
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDysfunction of vesicular storage in young-onset Parkinson's patient-derived dopaminergic neurons and organoids revealed by single cell electrochemical cytometry†
Wanying
Zhu‡
 a,
Mengdan
Tao‡
ad,
Yuan
Hong
a,
Shanshan
Wu
a,
Chu
Chu
a,
Zhilong
Zheng
b,
Xiao
Han
a,
Qian
Zhu
a,
Min
Xu
a,
Andrew G.
Ewing
*c,
Xing
Guo
*b and
Yan
Liu
*a
a,
Mengdan
Tao‡
ad,
Yuan
Hong
a,
Shanshan
Wu
a,
Chu
Chu
a,
Zhilong
Zheng
b,
Xiao
Han
a,
Qian
Zhu
a,
Min
Xu
a,
Andrew G.
Ewing
*c,
Xing
Guo
*b and
Yan
Liu
*a
aSchool of Pharmacy, Collaborative Innovation Center for Cardiovascular Disease Translational Medicine, State Key Laboratory of Reproductive Medicine, Nanjing Medical University, Nanjing 211166, China. E-mail: yanliu@njmu.edu.cn
bDepartment of Neurobiology, School of Basic Medical Sciences, State Key Laboratory of Reproductive Medicine, Nanjing Medical University, Nanjing 211166, China. E-mail: guox@njmu.edu.cn
cDepartment of Chemistry and Molecular Biology, University of Gothenburg, Gothenburg 412 96, Sweden. E-mail: andrewe@chem.gu.se
dSchool of Biological Science and Medical Engineering, Southeast University, Nanjing 210096, China
First published on 11th May 2022
Abstract
Electrochemical cytometry based on nano-tip microelectrodes was used to quantify the vesicular storage at the single-cell level in human neurons and midbrain organoids which acted as disease models of young-onset Parkinson's disease (YOPD). Human dopaminergic (DA) neurons and midbrain organoids were derived from an induced pluripotent stem cell line from one YOPD patient. We show a significant deficiency in vesicular catecholamine storage and a slower pore forming process on the surface of the microelectrode in the DA neurons derived from the YOPD patient. The upregulation of α-synuclein in both neurons and organoids derived from the YOPD patient is associated with vesicular storage dysfunction, revealing a correlation between the pathogenesis of YOPD and vesicular chemical storage deficiency, a novel chemical insight into the potential pathology of YOPD. Notably, efficacy evaluation and drug testing were performed with our platform to demonstrate that both amantadine, a clinical drug for Parkinson's disease (PD), and phorbol 12-myristate 13-acetate, an attractive candidate, ameliorate the dysfunction of vesicular storage in DA neurons derived from the YOPD patient. Our platform offers promising avenues for new drug discovery for PD and other neurodegenerative disorders.
Introduction
In the central nervous system, neuron–neuron communication is modulated by chemical signal transmission between synapses, which is accomplished by the transport of neurotransmitters in synaptic vesicles. During the resting stage, the neurotransmitter molecules are stored in the synaptic vesicle which is nearly the same in size and shape.1 Due to their critical involvement in cell communication, monitoring of vesicular neurotransmitter storage appears of great importance to better understand the occurrence and development of a variety of neurological diseases as well as the evaluation of therapeutic drugs.Parkinson's disease (PD) is a chronic neurodegenerative disease with movement disorder. The pathological hallmark of PD is characterized by the progressive loss of dopaminergic (DA) neurons in the substantia nigra of the midbrain.2 At present, the causes of PD are still poorly understood and whether it is related to the number of neurotransmitters in a single vesicle remains to be clarified. Therefore, it is important to establish a highly sensitive method for monitoring neurotransmitters in single vesicles to reveal individual vesicular storage related to the pathogenesis of PD at the single-cell level.
Intracellular vesicle impact electrochemical cytometry (IVIEC) first established by Ewing's group in 2015, has attracted increasing attention due to its high spatiotemporal resolution, high sensitivity, and minimum damage to cells.3,4 This method uses a conical nano-tip electrode to measure the contents of individual vesicles in situ, providing statistically quantitative information achievable by no other method. For example, the effect of DJ-1 protein deficiency caused by the PARK7 gene mutation on the vesicular storage and exocytosis of neurochemicals has been investigated using electrochemical methods with nano-tip electrodes.5 The cone-shaped carbon fiber nanoelectrodes were used to probe individual dopamine release events from the individual mouse DA neuron synapse.6 Intracellular electrochemical nano-measurements were applied to reveal that the fraction of the released vesicular octopamine from larval neuromuscular neuron of a living Drosophila is very small.7 Nevertheless, none of the studies used human neurons or tissues. Although animal models are an important tool for the study of disease mechanisms and clinical transformation; studies based on PD mouse models (e.g. Pink1 knockout mouse model) fail to show pathological features which are critical pathologies in PD patient's brain.8 The use of human DA neurons to carry out these chemical measurements is necessary to further disease research and effective drug discovery.
