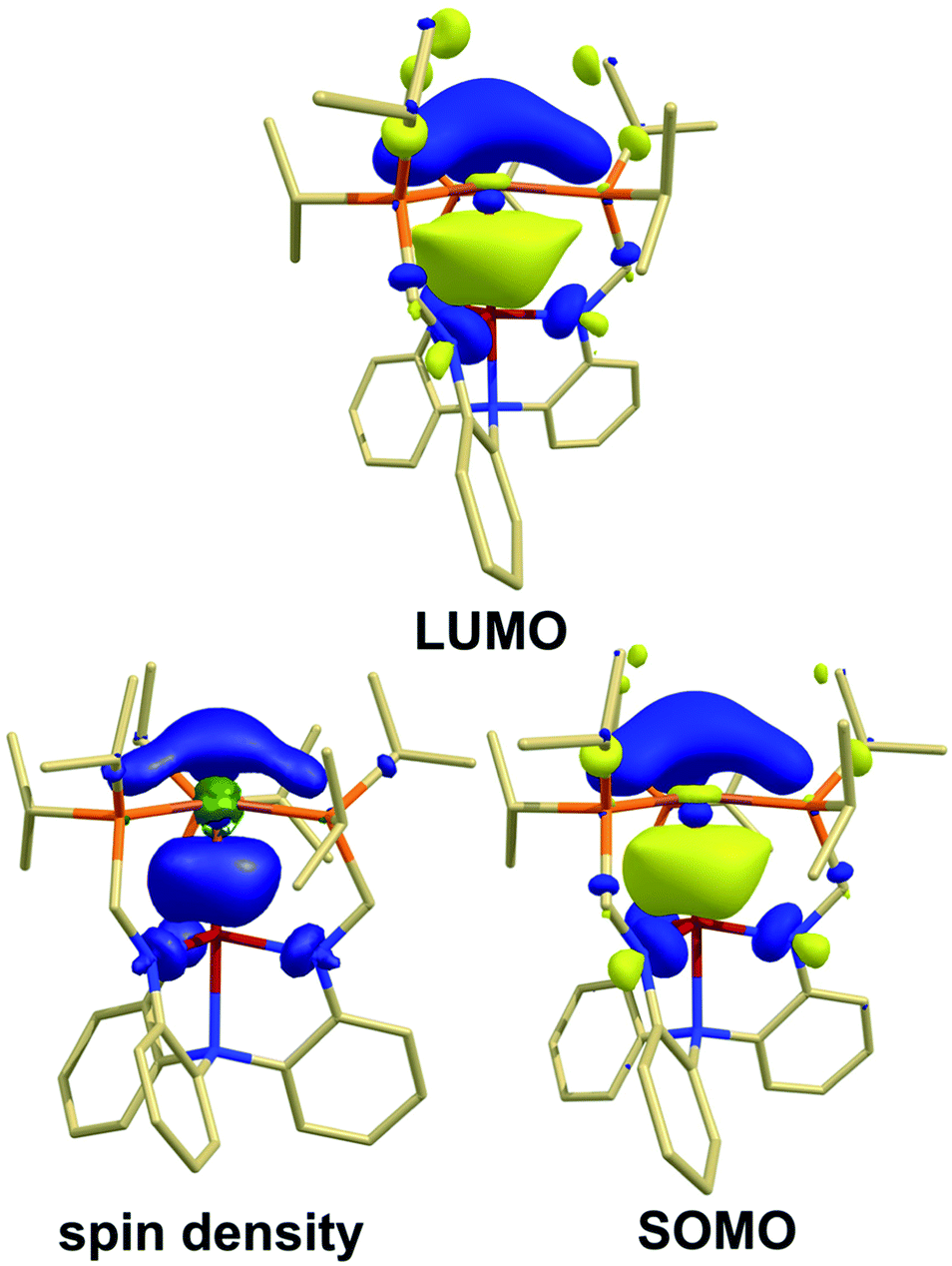 | ||
| Fig. 4 Contour plots for the LUMO for 2 and the SOMO and spin density for 2red. The corresponding contour plots for 1/1red are similar (see ESI†). | ||
The contributions of the Group 13 atoms to the SOMOs have significant s-character (ca. 19% for each species), while the Cu center only possesses contributions from the 4pz. The s/p-mixing predicted to be present at both Al and Ga likely contributes to the pyramidalization of the Group 13 center. This result is consistent with the greater hfc values observed in the EPR spectra (vide supra). Notably, the P contribution nearly doubles upon reduction (from ca. 17% to 35% for both 1red and 2red), further indicating the importance of the ancillary ligand in supporting the more electron-rich Cu center.
The Mulliken spin density of 1red and 2red revealed delocalization across Cu, Al/Ga, P, and N atoms as visualized through spin-density plots (Fig. 4, Table S11†). Comparison of spin densities between the two metal pockets CuP3:MN3 revealed a ratio of 1.0:1.2 suggesting the unpaired electron is almost equally delocalized across the two binding pockets. For 1red, spin densities were calculated to be 0.18 on Cu and 0.52 on Al, with a moderate amount distributed throughout the ancillary atoms (0.09 per P and 0.01 per Neq), and these values are in good agreement to those estimated by EPR spectroscopy (vide supra). The calculated spin densities for 2red were found to be similar to that of 1red with 0.24 on Cu, 0.44 on Ga, and the remainder on the supporting donor atoms (0.08 per P and 0.03 per Neq).
By considering the SOMOs and noting the extent of spin delocalization in the spin density plots, the radical species 1red and 2red can therefore best be described as possessing σ-bonding interactions between the Cu (4pz) and Al/Ga (npz/ns) orbitals, as well as a π-backbonding interaction between Cu and the three P atoms.53,54,69 The covalent nature of the σ-(Cu–M) bonds is supported by the significant contraction of the metal–metal bond lengths in the solid-state structures and the nearly equal spin delocalization between the two metal pockets, which matches well with the experimental EPR hfc values.
Conclusion
In summary, two cationic copper-Group 13 complexes were synthesized and their subsequent reduction provided access to unusual 17 e− {Cu–M}11 complexes. These radical species are characterized by short Cu–M bond lengths and exhibit significant hfc to Cu, M, and P atoms, collectively consistent with the highly covalent nature of SOMOs that involve both metal centers and the ancillary ligand donor atoms. These neutral radical {Cu–M}11 species possess rarely observed 2c/1e σ-bonds and are the first report of any such bond for the heavier elements Al and Ga. Along with the previously reported Cu(TPB), 1red and 2red complete a triad of copper-Group 13 complexes that demonstrate an increase in Cu bonding character and Cu–M covalency when traversing down the group in the order B < Al < Ga.27 Investigation of the metalloradical reactivity of these bimetallics is ongoing.Data availability
Additional data for synthesis, characterization, and DFT calculations are available in the ESI.† The combined XYZ coordinates of DFT-optimized geometries have been uploaded as ESI.†Author contributions
BJG and MVV performed the experimental work supervised by CCL. TRS performed the theoretical calculations supervised by LG. MJD assisted with the EPR data collection, and EB assisted with the EPR analysis and interpretation. VGY provided X-ray support. The original draft was written by BJG with guidance from CCL, and all authors edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is dedicated to Professor John E. Ellis on the occasion of his 79th birthday. The authors thank Prof. John Lipscomb and Melanie Rogers for access to the EPR spectrometer and Dr James T. Moore for collection of EPR data. The experimental and computational work was supported by the NSF (CHE-1954751 and CHE-2054723). TRS was supported by a NSF graduate fellowship. X-ray diffraction experiments were performed using a crystal diffractometer acquired through an NSF-MRI award (CHE-1229400). The computational resources were provided by the Minnesota Supercomputing Institute (MSI) at the University of Minnesota and the Chicago Center for Theoretical Chemistry (CCTCh) at the University of Chicago.References
- L. Pauling, J. Am. Chem. Soc., 1931, 53, 3225–3237 CrossRef CAS.
- N. C. Baird, J. Chem. Educ., 1977, 54, 291–293 CrossRef CAS.
- M. Goez, J. Rozwadowski and B. Marciniak, Angew. Chem., Int. Ed., 1998, 37, 628–630 CrossRef CAS PubMed.
- A. Abu-Raqabah and M. C. R. Symons, J. Am. Chem. Soc., 2002, 112, 8614–8615 CrossRef.
- K. D. Asmus, Acc. Chem. Res., 2002, 12, 436–442 CrossRef.
- D. Griller and K. U. Ingold, Acc. Chem. Res., 2002, 9, 13–19 CrossRef.
- H. Grutzmacher and F. Breher, Angew. Chem., Int. Ed., 2002, 41, 4006–4011 CrossRef CAS PubMed.
- P. P. Power, Chem. Rev., 2003, 103, 789–810 CrossRef CAS PubMed.
- C. Schoneich, D. Pogocki, G. L. Hug and K. Bobrowski, J. Am. Chem. Soc., 2003, 125, 13700–13713 CrossRef PubMed.
- X. Zheng, X. Wang, Z. Zhang, Y. Sui, X. Wang and P. P. Power, Angew. Chem., Int. Ed., 2015, 54, 9084–9087 CrossRef CAS PubMed.
- J. F. Berry, Acc. Chem. Res., 2016, 49, 27–34 CrossRef CAS PubMed.
- D. W. O. Sousa and M. A. C. Nascimento, Acc. Chem. Res., 2017, 50, 2264–2272 CrossRef CAS PubMed.
- D. W. O. de Sousa and M. A. C. Nascimento, Phys. Chem. Chem. Phys., 2019, 21, 13319–13336 RSC.
- W. Yang, L. Zhang, D. Xiao, R. Feng, W. Wang, S. Pan, Y. Zhao, L. Zhao, G. Frenking and X. Wang, Nat. Commun., 2020, 11, 3441 CrossRef CAS PubMed.
- T. G. Castner and W. Känzig, J. Phys. Chem. Solids, 1957, 3, 178–195 CAS.
- T. Drews and K. Seppelt, Angew. Chem., Int. Ed., 1997, 36, 273–274 CrossRef CAS.
- J. D. Hoefelmeyer and F. P. Gabbaï, J. Am. Chem. Soc., 2000, 122, 9054–9055 CrossRef CAS.
- R. L. Hudson and F. Williams, J. Am. Chem. Soc., 2002, 99, 7714–7716 CrossRef.
- Y. Canac, D. Bourissou, A. Baceiredo, H. Gornitzka, W. W. Schoeller and G. Bertrand, Science, 1998, 279, 2080–2082 CrossRef CAS PubMed.
- L. Cataldo, S. Choua, T. Berclaz, M. Geoffroy, N. Mezailles, L. Ricard, F. Mathey and P. Le Floch, J. Am. Chem. Soc., 2001, 123, 6654–6661 CrossRef CAS PubMed.
- A. Hübner, A. M. Diehl, M. Diefenbach, B. Endeward, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, Angew. Chem., Int. Ed., 2014, 126, 4932–4935 CrossRef.
- Two groups simultaneously isolated and characterized the π-radical [Al2{CH(SiMe3)2}4]˙−. See ref. 25 and 26.
- X. He, R. A. Bartlett, M. M. Olmstead, K. Ruhlandt-Senge, B. E. Sturgeon and P. P. Power, Angew. Chem., Int. Ed. Engl., 1993, 32, 717–719 CrossRef.
- C. Pluta, K.-R. Pörschke, C. Krüger and K. Hildenbrand, Angew. Chem., Int. Ed., 1993, 32, 388–390 CrossRef.
- W. Uhl, A. Vester, W. Kaim and J. Poppe, J. Organomet. Chem., 1993, 454, 9–13 CrossRef CAS.
- R. J. Wehmschulte, K. Ruhlandt-Senge, M. M. Olmstead, H. Hope, B. E. Sturgeon and P. P. Power, Inorg. Chem., 2002, 32, 2983–2984 CrossRef.
- M. E. Moret, L. Zhang and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 3792–3795 CrossRef CAS PubMed.
- E. Kusevska, M. M. Montero-Campillo, O. Mo and M. Yanez, Angew. Chem., Int. Ed., 2017, 56, 6788–6792 CrossRef CAS PubMed.
- M. V. Vollmer, J. Xie, R. C. Cammarota, V. G. Young, Jr., E. Bill, L. Gagliardi and C. C. Lu, Angew. Chem., Int. Ed., 2018, 57, 7815–7819 CrossRef CAS PubMed.
- M. Munakata, S. Kitagawa, H. Shimono and H. Masuda, Inorg. Chem., 1991, 30, 2610–2614 CrossRef CAS.
- M. Stricker, B. Oelkers, C. P. Rosenau and J. Sundermeyer, Chem. - Eur. J., 2013, 19, 1042–1057 CrossRef CAS PubMed.
- C. S. Gravatt, L. Melecio-Zambrano and T. P. Yoon, Angew. Chem., Int. Ed., 2021, 60, 3989–3993 CrossRef CAS PubMed.
- T. Tsuda, K. Watanabe, K. Miyata, H. Yamamoto and T. Saegusa, Inorg. Chem., 2002, 20, 2728–2730 CrossRef.
- M. Ohashi, T. Adachi, N. Ishida, K. Kikushima and S. Ogoshi, Angew. Chem., Int. Ed., 2017, 56, 11911–11915 CrossRef CAS PubMed.
- M. Brookhart, B. Grant and A. F. Volpe, Organometallics, 2002, 11, 3920–3922 CrossRef.
- Solutions of 1[BArF4] were shown to be unstable in the prescence of common electrolyte TBA[PF6]. The second reduction event of 1[BArF4] is likely beyond the TPA[BArF4] window (1.3 to –2.8 V), required for CV of this species precluding its observation .
- W. Gu and O. V. Ozerov, Inorg. Chem., 2011, 50, 2726–2728 CrossRef CAS PubMed.
- M. T. Whited, J. Zhang, S. Ma, B. D. Nguyen and D. E. Janzen, Dalton Trans., 2017, 46, 14757–14761 RSC.
- J. T. Moore, S. Chatterjee, M. Tarrago, L. J. Clouston, S. Sproules, E. Bill, V. Bernales, L. Gagliardi, S. Ye, K. M. Lancaster and C. C. Lu, Inorg. Chem., 2019, 58, 6199–6214 CrossRef CAS PubMed.
- Weak interaction may be indicated by the slight red shift in the visible adsorption peak when going from [Cu(LH3)] [BArF4] to 1[BArF4] to 2[BArF4] .
- B. Cordero, V. Gomez, A. E. Platero-Prats, M. Reves, J. Echeverria, E. Cremades, F. Barragan and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC.
- Multiple Bonds Between Metal Atoms, ed. F. A. Cotton, C. A. Murillo and R. A. Walton, Springer Science, New York, 2005, pp. 35–68 Search PubMed.
- P. A. Rudd, S. Liu, L. Gagliardi, V. G. Young, Jr. and C. C. Lu, J. Am. Chem. Soc., 2011, 133, 20724–20727 CrossRef CAS PubMed.
- R. C. Cammarota and C. C. Lu, J. Am. Chem. Soc., 2015, 137, 12486–12489 CrossRef CAS PubMed.
- M. V. Vollmer, J. Xie and C. C. Lu, J. Am. Chem. Soc., 2017, 139, 6570–6573 CrossRef CAS PubMed.
- C. Ganesamoorthy, J. Wessing, C. Kroll, R. W. Seidel, C. Gemel and R. A. Fischer, Angew. Chem., Int. Ed. Engl., 2014, 53, 7943–7947 CrossRef CAS PubMed.
- C. Jones, D. P. Mills, R. P. Rose, A. Stasch and W. D. Woodul, J. Organomet. Chem., 2010, 695, 2410–2417 CrossRef CAS.
- S. P. Green, C. Jones, D. P. Mills and A. Stasch, Organometallics, 2007, 26, 3424–3430 CrossRef CAS.
- K. L. Mears, C. R. Stennett, E. K. Taskinen, C. E. Knapp, C. J. Carmalt, H. M. Tuononen and P. P. Power, J. Am. Chem. Soc., 2020, 142, 19874–19878 CrossRef CAS PubMed.
- A sum of 360° corresponds to no pyramidalization and thus a trigonal planar geometry .
- J. R. Morton and K. F. Preston, J. Magn. Reson., 1978, 30, 577–582 CAS.
- This method cannot distinguish the pz and dz2 components. However, due to the computationally supported d10 nature of the starting Cu(I) ion and lack of d-character in the SOMO, the 4pz orbital is likely the major contributor.
- J. A. Howard, B. Mile, J. R. Morton, K. F. Preston and R. Sutcliffe, Chem. Phys. Lett., 1985, 117, 115–117 CrossRef CAS.
- M. Histed, J. A. Howard, H. A. Joly and B. Mile, Chem. Phys. Lett., 1990, 174, 411–415 CrossRef CAS.
- D. S. Weinberger, N. Amin Sk, K. C. Mondal, M. Melaimi, G. Bertrand, A. C. Stuckl, H. W. Roesky, B. Dittrich, S. Demeshko, B. Schwederski, W. Kaim, P. Jerabek and G. Frenking, J. Am. Chem. Soc., 2014, 136, 6235–6238 CrossRef CAS PubMed.
- J. W. Taylor, A. McSkimming, M. E. Moret and W. H. Harman, Inorg. Chem., 2018, 57, 15406–15413 CrossRef CAS PubMed.
- J. R. M. Giles and B. P. Roberts, J. Chem. Soc., Chem. Commun., 1981, 1167–1168 RSC.
- J. C. Brand and B. P. Roberts, J. Chem. Soc., Chem. Commun., 1984, 109–110 RSC.
- M. Nakamoto, T. Yamasaki and A. Sekiguchi, J. Am. Chem. Soc., 2005, 127, 6954–6955 CrossRef CAS PubMed.
- A. Begum, A. R. Lyons and M. C. R. Symons, J. Chem. Soc. A, 1971, 2290–2293 RSC.
- J. R. M. Giles and B. P. Roberts, J. Chem. Soc., Perkin Trans. 2, 1983, 1699–1711 Search PubMed.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- E. D. Hedegard, J. Kongsted and S. P. Sauer, J. Chem. Theory Comput., 2013, 9, 2380–2388 CrossRef CAS PubMed.
- M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029–5036 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- The LUMO of [Cu(LH3)]+ was found to contain Cu and P contributions only .
- J. A. Howard, B. Mile, J. R. Morton and K. F. Preston, J. Phys. Chem., 2002, 90, 2027–2029 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2076274–2076278, 2092926. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc01998a |
| This journal is © The Royal Society of Chemistry 2022 |