
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolution structure of a thrombin binding aptamer complex with a non-planar platinum(II) compound†
Bo-Chen
Zhu‡
a,
Juan
He‡
b,
Xiao-Yu
Xia‡
a,
Jingxing
Jiang
c,
Wenting
Liu
 *a,
Liu-Yi
Liu
a,
Bing-Bing
Liang
a,
Hua-Gang
Yao
b,
Zhuofeng
Ke
*c,
Wei
Xia
*a and
Zong-Wan
Mao
*a
*a,
Liu-Yi
Liu
a,
Bing-Bing
Liang
a,
Hua-Gang
Yao
b,
Zhuofeng
Ke
*c,
Wei
Xia
*a and
Zong-Wan
Mao
*a
aMOE Key Laboratory of Bioinorganic and Synthetic Chemistry, School of Chemistry, Sun Yat-sen University, Guangzhou 510275, China. E-mail: cesmzw@mail.sysu.edu.cn; liuwenting@mail.sysu.edu.cn; xiawei5@mail.sysu.edu.cn
bSchool of Pharmaceutical and Chemical Engineering, Guangdong Pharmaceutical University, Zhongshan 528458, China
cSchool of Materials Science and Engineering, PCFM Lab, School of Chemistry, Sun Yat-sen University, Guangzhou 510275, China. E-mail: kezhf3@mail.sysu.edu.cn
First published on 23rd June 2022
Abstract
Thrombin Binding Aptamer (TBA) is a monomolecular well-defined two G-tetrad antiparallel G-quadruplex DNA that inhibits the activity of human α-thrombin. In this report, we synthesized a quasi-cross-shaped platinum(II) compound (L′2LPt) with one cyclometalated and two carbene ligands. We found L′2LPt has selective affinity to bind the TBA G-quadruplex. A fibrinogen clotting assay revealed that L′2LPt can abrogate the inhibitory activity of TBA against thrombin. We solved the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 L′2LPt–TBA complex structure by NMR, which revealed a unique self-adaptive property of L′2LPt upon binding to TBA. In the complex, a carbene ligand of L′2LPt rotates to pair with the cyclometalated ligand to form a plane stacking over half of the TBA G-tetrad and covered by lateral TT loops. It is notable that the heavy atom Pt stays out of the G-tetrad. Meanwhile, the other carbene ligand remains relatively perpendicular and forms a hydrogen bond with a guanine to anchor the L′2LPt position. This structure exhibits a quasi-cross-shaped Pt(II) compound bound to the G-quadruplex with an unusual “wall-mounted” binding mode. Our structures provide insights into the specific recognition of antiparallel G-quadruplex DNA by a self-adaptive Pt(II) compound and useful information for the design of selective G-quadruplex targeting non-planar molecules.
:1 L′2LPt–TBA complex structure by NMR, which revealed a unique self-adaptive property of L′2LPt upon binding to TBA. In the complex, a carbene ligand of L′2LPt rotates to pair with the cyclometalated ligand to form a plane stacking over half of the TBA G-tetrad and covered by lateral TT loops. It is notable that the heavy atom Pt stays out of the G-tetrad. Meanwhile, the other carbene ligand remains relatively perpendicular and forms a hydrogen bond with a guanine to anchor the L′2LPt position. This structure exhibits a quasi-cross-shaped Pt(II) compound bound to the G-quadruplex with an unusual “wall-mounted” binding mode. Our structures provide insights into the specific recognition of antiparallel G-quadruplex DNA by a self-adaptive Pt(II) compound and useful information for the design of selective G-quadruplex targeting non-planar molecules.
Introduction
G-quadruplexes (G4s) are four-stranded nucleic acid structures formed by guanine-rich nucleic acids.1 Typically, four guanine bases can self-assemble through a cyclic array of Hoogsteen hydrogen bonds to form a G-quartet with a square planar structure, which is stabilized by cations, such as K+ or Na+, in the center of the tetrads.2–4 Two or more G-quartets are stacked together and interconnected by loops of different length to form a G4.5,6Aptamers are single-stranded DNA or RNA sequences that can selectively bind to a specific target, including proteins, nucleic acids, cells, tissues and even organisms.7–9 Aptamers with affinity for a desired target can be selected through a process called SELEX (Systematic Evolution of Ligands by EXponential enrichment).10 Previous studies have demonstrated that some aptamers can fold into a G4 structure.11 The best-known example is the thrombin-binding aptamer (TBA), a 15-mer DNA oligonucleotide with sequence of 5′-GGTTGGTGTGGTTGG-3′, which was discovered in 1992 for the recognition of thrombin and is still the best studied anti-thrombin aptamer.12 This aptamer is capable of inhibiting fibrin clot formation by binding to human thrombin with high selectivity and affinity.13–15 Therefore, TBA and its analogs were considered as promising anticoagulant agents and clinical trials have evaluated TBA as an anticoagulant for potential use in acute cardiovascular settings, such as coronary artery bypass graft (CABG) surgery.16,17 Due to the potential therapeutic application, many research studies focused on improving its pharmacological properties have been reported in recent years.16–18 Simultaneous antidote development is also crucial, as anticoagulant treatment can have side effects such as hemorrhagic complications.19,20
Previous NMR and X-ray structural studies have demonstrated that TBA adopts an antiparallel chair-like G4 structure, which consists of two G-tetrads connected by a TGT loop on one side and two TT loops on the opposite side.21–23 Typically, the two TT loops interact with human α-thrombin, which is the binding site of fibrinogen.23–27 The G4 structure is of great importance to the function of TBA. Hence, the TBA anticoagulant activity can be inhibited by hybridization with some antidotes, such as a complementary oligonucleotide or small-molecule ligands that can disrupt the conformation of G4.13,28 Up to now, only a few TBA G4-binding small molecules have been reported, including TMPyP4, thioflavin T (ThT) and N-methylmesoporphyrin IX (NMM).29,30 To recognize and stabilize G4 structures, a series of synthetic small molecules have been identified.31–33 In particular, small molecules that have (a) a π-delocalized system, (b) a partial positive charge in the centre of the molecular scaffold, and (c) a positively charged substituent to interact with the grooves, loops, and the negatively charged phosphate backbone prefer to bind to G4 structures.33–35 Moreover, small molecule ligands that stabilize the G4 structure could also be applied to regulate or detect intracellular G4 formation.36,37 Current structural studies of G4-binding small molecules have mainly focused on compounds with a large planar group, which can form π–π interactions with the guanine plane. Small molecules with non-planar central core structures are rarely studied.
We recently reported an electrically neutral and non-planar platinum compound (Pt1) with a freely rotating N-heterocyclic carbene ligand.36 This non-charged and non-planar platinum compound is converted into a positive monovalent planar structure upon binding to VEGF G4. The report highlights the G4 binding activated by adaptive ligand structural transformations and G4 selectivity controlled by binding kinetics, which is a new strategy for G4 selective binding. In this report, we synthesized and characterized an organometallic platinum(II) bis-N-heterocyclic carbene compound (L′2LPt, Fig. 1A). FRET and CD thermal melting assays revealed that the melting temperature increase of TBA G4 (Fig. 1B) after L′2LPt binding is clearly higher than that of other G4 DNAs, which is indicative of L′2LPt binding to TBA G4 with a higher affinity. The solution structure of the L′2LPt–TBA G4 complex was determined by NMR spectroscopy. A rotation of the carbene ligand was observed and the cross-shaped L′2LPt molecule bound to antiparallel TBA G4 in a unique half-ligand to half-G-tetrad “wall-mounted” mode with lateral loop interactions. Furthermore, we showed that L′2LPt could abrogate the interaction between the TBA aptamer and thrombin and thereby restore the activity of thrombin.