Recent advances in induced pluripotent stem cells (iPSCs)-derived culturing systems have opened new avenues to study human neurodegenerative diseases.9,10 Reprogramming patient-derived cells into iPSCs with subsequent differentiation into midbrain DA cultures provides a human tissue-specific model of PD.11,12 The iPSCs share the same genetic information with patients and it is conducive to maximizing simulated disease.13 The midbrain DA neurons (2D cultures) derived from patient iPSCs were used to study the origin and progression of PD.11,14,15 In addition, 3D cultures which are also referred to organoids,16,17 are composed of multiple cell types including progenitor cells, neurons, and glial cells, and are getting more and more attention since they can recapitulate complex cell–cell interaction and cell diversity to a great degree.18 Also, treated with different small molecules and growth factors, iPSCs can form various specific organoids representing certain brain regions, such as the cerebral cortex,16 forebrain,19 cerebellum,19 and midbrain.17 Hence, the use of iPSCs to model PD, including 2D and 3D cultures, provides an alternative model system to investigate cellular vesicular content and functional changes that may contribute to PD pathogenesis. Early onset of disease may produce a more pronounced phenotype in iPSC models. Therefore, an iPSC line was induced from a patient with young-onset Parkinson's disease (YOPD), which is defined as onset at the age less than 50 years and accounts for about ten percent of all cases of Parkinson's disease.14
In this work, a platform combining IVIEC and human midbrain-like cultures derived from human pluripotent stem cell (hPSC) lines from control and a YOPD patient was established to explore the link between neurotransmitter storage in single vesicles of human DA cultures and disease-specific phenotypes (Scheme S1†). We found that, in the DA neurons derived from the YOPD patient, the numbers of dopamine molecules located in each single vesicle significantly decreased and the pore forming process for vesicles adsorbed on the surface of nano-tip microelectrodes was prolonged. Additionally, using this platform, we evaluated the efficacy of amantadine (AMA) showing that it rescues the vesicle storage dysfunction of DA neurons. Also, phorbol 12-myristate 13-acetate (PMA) was efficient in increasing the storage of dopamine in the vesicles. Our approach suggests a novel potential mechanism underlying the pathology of PD.
Results and discussion
Differentiation of hPSCs to midbrain DA neurons
The iPSC line was generated from one YOPD patient with the PINK1 gene mutation at first (Fig. S1†). The iPSC line from a YOPD patient (PD52) and a standard control cell line (H9) were differentiated to midbrain DA neural cultures using a previously described protocol (Fig. 1A).20 In the early stage of differentiation (day 18), both the PD52 and H9 cell lines expressed the proliferation marker KI67, the neural precursor cell markers SOX2 and NESTIN, the immature neuronal marker DCX, mature neurons marker MAP2 (Fig. S2†). Notably, the midbrain markers FOXA2, OTX2, and CORIN (at day 24) and the DA neuron marker tyrosine hydroxylase (TH) (at day 45) were observed in the differentiated cultures (Fig. 1B). Immunostaining quantification showed the YOPD iPSC line (PD52) has a smaller number of FOXA2 and OTX2-expressing cells than the control line (H9) (Fig. 1C).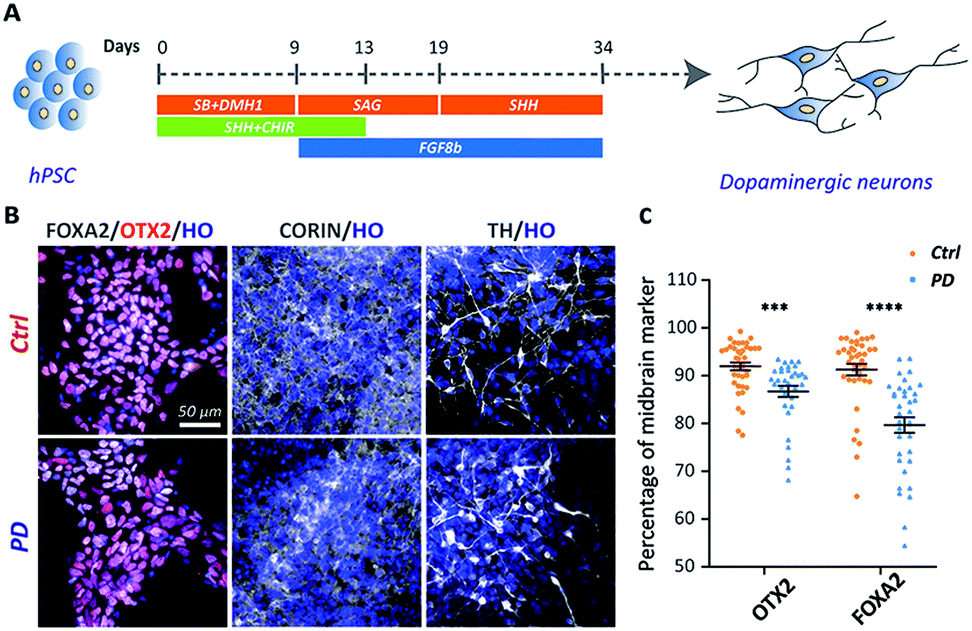 | ||
| Fig. 1 Differentiation of hPSCs to midbrain DA neurons. (A) Schematic diagrams illustrating the overall strategy to generate DA neurons. (B) Normal control (H9) or YOPD (PD52) derived DA neurons stained for the midbrain markers FOXA2 (floor plate progenitors' marker), OTX2 (midbrain intermediate progenitors' marker), and CORIN (at day 24) and the DA neuron marker tyrosine hydroxylase (TH) (DA neuron marker) (at day 45). (C) Quantification of the percentage of OTX2/HO and FOXA2/HO neurons at day 24. Neurons (n ≥ 30) from 3 independent biological replicate experiments were analysed for each group. Data represent mean ± SEM. ***: p < 0.001; ****: p < 0.0001. | ||
Vesicle content decreases in PD52 iPSC-derived midbrain DA neurons
The catecholamine storage of vesicles in PD52-derived DA neurons or normal controls was measured via IVIEC. The fabrication of the conical nano-tip electrodes is detailed in Section 2.4 of the ESI (Fig. S3).†Fig. 2A and B shows traces of release events obtained from neurons derived from H9 and PD52, where each current transient represents the total catecholamine content inside a single vesicle. After quantification with Faraday's equation (N = Q/nF), a normalized frequency histogram is shown in Fig. 2C describing the distribution of the number of molecules per vesicle, which provides a near-Gaussian logarithmic distribution and a decreased amount for the PD52-derived midbrain DA cultures.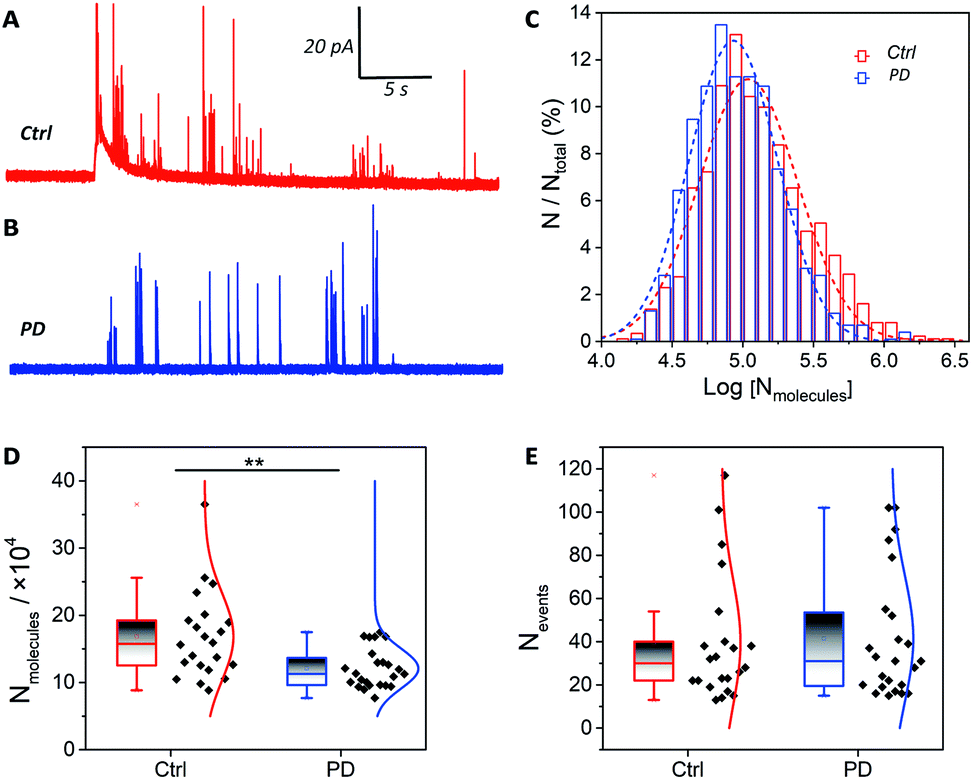 | ||
| Fig. 2 Vesicle content detection in H9 and PD52 derived midbrain DA neurons at 6–8 weeks. Typical traces of vesicle content in single neurons derived from H9 (A) and PD52 (B) cell lines. (C) Normalized frequency distribution for vesicular content from DA neurons derived from H9 (ctrl, red, n = 873 from 22 neurons from 3 independent biological replicate experiments (differentiations)) and PD52 (blue, n = 994 from 24 neurons from 3 independent biological replicate experiments (differentiations)). Gaussian fits are shown. Average numbers of catecholamine molecules per vesicle (D) and events detected in each neuron (E) for H9 and PD52 derived neurons with each point representing the average level from single neurons. Data represent mean ± SEM. **: p < 0.01. | ||
Previous studies of animal models showed a striking loss (50–70%) of DA neurons in the substantia nigra pars compacta in PD.21 However, direct measurements regarding the catecholamine levels in single human neurons, especially single vesicles, have not been reported. Our data indicate that catecholamine levels in single vesicles have a significant decline in DA neurons derived from PD52 compared with normal controls. Furthermore, the mean values of the average number of molecules from single neurons were compared in order to minimize the impact of cell-to-cell variation (Fig. 2D). The vesicular catecholamine content shows a significant decrease in the DA neurons derived from PD52 (ctrl: (16.82 ± 1.4) × 104, n = 22 vs. PD: (12.08 ± 0.6) × 104, n = 24) (p < 0.01), revealing an abnormal vesicular catecholamine storage ability of PD52-derived DA neurons. The numbers of events detected in each neuron were analyzed, and no significant differences in number of vesicles were found between the neurons from both groups (Fig. 2E). Thus, it appears that the number of vesicles from single neurons does not decrease in DA neural cultures derived from PD52. The single peaks were also analyzed, and plasticity difference was found between H9 and PD52 derived DA cultures (Fig. S4†).
Differentiation of hPSCs to midbrain organoids
Brain tissue is the most complex structure in the human body, and many nervous system diseases are difficult to reproduce in animals. Therefore, establishing an in vitro model of brain development appears to be extremely important for studying nervous system diseases. Brain organoids, which can mimic human brain cell composition and structure, have been widely used. In order to generate midbrain organoids, we referred to and modulated an established 2D culture protocol (Fig. 3A). By day 24, immunocytochemical analysis showed that both H9 and PD52 derived nascent neural organoids expressed KI67, SOX2, NESTIN, DCX, TUJ1, and MAP2 (Fig. S5A†). Besides, OTX2, FOXA2, and CORIN were heavily expressed in the organoids (Fig. 3B), indicating that the organoids have been correctly patterned into the VM-type embryonic brain region and composed of amounts of neural stem cells. Furthermore, the expression of OTX2 and FOXA2 in the early stage of the YOPD patient derived midbrain organoids was markedly reduced (OTX2: 75.75 ± 3.52% vs. 65.79 ± 1.97%; FOXA2: 74.55 ± 2.88% vs. 56.69 ± 2.51%) (Fig. 3B). After continuous culture for 21 days, abundant of cells expressed DA neural marker TH and postmitotic DA neural marker NURR1 and PITX3 (Fig. 3C and E). We then verified if the PD52-derived organoids had decreased DA neurons in our system. Immunocytochemical analysis and western blot showed that, in contrast to control, these markers were all significantly decreased in organoids derived from PD52 (Fig. 3C and D). These results suggested reduced differentiation efficiency in the PD52-derived organoids.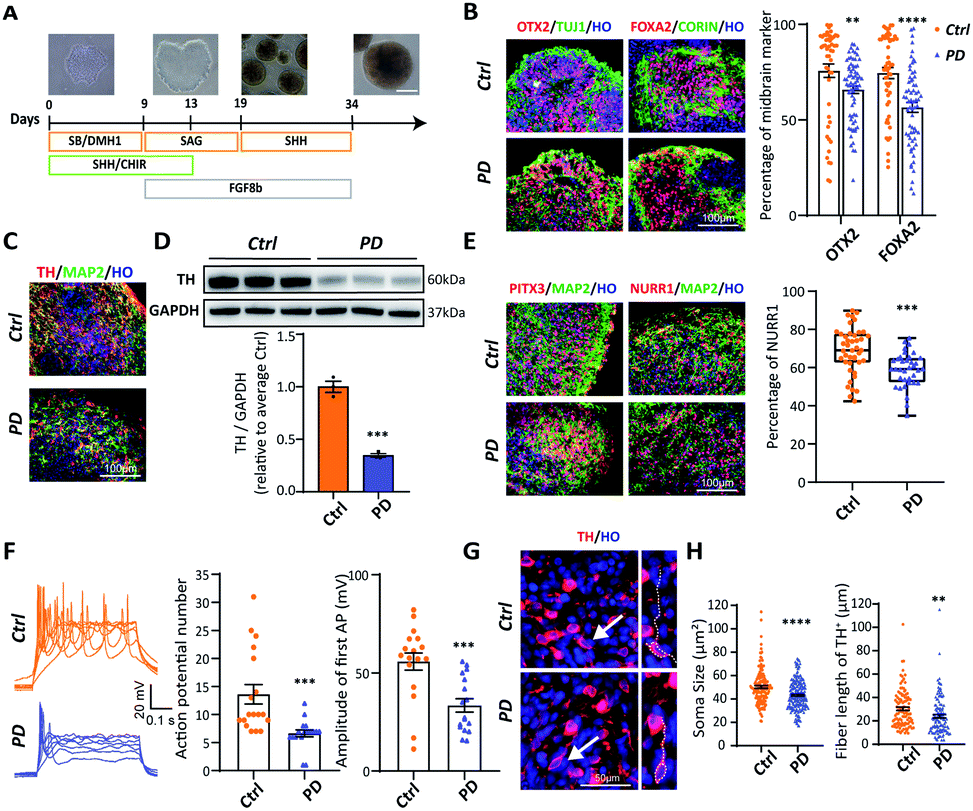 | ||
| Fig. 3 Generation and characterization of H9 and PD52 derived midbrain organoids. (A) Schematic diagrams illustrating the overall strategy to generate midbrain organoids. (B) Cryosection of midbrain organoids at day 24 and stained for FOXA2 and OTX2. Scale bar = 100 μm. Quantification of FOXA2+ cell and OTX2+ cell populations in H9 and PD52 derived midbrain organoids at day 24. (C) Cryosection of midbrain organoids at day 45 and stained for TH. (D) Western blots of day 45 midbrain organoids cultures for TH production and GAPDH as a housekeeping control and relative intensities from multiple western blots. Each point represents an average of about 20 organoids from an independent differentiation (ctrl, n = 3; PD, n = 3). Intensities are given relative to the average for the control lines. (E) Cryosection of midbrain organoids at day 45 and stained for PITX3 and NURR1 (post-mitotic DA neuron marker). Scale bar = 100 μm. Quantification of NURR1+ cell populations at day 45 post differentiation. (F) Representative traces of multiple APs in the midbrain organoids. Quantification of evoked AP number and amplitude inside midbrain organoids that were generated in response to a particular current pulse. (Two-tailed unpaired t-test.) (G) Representative images of TH+ cells in ctrl and PD52 derived midbrain organoids. Scale bar = 50 μm. (H) Quantification of soma size and dendrites length of TH+ cells in ctrl and PD52 derived midbrain organoids at day 45. Organoids (n ≥ 20) from 3 independent biological replicate experiments were analysed for each group. Data represent mean ± SEM. **: p < 0.01; ***: p < 0.001; ****: p < 0.0001. | ||
Next, to test whether the DA neurons in midbrain organoids were functionally mature, we performed whole-cell patch electrical experiments (Fig. S5B†). Both in the disease and control derived organoids, patched neurons showed similar large voltage-dependent sodium (Na+) and potassium (K+) currents (Fig. S5C and D†). Action potentials could be elicited upon current injection (Fig. 3F). Compared with control, decreased action potential (AP) numbers and amplitude were observed in the PD52-derived DA neurons, suggesting the PD52-derived neurons were less mature than H9-derived neurons.
To further confirm the authenticity of the PD52-derived organoid model, we explored apoptosis in the PD52-derived organoids. We first qualified and compared nuclear fragmentation of two groups, which represents cell death, and found that organoids derived from PD52 showed higher apoptosis rates (Fig. S5E†), which was consistent with previous autopsy data.22 We found that small and shrunken cell bodies with shortened neurites were exhibited in the PD52-derived organoids (Fig. 3G and H), a sign of neuronal cell degeneration.23 These data confirmed that DA neurons exhibited PD pathology in the 3D culture system.
The reduced vesicular storage in PD52 iPSC-derived midbrain organoids is reversed by the treatment of AMA
To further provide evidence of vesicular storage dysfunction in YOPD, IVIEC with nano-tip electrodes was used to quantify the dopamine storage in a single vesicle of DA neurons which located in midbrain organoids (Fig. 4A). Typical traces were obtained at the intracellularly placed electrode, and each of the current transients (spikes) corresponds to the current response from dopamine stored in one vesicle (Fig. 4B). The vesicular dopamine content (Nmolecules) was calculated according to Faraday's law and the histogram of relative event number vs. log molecular count resulted in a near Gaussian distribution (Fig. 4C). Again, as for the neurons in culture, a significant decrease amount of dopamine in the PD52-derived organoid vesicles ((15.22 ± 1.1) × 104, n = 30), in contrast to the vesicles from the control organoids ((27.64 ± 4.1) × 104, n = 24) was observed (Fig. 4D). In addition, we examined the numbers of spikes (Fig. 4E) and no significant difference was observed between H9 and PD52 derived midbrain organoids. Data from both cultured human cells and organoids illustrate vesicular storage dysfunction in midbrain-like cultures derived from PD52 while leaving the number of vesicles in the cells intact.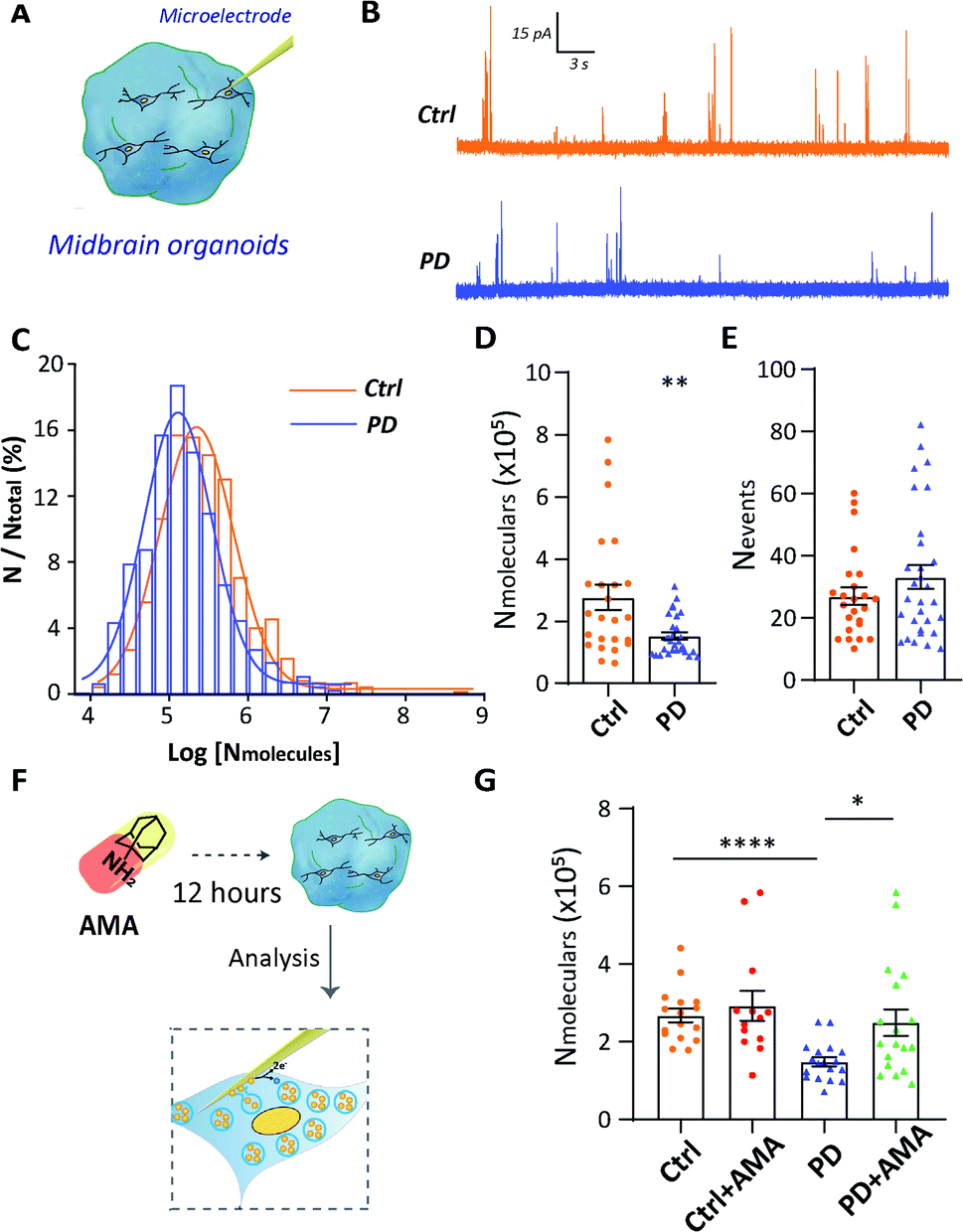 | ||
| Fig. 4 Reversal of reduced vesicular storage in PD52-derived midbrain organoids by the treatment of AMA. (A) Schematic diagram illustrating the analysis of vesicular storage in midbrain organoids derived from H9 and PD52. (B) Typical traces of vesicle content in both ctrl and PD cells. (C) Normalized frequency distribution for vesicular content in midbrain organoids derived from H9 (ctrl, orange, n = 649 from 24 cells from 3 independent biological replicate experiments (differentiations)) and PD52 (PD, blue, n = 995 from 30 cells from 3 independent biological replicate experiments (differentiations)). (D) Average number of dopamine molecules per vesicle and (E) average number of events in midbrain organoids derived from H9 and PD52, each point representing a cell. (F) Schematic diagram detailing AMA treatment experiment. (G) Average number of dopamine molecules per vesicle in midbrain organoids derived from H9 and PD52 before and after incubation with AMA, each point representing a cell. Three independent biological replicate experiments were conducted for each group. Data represent mean ± SEM. *: p < 0.05; **: p < 0.01; ****: p < 0.0001. | ||
Our technical platform for real-time detection of single vesicle content in situ allows us to evaluate drug efficacy and models. The efficacy of the clinical drug amantadine (AMA) in rescuing vesicular storage in PD52-derived organoids was evaluated. The mechanism of AMA in treating Parkinson's disease is still not fully clear. It may be related to that AMA promotes the release of dopamine from DA neurons, strengthening the role of dopamine and catecholamine in the central nervous system and increasing the dopamine content of neurons.24 Hence, we studied the effects of AMA on the single vesicle dopamine level from a single neuron (Fig. 4F). Organoids were treated with 10 μM AMA for 12 h before the single vesicle detection experiments. For both AMA-treated H9 and PD52 derived midbrain organoids, statistical analyses of the recorded individual current spikes showed that vesicular dopamine content followed normal distributions (Fig. S6†). As shown in Fig. 4G, compared to the untreated group (PD: (14.87 ± 1.1) × 104, n = 18), the number of dopamine content in single vesicles in AMA-treated PD52-derived organoids (PD with AMA: (24.96 ± 3.4) × 104, n = 18) increased significantly. No significant dopamine content change occurred in AMA-treated control group (ctrl: (26.80 ± 1.8) × 104, n = 16 vs. ctrl with AMA: (29.24 ± 3.9) × 104, n = 13) (Fig. 4G), clearly showing AMA rescues the vesicle storage defect in the organoids derived from PD52. As an added note, this observation implied feasibility and effectiveness to use our hPSC-based model to evaluate drug efficacy for clinical medication.
PMA alters vesicle content and α-synuclein accumulation in midbrain-like cultures
As a membrane protein localized to presynaptic terminals, α-synuclein (α-syn) is critical to the initiation and progression of PD.14 It has been reported that α-syn functions in the modulation of synaptic activity through regulating vesicle trafficking and neurotransmitter release and reuptake.25 However, the mechanism of α-syn influencing neurotransmission remains elusive. Thereby, we next determined whether α-syn was differentially expressed in midbrain-like cultures derived from PD52. Both immunofluorescent staining and western blot analyses showed that α-syn protein levels rose remarkably in PD52-derived midbrain DA neural cultures (Fig. S7†). Higher expression of α-syn was also observed in PD52-derived organoids based on immunocytochemical analysis and western blot results (Fig. 5A–C). These data show that midbrain DA cultures demonstrate a PD phenotype of α-syn protein accumulation. In addition, similarly to recent reports,26 we performed mean fluorescence intensity analysis to explore the differential expression of α-syn in single neurons between control and DA neurons derived from PD52. We found that DA neurons derived from PD52 exhibited significantly higher fluorescence intensity, which represents higher α-syn expression in PD52-derived single DA neurons (Fig. 5B). | ||
| Fig. 5 Alteration of vesicle content and α-syn accumulation in midbrain DA cultures induced by PMA. (A) Representative images of α-syn expression in H9 and PD52 derived midbrain organoids respectively. Scale bar = 100 μm. (B) Left: quantification of α-syn+ cell populations in H9 and PD52 derived midbrain organoids at day 45. Right: quantification of mean fluorescence intensity of α-syn relative to HO per cell. n = 67 cells for ctrl, 88 cells for PD. (C) Left: western blots of day 45 midbrain organoids cultures for α-syn production and GAPDH as a housekeeping control. Right: relative intensities from multiple western blots (n = 3). (D) Schematic diagram detailing PMA treatment experiment. (E) Average number of dopamine molecules per vesicle in midbrain organoids derived from ctrl and PD52 before and after incubation, each point representing a cell. (F) Western blots of day 45 midbrain organoids cultures with or without treatment of PMA for α-syn production, and GAPDH as a housekeeping control and relative intensities from multiple western blots (n = 3). Each point represents an average of about 20 organoids from an independent differentiation. Intensities are given relative to the average for the control lines. Three independent biological replicate experiments were analyzed for each group. Data represent mean ± SEM. *: p < 0.05; **: p < 0.01; ***: p < 0.001; ****: p < 0.0001. | ||
Phorbol 12-myristate 13-acetate (PMA), a compound that triggers α-syn reduction, might be able to enhance the level of dopamine neurotransmitter, and further maintain neuron survival.14 Hence, we next attempted to explore if our model could offer promising therapeutic avenues for exploring the effects of PMA on the number of molecules of dopamine per vesicle from single DA neurons. We treated both control and PD52-derived midbrain organoids with 50 μM PMA for 12 hours (Fig. 5D). Single vesicle detection experiments (Fig. 5E and S8†) showed that, in contrast to the untreated group (PD: (15.25 ± 0.8) × 104, n = 25), the dopamine content in vesicles was obviously improved in PD52-derived organoids after treated with PMA (PD with PMA: (33.48 ± 4.4) × 104, n = 17). Unlike the disease group, PMA has no significant effect on the normal group (ctrl: (26.57 ± 2.3) × 104, n = 20 vs. ctrl with PMA: (27.22 ± 4.0) × 104, n = 14). Next, we studied if the rescue of vesicle content by PMA in the PD52-derived organoids was associated with α-syn. Western blot showed that α-syn was greatly down-regulated in PMA-treated PD52-derived midbrain organoids (Fig. 5F).
A speculative chemical mechanism can be proposed for the loss of function by depleted vesicles in DA neurons and midbrain organoids derived from PD52 with upregulated α-syn and the recovery after PMA treatment. From the amperometric data, it is clear that the neurotransmitter (catecholamine) content in individual vesicles decreases significantly in DA neurons and organoids derived from PD52. The membrane protein α-syn has been reported to be involved in the regulation of synaptic activity through modulating vesicle trafficking and neurotransmitter release and reuptake.27 Overexpression of α-syn was found in our human disease model (both DA neurons and midbrain organoids derived from PD52). More importantly, upregulated α-syn in the PD52-derived single DA neurons was observed. α-Syn is a very sticky protein and has been reported to bind over hundreds of different proteins and lipids, particularly when overexpressed.28 These effects could for example be due to changes in binding to DAT, to synapsin, and even to disruption of vesicle membrane and proton gradient collapse which decreases dopamine storage.29 The prolongation of thalf is also associated with the excessive α-syn production in the midbrain-like cultures derived from PD52. Thus, we assert that the biophysical properties of the vesicle membrane, including bilayer curvature, strength, and plasticity might be governed by the excessive α-syn,30,31 which further affects the fusion pore formed on the nano-tip electrode during the electroporation in IVIEC. This change in vesicular structure might be involved in the dopamine dysfunction of intercellular communication in PD by altering exocytosis. Ultimately, PMA can significantly decrease the expression of α-syn and rescue the dysfunction of vesicular storage in DA neurons and organoids derived from PD52, further confirming the function of α-syn in modulation of vesicular storage. It is worth mentioning that PKC has been suggested to accelerate catecholamine vesicle exocytosis.32 PMA as a PKC agonist could activate PKC, which increases the quantal size of catecholamine release33 and changes characteristics of synaptic vesicle fusion.34 This might in turn promote the reuptake of vesicles and then affect the size of vesicles. This may be coupled with decreased α-syn expression to rescue the vesicle storage deficiency in PD-derived midbrain-like culture.
Conclusions
By establishing an iPSC-based model using midbrain DA neurons (2D) and midbrain organoids (3D) derived from a human YOPD patient, we have been able to investigate the role and function of vesicular transmitter storage in YOPD-derived midbrain-like cultures. Dopamine storage in individual vesicles was explored at the single-cell level via electrochemical cytometry with nano-tip microelectrodes. Singly important is that we observed a deficiency in vesicular catecholamine storage caused neurons and organoids derived from a YOPD patient with upregulated α-syn compared to the control. Furthermore, this loss of vesicular transmitter content is recovered by AMA, a drug for the treatment of PD.We note that overexpressed α-syn alters the decay time of pore formation on the microelectrode surface during IVIEC and we suggest this resulted from a change in vesicle membrane curvature influenced by the augmentation of α-syn. We thus propose a mechanism where this α-syn derived membrane curvature change is partially responsible for dopaminergic dysfunction in intercellular communication via exocytosis.
Applications for the iPSCs-derived neuron/organoid models and analysis platform we present here should largely contribute to the pathological research of YOPD or other neurodegenerative diseases. We provide the first evidence that dopamine levels decrease in the vesicles of midbrain-like cultures derived from a YOPD patient. Our work provides a simplified discovery testing platform for new therapeutic agents that target the underlying pathologic process in PD.
Ethical approval
The human sample was collected with informed consent under full ethical approval in accordance with the ethics committee at Nanjing Medical University ([2019] No. 485).Data availability
All experimental supporting data and procedures are available in the ESI.†Author contributions
W. Z. and Y. L. designed research; W. Z. and M. T. performed research with technical assistance from Y. H., S. W., C. C., Z. Z., M. X.; W. Z. and M. T. analyzed data with assistance from Y. H., X. H., Q. Z.; W. Z. and M. T. wrote the paper; A. E. and X. G. contributed intellectually to data interpretation and in intellectual discussions; Y. L., W. Z. and A. E. provided funding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Key Research and Development Program of China (grant no. 2021YFF1200800, 2021YFA1101800, and 2019YFA0802703), the Strategic Priority Research Program of the Chinese Academy of Sciences (grant no. XDA16010306), the National Natural Science Foundation of China Grants (grant no. 21904069, 81922022, 82022021, and 82171528), and the Natural Science Foundation of Jiangsu Province (grant no. BK20190653). AGE acknowledges support from the European Research Council (ERC Advanced grant project no. 787534 NanoBioNext), Knut and Alice Wallenberg Foundation, and the Swedish Research Council (VR Grants 2017-04366, 2017-05962 and 2020-00803).Notes and references
- J. R. Sanes and S. L. Zipursky, Cell, 2020, 181, 1434–1435 CrossRef CAS PubMed.
- P. P. Michel, E. C. Hirsch and S. Hunot, Neuron, 2016, 90, 675–691 CrossRef CAS PubMed.
- X. Li, S. Majdi, J. Dunevall, H. Fathali and A. G. Ewing, Angew. Chem., Int. Ed. Engl., 2015, 54, 11978–11982 CrossRef CAS PubMed.
- W. Zhu, C. Gu, J. Dunevall, L. Ren, X. Zhou and A. G. Ewing, Angew. Chem., Int. Ed. Engl., 2019, 58, 4238–4242 CrossRef CAS PubMed.
- Q. Yue, X. Li, F. Wu, W. Ji, Y. Zhang, P. Yu, M. Zhang, W. Ma, M. Wang and L. Mao, Angew. Chem., Int. Ed. Engl., 2020, 59, 11061–11065 CrossRef CAS PubMed.
- Y. Tang, X. K. Yang, X. W. Zhang, W. T. Wu, F. L. Zhang, H. Jiang, Y. L. Liu, C. Amatore and W. H. Huang, Chem. Sci., 2019, 11, 778–785 RSC.
- A. Larsson, S. Majdi, A. Oleinick, I. Svir, J. Dunevall, C. Amatore and A. G. Ewing, Angew. Chem., Int. Ed., 2020, 59, 6711–6714 CrossRef CAS PubMed.
- W. Yang, X. Guo, Z. Tu, X. Chen, R. Han, Y. Liu, S. Yan, Q. Wang, Z. Wang, X. Zhao, Y. Zhang, X. Xiong, H. Yang, P. Yin, H. Wan, X. Chen, J. Guo, X. X. Yan, L. Liao, S. Li and X. J. Li, Protein Cell, 2022, 13, 26–46 CrossRef CAS PubMed.
- J. Jo, L. Yang, H. D. Tran, W. Yu, A. X. Sun, Y. Y. Chang, B. C. Jung, S. J. Lee, T. Y. Saw, B. Xiao, A. T. T. Khoo, L. P. Yaw, J. J. Xie, H. Lokman, W. Y. Ong, G. G. Y. Lim, K. L. Lim, E. K. Tan, H. H. Ng and H. S. Je, Ann. Neurol., 2021, 90, 490–505 CrossRef CAS PubMed.
- J. Penney, W. T. Ralvenius and L. H. Tsai, Mol. Psychiatry, 2020, 25, 148–167 CrossRef PubMed.
- D. C. Schondorf, M. Aureli, F. E. McAllister, C. J. Hindley, F. Mayer, B. Schmid, S. P. Sardi, M. Valsecchi, S. Hoffmann, L. K. Schwarz, U. Hedrich, D. Berg, L. S. Shihabuddin, J. Hu, J. Pruszak, S. P. Gygi, S. Sonnino, T. Gasser and M. Deleidi, Nat. Commun., 2014, 5, 4028–4045 CrossRef PubMed.
- J. Yu, M. A. Vodyanik, K. Smuga-Otto, J. Antosiewicz-Bourget, J. L. Frane, S. Tian, J. Nie, G. A. Jonsdottir, V. Ruotti, R. Stewart, I. I. Slukvin and J. A. Thomson, Science, 2007, 318, 1917–1920 CrossRef CAS PubMed.
- W. K. Raja, A. E. Mungenast, Y. T. Lin, T. Ko, F. Abdurrob, J. Seo and L. H. Tsai, PLoS One, 2016, 11, e0161969 CrossRef PubMed.
- A. H. Laperle, S. Sances, N. Yucer, V. J. Dardov, V. J. Garcia, R. Ho, A. N. Fulton, M. R. Jones, K. M. Roxas, P. Avalos, D. West, M. G. Banuelos, Z. Shu, R. Murali, N. T. Maidment, J. E. Van Eyk, M. Tagliati and C. N. Svendsen, Nat. Med., 2020, 26, 289–299 CrossRef CAS PubMed.
- F. Soldner, D. Hockemeyer, C. Beard, Q. Gao, G. W. Bell, E. G. Cook, G. Hargus, A. Blak, O. Cooper, M. Mitalipova, O. Isacson and R. Jaenisch, Cell, 2009, 136, 964–977 CrossRef CAS PubMed.
- M. A. Lancaster, M. Renner, C. A. Martin, D. Wenzel, L. S. Bicknell, M. E. Hurles, T. Homfray, J. M. Penninger, A. P. Jackson and J. A. Knoblich, Nature, 2013, 501, 373–379 CrossRef CAS PubMed.
- J. Jo, Y. X. Xiao, A. X. Sun, E. Cukuroglu, H. D. Tran, J. Goke, Z. Y. Tan, T. Y. Saw, C. P. Tan, H. Lokman, Y. Lee, D. Kim, H. S. Ko, S. O. Kim, J. H. Park, N. J. Cho, T. M. Hyde, J. E. Kleinman, J. H. Shin, D. R. Weinberger, E. K. Tan, H. S. Je and H. H. Ng, Cell Stem Cell, 2016, 19, 248–257 CrossRef CAS PubMed.
- S. P. Pasca, Nature, 2018, 553, 437–445 CrossRef CAS PubMed.
- F. Birey, J. Andersen, C. D. Makinson, S. Islam, W. Wei, N. Huber, H. C. Fan, K. R. C. Metzler, G. Panagiotakos, N. Thom, N. A. O'Rourke, L. M. Steinmetz, J. A. Bernstein, J. Hallmayer, J. R. Huguenard and S. P. Pasca, Nature, 2017, 545, 54–59 CrossRef CAS PubMed.
- Y. J. Chen, M. Xiong, Y. Dong, A. Haberman, J. Y. Cao, H. S. Liu, W. H. Zhou and S. C. Zhang, Cell Stem Cell, 2016, 18, 817–826 CrossRef CAS PubMed.
- P. Jadiya, D. W. Kolmetzky, D. Tomar, A. Di Meco, A. A. Lombardi, J. P. Lambert, T. S. Luongo, M. H. Ludtmann, D. Pratico and J. W. Elrod, Nat. Commun., 2019, 10, 3885–3899 CrossRef PubMed.
- N. A. Tatton, Exp. Neurol., 2000, 166, 29–43 CrossRef CAS PubMed.
- N. Wulansari, W. H. W. Darsono, H. J. Woo, M. Y. Chang, J. Kim, E. J. Bae, W. Sun, J. H. Lee, I. J. Cho, H. Shin, S. J. Lee and S. H. Lee, Sci. Adv., 2021, 7, eabb1540 CrossRef CAS PubMed.
- O. Rascol, M. Fabbri and W. Poewe, Lancet Neurol., 2021, 20, 1048–1056 CrossRef CAS.
- J. C. Bridi and F. Hirth, Front. Neurosci., 2018, 12, 80–98 CrossRef PubMed.
- A. Gordon, S. J. Yoon, S. S. Tran, C. D. Makinson, J. Y. Park, J. Andersen, A. M. Valencia, S. Horvath, X. Xiao, J. R. Huguenard, S. P. Pasca and D. H. Geschwind, Nat. Neurosci., 2021, 24, 331–342 CrossRef CAS PubMed.
- H. A. Lashuel, C. R. Overk, A. Oueslati and E. Masliah, Nat. Rev. Neurosci., 2013, 14, 38–48 CrossRef CAS PubMed.
- L. M. A. Oliveira, T. Gasser, R. Edwards, M. Zweckstetter, R. Melki, L. Stefanis, H. A. Lashuel, D. Sulzer, K. Vekrellis, G. M. Halliday, J. J. Tomlinson, M. Schlossmacher, P. H. Jensen, J. Schulze-Hentrich, O. Riess, W. D. Hirst, O. El-Agnaf, B. Mollenhauer, P. Lansbury and T. F. Outeiro, NPJ Parkinsons Dis., 2021, 7, 65–88 CrossRef PubMed.
- E. V. Mosharov, R. G. Staal, J. Bove, D. Prou, A. Hananiya, D. Markov, N. Poulsen, K. E. Larsen, C. M. Moore, M. D. Troyer, R. H. Edwards, S. Przedborski and D. Sulzer, J. Neurosci., 2006, 26, 9304–9311 CrossRef CAS PubMed.
- W. S. Davidson, A. Jonas, D. F. Clayton and J. M. George, J. Biol. Chem., 1998, 273, 9443–9449 CrossRef CAS PubMed.
- T. Logan, J. Bendor, C. Toupin, K. Thorn and R. H. Edwards, Nat. Neurosci., 2017, 20, 681–689 CrossRef CAS PubMed.
- R. Borges, J. D. Machado, G. Betancor and M. Camacho, Ann. N. Y. Acad. Sci., 2002, 971, 184–192 CrossRef CAS PubMed.
- R. G. Staal, A. Hananiya and D. Sulzer, J. Neurochem., 2008, 105, 1635–1641 CrossRef CAS PubMed.
- R. G. Staal, E. V. Mosharov and D. Sulzer, Nat. Neurosci., 2004, 7, 341–346 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and additional spectra and imaging data. See https://doi.org/10.1039/d2sc00809b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |