
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced catalysis of 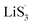 radical-to-polysulfide interconversion via increased sulfur vacancies in lithium–sulfur batteries†
radical-to-polysulfide interconversion via increased sulfur vacancies in lithium–sulfur batteries†
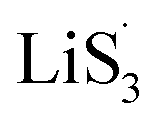
Rui
Xu‡
a,
Hongan
Tang‡
a,
Yuanyuan
Zhou
a,
Fangzheng
Wang
a,
Hongrui
Wang
a,
Minhua
Shao
 b,
Cunpu
Li
*a and
Zidong
Wei
*a
b,
Cunpu
Li
*a and
Zidong
Wei
*a
aThe State Key Laboratory of Power Transmission Equipment & System Security and New Technology, Chongqing Key Laboratory of Chemical Process for Clean Energy and Resource Utilization, School of Chemistry and Chemical Engineering, Chongqing University, Shazhengjie 174, Chongqing 400044, China. E-mail: lcp@cqu.edu.cn; zdwei@cqu.edu.cn
bDepartment of Chemical and Biological Engineering, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong
First published on 10th May 2022
Abstract
The practical application of lithium–sulfur (Li–S) batteries is seriously hindered by severe lithium polysulfide (LiPS) shuttling and sluggish electrochemical conversions. Herein, the Co9S8/MoS2 heterojunction as a model cathode host material is employed to discuss the performance improvement strategy and elucidate the catalytic mechanism. The introduction of sulfur vacancies can harmonize the chemisorption of the heterojunction component. Also, sulfur vacancies induce the generation of 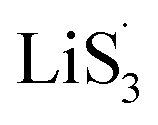 radicals, which participate in a liquidus disproportionated reaction to reduce the accumulation of liquid LiPSs. To assess the conversion efficiency from liquid LiPSs to solid Li2S, a new descriptor calculated from basic cyclic voltammetry curves, nucleation transformation ratio, is proposed.
radicals, which participate in a liquidus disproportionated reaction to reduce the accumulation of liquid LiPSs. To assess the conversion efficiency from liquid LiPSs to solid Li2S, a new descriptor calculated from basic cyclic voltammetry curves, nucleation transformation ratio, is proposed.
Introduction
With the growing demands of high-energy-storage equipment, traditional lithium-ion batteries (LIBs) could fail to meet the increasing requirements for future renewable energies. Based on the ultrahigh theoretical specific capacity (1675 mA h g−1) of sulfur cathodes, lithium–sulfur (Li–S) batteries are one of the most promising and practical candidates for next-generation energy storage systems.1–3 Nevertheless, the commercialized application of Li–S batteries is hampered by their low practical capacity and poor cyclic stability, hindering their commercialization due to the sluggish redox kinetics for lithium polysulfide (Li2Sx, 4 ≤ x ≤ 8) conversion and the notorious lithium polysulfide (LiPS) shuttling effect.4–7 To address the above problems, designed materials are reported based on physical confinement and chemisorption. To date, many host materials have been introduced for Li–S batteries.8–13 For example, porous carbon materials endow a porous structure to physically confine LiPS shuttling.14 Metal oxides have excellent chemisorption to alleviate LiPS shuttling.8,11 However, both of these strategies are passive methods to tie the LiPSs in interior or surrounding of the host materials. Driven by the concentration gradient of the soluble LiPSs, the notorious shuttling effect is still hard to avoid.15,16 Hence, LiPSs have gained wide interest to introduce catalysis into Li–S batteries, hoping this strategy will accelerate the conversion of LiPSs for sulfur reduction reactions and sulfur evolution reactions.17–21 Up to now, nitrides, carbides and sulfides have been widely recommended as catalysts.From recent studies, heterojunction host materials can endow composites with favorable physicochemical properties, one component to chemisorb LiPSs and the other to catalyze LiPS conversion. Although heterojunction host materials possess certain advantages to meet the requirements of Li–S batteries, they are trapped by the limitation of finite heterointerfaces and active sites, hardly achieving the expected rapid conversion of LiPSs.20,22 In addition, too strong chemisorption of LiPSs on one component will block soluble LiPSs from moving to the other component, where further electrochemical conversions of LiPSs take place (Scheme 1(a)). Moreover, on the liquid–solid interface, it is not easy for Li2S4 species to gain two electrons and convert themselves to solid-phase Li2S2 in the whole electrochemical conversion process.23–26 Thus, it is difficult to obtain suitable chemisorption and accelerate the conversions of LiPSs for Li–S batteries.15,25,27,28 Of note, defect engineering is often used to modulate the properties of materials without introducing other elements. In the case of the Co9S8/MoS2 heterojunction, sulfur vacancies can be naturally thought to tailor the chemisorption and catalytic properties.29 However, the catalytic mechanisms are usually discussed but do not expound clearly.
 | ||
Scheme 1 Catalytic mechanism by which sulfur-vacancy heterojunctions enhance the interconversion of LiPSs. (a) Conventional Co9S8/MoS2 heterojunction: Co9S8 was designed to adsorb liquid-phase LiPSs (orange-yellow liquid balls), while MoS2 was used to convert LiPSs to Li2S. However, liquid-phase LiPSs are strongly adsorbed by Co9S8 and therefore cannot be transferred to MoS2 to accomplish fast conversion. (b) By the introduction of sulfur vacancies, heterojunction materials can harmonize the chemisorption of components to uniformly adsorb LiPSs and produce abundant  radicals to facilitate the conversion of LiPSs to Li2S via a chemical nucleation route. radicals to facilitate the conversion of LiPSs to Li2S via a chemical nucleation route. | ||
Based on the abovementioned discussion, reducing the accumulation of liquid LiPSs is crucial for the performance of Li–S batteries. We chose Co9S8/MoS2 heterojunction composites with sulfur vacancies as a template sulfur host to demonstrate how sulfur vacancies modulate the behavior of the heterojunction composites with respect to chemisorption and LiPS conversions (Scheme 1(b)). With the introduction of sulfur vacancies, a large number of free radicals 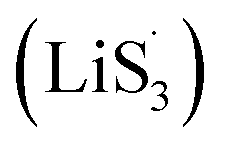 are generated on the sites of sulfur vacancies to promote the nucleation of Li2S via a liquidus disproportionated reaction. In addition, the harmonized chemisorption of heterojunctions can speed up LiPS transport between the two components and then maximize the catalytic effect to achieve rapid LiPS conversion. Furthermore, to assess the accumulation behavior of the liquid LiPSs, a new quantitative descriptor, Nucleation Transformation Ratio (NTR), is proposed. NTR is defined as the ratio of the die-out amount to the formation amount of liquid LiPSs, and can be calculated from basic cyclic voltammetry curves. Therefore, the transformation efficiency of S-related species in Li–S battery can be reflected quantitatively.
are generated on the sites of sulfur vacancies to promote the nucleation of Li2S via a liquidus disproportionated reaction. In addition, the harmonized chemisorption of heterojunctions can speed up LiPS transport between the two components and then maximize the catalytic effect to achieve rapid LiPS conversion. Furthermore, to assess the accumulation behavior of the liquid LiPSs, a new quantitative descriptor, Nucleation Transformation Ratio (NTR), is proposed. NTR is defined as the ratio of the die-out amount to the formation amount of liquid LiPSs, and can be calculated from basic cyclic voltammetry curves. Therefore, the transformation efficiency of S-related species in Li–S battery can be reflected quantitatively.
Experimental
Chemicals
Methyl-2-pyrrolidone (NMP, 99.5 wt%), polyvinylidene fluoride (PVDF, 99.5 wt%), elemental sulfur (99.5 wt%), lithium sulfide (Li2S, 99.98 wt%), 1,3-dioxolane (DOL, 99.8 wt%), 1,2-dimethoxyethane (DME, 99.5 wt%), tetraglyme (99 wt%), lithium bis (trifluoromethanesulfonyl) imide (LiTFSI, 99.95 wt%), lithium nitrate (LiNO3, 99.99 wt%), cobalt chloride hexahydrate (CoCl2·6H2O), potassium permanganate (KMnO4, 99.5 wt%), graphite powder, ammonium tetrathiomolybdate ((NH4)2MoS4, 99.95 wt%), reduced graphene oxide (rGO, chemically reduced) were purchased from Sigma-Aldrich. Graphene oxide (GO, >99 wt%) was purchased from Aladdin reagent. Hydrochloric acid (HCl, 37 wt%), sodium nitrate (NaNO3, 99 wt%), hydrogen peroxide (H2O2, 30 wt%) were purchased from Chuandong Reagent. Super P carbon (99.5 wt%) from Timcal were used as received. The carbon paper (HCP120, thickness ∼0.21 mm) was purchased from Shanghai HESEN Co., Ltd.Preparation of Co9S8/MoS2-rGO composite
The Co9S8/MoS2-rGO composites were synthesized through a simple hydrothermal synthesis and subsequent pyrolysis process. Briefly, 100 mg of graphene oxide (GO) powder was mixed with 45 mL deionized water, then ultrasonicated for 3 h to obtain the GO suspension. CoCl2·6H2O (5 mL; 8 mg mL−1) aqueous solution was then dropwise added into the prepared GO suspension under vigorous sonication for 30 min. Then, 35 mg (NH4)2MoS4 dispersed in 15 mL of deionized water similarly was added into the above-mentioned suspension drop by drop. The obtained suspension was transferred into a 100 mL Teflon-lined stainless-steel autoclave. The autoclave was moved to a oven to heat at 200 °C for 24 h. After annealing to room temperature, the solid product was collected by centrifugation and then washed several times with deionized water. The precursor was then freeze–dried at −70 °C. Finally, the precursor was annealed at 700 °C in N2 atmosphere using a heating rate of 2 °C min−1 to obtain the CMG-L composites.Preparation of CMG composites
The CMG-M and CMG-H composites with sulfur deficiency were formed by heating the CMG-L products in a H2/N2 (10%/90%) mixed gas. The reaction temperatures were chosen for 400 °C and 700 °C. All the composites are collectively called CMG.Preparation of sulfur composite cathode materials
The above CMG composite powders and sublimed sulfur with a weight ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 were mixed and ground. Then the mixture was heated to 155 °C and kept for 12 h in a tube furnace under an N2 atmosphere to obtain the sulfur composite cathode materials.
:7 were mixed and ground. Then the mixture was heated to 155 °C and kept for 12 h in a tube furnace under an N2 atmosphere to obtain the sulfur composite cathode materials.
Materials characterization
The prepared materials was characterized with various morphology and spectroscopy methods. The scanning electron microscopy (SEM) images were collected with a JEOL JSM-7800F. The transmission electron microscope (TEM) and high resolution transmission electron microscopy (HR-TEM) observations were performed with Tecnai G2F20 TWIN and JEM-2100F, respectively. X-ray diffraction (XRD) were performed with Rigaku D/max 2200 pc diffractometer under 40 kV and 40 mA with monochromatic Cu (Kα) radiation (λ = 1.54 Å). Raman spectra, ultraviolet-visible (UV-Vis) spectra, and X-ray photoelectron spectroscopy (XPS) were collected with Labram HR800 (HORIBA Jobin Yvon)), Lambda 750 spectrophotometer (PerkinElmer), and ESCALAB250Xi (Thermo Scientific instrument) with Al (Kα) (1486.6 eV) radiation, respectively. Thermo gravimetric analysis (TGA) was carried out by a Mettler Toledo TGA in the temperature range of 25 to 600 °C at a heating rate of 10 °C min−1 under N2 atmosphere. The pore structure and distribution was analyzed with Brunauer–Emmett–Teller (BET) method using a Micromeritics ASAP 2460 BET analyzer.Visualized adsorption test
10 mg of rGO, CMG-L, CMG-M and CMG-H were added into 2000 μL Li2S6 solution with ultrasonic dispersion for 1 min, followed by static adsorption in Ar-filled glovebox. The used Li2S6 solution was prepared by mixing lithium sulfide and element sulfur into the electrolyte based on the mixing solvent of DOL/DME with a volume ratio of 1:1 to form saturated 1 mM Li2S6 solution. All the operations were performed in an Ar-filled glovebox. After absorption for 12 h, the supernatant solution was poured into the cuvette for UV-Vis spectrum test on a Lambda 750 spectrometer.
Li2S nucleation tests
Elemental sulfur and Li2S was vigorous mixed for 24 h with a molar ratio of 7:1 in tetraglyme to obtain the 0.20 mol L−1 Li2S8 electrolyte. Carbon papers were punched into 12 mm circle disks to load CMG composites. The loading was controlled to be 1.0 mg cm−2. Lithium foils and the obtained CMG loaded carbon paper were used as the anode and cathode, respectively. LIR2032 coin cells was assembled with Celgard 2400 separator. The cathode was firstly be wetted with the previously prepared Li2S8 electrolyte, and the other 20 μL of LiTFSI (1.0 mol L−1) was added into the LIR2032 coin cell. These cells should be firstly galvanostatically discharged to 2.06 V, and then switched to 2.05 V potentiostatically test until the current below 10−5 A. These procedure to guarantee the fully precipitation of Li2S.30
Symmetric cell assembly and measurement
Typically, Co9S8/MoS2-rGO or CMG composites were mixed with PVDF and carbon black (with a mass ratio of 3:1:1) in NMP solvent. Then the mixtures were uniformly coated onto carbon papers. The average mass loading of electrodes were controlled at about 1 mg cm−2. Two identical electrodes as working and counter electrodes were assembled into a standard LIR2032 cell with a Celgard 2400 separator in an Ar-filled glovebox. 0.1 mol L−1 Li2S6 in 40.0 μL of DOL/DME (with a volume ratio of 1:1) was used as electrolyte, which also contained 1.0 M LiTFSI. Cyclic voltammetry (CV) tests were carried out on an electrochemical workstation (CHI660D, Shanghai Chenhua) at a scan rate of 1.0 mV s−1. The voltage range of CV measurement was −0.8 to 0.8 V. The symmetric cell with Li2S6-free electrolyte was also tested as a reference.
Electrochemical tests
Electrochemical tests of these electrode materials were performed using coin-type (LIR2032) cells. The cells were assembled with the prepared sulfur composite cathodes (active material:carbon black:PVDF = 8:1:1), lithium anodes, electrolyte and Celgard 2400 in an argon filled glove box with extremely low H2O and O2 concentrations (<1.0 ppm). For compatibility of sulfur cathode and lithium anode, DOL/DME formulation which possess moderate solvating capability for LiPSs was chosen as the solvent.31 The used electrolyte was 1 M LiTFSI dissolved in a mixed solvent of DOL/DME (volume ratio of 1:1). For each composite cathode, the average areal loading was around 3.2 mg cm−2 and sulfur content was about 68 wt%, with an electrolyte volume of 50 μL in full cells (the diameter of cathode is 12 mm, the electrolyte-to-sulfur ratio was 13.8 μL mg−1). The mass of the corresponding carbon paper was measured to be around 17.7 mg. The galvanostatic charge–discharge (GCD) tests were conducted on LAND CT2001A between 1.6–2.8 V (vs. Li/Li+). Galvanostatic intermittent titration technique (GITT) tests were performed on Land battery test system with discharging current of 167.2 mA g−1 for 0.5 h and resting for 2.5 h. The specific capacity and current rates (1C = 1672 mA h g−1) were calculated on the basic of the sulfur weight in the cathode. The CV tests were performed with an electrochemical workstation CHI660D with the cut-off voltage of 1.6–2.8 V. To evaluate the conductivities of the samples, CMG samples with different sulfur vacancies were collected after annealing process. Subsequently, under the press of 10 MPa, the obtained powders (about 100 mg) were compacted via an infrared spectroscopy tablet mould to obtain corresponding compact sheets. It should note that the diameter of the tablet mould is 12 mm and the sheet thickness was measured with an micrometer. The obtained compact sheets were assembled in LIR2032 coin-type cells without electrolyte for conductivity tests. Electrochemical impedance spectroscopy (EIS) data were recorded by applying a sine wave with 5 mV fluctuation from a frequency range of 100 mHz to 103 kHz (Princeton 1260A impedance analyser). Besides, the conductivity formula is as follows:
where R is the resistance, ρ is the specific resistance, L is the thickness of sheet, S is the area of the sheet, and σ is the electronic conductivity, respectively. All the electrochemical tests were performed at room temperature.
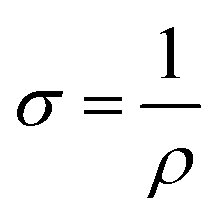
Theoretical calculation
Vienna ab initio simulation package (VASP) software was used to perform the density functional theory (DFT) theoretical calculations. The DFT+U calculation was implemented with the Perdew–Burke–Ernzerhof (PBE) functional within the generalized gradient approximation (GGA).32,33 To assess the adsorption behavior of LiPS and base materials, a 15 Å vacuum layers was set in the z-direction to avoid inter-layer interactions. For all the structural optimization processes, the bottom three layers were fixed and the other atoms were fully relaxed to reach a thermodynamical stable state. The cut-off energy was set to be 500 eV. 3 × 2 × 1 and 2 × 2 × 1 k-point sampling was provided for Co9S8 and MoS2, respectively. The convergence criterion was set to be 0.02 eV Å−1 for force on each atom and 10−5 eV for total energies during the geometry optimization calculations. The adsorption energy (ΔEads) of the species on base surface was determined by the following equation:33| ΔEads = Eads/base − Eads − Ebase |
Results and discussion
From previous studies, Co9S8 endows strong chemisorption to alleviate LiPS shuttling.34 In addition, MoS2 possesses weak chemisorption yet can significantly enhance the LiPSs conversion kinetics.35 Herein, we designed a sulfur-vacancy heterojunction material based on Co9S8 and MoS2 loaded with rGO to explore the promotion mechanism. The synthetic process of CMG with different sulfur vacancy densities was prepared following a hydrothermal and annealing method in different atmospheres, as illustrated in Fig. 1(a). Consequently, we defined the obtained CMG with a low sulfur vacancy as CMG-L, a middle sulfur vacancy as CMG-M and a high sulfur vacancy as CMG-H. The characteristic peaks in XRD spectra revealed the presence of two crystal structures: MoS2 and Co9S8 (Fig. S1†). XPS was used to confirm the heterojunction structure of CMG-L and MoS2-rGO (Fig. S2†). The Mo 3d binding energy (BE) indicates that the BE peaks at 228.9 eV and 231.9 eV are pointed to Mo4+ for CMG-L. Compared with MoS2-rGO, the 0.17 eV negative shift of the Mo4+ BE peak of CMG-L indicates the strong interaction and electron transfer between MoS2 and Co9S8. This observation implies that the heterojunction structure of Co9S8 and MoS2 were successfully obtained.36,37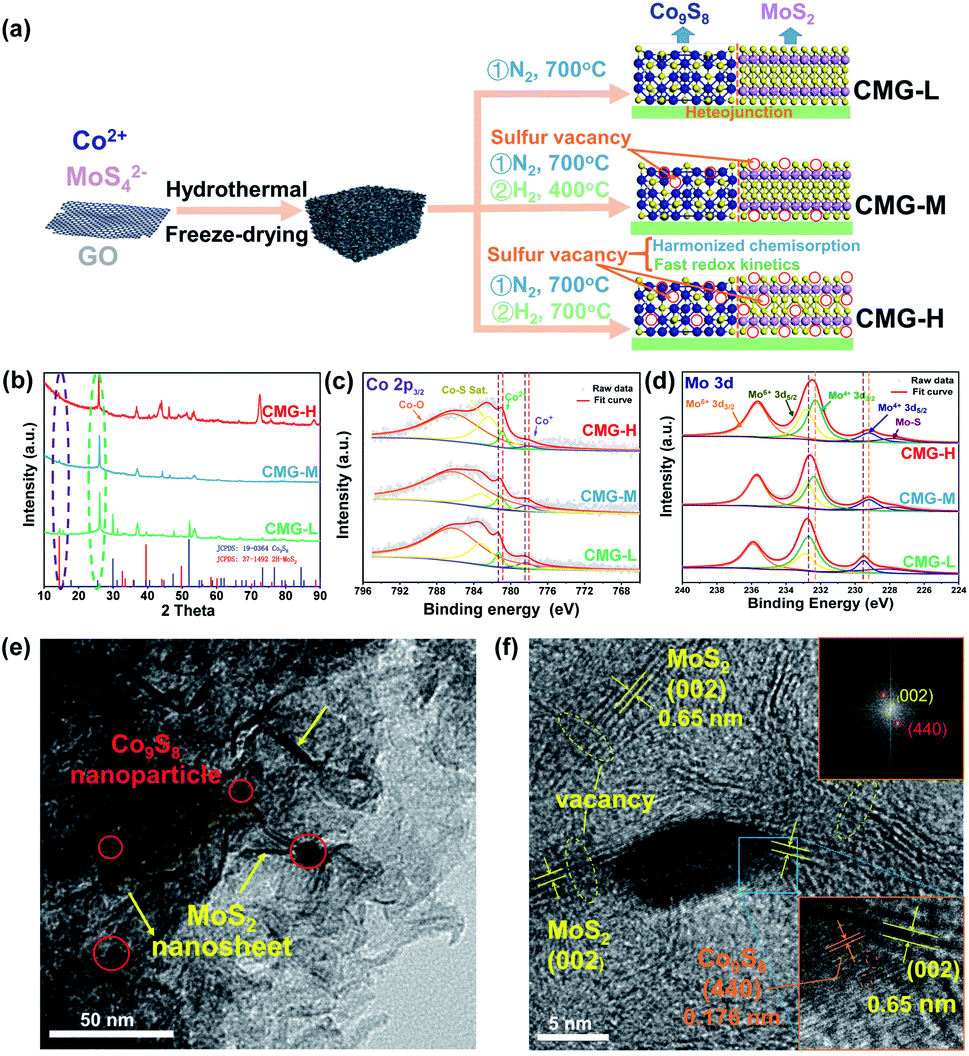 | ||
| Fig. 1 (a) Schematic of the synthesis of the CMG composite; (b) XRD spectra of CMG-L, CMG-M and CMG-H; high-resolution XPS spectra of Co 2p (c) and Mo 3d (d) for CMG-L, CMG-M and CMG-H; TEM (e) and selected area electron diffraction (SAED) images (f) of CMG-H. | ||
Based on previous research, the annealing atmosphere and temperatures were regulated, resulting in different sulfur vacancy concentrations.34,35,38 SEM images of the CMG composites revealed a porous, sponge-like morphology consisting of countless erect sheets with a lacunose interconnected structure (Fig. S3†). Moreover, with increasing sulfur vacancy concentration, CMG-H exhibited the largest specific surface area via N2 adoption–desorption isotherms, which displayed good exposure of sulfur vacancies on the catalyst surface for LiPS interconversions (Fig. S4†).
As shown in Fig. 1(b), the XRD peaks at the same location were in good agreement with Co9S8 and MoS2 for the CMG samples with different sulfur vacancy concentrations. With the increasing of the sulfur vacancy concentration, the negative shift of the 2θ degree peak at ∼14° (MoS2 (002)) and ∼26° (Co9S8 (220)) in the expanded view (Fig. S5†) indicated lattice expansions, which were mostly caused by the removal of sulfur by hydrogen.34,35 In addition, the XPS binding energy of Mo and Co binding energy shifted to a lower position, which also proved the partial reduction of Co9S8 and MoS2 (Fig. 1(c and d)).38 The sulfur vacancy concentrations can be calculated from the S ratio loss (from 0.09% to 0.51%) from Table S1,† which is consistent with the above analyzed fine spectra.
The sulfur-vacancy heterojunction material CMG-H was further investigated by TEM. As shown in Fig. 1(e) and S6,† the heterojunction is anchored on the reduced graphene oxide. Furthermore, Co9S8 exists in the form of nanoparticles, and MoS2 exists in the form of nanosheets. HR-TEM images of the Co9S8/MoS2 heterogeneous interface are shown in Fig. 1(f). Lattice fringes with spacings of 0.174 nm and 0.65 nm were indexed to the (440) plane of Co9S8 and the (002) plane of MoS2, respectively. The corresponding SEAD pattern (inset of Fig. 1(f)) also consistent with both planes. Moreover, the interface between Co9S8 (440) and MoS2 (002) can be observed, and discontinuous (002) and (440) facet crystal fringes also emerged on account of the presence of abundant sulfur vacancies.
It is well known that the chemisorption ability plays a significant role in inhibiting LiPS shuttling during cycling. Hence, to investigate the influence of different sulfur vacancy concentrations on chemisorption ability, first-principal calculations based on density functional theory (DFT) were used to probe the chemical adsorption energies between the components of the heterojunction and the representative soluble LiPS – Li2S6. As shown in Fig. 2(a), the adsorption energy between Li2S6 and Co9S8 (ΔEads = −3.84 eV) is much stronger than that between Li2S6 and MoS2 (ΔEads = −0.23 eV). From the view of thermodynamics, the results demonstrated that MoS2 is relatively easily adsorbed by Li2S6; and Co9S8 is more favorable for Li2S6 desorption. After the introduction of sulfur vacancies, Co9S8−x presented a weaker ΔEads (−3.55 eV) than Co9S8 (−3.84 eV). However, the plane of MoS2−y (002) showed a distinctly stronger ΔEads (−2.53 eV) than that of MoS2 (−0.23 eV) for Li2S6 adsorption. In this regard, the ΔEads difference for Co9S8−x and MoS2−y was significantly reduced after the introduction of sulfur vacancies. The chemisorptions strength between the heterojunction components are therefore harmonized. It is interesting that for Co9S8−x and MoS2−y, one of the S atoms in the chemisorbed Li2S6 will be unsymmetrical adsorbed to the sulfur vacancy, then leave a relatively reactive Li2S5 species, which may regulate the subsequent LiPS interconversions.
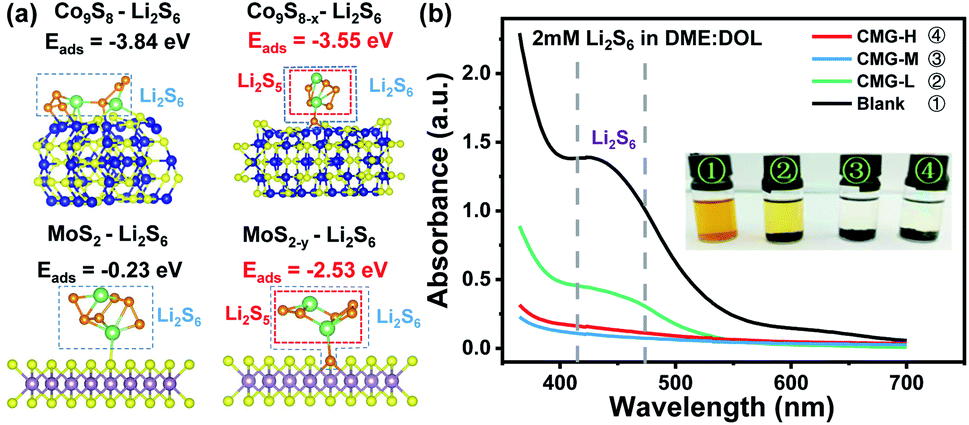 | ||
| Fig. 2 (a) Calculated adsorption energies (ΔEads) of Li2S6 on the (002) planes of MoS2 and MoS2−y crystals as well as (220) planes of Co9S8 and Co9S8−x crystals; (b) the adsorption abilities of three host materials with Li2S6 as the representative lithium polysulfides. | ||
The harmonized chemisorption behavior was further verified from the visualized adsorption tests. Visualized adsorption tests were adopted via heterojunction materials soaked in Li2S6 solution (inset of Fig. 2(b)). CMG-L can slightly lighten the color of the Li2S6 solution. The CMG-M and CMG-H composites can fully decolor the Li2S6 solution, suggesting their superior adsorption abilities for LiPSs. UV-Vis absorption tests provided quantitative comparisons to the chemisorption abilities of the materials. From Fig. 2(b), compared with the blank group and the CMG-L material, the CMG-M and CMG-H materials rarely showed no adsorption peaks in the 400–500 nm region for Li2S6.4,39 Also, as expected from the DFT calculation, the absorbance of CMG-M material with moderate sulfur vacancies exhibited the lowest Li2S6 signal, which can be attributed to the harmonized chemisorption – these exists an adsorption extremum for the heterojunction materials, because the different adsorption-energy shift directions for Co9S8−x and MoS2−y.
Commonly, a higher chemisorption ability corresponds to a better electrochemical performance, owing to the suppression of LiPS shuttling. However, although the chemisorption ability does not monotonously increase with sulfur vacancies, the electrochemical and battery performances nevertheless increase with sulfur vacancies. Sulfur composite cathode materials were made by the melt diffusion method (Fig. S7†) for CMG-L, CMG-M, and CMG-H. The sulfur loading was controlled to ∼3.2 mg cm−2, and the proportion was around 68% in the sulfur composite cathodes (Fig. S7†). Full cells were fabricated using lithium metal as the anode. From Fig. 3(a), full cells using the CMG-H electrode delivered the highest initial capacity (1129 mA h g−1) and lowest capacity decay rate (0.108% per cycle, after 300 cycles) at 0.5C. Of note, the high sulfur-vacancy composite cathodes CMG-H battery delivered the best charge/discharge specific capacity (1308 mA h g−1). Moreover, the plateau gap of CMG-H (190 mV) between charge/discharge is much smaller than that of CMG-L (221 mV) and CMG-M (269 mV), indicating that CMG-H possesses better kinetics during the charge/discharge process (Fig. 3(b)). In addition, as shown in Fig. 3(c), the CMG-H batteries exhibited the best rate performances, delivering discharge capacities of 1534, 1215, 1024, 938, and 816 mA h g−1 at rates of 0.1C, 0.2C, 0.5C, 1C, and 2C, respectively. When the rate gradually shifted back from 2C to 0.1C, the CMG-H battery also exhibited the best reversibility and excellent stability. It was extraordinary that CMG-M, with the strongest chemisorption, cannot express the best charge/discharge performance.
 | ||
| Fig. 3 (a) Long-term cycling performances of CMG-L, CMG-M and CMG-H at 0.5C. The sulfur loading was 3.2 mg cm−2; (b) galvanostatic charge/discharge profiles of CMG-L, CMG-M and CMG-H at 0.2C; (c) rate capability performance of CMG-L, CMG-M and CMG-H. | ||
The electron-transport ability difference could be one contribution factor to this phenomenon. It can be found that the electronic conductivities of CMG gradually improved with the increase of sulfur vacancies, which gives rise to fast transport of electrons (Fig. S8†).30,40 Besides, EIS measurements also registered the smallest charge transfer resistance (the size of the high-frequency semicircle in the Nyquist plot) for the CMG-H electrode (Fig. S9 and Table S2†). These results suggested that the CMG-H electrode can possess better conductivity ability, meaning that the adsorbed LiPSs can gain electrons more easily to convert to solid phase Li2S. However, except for the conductivity difference, the reaction kinetics should also be discussed to understand the difference between CMG-M and CMG-H.
With the increase of sulfur vacancies, faster reaction kinetics could be beneficial to improve the electrochemical performance of the battery. Therefore, it is worth further to determine the reason that the slightly weaker chemisorption of the high sulfur-vacancy heterojunction material CMG-H exhibited better electro-chemical performance than the low sulfur-vacancy material. Hence, it is meaningful to evaluate the role of sulfur vacancies on the interconversion reactions of LiPSs. CV tests were performed within a voltage window of −0.8 V to 0.8 V for symmetrical cells assembled by heterojunction materials with different sulfur vacancies (Fig. 4(a and b)).41 The CMG-L materials exhibited two pairs of broad peaks at −0.209/0.199 V and −0.083/0.083 V, which can be assigned to the conversion between Li2S6 and Li2Sx (x < 6), as well as the conversion between S8 and Li2S6, respectively.42 Moreover, as the sulfur vacancies increased, the peak current densities increased and the voltage hysteresis between the cathodic peaks and anodic peaks gradually decreased, indicated that the sulfur vacancies could dynamically accelerate the electrochemical reactions of LiPSs.43 The appearance of staged peaks at negative potentials, which correspond to the relatively short-chain polysulfide anion (S2−4) or free radical 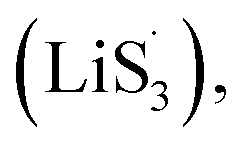 is worth comprehensively discussing to understand the role of sulfur vacancies.18,20,34,35,39,41,44 The appearance of sulfur radicals
is worth comprehensively discussing to understand the role of sulfur vacancies.18,20,34,35,39,41,44 The appearance of sulfur radicals 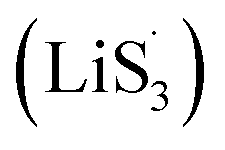 promoted by sulfur vacancies was tested by UV-Vis spectroscopy in a 1 mM Li2S8 solution. Li2S8 solution was used to simulate the active sulfur source, and dimethyl sulfoxide (DMSO) was used as the solvent because it could stabilize
promoted by sulfur vacancies was tested by UV-Vis spectroscopy in a 1 mM Li2S8 solution. Li2S8 solution was used to simulate the active sulfur source, and dimethyl sulfoxide (DMSO) was used as the solvent because it could stabilize  From Fig. 4(c), it is obvious that more sulfur vacancies correspond to more sulfur radicals
From Fig. 4(c), it is obvious that more sulfur vacancies correspond to more sulfur radicals 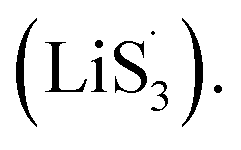 Sulfur radical was believed to be generated from Li2S5, which was produced from the sulfur-vacancy sites and Li2S6 (Fig. 2(a)). The formed Li2S5 is extremely unstable and easily converts to other LiPS intermediates, such as
Sulfur radical was believed to be generated from Li2S5, which was produced from the sulfur-vacancy sites and Li2S6 (Fig. 2(a)). The formed Li2S5 is extremely unstable and easily converts to other LiPS intermediates, such as  radicals.
radicals.
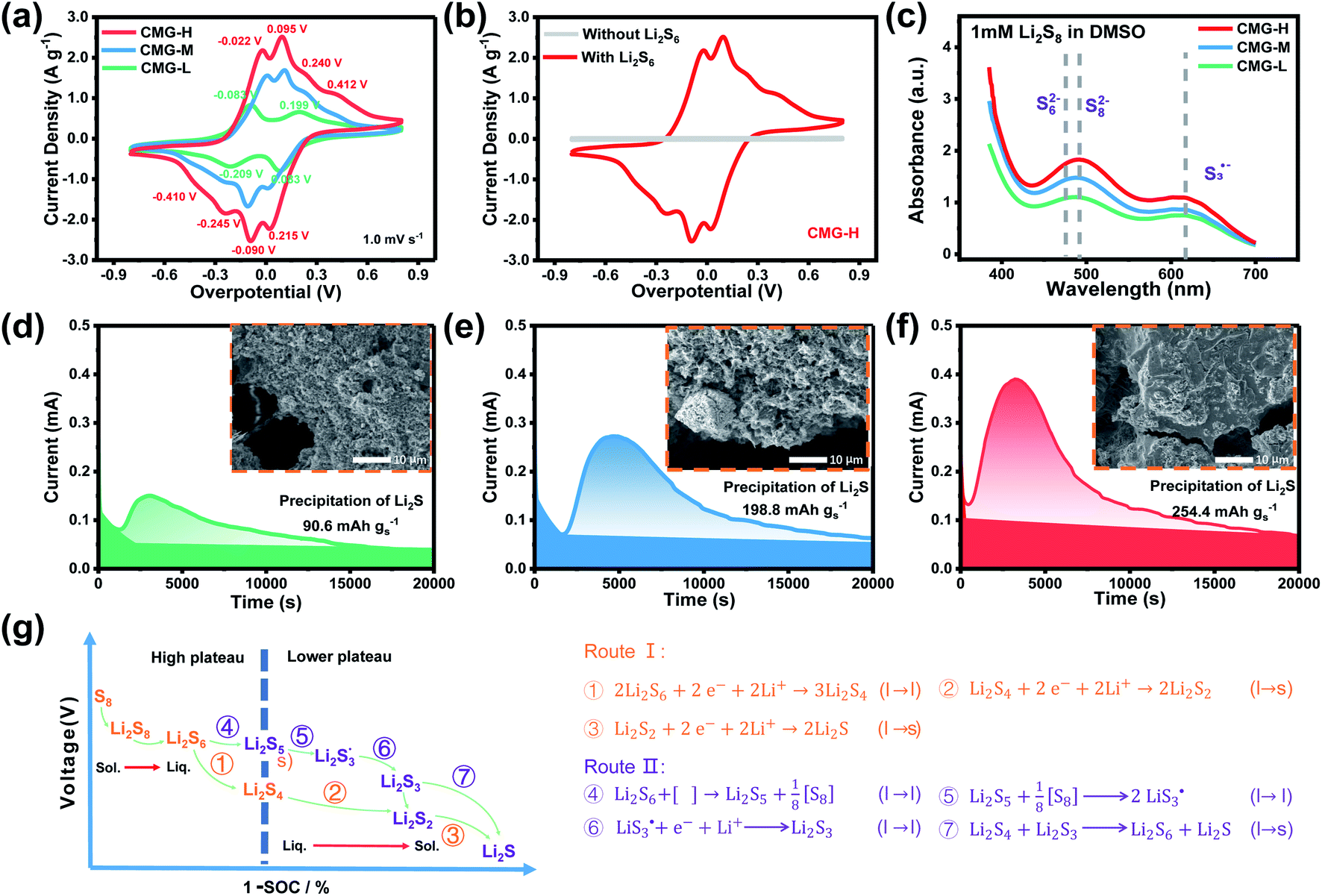 | ||
| Fig. 4 (a) Cyclic voltammograms of symmetric cells at 1 mV s−1; (b) cyclic voltammograms of symmetric cells with and without Li2S6 solution; (c) the variation in UV absorbance with increasing sulfur vacancy concentration (d–f) potentiostatic discharging curves of Li2S8/tetraglyme solution at 2.05 V and the corresponding SEM images after Li2S deposition; (g) proposed sulfur reduction reaction routes for the Li–S battery with the sulfur-vacancy heterojunction material. | ||
It is known that 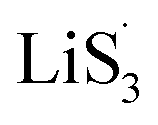 could accelerate the interconversion of LiPSs by providing additional chemical pathways, particularly for Li2S deposition.20,39,45 Therefore, to further investigate the role of sulfur vacancies, Li2S precipitation experiments were carried out with the above three host materials as electrodes (Fig. 4(d–f)). Galvanostatic discharge was conducted to 2.06 V, and then the voltage was kept at 2.05 V until the current was below 10−5 A.46 Nucleation experiments show that the capacity of precipitated Li2S on CMG-H (254.4 mA h gs−1) is much higher than those of the two other heterojunction materials (90.6 mA h gs−1 and 198.8 mA h gs−1).
could accelerate the interconversion of LiPSs by providing additional chemical pathways, particularly for Li2S deposition.20,39,45 Therefore, to further investigate the role of sulfur vacancies, Li2S precipitation experiments were carried out with the above three host materials as electrodes (Fig. 4(d–f)). Galvanostatic discharge was conducted to 2.06 V, and then the voltage was kept at 2.05 V until the current was below 10−5 A.46 Nucleation experiments show that the capacity of precipitated Li2S on CMG-H (254.4 mA h gs−1) is much higher than those of the two other heterojunction materials (90.6 mA h gs−1 and 198.8 mA h gs−1).
As shown in the insets of Fig. 4(d–f), after the deposition of Li2S, the surface of CMG-H is smoother and more uniform than the two other materials. It is interesting that the Li2S peak appeared much earlier for CMG-H than the others during the galvanostatic discharge process. This observation implies that the reduction reactions of LiPSs to Li2S occur much easier in CMG-H cathode, which can contribute to the detected electric current earlier. We named this current generated from all the S-related species involved reactions as the hybrid current. The earlier occurrence of the Li2S deposition current suggests that the liquid LiPSs will not accumulate but fastly be reduced to solid Li2S. Therefore, in the hybrid current, a greater contribution from LiPSs conversion to Li2S will decrease LiPSs shuttling to increase the stability of Li–S battery.
Based on the above discussion of the relationship of chemisorption and sulfur vacancies, the sulfur vacancies in the heterojunction could not only to harmonize the chemisorption, but also to adjust the redox kinetics process of LiPSs as follows. As illustrated in Fig. 4(g), harmonization of the chemisorption could hold back the LiPS shuttling. In the meantime, without the participation of  only the conventional electrochemical nucleation route (route I) can accomplish liquid–solid transformation. As the electrochemical requirement is crucial for liquid-phase Li2S4 to gain two electrons on the liquid–solid interface of the cathode to be converted to solid-phase Li2S2.43,47 Li2S and Li2S2 deposition is controlled by the sparsely distributed nucleation sites on the liquid–solid interface of the cathode. In comparison, in addition to the route I, sulfur vacancies can promote the formation of sulfur radicals
only the conventional electrochemical nucleation route (route I) can accomplish liquid–solid transformation. As the electrochemical requirement is crucial for liquid-phase Li2S4 to gain two electrons on the liquid–solid interface of the cathode to be converted to solid-phase Li2S2.43,47 Li2S and Li2S2 deposition is controlled by the sparsely distributed nucleation sites on the liquid–solid interface of the cathode. In comparison, in addition to the route I, sulfur vacancies can promote the formation of sulfur radicals 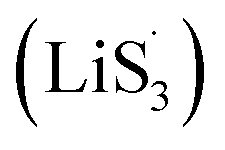 to accelerate liquid-phase conversion (chemical nucleation route, route II). Taking the discharge process as an example, sulfur vacancies chemisorb Li2S6 to form reactive Li2S5, and then Li2S5 reacts with solid S8 to form
to accelerate liquid-phase conversion (chemical nucleation route, route II). Taking the discharge process as an example, sulfur vacancies chemisorb Li2S6 to form reactive Li2S5, and then Li2S5 reacts with solid S8 to form 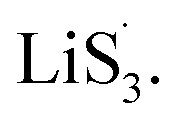 47–50 Liquid-phase
47–50 Liquid-phase  gains one electron and one Li+ to convert to liquidus Li2S3 (reaction ⑦). Then, Li2S3 may react with Li2S4 to convert to Li2S6 and solid-phase Li2S via a spontaneous liquid-phase disproportionated reaction (ΔH = −1.31 eV), simultaneously achieving the formation of homogeneous nucleation sites.
gains one electron and one Li+ to convert to liquidus Li2S3 (reaction ⑦). Then, Li2S3 may react with Li2S4 to convert to Li2S6 and solid-phase Li2S via a spontaneous liquid-phase disproportionated reaction (ΔH = −1.31 eV), simultaneously achieving the formation of homogeneous nucleation sites.
As the function of  is crucial for the deposition of Li2S and the formation of the hybrid current, the redox kinetics of heterojunctions with different sulfur vacancies were further studied by CV (Fig. 5(a–c)).51 Two cathodic peaks, located at 2.20–2.30 V (peak 1) and 1.80–2.05 V (peak 2) should correspond to the reduction of sulfur into high-order LiPSs (Li2Sx, 4 ≤ x ≤ 8) and further LiPSs to low-order Li2S2/Li2S.24–26,52 Simultaneously, the broad anodic peaks should be assigned to the oxidation of Li2S2/Li2S to Li2S6/S8via the formation of intermediate LiPSs.53 Compared with the CMG-L and CMG-M electrodes, the CMG-H electrode showed distinguished, smaller polarization, demonstrating that peak 1 and peak 2 of CMG-H shifted 0.03 V and 0.07 V, respectively, with the scan rate increase (Fig. 5(a–c)).
is crucial for the deposition of Li2S and the formation of the hybrid current, the redox kinetics of heterojunctions with different sulfur vacancies were further studied by CV (Fig. 5(a–c)).51 Two cathodic peaks, located at 2.20–2.30 V (peak 1) and 1.80–2.05 V (peak 2) should correspond to the reduction of sulfur into high-order LiPSs (Li2Sx, 4 ≤ x ≤ 8) and further LiPSs to low-order Li2S2/Li2S.24–26,52 Simultaneously, the broad anodic peaks should be assigned to the oxidation of Li2S2/Li2S to Li2S6/S8via the formation of intermediate LiPSs.53 Compared with the CMG-L and CMG-M electrodes, the CMG-H electrode showed distinguished, smaller polarization, demonstrating that peak 1 and peak 2 of CMG-H shifted 0.03 V and 0.07 V, respectively, with the scan rate increase (Fig. 5(a–c)).
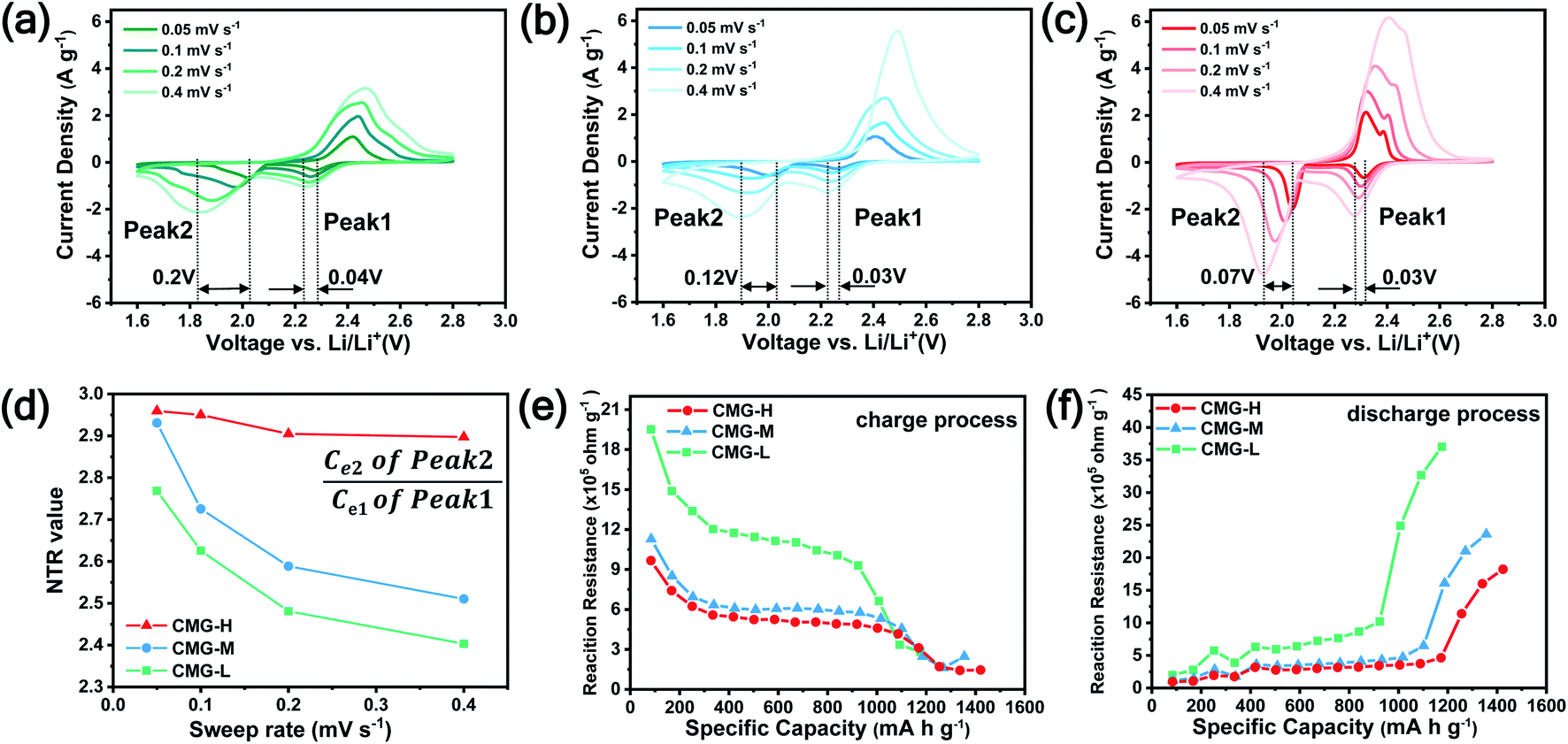 | ||
| Fig. 5 CV curves within the voltage range of 1.6–2.8 V at different sweep rates: (a) CMG-L. (b) CMG-M. (c) CMG-H. (d) The NTR values of CMG-L, CMG-M and CMG-H at different sweep rates; in situ reaction resistances during discharge (e) and charge (f) processes of CMG-L, CMG-M and CMG-H during GITT measurement. | ||
High-efficiency Li2S deposition was quantitatively analysed via CV curves. The discharge currents for peak 1 and peak 2 were integrated with time as the quantities of charge for the corresponding reactions. Herein, the quantity of electron transfer was defined for the three heterojunctions based on the following equation:
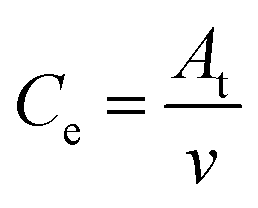 | (1) |
As illustrated in Table 1, 1 mol of solid-phase S8 molecules will obtain 4 mol of electrons to form 2 mol of liquid-phase Li2S4, which corresponds to peak 1 (reaction (1) in Table 1). These Li2S4 molecules will gain 12 mol of electrons to yield 8 mol of Li2S (peak 2, reaction (2) in Table 1). Therefore, a quantitative descriptor, named as Nucleation Transformation Ratio (NTR), to assess the kinetics behaviors of the cathode reactions can be defined:
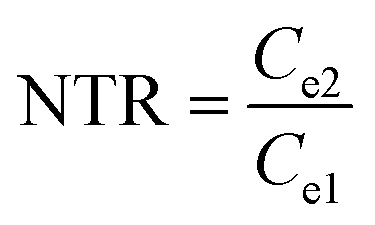 | (2) |
 followed by the disproportionated nucleation reaction. The fastest Li+ ion diffusion also confirmed the rapid conversion via the Randles–Sevcik equation, which validated our introduced NTR in the peak current vision (see further Discussion in ESI and Fig. S11 and S12†). In addition, the GITT measurements were performed. A constant current density of 167.2 mA g−1 (the theoretical 0.1C current for 1 g of sulfur) was applied for 1 h, and a pulse duration of 2.5 h was applied to collect the potential response after the first active cycle, as presented in Fig. 5(e), (f) and S13.†54 CMG-H consistently expressed the lowest reaction resistances in both charge/discharge processes, suggesting a minimum LiPS interconversion barrier.
followed by the disproportionated nucleation reaction. The fastest Li+ ion diffusion also confirmed the rapid conversion via the Randles–Sevcik equation, which validated our introduced NTR in the peak current vision (see further Discussion in ESI and Fig. S11 and S12†). In addition, the GITT measurements were performed. A constant current density of 167.2 mA g−1 (the theoretical 0.1C current for 1 g of sulfur) was applied for 1 h, and a pulse duration of 2.5 h was applied to collect the potential response after the first active cycle, as presented in Fig. 5(e), (f) and S13.†54 CMG-H consistently expressed the lowest reaction resistances in both charge/discharge processes, suggesting a minimum LiPS interconversion barrier.
| Peak | Reaction | Phase | Electron transfer number | Reaction |
|---|---|---|---|---|
| 1 | S8 + 4e− + 4Li+ → 2Li2S4 | Solid → liquid | 4 | (1) |
| 2 | 2Li2S4 + 12e− + 12Li+ → 8Li2S | Solid → liquid | 12 | (2) |
Conclusions
In summary, we introduced sulfur vacancies into heterojunction materials and conducted a systematic investigation of the chemisorption and kinetics of heterojunctions with increasing sulfur vacancies in Li–S cells. Chemisorption can be harmonized to realize a uniform distribution of LiPSs in the heterojunction. The introduction of sulfur vacancies can generate a large amount of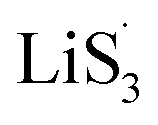 radicals, which can promote the nucleation of Li2S via a spontaneous disproportionated reaction. The formation of Li2S can participate in the hybrid current in the early discharge stage to reduce the accumulation of liquid LiPSs. Additionally, the defined descriptor nucleation transformation ratio was applied to quantitatively elucidate the kinetic behaviors of the materials and understand Li–S full battery performance. The catalytic mechanisms were therefore elucidated and paved the way for material design and theoretical direction.
radicals, which can promote the nucleation of Li2S via a spontaneous disproportionated reaction. The formation of Li2S can participate in the hybrid current in the early discharge stage to reduce the accumulation of liquid LiPSs. Additionally, the defined descriptor nucleation transformation ratio was applied to quantitatively elucidate the kinetic behaviors of the materials and understand Li–S full battery performance. The catalytic mechanisms were therefore elucidated and paved the way for material design and theoretical direction.
Author contributions
C. L. and Z. W. directed the project. H. Tang and R. Xu performed the main experimental works. Y. Z., F. W. and H. W. participate in some of experimental works. R. X. analysed the data and wrote the manuscript. R. X. and H. T. contributed equally. All the authors discussed the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research work was financially sponsored by National Natural Science Foundation of China (Grant No: 22075033, U21A20312, and 91834301).Notes and references
- R. Gan, N. Yang, Q. Dong, N. Fu, R. Wu, C. Li, Q. Liao, J. Li and Z. Wei, J. Mater. Chem. A, 2020, 8, 7253–7260 RSC.
- Q. Dong, R. Shen, C. Li, R. Gan, X. Ma, J. Wang, J. Li and Z. Wei, Small, 2018, 14, 1804277 CrossRef PubMed.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS PubMed.
- W. Chen, T. Lei, W. Lv, Y. Hu, Y. Yan, Y. Jiao, W. He, Z. Li, C. Yan and J. Xiong, Adv. Mater., 2018, 30, 1804084 CrossRef PubMed.
- Z. Cheng, H. Pan, J. Chen, X. Meng and R. Wang, Adv. Energy Mater., 2019, 9, 1901609 CrossRef.
- Q. Dong, T. Wang, R. Gan, N. Fu, C. Li and Z. Wei, ACS Appl. Mater. Interfaces, 2020, 12, 20596–20604 CrossRef CAS PubMed.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS PubMed.
- E. H. M. Salhabi, J. Zhao, J. Wang, M. Yang, B. Wang and D. Wang, Angew. Chem., 2019, 131, 9176–9180 CrossRef.
- D.-R. Deng, F. Xue, Y.-J. Jia, J.-C. Ye, C.-D. Bai, M.-S. Zheng and Q.-F. Dong, ACS Nano, 2017, 11, 6031–6039 CrossRef CAS PubMed.
- Z. Sun, J. Zhang, L. Yin, G. Hu, R. Fang, H.-M. Cheng and F. Li, Nat. Commun., 2017, 8, 1–8 CrossRef PubMed.
- Z. Li, J. Zhang and X. W. Lou, Angew. Chem., 2015, 127, 13078–13082 CrossRef.
- Z. Yuan, H.-J. Peng, T.-Z. Hou, J.-Q. Huang, C.-M. Chen, D.-W. Wang, X.-B. Cheng, F. Wei and Q. Zhang, Nano Lett., 2016, 16, 519–527 CrossRef CAS PubMed.
- F. Zhou, Z. Li, X. Luo, T. Wu, B. Jiang, L.-L. Lu, H.-B. Yao, M. Antonietti and S.-H. Yu, Nano Lett., 2018, 18, 1035–1043 CrossRef CAS PubMed.
- H. Pan, Z. Cheng, Z. Xiao, X. Li and R. Wang, Adv. Funct. Mater., 2017, 27, 1703936 CrossRef.
- L. Peng, Z. Wei, C. Wan, J. Li, Z. Chen, D. Zhu, D. Baumann, H. Liu, C. S. Allen and X. Xu, et al. , Nat. Catal., 2020, 3, 762–770 CrossRef CAS.
- X. Ding, S. Yang, S. Zhou, Y. Zhan, Y. Lai, X. Zhou, X. Xu, H. Nie, S. Huang and Z. Yang, Adv. Funct. Mater., 2020, 30, 2003354 CrossRef CAS.
- T. Zhou, W. Lv, J. Li, G. Zhou, Y. Zhao, S. Fan, B. Liu, B. Li, F. Kang and Q.-H. Yang, Energy Environ. Sci., 2017, 10, 1694–1703 RSC.
- Y. Song, W. Zhao, L. Kong, L. Zhang, X. Zhu, Y. Shao, F. Ding, Q. Zhang, J. Sun and Z. Liu, Energy Environ. Sci., 2018, 11, 2620–2630 RSC.
- L. Zhang, Y. Liu, Z. Zhao, P. Jiang, T. Zhang, M. Li, S. Pan, T. Tang, T. Wu and P. Liu, et al. , ACS Nano, 2020, 14, 8495–8507 CrossRef CAS PubMed.
- M. Zhang, W. Chen, L. Xue, Y. Jiao, T. Lei, J. Chu, J. Huang, C. Gong, C. Yan and Y. Yan, et al. , Adv. Energy Mater., 2020, 10, 1903008 CrossRef CAS.
- H. Ye, J. Sun, S. Zhang, H. Lin, T. Zhang, Q. Yao and J. Y. Lee, ACS Nano, 2019, 13, 14208–14216 CrossRef CAS PubMed.
- X. Liu, J.-Q. Huang, Q. Zhang and L. Mai, Adv. Mater, 2017, 29, 1601759 CrossRef PubMed.
- Z. Liu, P. B. Balbuena and P. P. Mukherjee, J. Phys. Chem. Lett., 2017, 8, 1324–1330 CrossRef CAS PubMed.
- C.-S. Cheng and S.-H. Chung, Chem. Eng. J., 2022, 429, 132257 CrossRef CAS.
- Y.-J. Yen and S.-H. Chung, ACS Appl. Mater. Interfaces, 2021, 13, 58712–58722 CrossRef CAS PubMed.
- C. Geng, W. Hua, D. Wang, G. Ling, C. Zhang and Q.-H. Yang, SusMat, 2021, 1, 51–65 CrossRef.
- Y. Wei, B. Wang, Y. Zhang, M. Zhang, Q. Wang, Y. Zhang and H. Wu, Adv. Funct. Mater., 2021, 31, 2006033 CrossRef CAS.
- C. Zu, L. Li, J. Guo, S. Wang, D. Fan and A. Manthiram, J. Phys. Chem. Lett., 2016, 7, 1392–1399 CrossRef CAS PubMed.
- J. Qian, S. Wang, Y. Li, M. Zhang, F. Wang, Y. Zhao, Q. Sun, L. Li, F. Wu and R. Chen, Adv. Funct. Mater., 2021, 31, 2006950 CrossRef CAS.
- Y. Zhang, P. Chen, Q. Wang, Q. Wang, K. Zhu, K. Ye, G. Wang, D. Cao, J. Yan and Q. Zhang, Adv. Energy Mater., 2021, 11, 2101712 CrossRef CAS.
- Y. Liu, Y. Elias, J. Meng, D. Aurbach, R. Zou, D. Xia and Q. Pang, Joule, 2021, 5, 2323–2364 CrossRef CAS.
- M. Bajdich, M. García-Mota, A. Vojvodic, J. K. Nørskov and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 13521–13530 CrossRef CAS PubMed.
- C. Huang, Z.-Q. Wang and X.-Q. Gong, Chin. J. Catal., 2018, 39, 1520–1526 CrossRef CAS.
- H. Lin, S. Zhang, T. Zhang, S. Cao, H. Ye, Q. Yao, G. W. Zheng and J. Y. Lee, ACS Nano, 2019, 13, 7073–7082 CrossRef CAS PubMed.
- H. Lin, L. Yang, X. Jiang, G. Li, T. Zhang, Q. Yao, G. W. Zheng and J. Y. Lee, Energy Environ. Sci., 2017, 10, 1476–1486 RSC.
- L. Diao, B. Zhang, Q. Sun, N. Wang, N. Zhao, C. Shi, E. Liu and C. He, Nanoscale, 2019, 11, 21479–21486 RSC.
- J. Ru, T. He, B. Chen, Y. Feng, L. Zu, Z. Wang, Q. Zhang, T. Hao, R. Meng and R. Che, et al. , Angew. Chem., 2020, 132, 14729–14735 CrossRef.
- W.-G. Lim, C. Jo, A. Cho, J. Hwang, S. Kim, J. W. Han and J. Lee, Adv. Mater., 2019, 31, 1806547 CrossRef PubMed.
- Q. Zou and Y.-C. Lu, J. Phys. Chem. Lett., 2016, 7, 1518–1525 CrossRef CAS PubMed.
- X. Zhang, Q. Liao, Z. Kang, B. Liu, X. Liu, Y. Ou, J. Xiao, J. Du, Y. Liu and L. Gao, et al. , Adv. Mater., 2021, 33, 2007051 CrossRef CAS PubMed.
- R. Li, H. Peng, Q. Wu, X. Zhou, J. He, H. Shen, M. Yang and C. Li, Angew. Chem., Int. Ed., 2020, 59, 12129–12138 CrossRef CAS PubMed.
- S. Rehman, T. Tang, Z. Ali, X. Huang and Y. Hou, Small, 2017, 13, 1700087 CrossRef PubMed.
- Z. Du, X. Chen, W. Hu, C. Chuang, S. Xie, A. Hu, W. Yan, X. Kong, X. Wu and H. Ji, et al. , J. Am. Chem. Soc., 2019, 141, 3977–3985 CrossRef CAS PubMed.
- H. Lin, S. Zhang, T. Zhang, H. Ye, Q. Yao, G. W. Zheng and J. Y. Lee, Adv. Energy Mater., 2018, 8, 1801868 CrossRef.
- M. Cuisinier, C. Hart, M. Balasubramanian, A. Garsuch and L. F. Nazar, Adv. Energy Mater., 2015, 5, 1401801 CrossRef.
- F. Y. Fan, W. C. Carter and Y.-M. Chiang, Adv. Mater, 2015, 27, 5203–5209 CrossRef CAS PubMed.
- P. Schön, F. Hintz and U. Krewer, Electrochim. Acta, 2019, 295, 926–933 CrossRef.
- K. H. Wujcik, T. A. Pascal, C. Pemmaraju, D. Devaux, W. C. Stolte, N. P. Balsara and D. Prendergast, Adv. Energy Mater., 2015, 5, 1500285 CrossRef.
- Q. Wang, J. Zheng, E. Walter, H. Pan, D. Lv, P. Zuo, H. Chen, Z. D. Deng, B. Y. Liaw and X. Yu, et al. , J. Electrochem. Soc., 2015, 162, A474 CrossRef CAS.
- A. Kumar, A. Ghosh, M. Forsyth, D. R. MacFarlane and S. Mitra, ACS Energy Lett., 2020, 5, 2112–2121 CrossRef CAS.
- J.-P. Jones, S. C. Jones, F. C. Krause, J. Pasalic and R. Bugga, J. Phys. Chem. Lett., 2018, 9, 3751–3755 CrossRef CAS PubMed.
- X. Gao, X. Yang, M. Li, Q. Sun, J. Liang, J. Luo, J. Wang, W. Li, J. Liang and Y. Liu, et al. , Adv. Funct. Mater., 2019, 29, 1806724 CrossRef.
- B. Li, Q. Su, L. Yu, J. Zhang, G. Du, D. Wang, D. Han, M. Zhang, S. Ding and B. Xu, ACS Nano, 2020, 14, 17285–17294 CrossRef CAS PubMed.
- G. Liu, C. Yuan, P. Zeng, C. Cheng, T. Yan, K. Dai, J. Mao and L. Zhang, J. Energy Chem., 2022, 67, 73–81 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc01353c |
| ‡ Rui Xu and Hongan Tang contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2022 |