
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHarnessing natural-product-inspired combinatorial chemistry and computation-guided synthesis to develop N-glycan modulators as anticancer agents†
Wei-An
Chen
 a,
Yu-Hsin
Chen
a,
Chiao-Yun
Hsieh
a,
Pi-Fang
Hung
a,
Chiao-Wen
Chen
a,
Chien-Hung
Chen
a,
Jung-Lee
Lin
a,
Ting-Jen R.
Cheng
a,
Tsui-Ling
Hsu
a,
Ying-Ta
Wu
a,
Chia-Ning
Shen
*a and
Wei-Chieh
Cheng
*abcd
a,
Yu-Hsin
Chen
a,
Chiao-Yun
Hsieh
a,
Pi-Fang
Hung
a,
Chiao-Wen
Chen
a,
Chien-Hung
Chen
a,
Jung-Lee
Lin
a,
Ting-Jen R.
Cheng
a,
Tsui-Ling
Hsu
a,
Ying-Ta
Wu
a,
Chia-Ning
Shen
*a and
Wei-Chieh
Cheng
*abcd
aGenomics Research Center, Academia Sinica, 128, Section 2, Academia Road, Taipei, 11529, Taiwan. E-mail: wcheng@gate.sinica.edu.tw
bDepartment of Chemistry, National Cheng-Kung University, 1, University Road, Tainan 701, Taiwan
cDepartment of Applied Chemistry, National Chiayi University, 300, Xuefu Rd, East Dist., Chiayi 600, Taiwan
dDepartment of Medicinal and Applied Chemistry, Kaohsiung Medical University, 100 Shih-Chuan 1st Rd, Kaohsiung 807, Taiwan
First published on 19th April 2022
Abstract
Modulation of N-glycosylation using human Golgi α-mannosidase II (α-hGMII) inhibitors is a potential anticancer approach, but the clinical utility of current α-hGMII inhibitors is limited by their co-inhibition of human lysosomal α-mannosidase (α-hLM), resulting in abnormal storage of oligomannoses. We describe the synthesis and screening of a small library of novel bicyclic iminosugar-based scaffolds, prepared via natural product-inspired combinatorial chemistry (NPICC), which resulted in the identification of a primary α-hGMII inhibitor with 13.5-fold selectivity over α-hLM. Derivatization of this primary inhibitor using computation-guided synthesis (CGS) yielded an advanced α-hGMII inhibitor with nanomolar potency and 106-fold selectivity over α-hLM. In vitro studies demonstrated its N-glycan modulation and inhibitory effect on hepatocellular carcinoma (HCC) cells. In vivo studies confirmed its encouraging anti-HCC activity, without evidence of oligomannose accumulation.
Introduction
Protein glycosylation is implicated in numerous biological functions such as cellular development, movement, and recognition;1,2 and the aberrant distribution patterns of N-glycans found in tumor cells are associated with cancer growth and metastasis.3,4 Human Golgi α-mannosidase II (α-hGMII, EC 3.2.1.114) plays a key role in the modulation of N-glycan processing (as part of the N-glycan biosynthetic pathway), and its inhibition is known to induce tumor repression.5–7 For example, Drosophila α-GMII (α-dGMII) catalyzes the sequential hydrolysis of two mannoses from GlcNAcMan5GlcNAc2 to produce GlcNAcMan3GlcNAc2 – the main precursor of complex type N-glycans including the branched N-glycans associated with cancer and abnormal cell behavior.8–11The indolizidine alkaloid swainsonine (sw) is a potent but poorly selective α-GMII inhibitor, also inhibiting human lysosomal α-mannosidase (α-hLM, EC 3.2.1.24), causing the accumulation of undegraded oligomannoses in cells or tissues (swainsonine-induced mannosidosis) and limiting its further development (Fig. 1a).7,12 Although sw derivatives such as 5-substituted sw analogues and other monocylic iminosugars incorporating a pyrrolidine or pyrrolidone skeleton have been investigated, their inhibitory potency and selectivity were all found to be unsatisfactory;13–17 and crystal structure studies using α-dGMII and bovine lysosomal α-mannosidase (α-bLM) concluded that the key residues in the active site pocket and the zinc coordination involved in sw binding are highly conserved in both enzymes, confounding a simple solution to this vexing selectivity problem.8,14,18
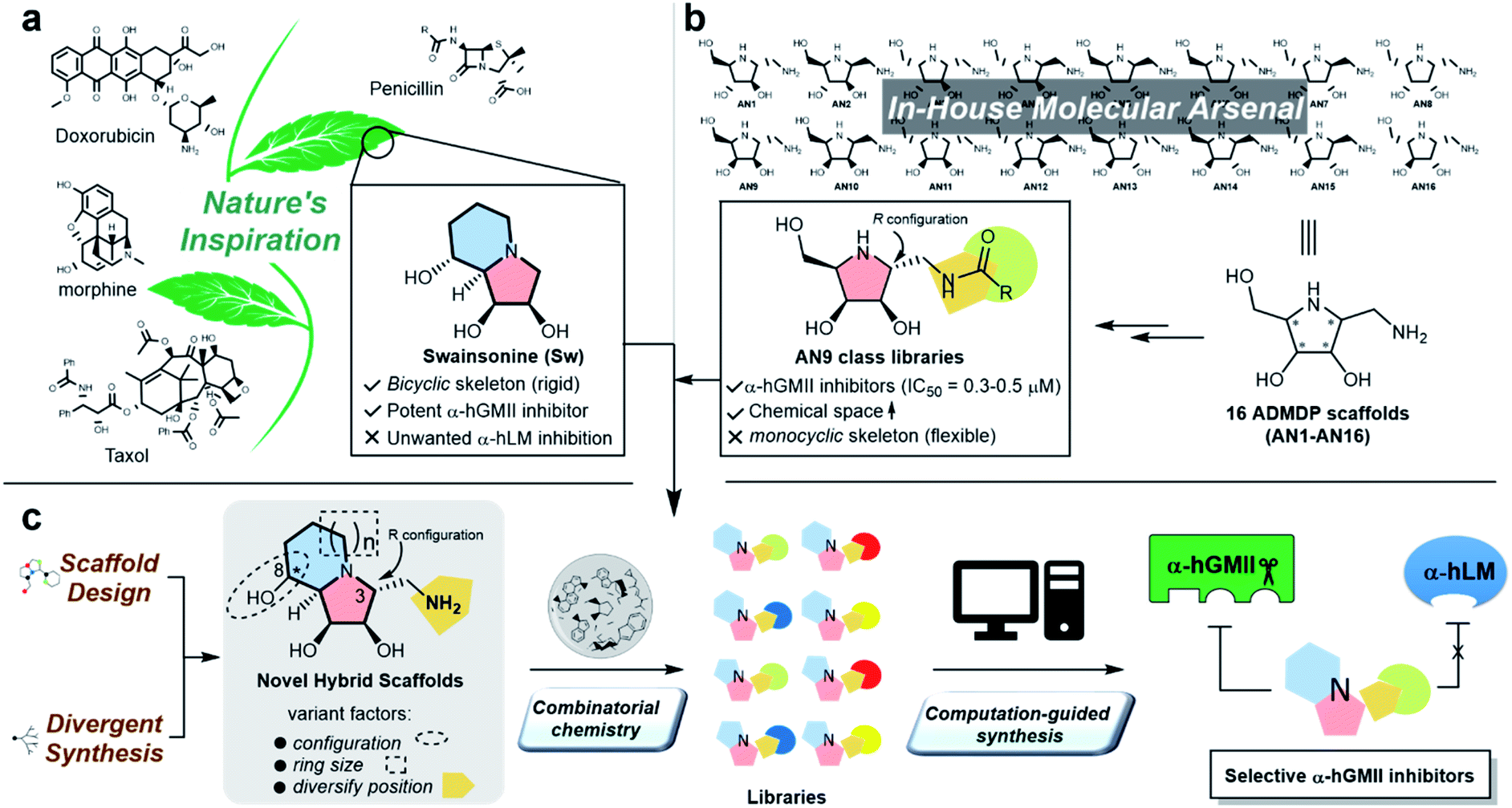 | ||
| Fig. 1 The natural product-inspired discovery of new selective α-hGMII inhibitors. Inspired by (a) sw and (b) AN9-type molecules from our in-house molecular arsenal to (c) design and synthesize novel scaffolds with core-, configuration- and substituent diversity in a divergent manner for exploration of selective α-hGMII inhibitors assisted by combinatorial chemistry and computation-guided synthesis. | ||
One of our research interests is the expeditious development of inhibitors and activators of disease-associated glyco-processing enzymes.15,19–21 In previous work, we identified pyrrolidine AN9 incorporating the 2R,3R,4S,5R configuration pattern as a potential α-hGMII-inhibiting scaffold, whose C-2 aminomethyl moiety could be well-suited to diversity exploration (Fig. 1b). Herein, we describe the design and synthesis of a series of new bicyclic iminosugar-based scaffolds bearing a diversity position on the polyhydroxylated pyrrolidine substructure, followed by the development of novel α-hGMII inhibitors guided by natural product-inspired combinatorial chemistry (NPICC) and computation-guided synthesis (CGS). The resulting inhibitors were assessed for their in vitro N-glycan modulation, anti-hepatocellular carcinoma (HCC) activity, cell migration and invasion studies, and in vivo anti-HCC activity.
Results and discussion
General chemistry strategy and new molecular scaffold design
Inspired by the skeletons of sw and AN9, a new bicyclic scaffold bearing a C3 aminomethyl group with the R configuration (Fig. 1c) was adopted as the basic structure for the development of a novel and selective α-hGMII inhibitor. A bicyclic-based scaffold was selected due to its conformational constraints, which were anticipated to decrease internal molecular rotations and hence improve the binding potency or selectivity.21 Scaffolds incorporating both C8 epimers and alternately sized rings were prepared and assessed for their inhibitory selectivity. To efficiently synthesize molecules for testing, a divergent synthetic approach incorporating several common intermediates was planned.22 The C3 aminomethyl moiety of these scaffolds was anticipated to allow rapid substituent diversity via the amide or (thio)urea formation, and conjugation with a structurally diverse carboxylic acid or isocyanate library collection, increasing the chemical space for further enzyme inhibition and selectivity studies. Further structural modifications were guided by computational analysis and docking studies to efficiently create the advanced α-hGMII inhibitors (Fig. 1c).Preparation and bioevaluation of scaffolds 8a, 8b, 10a, and 10b
Our synthesis of desired scaffolds 8a, 8b, 10a, and 10b commenced with cyclic nitrone 1, prepared from L-ribose according to previous reports (Fig. 2a).19,23 Selective nucleophilic addition of TMSCN to 1 followed by (i) our previously reported, one-pot N–O and nitrile reduction in the presence of Boc2O using RANEY® Ni/H2,19 (ii) selective deprotection of the trityl group under mild acidic conditions (formic acid),24 and (iii) Dess–Martin periodinane (DMP) oxidation led to aldehyde 3 (52% yield over 4 steps). Notably, 3 could be prepared on a multigram scale (>10 g), qualifying it as a common intermediate for our divergent synthetic application. Aldehyde 3 was reacted with AllylMgBr to afford the homoallylic alcohol 4a.20 The high diastereoselectivity of this transformation can be explained using the Felkin–Ahn model;25 the stereochemistry of 4a was confirmed by X-ray crystallographic analysis (Fig. 2b). | ||
| Fig. 2 Pathways for synthesis of target scaffolds 8a, 8b, 10a and 10b. Reagents and conditions: (a) TMSCN, MeOH, 50 °C, 2 h, 90%. (b) (1) RANEY® Ni, H2, Boc2O, MeOH, 8 h, rt, 74%; (2) HCOOH, Et2O, rt, 2 h, 78%. (c) DMP, CH2Cl2, rt, 2 h, 99%. (d) AllylMgCl, THF, 0 °C to rt, 1 h, 64%. (e) BH3-THF, THF, rt, 1 h, then, NaOH(aq), H2O2, rt, 1 h, 75%. (f) (1) DMP (1.1 eq.), CH2Cl2, rt, 2 h. (2) TFA, CH2Cl2, 0 °C, 1 h. (3) Pd/C, H2, MeOH, rt, o.n. (g) 3-butenylMgBr, THF, 0 °C to rt, 1 h, 78%. (h) (1) ZnBr2, CH2Cl2, rt, 12 h; (2) CbzCl, NaHCO3(aq), THF, rt, 3 h, 69% (7a) and 76% (9a) over 2 steps. (i) (1) OsO4, NaIO4, 2,6-lutidine, dioxane/H2O, rt, 8 h; (2) Pd/C, H2, MeOH, rt, o.n. (3) HCl, MeOH, rt, 8 h, 80% (8a), 90% (8b), 84% (10a) and 78% (10b) over 3 steps. (j) Tf2O, py, CH2Cl2, 0 °C to rt, 3 h, 73% (6b) and 75% (9b). (k) (1) LiOH, EtOH, H2O, 90 °C, 12 h; (2) CbzCl, NaHCO3(aq), THF, rt, 3 h, 69% (7b) and 73% (S17) over 2 steps. HRMS refers to high resolution mass. | ||
Initial attempts to prepare scaffold 8a from 4a by either (i) SN2-type cyclization,20 or (ii) reductive amination/cyclization resulted in intractable mixtures of products containing only very low yields of 8a (Fig. 2b and ESI S1†). Accordingly, we re-designed the synthetic route (Fig. 2c). Following the similar conversion from 3 to 4a, nucleophilic addition of 3-butenylMgBr to 3 gave 6a as a single isomer (78%). Removal of the two N-Boc groups of 6a using ZnBr2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 26,30 followed by N-Cbz protection furnished bis-N-Cbz 7a (69% over 2 steps). Lemieux–Johnson oxidation of 7a27 followed by reductive amination (H2, Pd/C) and acetonide deprotection resulted in an excellent yield of the first scaffold 8a (80% over 3 steps), the configuration of which was confirmed by 2D NOESY analysis (Fig. 2c).
26,30 followed by N-Cbz protection furnished bis-N-Cbz 7a (69% over 2 steps). Lemieux–Johnson oxidation of 7a27 followed by reductive amination (H2, Pd/C) and acetonide deprotection resulted in an excellent yield of the first scaffold 8a (80% over 3 steps), the configuration of which was confirmed by 2D NOESY analysis (Fig. 2c).
Our attention then turned to the synthesis of scaffold 8b, the C8-epimer of 8a (Fig. 2d). Intermediate 6a was reacted with Tf2O to generate cyclic carbamate 6b (incorporating the appropriately inverted chiral center) in 73% yield.28 The stereochemistry of 6b was determined by 2D NOESY NMR analysis. Hydrolysis of 6b followed by N-Cbz protection gave alcohol 7b in 69% yield over 2 steps. Similarly, following the sequence of transformations from 7a to 8a, the second desired scaffold 8b was synthesized from 7b in 90% over 3 steps.
The preparation of the two ring-contracted scaffolds 10a and 10b is depicted in Fig. 2e. Compound 4a was subjected to similar conditions as that for 6a, and then two new scaffolds 10a (64% over 5 steps) and 10b (43% over 6 steps) were obtained. Notably, 10a was also obtained from 4a using our established protocol via the SN2 type cyclization.20 Chemically, we successfully developed synthetic routes to prepare common intermediates 3, 4a, and 6a, which play a key role in building our desired scaffolds.
With four scaffolds 8a, 8b, 10a, and 10b in hand, their inhibitory activity against α-hGMII was evaluated. As shown in Fig. 2, 8a was less active (IC50 = 48 μM) compared to 8b (IC50 = 0.65 μM), but both 8a and 8b (5-6 ring fused analogues) were found to have better activities compared to their ring-contracted analogues 10a and 10b (5-5 fused analogues), which showed nearly no α-hGMII inhibition (IC50 > 200 μM). These findings suggested that the ring size of the second ring and the orientation of the C8 hydroxyl group significantly influence the inhibitory potency. Based on these results and the incorporation of a pendent amino methylene moiety considered suitable for structural diversity, 8b was selected for further development.
Discovery of selective α-hGMII inhibitors via NPICC and CGS
Scaffold 8b was coupled with a random selection of 96 iso(thio)cyanates to generate the corresponding (thio)urea derivatives via combinatorial parallel synthesis (ESI Fig. S2†).29 After 24 h, 8b was consumed and formation of the corresponding product was monitored by TLC or MS. The reaction was quenched by adding an aqueous buffer (see ESI methods†). The products were directly evaluated without further purification in an enzyme-based assay, as previously reported.15 Only 8b-1 was found to exhibit a high relative inhibition ratio (RIR) of around 3.7 (the RIR values of most analogues including sw and scaffold 8b were in the range of 0.36 to 1.06). Compound 8b-1 was therefore considered to be a promising hit worthy of further investigation (ESI Fig. S3†).After re-synthesis of 8b-1 and its truncated form 8b-2 (for comparison), their inhibition constant (Ki) and selectivity were examined (Fig. 3b and S5 ESI†). The selectivity index (SI) of 8b-1, 8b-2, scaffold 8b, and sw was 13.5, 3.1, 3.7, and 4.6, respectively. The α-hGMII inhibitory activities of 8b-1 (Ki = 0.35 μM, competitive) and 8b-2 (Ki = 0.2 μM, competitive) were similar, indicating that the 4-hexylcyclohexyl moiety of 8b-1 does not significantly contribute to its potency. In contrast, 8b-1 (Ki = 4.72 μM, competitive) was 8-fold less potent than 8b-2 (Ki = 0.61 μM, competitive) against α-hLM; presumably binding of the hydrophobic substituent of 8b-1 was not favorable in the α-hLM binding pocket, but its attenuation of the α-hLM enzyme activity increased the SI value (8b-1vs.8b-2). Therefore, 8b-1 was selected for further development.
 | ||
| Fig. 3 Synthesis and evaluation of a compound library. (a) Parallel synthesis of library 8b followed by in situ enzyme-based inhibition evaluation against α-hGMII and α-hLM to discover potentially selective α-hGMII inhibitors. (b) Table of α-hLM, α-hGMII inhibition data of sw, 8b, 8b-1 and 8b-2. Computational modeling of the complex between (c) α-hGMII and 8b-3 and (d) α-hLM and 8b-3. (e) Molecular structure of 8b-4. (f) Table of α-hLM, α-hGMII inhibition data of 8b-3 and 8b-4. (g) Table of Y354A α-hGMII inhibition data of sw and 8b-3. Assays were conducted using Man-α-4MU as the substrate (5 mM for wild-type α-hGMII; 2.5 mM for α-hLM and Y354A α-hGMII) and are described in the ESI.† The Km of α-hLM, α-hGMII and Y354A α-hGMII toward 4MU-α-Man is 0.56, 1.22 and 0.63 mM, respectively (ESI Fig. S4†). RIR: Relative inhibition ratio = inhibition percentage against α-hGMII/inhibition percentage against α-hLM at certain concentrations; Ki: inhibition constant; SI: selectivity index = [(α-hLM Ki)/(α-hGMII Ki)]; data are the mean of three determinations. | ||
To date, no structure of α-hLM and α-hGMII is available. In contrast, both α-dGMII and α-bLM display high sequence identity with their corresponding α-hGMII and α-hLM, respectively, (ESI Fig. S6†) and their crystal structures are accessible, allowing us to apply them as the 3D templates to build the human enzyme structures for molecular docking (ESI Fig. S7†).18,30 In our study, the residues of the indolizidine-binding pocket in both enzymes were highly conserved, and the binding modes of the indolizidine moiety of sw and 8b-1 were similar, implying that the selectivity of 8b-1 is mainly due to its substituents. The thiourea and phenyl moieties of 8b-1 were found to create two new hydrogen bonding interactions and a π–π interaction with Tyr 354; the 4-hexylcyclohexyl moiety of 8b-1 might undergo extra sigma–π interactions with Tyr 352 and His 358, and hydrophobic interactions with the nonpolar side chain of Gln 150 and Tyr 316 of α-hGMII (ESI Fig. S8a†). However, the 4-hexylcyclohexyl moiety of 8b-1 did not show sigma–π or hydrophobic interaction in the α-hLM model. Furthermore, the active site cleft of α-hLM is much wider than that of α-hGMII (13.8 Å vs. 8.7 Å), indicating that binding between 8b-1 and α-hLM is disfavored since the hydrophobic moiety of 8b-1 precludes its binding to the hydrophilic region in α-hLM (ESI Fig. S8b†).
To assist in the design of 8b-1 derivatives, several molecules were proposed and their α-hGMII binding energies were calculated (ESI Table S1†). Among them, 8b-3 incorporating a 4-propylcyclohexyl moiety was selected for investigations and its molecular models with α-hGMII and α-hLM were built (Fig. 3c and d). The binding mode of 8b-3 with α-hGMII showed that the 4-propylcyclohexyl moiety could undergo more sigma–π interactions with His 358 and Tyr 352 and offers less steric hindrance with protein residues, compared to 8b-1 bearing the longer hexyl chain (Fig. 3c and ESI S8c†). In contrast, the hydrophobicity of 8b-3, like 8b-1, disfavored its binding with α-hLM (Fig. 3d). Thus, we were hopeful that 8b-3 would be a potent and selective α-hGMII inhibitor.
Thiourea 8b-3 was synthesized, along with amide 8b-4 (for comparison purposes, Fig. 3e). Results showed that 8b-3 exhibited nanomolar potency against α-hGMII (Ki = 0.03 μM, competitive), a tenfold increase of α-hGMII inhibition compared to 8b-1, with the SI up to 106 (Fig. 3f, ESI S5†). As predicted, 8b-4 was much less potent than 8b-3 against α-hGMII and much less selective (SI value = 9.3), indicating that the thiourea linkage is better than the amide moiety. We also undertook a mutagenesis study; the activity of 8b-3 against Y354A α-hGMII was reduced by about 64-fold (Ki = 1.92 μM, competitive) compared to wild-type α-hGMII, suggesting that the phenyl ring of 8b-3 and the aromatic residue of Tyr 354 in α-hGMII significantly interact (π–π interactions) (Fig. 3g, S5 ESI†). In contrast, the Ki values of sw for Y354A and wild-type were similar, presumably because sw lacks substituents.
Studies of inhibitors with natural substrate-like oligosaccharides as enzyme substrates
In the preceding bioassays, the fluorogenic Man-α-4MU was selected as an easy and accessible substrate for both enzymes. However, the natural substrates of both enzymes are quite structurally different. Accordingly, we sought to prepare glycan bio-substrates more closely resembling those found in vivo, to further investigate the inhibition potency and selectivity of qualified inhibitors such as 8b-3 in an in vitro enzyme activity platform. In our UPLC-based assay platform, fluorescence labelling of glycan substrates with 2-aminobenzamide (2AB) was required to increase the signal sensitivity to easily monitor the enzyme activity or inhibitor activity through the peak area ratio of the remaining substrate to the internal standard.31 Based on our design, GlcNAcMan5GlcNAc2-2AB and Man5GlcNAc2-2AB32 were chosen as the substrate of α-hGMII and α-hLM, respectively. Gal-β-1,4-GlcNAc-β-1,3-Gal-β-1,4-Glc-2AB (LNnT-2AB) and (Gal-GlcNAc)2Man3(GlcNAc)2-2AB (NA2-2AB) were chosen as the internal standard for the α-hGMII assay and α-hLM assay, respectively. Notably, GlcNAcMan5GlcNAc2-2AB was prepared from Man5GlcNAc-2AB by the addition of a β-1,2-N-acetylglucosamine via the GnT-I catalyzed enzymatic synthesis (ESI Fig. S9†).33 As shown in UPLC spectra under varied conditions (Fig. 4 and S10, ESI†), Man5GlcNAc2-2AB was hydrolyzed by α-hLM into possible products including Man4-, Man3-and Man2-GlcNAc2-2AB.34 Likewise, GlcNAcMan5GlcNAc2-2AB was digested by α-hGMII into two possible products, GlcNAcMan4GlcNAc2-2AB and GlcNAcMan3GlcNAc2-2AB.8,9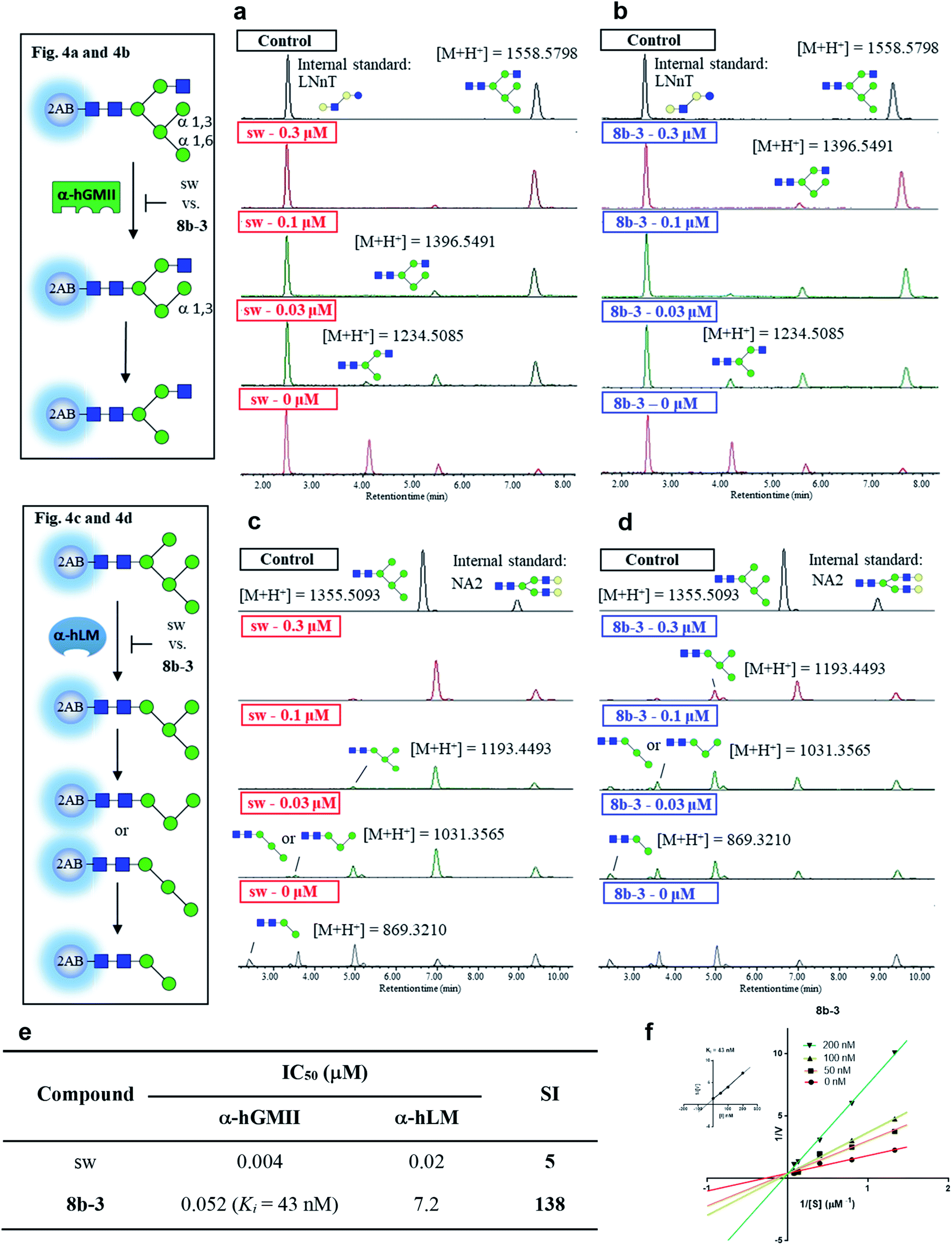 | ||
| Fig. 4 Study of selectivity index of 8b-3 for α-hLM vs. α-hGMII inhibition with oligosaccharides as a natural substrate. The α-hGMII and the glycan substrate (GlcNAcMan5GlcNAc2-2AB, also called GlcNAcMan5-2AB) were treated with (a) sw and (b) 8b-3 in a dose-dependent manner (0, 0.3, 0.1, 0.03 μM) for 24 h at 37 °C using Gal-β-1,4-GlcNAc-β-1,3-Gal-β-1,4-Glc-2AB (LNnT-2AB) as the internal standard based on the peak area ratio to determine the inhibition activity. The α-hLM and the glycan substrate (Man5GlcNAc2-2AB, also called Man5-2AB) were treated with (c) sw and (d) 8b-3 in a dose-dependent manner (0, 0.3, 0.1, 0.03 μM) for 24 h at 37 °C using (Gal-GlcNAc)2Man3(GlcNAc)2-2AB (NA2-2AB) as the internal standard based on the peak area ratio to determine the inhibition activity. (e) Glycan based α-hLM and α-hGMII inhibition data (ESI Fig. S11†), and selectivity index (SI) of sw and 8b-3. (f) Lineweaver–Burk plot for Ki determination of inhibitor 8b-3 against α-hGMII. Assays were conducted as described in the ESI.† IC50: half-maximal inhibitory concentration; SI: selectivity index = [(α-hLM IC50)/(α-hGMII IC50)]; data are the mean of three determinations. | ||
Both 8b-3 and sw exhibited dose-dependent inhibition (ESI Fig. S11†). Importantly, the SI of 8b-3 was around 138, while sw showed a much poorer selectivity (SI = 5) (Fig. 4e). Furthermore, the enzyme kinetics showed that 8b-3 is a potent competitive inhibitor (Ki = 43 nM) against α-hGMII (Fig. 4e and f). This reassuring finding indicates that the inhibition behavior of 8b-3 is similar in two assay systems without substrate dependence. Through this unprecedented inhibition and selectivity study, our exciting results established that 8b-3 is not only an effective α-hGMII inhibitor but also a selective inhibitor, worthy of further biochemistry experiments for anticancer drug discovery.
Modulation of cellular N-glycans with 8b-3
Modulation of N-glycans with 8b-3 was explored by monitoring the lysosomal catabolic and biosynthetic pathways in cell culture (Fig. 5a). To expedite the analysis, LC-MS was extensively utilized instead of the conventional TLC methods reported by Dr Winchester.35,36 | ||
| Fig. 5 Cellular effect of sw and 8b-3 in Golgi and lysosome. (a) Schematic representation of the effect of inhibition of sw and 8b-3 against α-LM and α-GMII in the cell. (b) Histogram showing LC-MS analysis of total accumulated oligomannose substrate Man2-9GlcNAc(M2-9G) and (c) accumulation of Man2-9GlcNAc in human normal fibroblast (08C0015) in the presence of mannosidase inhibitors sw or 8b-3 from 5 to 20 μM compared to the control culture (accumulated substrate in the cell treated with 20 μM sw is denoted as 100%). (d) Relative N-glycan abundance of HepG2 and Huh7 treated with 8b-3 in a dose-dependent manner compared to the control culture was analyzed by LC-MS. Mean values of glycan abundance (%) are shown as means of two independent experiments. Microscope images of untreated and 8b-3-treated HepG2 cells stained with (e) FITC conjugated concanavalin (ConA-FITC, green) and (f) Alexa Fluor 555 conjugated Galectin 1 (Gal1-555, red), and Huh7 cells stained with (g) ConA-FITC and (h) Gal1-555. *P < 0.05, **P < 0.01, ***P < 0.001, P < 0.05 is considered statistically significant. | ||
Accumulation of Man2-9GlcNAc in human fibroblasts caused by mannosidase inhibitors
Based on the N-glycan catabolism in the lysosomes, the biomarkers of α-LM inhibition are the mannose-rich oligosaccharides possessing only one GlcNAc (Man2-9GlcNAc) at the reducing end after the lysosomal chitobiase specifically cleaving glycoprotein carbohydrate chains, allowing us to easily monitor the specific lysosomal N-glycans by LC-MS without complex pre-treatments like enzymatic digestion.37 As shown in Fig. 5b and c, as expected, after 24 h treatment with sw, the relative intensity of overall Man2-9GlcNAc increased by about 5-fold compared to the control group indicating that the Man2-9GlcNAc substrate obviously accumulated caused by α-LM inhibition. In contrast, the oligomannosidic accumulation caused by 8b-3 is milder than that caused by sw (about 30% decrease at 20 μM), consistent with the results of the α-hLM inhibition study in Fig. 3 and 4 (ESI Table S2†). This finding suggests that its clinical use may be associated with fewer side effects resulting from the accumulation of undegraded oligosaccharides in lysosome compared to sw.Treatment of 8b-3 to alter the N-glycosylation of liver cancer cells
To understand the effect of α-hGMII inhibitor 8b-3 on the pattern of cellular N-glycosylation, HepG2 and Huh7 cells were treated with 8b-3 at varying concentrations (0–10 μM) and, after 72 h, their glycan profiles analyzed by LC-MS (see ESI methods†).38 As shown in Fig. 5d, the N-glycans of untreated HepG2 and Huh7 were mostly complex-type glycans incorporating sialic acids and fucoses, with relatively few high-mannose or hybrid-type glycans (ESI Table S3 and 4†). In contrast, treatment with 8b-3 shifted the N-glycopatterns from complex- to hybrid-type glycans in a dose dependent manner (Fig. 5d).Subsequently, the cell surface binding of fluorogenic lectins was investigated in the presence or absence of 8b-3 (Fig. 5e–h). Binding of FITC-labeled concanavalin A (ConA-FITC), a mannose binding lectin, markedly increased on these two cells treated with 8b-3 (10 μM) for 48 h.39 This indicated that more mannose-containing glycans (both high-mannose type and newly abundant hybrid type N-glycans) were detected on the cell surface, upregulated by α-hGMII inhibition of 8b-3 (Fig. 5e and g). Likewise, after treatment with 8b-3, binding of fluorogenic galectin-1 (Gal1-555) substantially decreased because of the lower abundance of β-galactose containing moieties such as N-acetyl lactosamine which are normally abundant in complex type N-glycans (Fig. 5f and h).40,41 Thus, inhibition of α-hGMII with 8b-3 altered the N-linked glycosylation process, demonstrating that both lectin binding results are complementary.
Taken together, we established that 8b-3 is a potent α-hGMII inhibitor capable of reducing the biosynthesis of complex type N-glycans, and, unlike sw, does not significantly impede α-hLM bioactivity which can result in the storage of oligomannose glycans in the lysosomes. These very promising outcomes encouraged us to subject 8b-3 to further anticancer activity studies and in vivo study of small molecule-induced storage of oligomannose glycans in serum.
Studying the in vitro and in vivo anticancer activity of 8b-3
Finally, the cytotoxicity of 8b-3 towards HCC cells including HepG2 and Huh7 cells was examined using sorafenib, a first-line therapy for advanced HCC, as a control.42 Interestingly, both 8b-3 and sorafenib exhibited low micromolar inhibition and anti-proliferation activities (Fig. 6a). For testing the potentially toxic side effects of 8b-3, the viability of primary human hepatocytes (HPH) was primarily evaluated and the MTT results revealed that 8b-3 has little cytotoxicity, even up to 400 μM. Significantly, 8b-3 suppressed cell migration and invasion of both HCC cell lines in a dose-dependent manner (Fig. 6b and c). α-hGMII inhibition causing a reduction in the synthesis of complex type glycans is consistent with these results. | ||
| Fig. 6 Iminosugar 8b-3 inhibits HCC in vitro and in vivo compared with sorafenib. (a) Table of cell viability data (MTT) of HepG2, Huh7, and human primary hepatocyte treated with 8b-3 and sorafenib for 72 h. The assay was conducted as described in the ESI.† NI: no inhibition. IC50: half-maximal inhibitory concentration; data are the mean of three determinations. The effects of 8b-3 on cell (b) migration and (c) invasion of HepG2 and Huh7 cells were tested by the Transwell assay. (d) Animal experimental schedule. (e) The bodyweights of mice were monitored. (f) The tumor volume (mm3) was recorded. (g) The final weights of the subcutaneous xenograft tumors. Assessment of the hepatorenal toxicity of the drugs by analyzing (h) the aspartate aminotransferase (AST), alanine aminotransferase (ALT), total bilirubin (TBIL), creatinine (CRE), and blood urea nitrogen (BUN) levels. (i) Histogram showing the LC-MS analysis of total accumulated oligomannose substrate (Man2-9GlcNAc) in the serum sample from control and 8b-3 treated mice (accumulated substrate in the serum from mice treated with 8b-3 is denoted as 100%). *P < 0.05, **P < 0.01, ***P < 0.001, P < 0.05 is considered statistically significant, n. s. refers to not significant. | ||
Next, the in vivo anticancer activity of 8b-3 was studied. In a typical procedure, xenograft tumors were generated by implanting Huh7 cells (5 × 106) subcutaneously into the left and right flanks of NOD-SCID mice. Starting on the 7th day after the inoculation of Huh7 cells, the mice were individually injected with either 8b-3 or sorafenib intraperitoneally (ip.) at a dose of 30 mg kg−1 and twice a week for 34 days (Fig. 6d). During treatment, neither mice injected with 8b-3 nor sorafenib exhibited signs of harm or decreased body weight (Fig. 6e). Our animal results (Fig. 6f and g) showed a significant reduction in tumor growth when administered with 8b-3 and sorafenib (P < 0.05) compared to the control. In addition, no serious side effects including hepatotoxicity and nephrotoxicity were observed (Fig. 6h and ESI Table S5†). Importantly, no significant accumulation of oligomannose glycans was detected in the serum of 8b-3-treated mice compared to the control (Fig. 6i and ESI Table S6†). These promising in vivo results demonstrate that 8b-3 is a selective α-hGMII inhibitor exhibiting anti-tumor activity against HCC cells, without affecting the storage of oligomannoses in the serum.
Conclusions
Inhibition of α-hGMII is known to alter N-glycan distribution patterns, suppressing cancer growth and metastasis, but most known inhibitors are poorly selective for α-hGMII over α-hLM (inhibition of which leads to abnormal storage of oligomannoses), limiting their further therapeutic potential. In this study, we successfully developed a new class of α-hGMII inhibitor 8b-3, with nanomolar potency against α-hGMII but little activity against α-hLM. This discovery was greatly accelerated by the exploration of new divergent synthetic approaches to build common intermediates and desired scaffolds, as well as adoption of NPICC and CGS.Notably, no toxic side-effects of 8b-3 toward HPH were observed, and the in vivo anticancer study showed that 8b-3 is an anti-HCC agent. No abnormal storage of oligomannoses was detected in the serum compared with normal controls. Moreover, our preliminary results showed that 8b-3 has similar anti-HCC potencies with the FDA approved drug sorafenib, and it is worthy of future systematic studies toward HCC or other tumors, as well as immunomodulatory studies.7,43–45
In the course of this work, 8b-3 was evaluated in a series of cell-based assay platforms. Notably, this is the first report describing the use of natural substrate-like oligosaccharides to characterize the potency and selectivity of inhibitors against α-hGMII and α-hLM in a UPLC study, as well as how to use LC-MS to analyze the accumulation of oligomannose glycans (Man2-9GlcNAc) in treated cells.
Finally, our synthetic chemistry strategy for efficiently developing selective inhibitors should be readily applicable to other targets or sugar processing enzymes. Additional studies are ongoing, and the results will be reported in due course.
Data availability
Experimental and computational data associated with this work are provided in the accompanying ESI†.Author contributions
W.-C. C. and W.-A. C. designed the experiments. W.-A. C. performed most of the experiments. W.-A. C. synthesized compounds with support from C.-W. C. W.-A. C. performed molecular libraries, in situ screening, compounds' resynthesis and enzyme kinetic studies. T.-J. R. C. provided α-hGMII and α-hLM. Y.-T. W. and W.-A. C. performed SAR and computational studies. Y.-H. C. and T.-L. H. performed enzyme kinetic studies using oligosaccharides as the natural substrate. W.-A. C. and T.-L. H. performed cell-based N-glycan profiling and oligomannose accumulation analysis by LC-MS assisted by C.-H. C. and J.-L. L. W.-A. C. performed fluorescence imaging. W.-A. C, C.-Y. H. and P.-F. H. performed cellular viability, migration and invasion assays. C.-N. S. designed in vivo studies, and C.-Y. H. and P.-F. H. performed the experiments. W.-A. C. wrote the manuscript and compiled the ESI.† T.-J. R. C., Y.-T. W., T.-L. H., J.-L. L. and C.-N. S. provided scientific input. W.-A. C. and W.-C. C analyzed the data, discussed the results, and contributed to the editing of the manuscript. W.-C. C. and C.-N. S. supervised the entire project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Academia Sinica and the Ministry of Science and Technology for financial support and the Core Facilities of Translational Medicine of BioTReC (National Biotechnology Research Park, Academia Sinica, Taiwan) for the technical support and data analysis.Notes and references
- K. Ohtsubo and J. D. Marth, Cell, 2006, 126, 855–867 CrossRef CAS PubMed.
- G. W. Hart and R. J. Copeland, Cell, 2010, 143, 672–676 CrossRef CAS PubMed.
- S. S. Pinho and C. A. Reis, Nat. Rev. Cancer, 2015, 15, 540–555 CrossRef CAS PubMed.
- C. Reily, T. J. Stewart, M. B. Renfrow and J. Novak, Nat. Rev. Nephrol., 2019, 15, 346–366 CrossRef PubMed.
- M. M. Fuster and J. D. Esko, Nat. Rev. Cancer, 2005, 5, 526–542 CrossRef CAS PubMed.
- J. M. H. van den Elsen, D. A. Kuntz and D. R. Rose, EMBO J., 2001, 20, 3008–3017 CrossRef CAS PubMed.
- P. E. Goss, M. A. Baker, J. P. Carver and J. W. Dennis, Clin. Cancer Res., 1995, 1, 935 CAS.
- N. Shah, D. A. Kuntz and D. R. Rose, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9570 CrossRef CAS PubMed.
- D. R. Rose, Curr. Opin. Struct. Biol., 2012, 22, 558–562 CrossRef CAS PubMed.
- S. R. Stowell, T. Ju and R. D. Cummings, Annu. Rev. Phytopathol., 2015, 10, 473–510 CrossRef CAS PubMed.
- D. A. Kuntz, C. A. Tarling, S. G. Withers and D. R. Rose, Biochemistry, 2008, 47, 10058–10068 CrossRef CAS PubMed.
- Z. Armstrong, C.-L. Kuo, D. Lahav, B. Liu, R. Johnson, T. J. M. Beenakker, C. de Boer, C.-S. Wong, E. R. van Rijssel, M. F. Debets, B. I. Florea, C. Hissink, R. G. Boot, P. P. Geurink, H. Ovaa, M. van der Stelt, G. M. van der Marel, J. D. C. Codée, J. M. F. G. Aerts, L. Wu, H. S. Overkleeft and G. J. Davies, J. Am. Chem. Soc., 2020, 142, 13021–13029 CrossRef CAS PubMed.
- T. Fujita, H. Nagasawa, Y. Uto, T. Hashimoto, Y. Asakawa and H. Hori, Org. Lett., 2004, 6, 827–830 CrossRef CAS PubMed.
- D. A. Kuntz, S. Nakayama, K. Shea, H. Hori, Y. Uto, H. Nagasawa and D. R. Rose, ChemBioChem, 2010, 11, 673–680 CrossRef CAS PubMed.
- T.-J. R. Cheng, T.-H. Chan, E.-L. Tsou, S.-Y. Chang, W.-Y. Yun, P.-J. Yang, Y.-T. Wu and W.-C. Cheng, Chem.–Asian J., 2013, 8, 2600–2604 CrossRef CAS PubMed.
- S. Šesták, M. Bella, T. Klunda, S. Gurská, P. Džubák, F. Wöls, I. B. H. Wilson, V. Sladek, M. Hajdúch, M. Poláková and J. Kóňa, ChemMedChem, 2018, 13, 373–383 CrossRef PubMed.
- T. Klunda, S. Šesták, J. Kóňa and M. Poláková, Bioorg. Chem., 2019, 83, 424–431 CrossRef CAS PubMed.
- P. Heikinheimo, R. Helland, H.-K. S. Leiros, I. Leiros, S. Karlsen, G. Evjen, R. Ravelli, G. Schoehn, R. Ruigrok, O.-K. Tollersrud, S. McSweeney and E. Hough, J. Mol. Biol., 2003, 327, 631–644 CrossRef CAS PubMed.
- E.-L. Tsou, Y.-T. Yeh, P.-H. Liang and W.-C. Cheng, Tetrahedron, 2009, 65, 93–100 CrossRef CAS.
- W.-A. Chen, A. Sayyad, C.-W. Chen, Y.-H. Chen, T.-J. R. Cheng and W.-C. Cheng, Asian J. Org. Chem., 2019, 8, 2233–2242 CrossRef CAS.
- W.-C. Cheng, C.-W. Guo, C.-K. Lin and Y.-R. Jiang, Isr. J. Chem., 2015, 55, 403–411 CrossRef CAS.
- L. Li, Z. Chen, X. Zhang and Y. Jia, Chem. Rev., 2018, 118, 3752–3832 CrossRef CAS PubMed.
- J.-S. Zhu, S. Nakagawa, W. Chen, I. Adachi, Y.-M. Jia, X.-G. Hu, G. W. J. Fleet, F. X. Wilson, T. Nitoda, G. Horne, R. van Well, A. Kato and C.-Y. Yu, J. Org. Chem., 2013, 78, 10298–10309 CrossRef CAS PubMed.
- M. Bessodes, D. Komiotis and K. Antonakis, Tetrahedron Lett., 1986, 27, 579–580 CrossRef CAS.
- A. Mengel and O. Reiser, Chem. Rev., 1999, 99, 1191–1224 CrossRef CAS PubMed.
- S. C. Nigama, A. Mann, M. Taddei and C.-G. Wermutha, Synth. Commun., 1989, 19, 3139–3142 CrossRef.
- W. Yu, Y. Mei, Y. Kang, Z. Hua and Z. Jin, Org. Lett., 2004, 6, 3217–3219 CrossRef CAS PubMed.
- R. Singh, M. K. Parai, S. Mondal and G. Panda, Synth. Commun., 2013, 43, 253–259 CrossRef CAS.
- C.-Y. Wu, C.-F. Chang, J. S.-Y. Chen, C.-H. Wong and C.-H. Lin, Angew. Chem., Int. Ed., 2003, 42, 4661–4664 CrossRef CAS PubMed.
- H. Fiaux, D. A. Kuntz, D. Hoffman, R. C. Janzer, S. Gerber-Lemaire, D. R. Rose and L. Juillerat-Jeanneret, Bioorg. Med. Chem., 2008, 16, 7337–7346 CrossRef CAS PubMed.
- T. Kuribara, M. Hirano, G. Speciale, S. J. Williams, Y. Ito and K. Totani, ChemBioChem, 2017, 18, 1027–1035 CrossRef CAS PubMed.
- J. C. Bigge, T. P. Patel, J. A. Bruce, P. N. Goulding, S. M. Charles and R. B. Parekh, Anal. Biochem., 1995, 230, 229–238 CrossRef CAS PubMed.
- R. Chen, M. A. Pawlicki, B. S. Hamilton and T. J. Tolbert, Adv. Synth. Catal., 2008, 350, 1689–1695 CrossRef CAS.
- S. al Daher, R. de Gasperi, P. Daniel, N. Hall, C. D. Warren and B. Winchester, Biochem. J., 1991, 277, 743–751 CrossRef CAS PubMed.
- I. Cenci di Bello, P. Dorling and B. Winchester, Biochem. J., 1983, 215, 693–696 CrossRef CAS PubMed.
- I. Cenci di Bello, G. Fleet, S. K. Namgoong, K. I. Tadano and B. Winchester, Biochem. J., 1989, 259, 855–861 CrossRef CAS PubMed.
- J.-C. Michalski and A. Klein, Biochim. Biophys. Acta, Mol. Basis Dis., 1999, 1455, 69–84 CrossRef CAS.
- S. Abdul Rahman, E. Bergström, C. J. Watson, K. M. Wilson, D. A. Ashford, J. R. Thomas, D. Ungar and J. E. Thomas-Oates, J. Proteome Res., 2014, 13, 1167–1176 CrossRef CAS PubMed.
- R. D. Cummings, A. G. Darvill, M. E. Etzler, et al., Essentials of Glycobiology, ed. A. Varki, R. D. Cummings, J. D. Esko, et al., Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY), 3rd edn, 2015, ch. 48 Search PubMed.
- D. O. Croci, J. P. Cerliani, T. Dalotto-Moreno, S. P. Méndez-Huergo, I. D. Mascanfroni, S. Dergan-Dylon, M. A. Toscano, J. J. Caramelo, J. J. García-Vallejo, J. Ouyang, E. A. Mesri, M. R. Junttila, C. Bais, M. A. Shipp, M. Salatino and G. A. Rabinovich, Cell, 2014, 156, 744–758 CrossRef CAS PubMed.
- S. K. Patnaik, B. Potvin, S. Carlsson, D. Sturm, H. Leffler and P. Stanley, Glycobiology, 2005, 16, 305–317 CrossRef PubMed.
- A. Huang, X.-R. Yang, W.-Y. Chung, A. R. Dennison and J. Zhou, Signal Transduction Targeted Ther., 2020, 5, 146 CrossRef PubMed.
- C.-W. Li, S.-O. Lim, E. M. Chung, Y.-S. Kim, A. H. Park, J. Yao, J.-H. Cha, W. Xia, L.-C. Chan, T. Kim, S.-S. Chang, H.-H. Lee, C.-K. Chou, Y.-L. Liu, H.-C. Yeh, E. P. Perillo, A. K. Dunn, C.-W. Kuo, K.-H. Khoo, J. L. Hsu, Y. Wu, J.-M. Hsu, H. Yamaguchi, T.-H. Huang, A. A. Sahin, G. N. Hortobagyi, S. S. Yoo and M.-C. Hung, Cancer Cell, 2018, 33, 187–201 CrossRef CAS PubMed.
- S. Shi, S. Gu, T. Han, W. Zhang, L. Huang, Z. Li, D. Pan, J. Fu, J. Ge, M. Brown, P. Zhang, P. Jiang, K. W. Wucherpfennig and X. S. Liu, Clin. Cancer Res., 2020, 26, 5990 CrossRef CAS PubMed.
- L. Sun, C.-W. Li, E. M. Chung, R. Yang, Y.-S. Kim, A. H. Park, Y.-J. Lai, Y. Yang, Y.-H. Wang, J. Liu, Y. Qiu, K.-H. Khoo, J. Yao, J. L. Hsu, J.-H. Cha, L.-C. Chan, J.-M. Hsu, H.-H. Lee, S. S. Yoo and M.-C. Hung, Cancer Res., 2020, 80, 2298 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC [2116295-2116296]. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d1sc05894k |
| This journal is © The Royal Society of Chemistry 2022 |