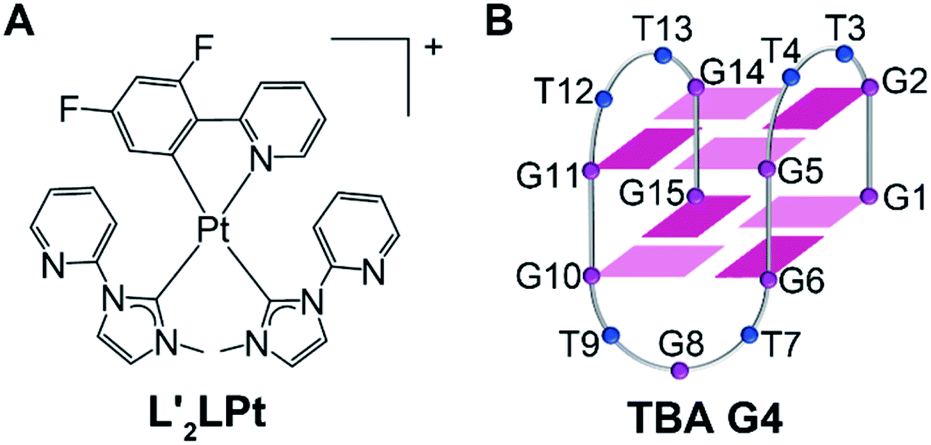 | ||
| Fig. 1 (A) Schematic diagram of the chemical structure of L′2LPt. (B) The folding topology of TBA G4. Deep red box = (anti)-guanine, light red box = (syn)-adenine, blue ball = thymine. | ||
Results and discussion
Binding of aptamer TBA G4 by L′2LPt
The non-planar platinum compound L′2LPt, which is coordinated by a cyclometalated ligand (L) and two rotated N-heterocyclic carbene ligands (L′), was synthesized and characterized (Fig. 1A, Scheme S1, Fig. S1–S3 and Table S1†). FRET and CD melting assays were first applied to investigate the change in L′2LPt-induced thermal stability of various DNA oligonucleotides (Fig. S4 and Table S2†), including three three-G-tetrad telomeric G4s (Tel26,38 wtTel26,39 and wtTel22 (ref. 40)), five three-G-tetrad promoter G4s (c-myc,41 Bcl2,42 P1G4T,42 VEGF43 and c-kit87up44), two three-G-tetrad intron G4s (MYT1L45 and CHL1 (ref. 46)), two two-G-tetrad promoter G4s (c-kit* (ref. 47) and HIV-PRO1 (ref. 48)), one two-G-tetrad aptamer G4 (TBA) and one duplex DNA (F10T). The melting curves and melting temperature change values (ΔTm) of DNA after the addition of 1.0 equivalent of L′2LPt were recorded (Fig. 2A, S5, and S6, and Tables S3 and S4†).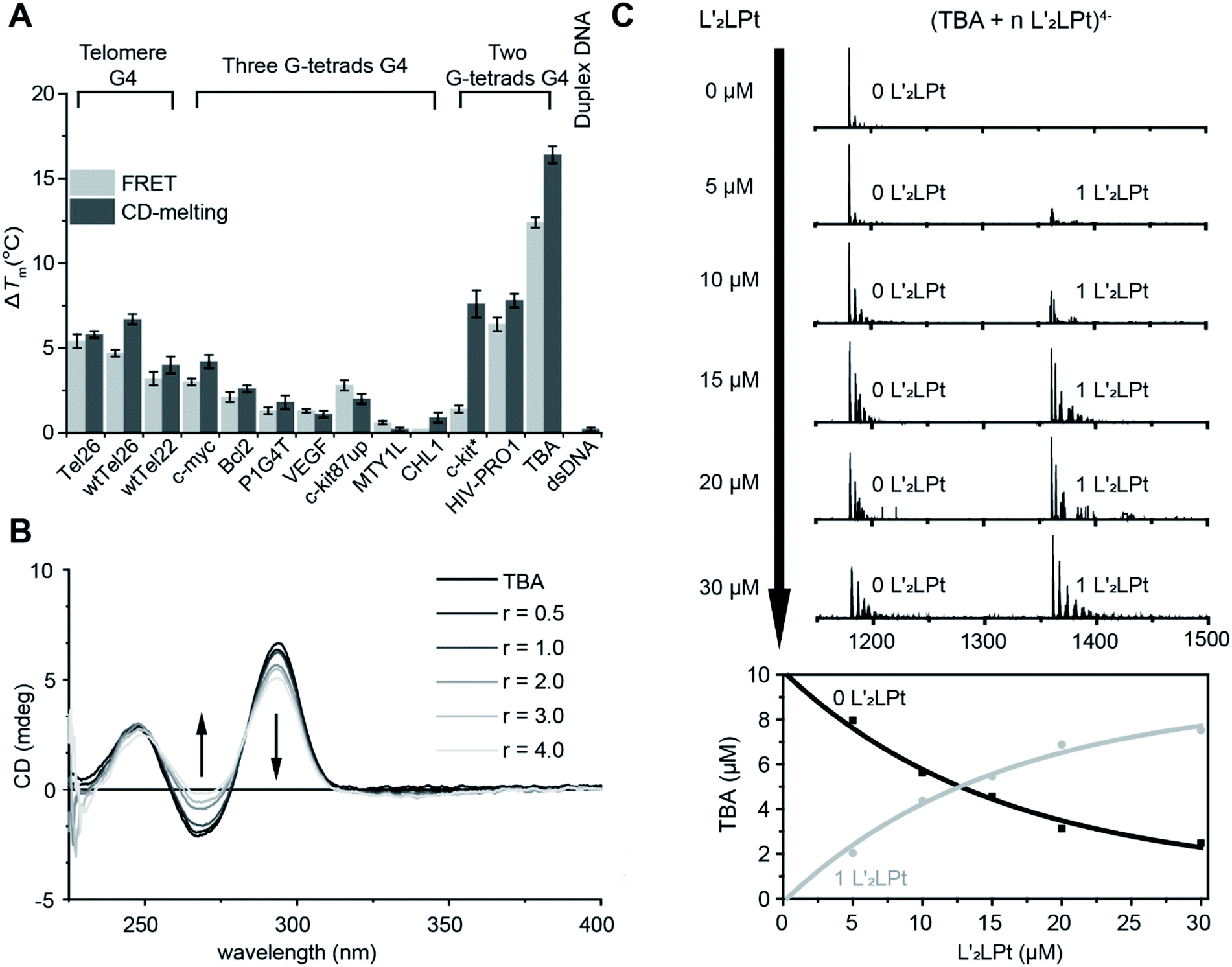 | ||
| Fig. 2 (A) ΔTm value from FRET and CD melting of different G4s and duplex DNA. FRET data were tested in 0.40 μM DNA interacting with 0.40 μM L′2LPt in 10 mM tris–HCl buffer (pH 7.4) containing 60 mM potassium cacodylate. CD melting data were tested in 3 μM DNA interacting with 3 μM L′2LPt in 10 mM tris–HCl and 100 mM KCl buffer (pH 7.4). (B) CD spectra of L′2LPt titrated into TBA G4 DNA solution at different L′2LPt/TBA ratios. Conditions: pH 7.4, 10 mM tris–HCl, 100 mM KCl solution. (C) Top: ESI-MS titrations of 10 μm TBA with L′2LPt in 150 mM ammonium acetate (CH3CO2NH4) buffer (pH 7.4) showing a zoom-in of the 4− charged state. Bottom: Quantification and fitting of the ESI-MS data based on the number of bound ligands (0 L′2LPt and 1 L′2LPt). | ||
We found that the stabilization temperatures of DNAs after adding L′2LPt were consistent between the FRET and CD experiments, except for c-kit* G4. The lower FRET ΔTm of c-kit* G4 is probably related to the labelled fluorophores in the c-kit* DNA sequence, which affect its G4 structure. The results showed that TBA G4 exhibited the largest ΔTm value (12.4 °C in FRET, 16.4 °C in CD), Tel26, wtTel26, c-kit*, HIV-PRO1 and wtTel22 exhibited moderate ΔTm values, and other G4s exhibited rather low ΔTm values. To enable greater comparability, we conducted FRET experiments at different concentrations of K+ buffer to make each DNA display similar initial Tm values in the range of 63 ± 2 °C (Fig. S7 and S8, and Tables S5 and S6†). The results of ΔTm value remained similar. These results indicated that L′2LPt can better stabilize TBA G4 than other G4s. Moreover, the ΔTm value of duplex DNA was very low (0.0 °C in FRET, 0.2 °C in CD), which is indicative of low affinity and no specific binding of duplex DNA with L′2LPt. By comparing the conformations of the G4s used in these experiments, we found that the G4s with lateral loops are more likely to be stabilized by L′2LPt. In particular, the two-G-tetrad antiparallel G4s with two lateral loops on the same side have much higher ΔTm values than those of other G4s.
FRET competitive experiments were carried out to explore the selectivity of L′2LPt between TBA and different kinds of DNAs (Fig. S9 and Table S7†). The results showed that after the addition of 50 equivalents of duplex DNA (ds26) and non-lateral-loop G4 DNA (VEGF), the stabilization of L′2LPt to TBA can still be maintained at 75.8% and 58.4%. In contrast, the G4 DNAs with lateral-loops (Tel26 and HIV-PRO1), especially the two-G-tetrad G4 DNA with double lateral-loops (HIV-PRO1), have a significant effect on the interaction of L′2LPt and TBA G4.
To determine the secondary structural transformation and stability of TBA G4 upon L′2LPt binding, CD spectra were acquired for TBA G4 alone or in the presence of different amounts of L′2LPt (Fig. 2B and S10†). The CD spectrum of TBA had two positive bands at 250 and 295 nm, and a negative band at 270 nm, which are characteristic of typical antiparallel G4 folding. No significant change in the CD spectra was observed when the L′2LPt/TBA molar ratio was below 1:1. The CD spectrum band intensities at 295 nm and 270 nm slightly decreased with higher concentrations of L′2LPt, suggesting a partial denaturation of TBA. However, the thermal stability of TBA increased when the ratio of L′2LPt/TBA ranged from 0 to 1, and basically remained unchanged at higher ratios. The CD experiment results implied that the stoichiometry of the binding between L′2LPt and TBA G4 is 1:1. We also conducted UV-Vis spectroscopy titration experiments (Fig. S11†). With the addition of TBA G4, we observed a slight decrease in the UV/Vis spectrum of L′2LPt at 318 nm.
ESI-MS was used to study the stoichiometries of the complexes and determine their individual Kapp values.49,50 As shown in the results (Fig. 2C and S12†), the ESI-MS spectra exhibited clear ion peaks for free TBA G4 with different electrical charges. After titrating L′2LPt into the TBA G4 solution, clear peaks for the 1:1 L′2LPt–TBA G4 complex were observed. We zoomed in on the 4− charged state of the free TBA G4 and the 1:1 L′2LPt–TBA G4 complex for the curve fitting procedure. The ESI-MS spectra of TBA and L′2LPt demonstrated that L′2LPt binds TBA G4 and forms a stable 1:1 complex with an apparent DNA binding constant (Kapp) value of 9.30 × 10−6 mol−1.
Moreover, ITC titration was also employed to examine the interactions among the TBA aptamer, L′2LPt and thrombin (Fig. 3). The combination ratio results verified that both the binding ratios of L′2LPt–TBA and TBA–thrombin were nearly 1.0, and the Ka values of the dual system with L′2LPt–TBA and TBA–thrombin were 1.83 × 107 mol−1 and 8.46 × 106 mol−1, respectively. We then added L′2LPt to the TBA–thrombin complex and set N = 1 and Ka-Pt = 1.83 × 107 mol−1. The result of Ka-TBA = 6.75× 105 mol−1 is much lower than its original intrinsic affinity. Therefore, L′2LPt can competitively bind TBA G4 against the TBA/thrombin system. The difference in enthalpy changes between the binary system and ternary system indicated that L′2LPt caused a thermodynamic change after addition to the TBA/thrombin complex. These results indicate that L′2LPt could compete with thrombin for the binding of TBA G4.
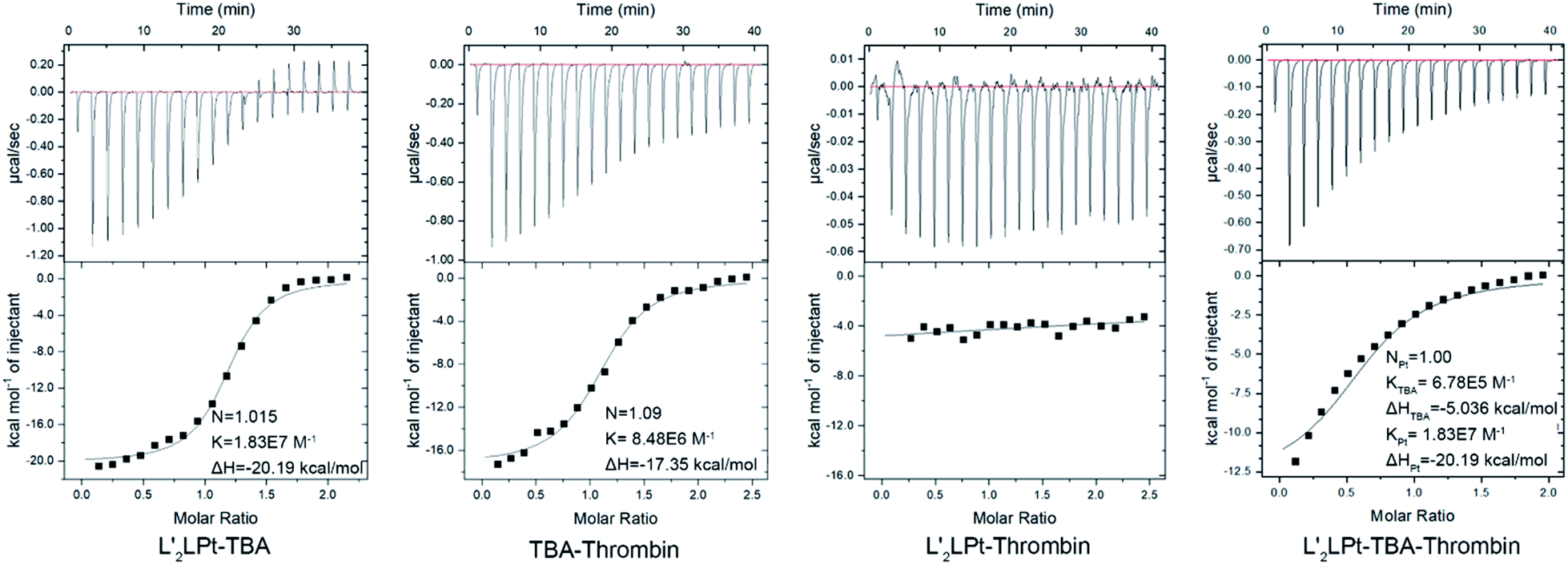 | ||
| Fig. 3 ITC data and fitting curves of different titrations of L′2LPt, TBA and thrombin at a cell concentration of 6 μM interacting with a 100 μM syringe in PBS buffer (pH 7.4) at 25 °C. | ||
NMR studies of L′2LPt interaction with TBA G4
Subsequently, to further explore the binding mechanism, the interaction between the organometallic compound L′2LPt and TBA G4 was analysed by NMR spectroscopy under physiologically relevant K+ conditions. 1D 1H NMR titration experiments were then executed (Fig. 4A). In the 1D NMR spectra, eight imino signals were observed for free TBA G4 and the signals were assigned (labelled in black) based on the reported TBA G4 structure.21 As L′2LPt was titrated into the TBA sample, the chemical shifts of TBA G4 imino protons gradually changed and a new set of distinct imino proton peaks (labelled in blue) appeared until the L′2LPt/TBA ratio reached 1:1. Even after adding more equivalents of L′2LPt compound at L′2LPt/TBA ratios of 2:1 and 3:1, the spectra retained the same pattern as for the 1:1 ratio. The results indicate that L′2LPt binds TBA G4 and can form a 1:1 stable complex, which is consistent with the ESI-MS data (Fig. 2C and S12†).
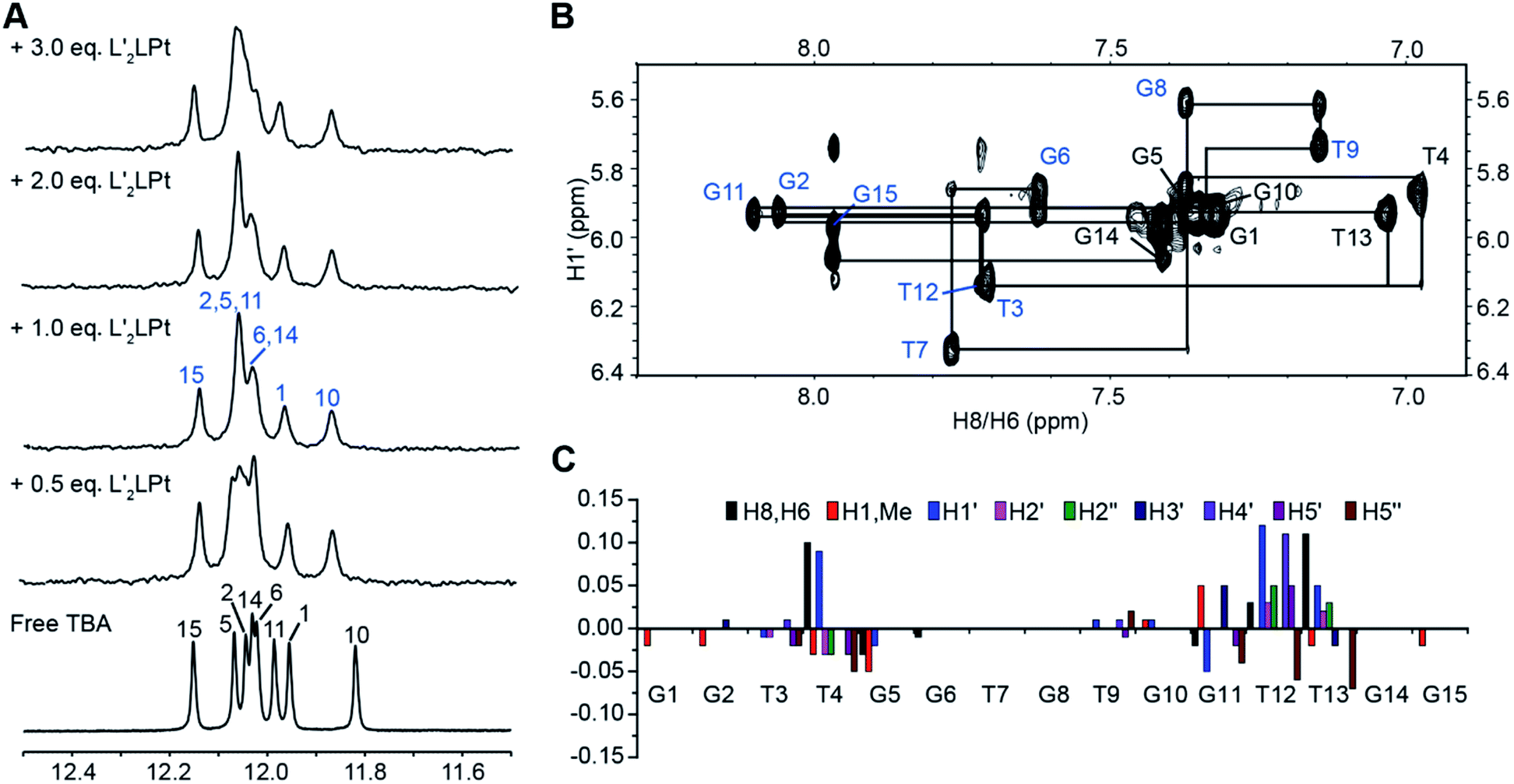 | ||
| Fig. 4 (A) Imino proton regions of the 1D 1H NMR titration spectra of TBA interacting with L′2LPt. The imino proton assignments are labeled. (B) The H1′–H6/H8 region of the 2D NOESY spectrum of the TBA G4 bond to 1.0 equivalents of L′2LPt. The complete sequential assignment pathway is shown. Residues with syn glycosidic conformations are marked in black, and those with anti glycosidic conformations are marked in blue. (C) The chemical shift difference of TBA G4 protons between the free TBA and the 1:1 L′2LPt–TBA complex. Conditions: 25 mM K-phosphate, 70 mM KCl solution, pH 7.0, 25 °C. | ||
As the well-resolved 1D NMR spectra indicated that the L′2LPt–TBA G4 complex was suitable for high resolution structural analysis, we decided to solve the solution structure of the complex to understand how L′2LPt interacts with TBA G4. A full set of 2D-NOESY, TOCSY and COSY NMR data on the 1:1 L′2LPt–TBA complex were recorded in 25 mM K-phosphate and 70 mM KCl buffer supplemented with 10% D2O (Fig. 4B and S13–S16†). The proton assignments of the H1s and H8/H6s of the free and bound TBA G4 were based on the previously reported NMR structure of TBA G4,21 as displayed by the H1′–H6/H8 (Fig. S13†) and H1–H6/H8/H1′/Me (Fig. S16†) regions. We completely assigned all of the protons of TBA G4 by assigning H1s and H8/H6s, followed by the sequential connectivity of H8/H6-sugar protons and the sequential assignment method (Tables S8 and S9†). The protons of bound L′2LPt were assigned using TOCSY (Fig. S14†) and NOESY spectra.
Similar to the free TBA G4, the G1, G5, G10, and G14 residues adopted a syn glycosidic conformation, which was reflected by the high intensities of the H1′–H8 NOE cross-peaks (Fig. S17†). Moreover, the 1:1 bound TBA G4 retained the same antiparallel folding as indicated by the similar characteristic guanine H1–H8 proton connectivity patterns of the free TBA G4 (Fig. S16†). Based on the complete NMR assignments, the proton chemical shift differences between the free TBA G4 and the 1:1 L′2LPt–TBA G4 complex were calculated (Fig. 4C). Significant proton chemical shift changes were found for the T4, G5, G11, T12 and T13 residues, illustrating that these residues are involved in L′2LPt binding and that large conformational changes of the T4 and T12–T13 loop regions are induced. The intermolecular NOE cross-peaks between L′2LPt and TBA G4 were observed and summarized (Tables S10 and S11†), revealing specific binding of L′2LPt with TBA G4.
Solution structure of the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1 L′2LPt–TBA G4 complex
:1 L′2LPt–TBA G4 complex
We calculated the solution structure of the 1:1 L′2LPt–TBA complex using NMR NOE restraints (Table S12†). The ensemble of the 10 lowest-energy structures is presented in stereo view (Fig. 5A). A representative structure is also shown (Fig. 5B). Both the L′2LPt compound and TBA G4 undergo large structural changes to form a stable complex structure.
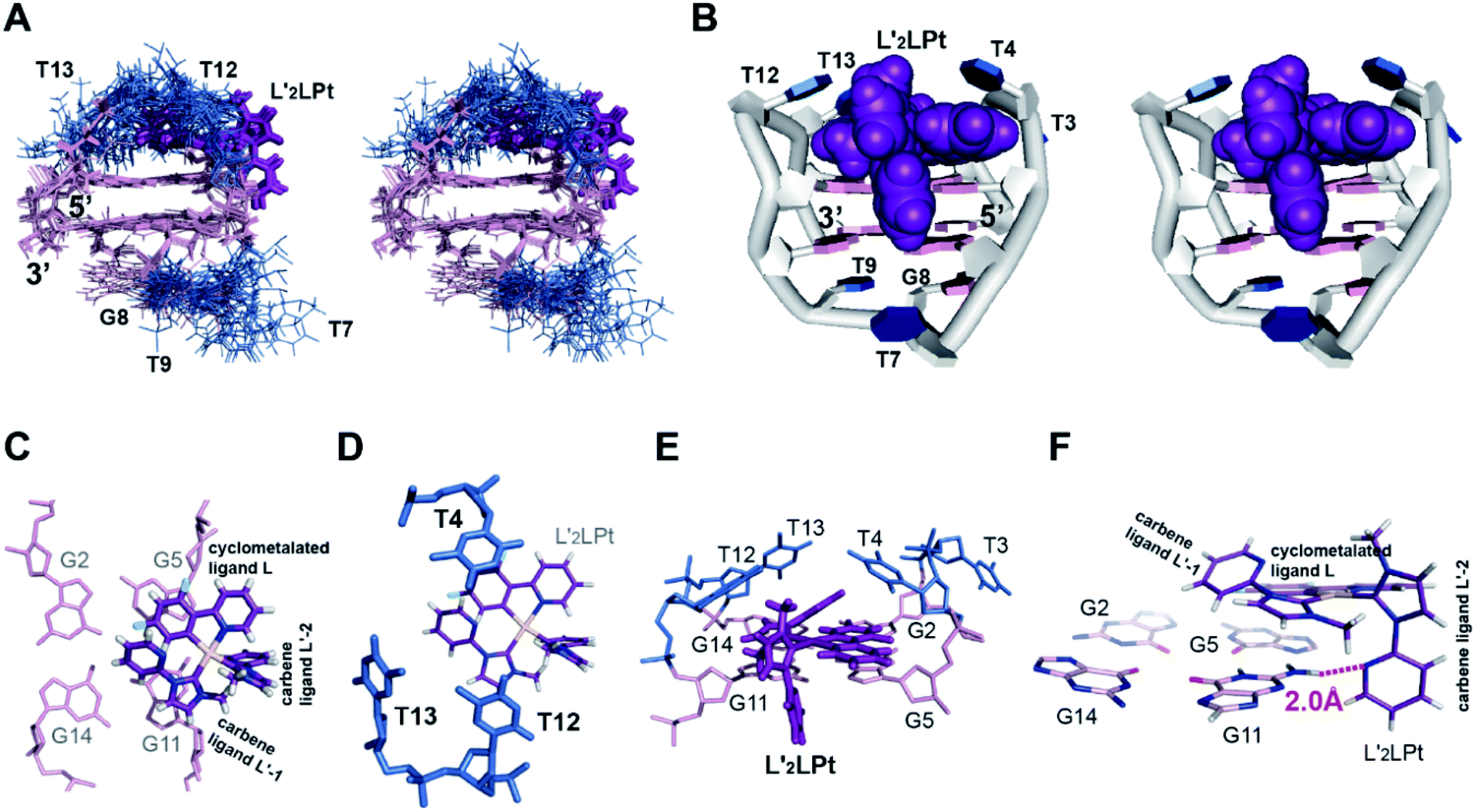 | ||
| Fig. 5 (A) The ensemble of the superimposed 10 NMR restraint-refined structures of the 1:1 L′2LPt–TBA complex in the stereo view (PDB ID 7V3T). (B) A representative structure of the 1:1 complex shown in stereo view. (C–E) Different views of the L′2LPt-induced binding pockets. (F) Potential hydrogen bonding between the L′2LPt carbene ligand L′-2 and G11 of TBA G4. The distance between the N atom on the pyridine of the carbene ligand L′-2 and the H atom on the NH2 of the G11 base is 2.0 Å. | ||
As shown in the complex (Fig. 5B), the L′2LPt compound adopts a non-planar cross-shape conformation. The carbene ligand L′-1 of L′2LPt rotates to form a plane with the cyclometalated ligand L (Fig. 5C). This cyclometalated-carbene ligand plane is embedded and stacks on G5 and G11, covering half of the external G-tetrad of TBA G4 (Fig. 5C), which is further covered by the lateral T4, T12 and T13 residues (Fig. 5D and E). In particular, the cyclometalated ligand L of L′2LPt stacks over G5, whereas the rotated carbene ligand L′-1 of L′2LPt stacks over the G11 residue with the pyridine of the carbene ligand tilting. It is notable that the heavy atom Pt stays out of the G-tetrad, which is disadvantageous for ligand binding. Intriguingly, the other carbene ligand L′-2 is relatively perpendicular to the L–L′-1 plane and stretches along the G4 groove forming a hydrogen bond between the N atom of pyridine and the NH2 group of G11 in the external G-tetrad (Fig. 5F), which anchors the L′2LPt position to achieve strong binding to G4. Therefore, the cross-shape organometallic compound L′2LPt adaptively adopts an unusual half-ligand to half-G-tetrad “wall-mounted” mode bound to antiparallel TBA G4 through both π-stacking and hydrogen bonding with the same G-tetrad.
The lateral loops T12–T13 and T3–T4 of TBA G4 also display important conformational changes induced by L′2LPt binding. In the NMR structure of free TBA G4 (ref. 21) (Fig. 6A), the T4 and T13 residues form hydrogen bonded T4–T13 base pair stacks on top of the G-tetrad and the T3 and T12 residues are more dynamic in conformation. Upon binding to L′2LPt, the T4–T13 base pair is destroyed and the lateral TT loops are reoriented to form a new binding pocket. In the complex structure (Fig. 5D and E), the T12–T13 residues cover and stack with the carbene ligand L′-1 and the T4 residue stacks on the cyclometalated ligand L to stabilize the entire L′2LPt–TBA G4 complex structure, whereas the T3 residue is flexible toward the outside of the G-tetrad.
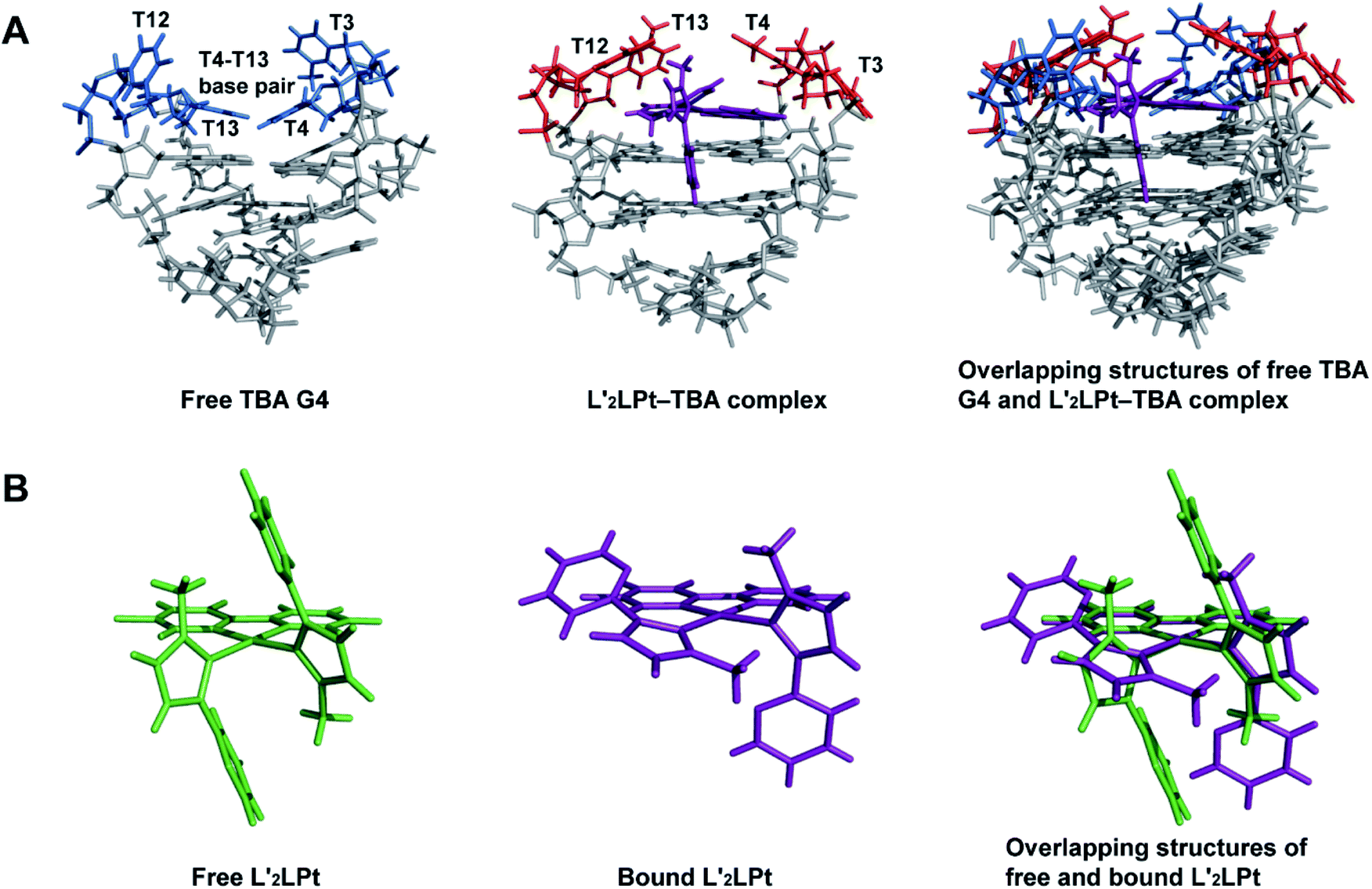 | ||
| Fig. 6 (A) Overlapping structures of free TBA G4 (PDB ID 148D) and 1:1 L′2LPt–TBA G4 complex (PDB ID 7V3T) showing the structural changes of TBA G4. (B) Overlapping structures of free and bound L′2LPt showing structural changes upon TBA G4 binding. | ||
“Wall-mounted” binding mode of L′2LPt to antiparallel TBA G4
In the 1:1 L′2LPt–TBA complex structure, the orientation and position of the L′2LPt compound is well defined. The L′2LPt molecule is coordinated by a cyclometalated ligand and two carbene ligands. This two carbene ligands can be rotated, which is quite different from the unchanged or slightly changed small molecules in most previous reports.51–53 Through structure calculation and optimization, the L′2LPt compound was found to have a non-planar structure (Fig. 6B), which is not a traditional ligand conformation for strong G4 binding. However, L′2LPt adjusts to a cross-shape conformation to bind TBA G4 and form a well-defined complex structure. This conformational adjustment upon G4 binding is significant for specific G4 interactions.
It is worth noting that TBA has an approximate 2-fold symmetry coincident with the helix axis apart from the TGT loop. Because the TGT loop does not interact with L′2LPt, it seems that two practically equivalent binding modes of the ligand to TBA are possible. However, in the free TBA G4 solution structure, the conformations of these two binding sites are different, which results in a large difference in the binding affinity. In the solution structure of TBA,21 the two TT loops (T3–T4, T12–T13) are not in the same conformation (Fig. S18A and B†). T4 and T13 are well-defined and stacked on the G2–G5–G11–G14 G-tetrad, and T3 is on top of G2 and T4, whereas T12 shows much flexibility. From the top view (Fig. S18B†), we see that the G2–G14 side is more buried and covered by T3, T12 and T13. In contrast, the G5–G11 side is exposed, so L′2LPt has greater potential for interacting and stacking with G5 and G11 (Fig. S18C and D†). Moreover, the TGT loop is on the G5–G11 side. Because of the topological and steric effect of the TGT loop, the groove on the G5–G11 side is wider than that on the G2–G14 side (Fig. S18B†). As a result, the G5–G11 side has a wider groove to accommodate the L′2LPt ligand and is more exposed, which creates a higher affinity for ligand binding than the G2–G14 side. Importantly, after binding one L′2LPt compound, it is difficult to bind another L′2LPt compound on the G2–G14 side because of steric hindrance by the G2–G5–G11–G14 G-tetrad. Consequently, we found one strong binding and obtained only one stable L′2LPt–TBA solution structure.
This is the first solution structure of small molecules binding to antiparallel DNA G4 with two G-tetrad layers. In particular, the NMR solution structure revealed that L′2LPt binds to antiparallel TBA in a very unique half-ligand to half-G-tetrad “wall-mounted” binding mode, which is significantly different from the previously reported π-delocalized molecules. The half-G-tetrad binding structure is reported in the solution structure of a 2:1 quindoline–c-MYC complex.41 The quindolines are stacked with two of the four guanines from each external tetrad with crescent-shaped cyclic fused rings. However, L′2LPt interacted with two guanines with its two separated ligands, i.e. the cyclometalated ligand and carbene ligand stacked with G5 and G11 residues respectively in the external G-tetrad.
The common strategy for targeting G4 is to design small molecules with a planar aromatic central core. From the obtained complex structure, we found that non-planar small molecules could also bind G4 in an induced-fit manner through a “wall-mounted” binding mode, different from most G4 ligands. Previous reports have shown a similar “wall-mounted” binding mode that is formed by the interactions of the ruthenium polypyridyl compounds with telomeric G4s in two crystal structures.55,56 Both reports utilize a large planar automatic ligand to bind to the G-tetrad via π–π stacking, whereas other ligands and metallic ruthenium centers are positioned on the G4 groove, without hydrogen bonding. No structural changes of these ruthenium compounds were observed. The ruthenium centers are also outside of the G-tetrad, similar to our “wall-mounted” mode. Thus, in future development, we believe that the non-planar small molecules will provide a complementary design principle for compounds targeting G4 or similar biomolecules, and the “wall-mounted” mode could be a possible interaction for these ligands and G4s.
Competitive binding of the platinum compound and thrombin to the TBA aptamer
TBA G4 is a potent inhibitor of thrombin, and it has been reported that the anti-thrombin effects of aptamers can be antagonized.54 We then preliminarily explored the effects of L′2LPt-binding on the inhibitory activity of TBA G4 against thrombin.As thrombin could induce fibrinogen to coagulate, we conducted a fibrinogen clotting assay using a thrombin, TBA and L′2LPt ternary system (Fig. 7 and Table S13†). As shown in the results, the fibrinogen clotting time is approximately 15 s in the system with only thrombin. The clotting time substantially increased to 52.7 ± 2.3 s, 97.4 ± 2.4 s and 133.8 ± 3.1 s when adding 200, 500, and 1000 nM TBA, respectively, which is indicative of the inhibition of thrombin activity by TBA G4. After incubation with L′2LPt, the fibrinogen clotting time was significantly reduced, implying that the recovery of thrombin activity was influenced by L′2LPt. Typically, the reduction in fibrinogen clotting time is correlated with the increasing amount of L′2LPt. Under incubation with a 3.0 L′2LPt/TBA ratio of L′2LPt, the thrombin clotting time decreased to 29.3 ± 0.9 s, 38.8 ± 3.0 s and 55.6 ± 3.9 s respectively, which indicated that the thrombin activity recovered by almost 50%. We also used two typical G4 binders TMPyP4 and PDS as control in this experiment. The result showed that the TMPyP4 had less effect on clotting time than PDS and the recovery of coagulation time of both TMPyP4 and PDS is weaker than L′2LPt. This may be due to the fact that these two typical G4 binders interact with the G-tetrad plane and have fewer interactions with the TT loop structure of TBA, which is the binding site of TBA and thrombin. These data clearly demonstrated that L′2LPt-binding abrogates the thrombin–TBA interaction and releases TBA-inactivated thrombin.
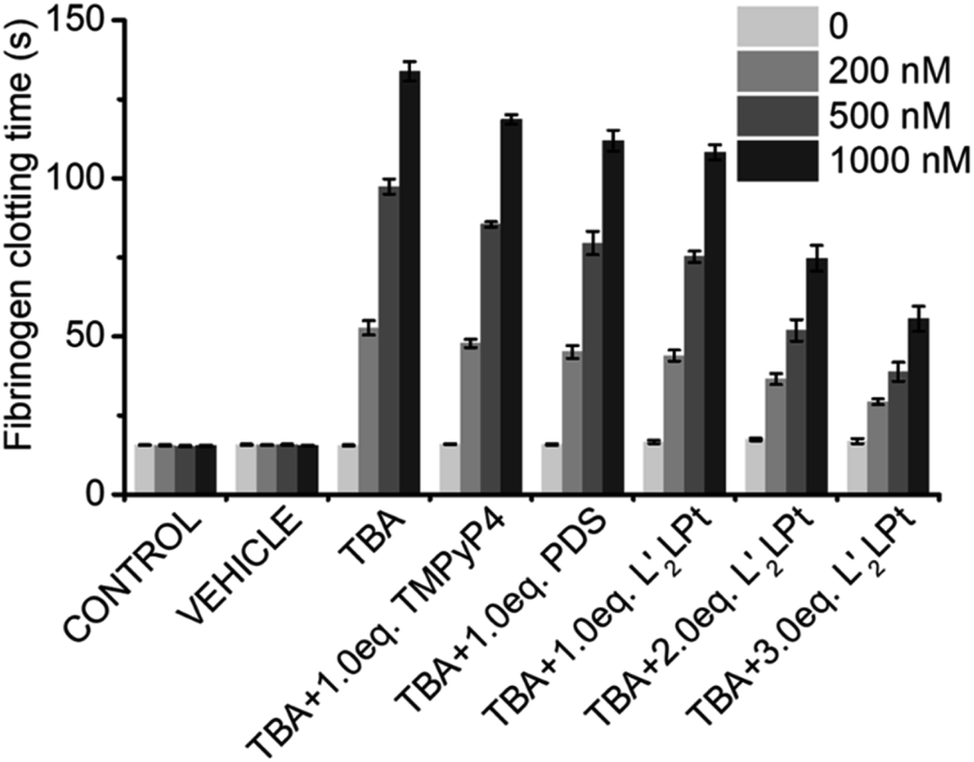 | ||
| Fig. 7 Fibrinogen clotting time of thrombin alone (1.0 ml of PBS containing 2.0 mg ml−1 of fibrinogen added to 100 μl/10 NIH per ml of human thrombin) or in the presence of 0, 200, 500 and 1000 nM concentrations of TBA and different equivalents of L′2LPt. TMPyP4 and PDS were used as positive control. The bars of the control and vehicle represent the fibrinogen clotting time values of the system in the absence of any L′2LPt/TBA and diluted with the buffer alone, respectively. | ||
Conclusion
In this study, we identified a quasi-cross-shaped Pt compound, L′2LPt, with a non-planar central core, which has a strong affinity for the DNA G4 aptamer TBA. The identification and characterization of this new half-ligand to half-G-tetrad “wall-mounted” binding mode between the small non-planar molecule L′2LPt and the antiparallel TBA G4 are of great importance. Currently, the structure-based design of small molecules interacting with unimolecular G4 is focused on the development of ligands with planar cyclic fused rings that interact with the external G-tetrads. Here we found that the non-planar small molecules could also bind G4 with high selectivity. The obtained L′2LPt–TBA complex structure is not only the first solution complex structure of a small molecule bound to intramolecular antiparallel G4 through both π-stacking and hydrogen bonding within a G-tetrad, but also provides a new paradigm for designing G4-specific targeting non-planar small molecules.Data availability
Experimental data associated with the article can be found in the ESI.† The coordinates for the structure of the 1:1 L′2LPt–TBA G4 complex (accession code 7V3T) have been deposited in the Protein Data Bank.
Author contributions
Z.-W. M. and W. L. conceived and directed the project. J. H. and B.-B. L. designed, synthesized and characterized the L′2LPt compound. H.-G. Y. analyzed the crystal structure of L′2LPt. B.-C. Z. and X.-Y. X. performed FRET, CD, ESI-MS, and NMR experiments, fibrinogen clotting and assigned NMR spectra. W. L., L.-Y. L. and J. J. did structure refinements. B.-C. Z. wrote the paper with the help of Z.-W. M., W. L., X. W. and Z. K.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was funded by the National Natural Science Foundation of China (No. 21837006, 22007103, 91953117, 21572282), the Ministry of Education of China (IRT-17R111), and Science and Technology Planning Project of Guangdong Province (No. 207999 and 2020A1515011439). We thank Dr Zhihui Xiao and Dr Xiaohong Zheng (Equipment Public Service Center, South China Sea Institute of Oceanology, Chinese Academy of Sciences) for their support with the NMR experiments.Notes and references
- M. Gellert, M. N. Lipsett and D. R. Davies, Proc. Natl. Acad. Sci., USA, 1962, 48, 2013–2018 CrossRef CAS PubMed.
- J. Zhou, S. Amrane, F. Rosu, G. F. Salgado, Y. Bian, H. Tateishi-Karimata, E. Largy, D. N. Korkut, A. Bourdoncle, D. Miyoshi, J. Zhang, H. Ju, W. Wang, N. Sugimoto, V. Gabelica and J.-L. Mergny, J. Am. Chem. Soc., 2017, 139, 7768–7779 CrossRef CAS PubMed.
- D. Varshney, J. Spiegel, K. Zyner, D. Tannahill and S. Balasubramanian, Nat. Rev. Mol. Cell Biol., 2020, 21, 459–474 CrossRef CAS PubMed.
- L. Stefan and D. Monchaud, Nat. Rev. Chem., 2019, 3, 650–668 CrossRef.
- M. Marušič, P. Šket, L. Bauer, V. Viglasky and J. Plavec, Nucleic Acids Res., 2012, 40, 6946–6956 CrossRef PubMed.
- J. Spiegel, S. Adhikari and S. Balasubramanian, Trends Chem., 2020, 2, 123–136 CrossRef CAS PubMed.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS PubMed.
- P. Röthlisberger and M. Hollenstein, Adv. Drug Deliv. Rev., 2018, 134, 3–21 CrossRef PubMed.
- A. B. Iliuk, L. Hu and W. A. Tao, Anal. Chem., 2011, 83, 4440–4452 CrossRef CAS PubMed.
- Y. X. Wu and Y. J. Kwon, Methods, 2016, 106, 21–28 CrossRef CAS PubMed.
- G. W. Collie and G. N. Parkinson, Chem. Soc. Rev., 2011, 40, 5867–5892 RSC.
- L. C. Bock, L. C. Griffin, J. A. Latham, E. H. Vermaas and J. J. Toole, Nature, 1992, 355, 564–566 CrossRef CAS PubMed.
- C. Riccardi, E. Napolitano, C. Platella, D. Musumeci and D. Montesarchio, Pharmacol. Therapeut., 2021, 217, 107649 CrossRef CAS PubMed.
- B. Deng, Y. Lin, C. Wang, F. Li, Z. Wang, H. Zhang, X.-F. Li and X. C. Le, Anal. Chim. Acta, 2014, 837, 1–15 CrossRef CAS PubMed.
- R. F. Macaya, P. Schultze, F. W. Smith, J. A. Roe and J. Feigon, Proc. Natl. Acad. Sci., USA, 1993, 90, 3745–3749 CrossRef CAS PubMed.
- A. Virgilio, L. Petraccone, V. Vellecco, M. Bucci, M. Varra, C. Irace, R. Santamaria, A. Pepe, L. Mayol, V. Esposito and A. Galeone, Nucleic Acids Res., 2015, 43, 10602–10611 CrossRef CAS PubMed.
- L. Martino, A. Virno, A. Randazzo, A. Virgilio, V. Esposito, C. Giancola, M. Bucci, G. Cirino and L. Mayol, Nucleic Acids Res., 2006, 34, 6653–6662 CrossRef CAS PubMed.
- I. Russo Krauss, V. Spiridonova, A. Pica, V. Napolitano and F. Sica, Nucleic Acids Res., 2016, 44, 3969 CrossRef PubMed.
- H.-L. Bao, T. Ishizuka, A. Yamashita, E. Furukoji, Y. Asada and Y. Xu, J. Med. Chem., 2021, 64, 711–718 CrossRef CAS PubMed.
- M. Mahmoud, C. Ruppert, S. Rentschler, S. Laufer and H.-P. Deigner, Sens. Actuators, B, 2021, 333, 129246 CrossRef CAS.
- P. Schultze, R. F. Macaya and J. Feigon, J. Mol. Biol., 1994, 235, 1532–1547 CrossRef CAS PubMed.
- J. A. Kelly, J. Feigon and T. O. Yeates, J. Mol. Biol., 1996, 256, 417–422 CrossRef CAS PubMed.
- I. Russo Krauss, A. Merlino, A. Randazzo, E. Novellino, L. Mazzarella and F. Sica, Nucleic Acids Res., 2012, 40, 8119–8128 CrossRef CAS PubMed.
- I. Russo Krauss, A. Merlino, C. Giancola, A. Randazzo, L. Mazzarella and F. Sica, Nucleic Acids Res., 2011, 39, 7858–7867 CrossRef CAS PubMed.
- A. Pica, I. Russo Krauss, A. Merlino, S. Nagatoishi, N. Sugimoto and F. Sica, FEBS J., 2013, 280, 6581–6588 CrossRef CAS PubMed.
- R. Dolot, C. H. Lam, M. Sierant, Q. Zhao, F.-W. Liu, B. Nawrot, M. Egli and X. Yang, Nucleic Acids Res., 2018, 46, 4819–4830 CrossRef CAS PubMed.
- I. Smirnov, N. Kolganova, R. Troisi, F. Sica and E. Timofeev, Mol. Ther.--Nucleic Acids, 2021, 23, 863–871 CrossRef CAS PubMed.
- C. Roxo, W. Kotkowiak and A. Pasternak, Molecules, 2019, 24, 3781 CrossRef CAS PubMed.
- A. Joachimi, G. Mayer and J. S. Hartig, J. Am. Chem. Soc., 2007, 129, 3036–3037 CrossRef CAS PubMed.
- D. Zhao, X. Dong, N. Jiang, D. Zhang and C. Liu, Nucleic Acids Res., 2014, 42, 11612–11621 CrossRef CAS PubMed.
- R. Vilar, Metal ions in life sciences, 2018, vol. 18 Search PubMed.
- Q. Cao, Y. Li, E. Freisinger, P. Z. Qin, R. K. O. Sigel and Z.-W. Mao, Inorg. Chem. Front., 2017, 4, 10–32 RSC.
- E. Ruggiero and S. N. Richter, Nucleic Acids Res., 2018, 46, 3270–3283 CrossRef CAS PubMed.
- S. Neidle, J. Med. Chem., 2016, 59, 5987–6011 CrossRef CAS PubMed.
- S. Neidle, Nat. Rev. Chem., 2017, 1, 0041 CrossRef CAS.
- B.-C. Zhu, J. He, W. Liu, X.-Y. Xia, L.-Y. Liu, B.-B. Liang, H.-G. Yao, B. Liu, L.-N. Ji and Z.-W. Mao, Angew. Chem., Int. Ed., 2021, 60, 15340–15343 CrossRef CAS PubMed.
- D. R. Calabrese, X. Chen, E. C. Leon, S. M. Gaikwad, Z. Phyo, W. M. Hewitt, S. Alden, T. A. Hilimire, F. He, A. M. Michalowski, J. K. Simmons, L. B. Saunders, S. Zhang, D. Connors, K. J. Walters, B. A. Mock and J. S. Schneekloth, Nat. Commun., 2018, 9, 4229 CrossRef PubMed.
- J. Dai, C. Punchihewa, A. Ambrus, D. Chen, R. A. Jones and D. Yang, Nucleic Acids Res., 2007, 35, 2440–2450 CrossRef CAS PubMed.
- J. Dai, M. Carver, C. Punchihewa, R. A. Jones and D. Yang, Nucleic Acids Res., 2007, 35, 4927–4940 CrossRef CAS PubMed.
- Y. Wang and D. J. Patel, Structure, 1993, 1, 263–282 CrossRef CAS PubMed.
- J. Dai, M. Carver, L. H. Hurley and D. Yang, J. Am. Chem. Soc., 2011, 133, 17673–17680 CrossRef CAS PubMed.
- B. Onel, M. Carver, G. Wu, D. Timonina, S. Kalarn, M. Larriva and D. Yang, J. Am. Chem. Soc., 2016, 138, 2563–2570 CrossRef CAS PubMed.
- P. Agrawal, E. Hatzakis, K. Guo, M. Carver and D. Yang, Nucleic Acids Res., 2013, 41, 10584–10592 CrossRef CAS PubMed.
- A. T. Phan, V. Kuryavyi, S. Burge, S. Neidle and D. J. Patel, J. Am. Chem. Soc., 2007, 129, 4386–4392 CrossRef CAS PubMed.
- K. W. Lim, P. Jenjaroenpun, Z. J. Low, Z. J. Khong, Y. S. Ng, V. A. Kuznetsov and A. T. Phan, Nucleic Acids Res., 2015, 43, 5630–5646 CrossRef CAS PubMed.
- V. Kuryavyi and D. J. Patel, Structure, 2010, 18, 73–82 CrossRef CAS PubMed.
- A. Kotar, R. Rigo, C. Sissi and J. Plavec, Nucleic Acids Res., 2018, 47, 2641–2653 CrossRef PubMed.
- S. Amrane, A. Kerkour, A. Bedrat, B. Vialet, M.-L. Andreola and J.-L. Mergny, J. Am. Chem. Soc., 2014, 136, 5249–5252 CrossRef CAS PubMed.
- G. Yuan, Q. Zhang, J. Zhou and H. Li, Mass Spectrom. Rev., 2011, 30, 1121–1142 CrossRef CAS PubMed.
- A. Marchand, D. Strzelecka and V. Gabelica, Chem.–Eur. J., 2016, 22, 9551–9555 CrossRef CAS PubMed.
- A. T. Phan, V. Kuryavyi, H. Y. Gaw and D. J. Patel, Nat. Chem. Biol., 2005, 1, 167–173 CrossRef CAS PubMed.
- W. Liu, Y.-F. Zhong, L.-Y. Liu, C.-T. Shen, W. Zeng, F. Wang, D. Yang and Z.-W. Mao, Nat. Commun., 2018, 9, 3496 CrossRef PubMed.
- L.-Y. Liu, W. Liu, K.-N. Wang, B.-C. Zhu, X.-Y. Xia, L.-N. Ji and Z.-W. Mao, Angew. Chem., Int. Ed., 2020, 59, 9719–9726 CrossRef CAS PubMed.
- M. Kovačič, P. Podbevšek, H. Tateishi-Karimata, S. Takahashi, N. Sugimoto and J. Plavec, Nucleic Acids Res., 2020, 48, 3975–3986 CrossRef PubMed.
- K. McQuaid, H. Abell, S. P. Gurung, D. R. Allan, G. Winter, T. Sorensen, D. J. Cardin, J. A. Brazier, C. J. Cardin and J. P. Hall, Angew. Chem., Int. Ed., 2019, 58, 9881–9885 CrossRef CAS PubMed.
- K. T. McQuaid, S. Takahashi, L. Baumgaertner, D. J. Cardin, N. G. Paterson, J. P. Hall, N. Sugimoto and C. J. Cardin, J. Am. Chem. Soc., 2022, 144, 5956–5964 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and methods, supporting scheme, figures and tables. See https://doi.org/10.1039/d2sc01196d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |