
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric synthesis of aryl/vinyl alkyl carbinol esters via Ni-catalyzed reductive arylation/vinylation of 1-chloro-1-alkanol esters†
Deli
Sun
 a,
Xianghua
Tao
b,
Guobin
Ma
*b,
Jifen
Wang
*a and
Yunrong
Chen
*b
a,
Xianghua
Tao
b,
Guobin
Ma
*b,
Jifen
Wang
*a and
Yunrong
Chen
*b
aSchool of Resources and Environmental Engineering, Shanghai Polytechnic University, 2360 Jinhai Road, Shanghai 201209, P. R. China. E-mail: wangjifen@sspu.edu.cn
bCenter for Supramolecular Chemistry and Catalysis and Department of Chemistry, College of Sciences, Shanghai University, 99 Shangda Road, Shanghai, 200444 P. R. China. E-mail: yr_chen@shu.edu.cn; maguobin@shu.edu.cn
First published on 27th June 2022
Abstract
We report herein an asymmetric Ni-catalyzed cross-electrophile coupling approach to prepare enantioenriched aryl/vinyl alkyl carbinol esters through arylation/vinylation of easily accessible racemic 1-chloro-1-alkanol esters with aryl/vinyl electrophiles. The method features a broad substrate scope as demonstrated by more than 60 examples including the challenging chiral allylic esters. It tolerates a wide array of functional groups including alkenyl, carbonyl and free hydroxyl groups that may not survive in conventional carbonyl reduction and addition methods. The synthetic utility of the present work was showcased by facile preparation of a few key intermediates and the modification of chiral drugs and naturally occurring compounds. Finally, we describe an efficient one-pot procedure for this method.
Introduction
Optically pure benzylic and allylic secondary alcohols and their derivatives represent an important class of carbinol structural units that are widely found in natural products and drug molecules (Scheme 1A), and serve as building blocks for diverse organic transformations.1,2 Therefore, considerable efforts have been devoted to the catalytic asymmetric preparation of these chiral secondary alcohols. A typical strategy is the transformation or resolution of achiral or racemic secondary alcohols through desymmetrization,3 dynamic kinetic/kinetic resolution,4 and deracemisation5 methods. Another strategy is the conversion of other organic compounds into alcohols, such as the classic asymmetric (transfer) hydrogenation of ketones6 and asymmetric addition of aldehydes.7 In addition, several impressive advancements of these strategies and other new methods have been achieved recently.8 The Zhou group developed a delicate method for asymmetric hydrogenation of dialkyl ketones and aliphatic cyclic ketones enabled by iridium catalysis, in which a bulky phosphine ligand was used.9 Transition metal-catalyzed asymmetric addition10 and reductive arylation11 of aldehydes are other powerful methods for the preparation of diaryl carbinols. Just a few days ago, the Xiao group reported a highly enantioselective photoassisted cobalt-catalyzed reductive arylation/vinylation of aldehydes, which efficiently affords diaryl, aryl/vinyl and aryl/alkyl carbinols.12 In addition, the use of transition metal-catalyzed carbon–carbon bond coupling protocols that bypass carbonyl additions has received increasing attention in recent years. A very notable development in the field lies in the asymmetric synthesis of enantioenriched dialkyl carbinols based on NiH-catalyzed enantioconvergent coupling of racemic alkyl halides with α-olefins developed by Fu,13 and hydroalkylation of enol esters with alkyl halides reported by Shu14 and Fu15 (Scheme 1B).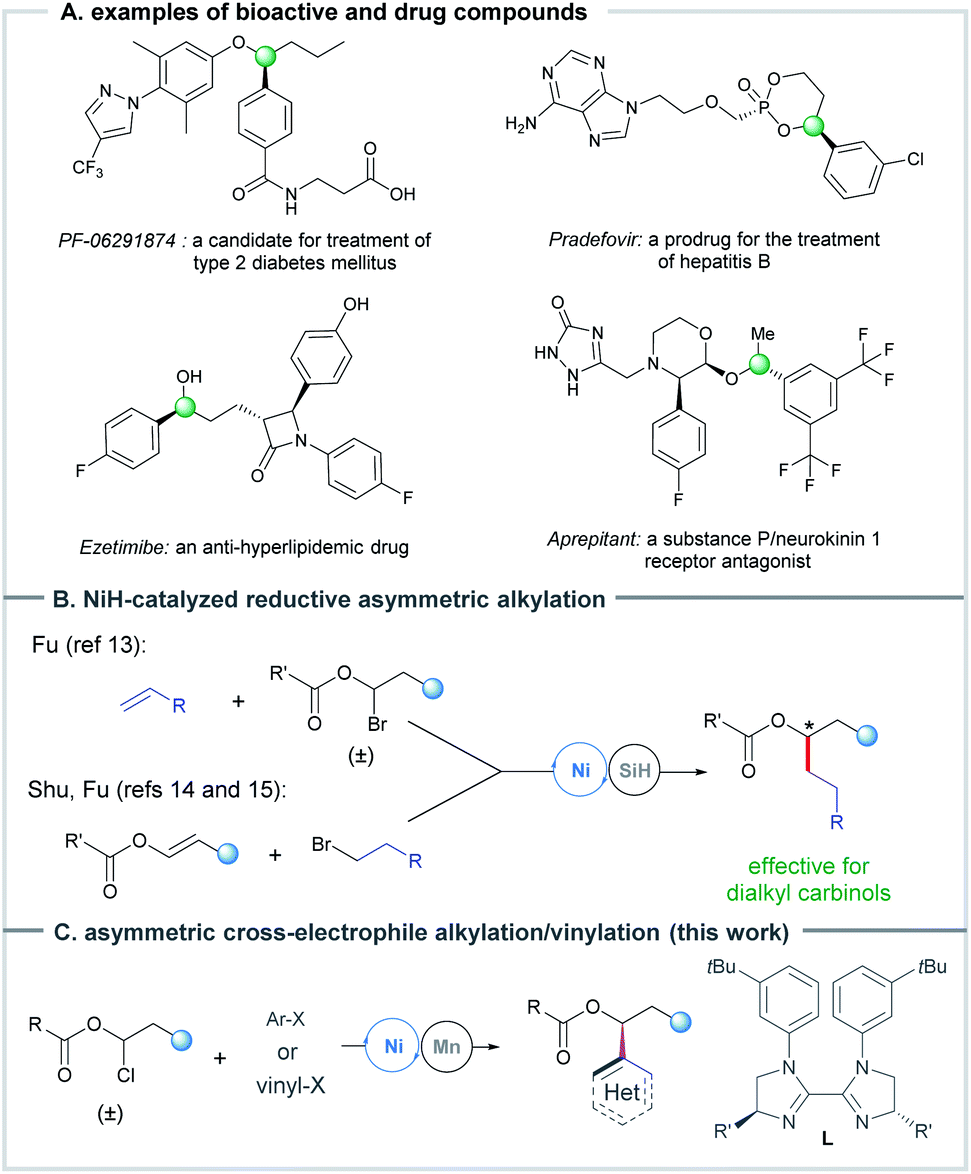 | ||
| Scheme 1 Ni-catalyzed asymmetric reductive alkylation to access chiral alkyl carbinols and selected examples of bioactive aryl alkyl carbinols. | ||
In line with the rapid growth of Ni-catalyzed enantioselective cross-electrophile coupling reactions over the past decade,16,17 we wondered the feasibility of the preparation of chiral secondary alcohols via Ni-catalyzed asymmetric cross-coupling of the readily available 1-halo-1-alkanol esters13,18 with C(sp2)-electrophiles. The Glorius group has demonstrated that a Ni-catalyzed reductive coupling of 1-bromo-1-alkanol ester intermediates with aryl bromides could furnish racemic aryl alkyl carbinols.18 Herein, we report a strategy of Ni-catalyzed reductive coupling of a diverse set of racemic 1-chloro-1-alkanol esters with aryl and vinyl halides/triflates using a chiral bisimidazoline (BiIm) ligand, affording a facile access to enantioenriched aryl/vinyl alkyl carbinols (Scheme 1C). This method displays a broad substrate scope and excellent functional group compatibility for the efficient preparation of chiral aryl/vinyl alkyl carbinols. The mild conditions are compatible with many sensitive functional groups, e.g., formyl, alkenyl, 2-halo-pyridinyl and free hydroxyl groups, that are usually not tolerated in carbonyl reduction or addition reactions. Its utility was showcased by a number of examples of the functionalization and preparation of key intermediates of drug compounds.
Results and discussion
Our investigations commenced with the coupling between racemic 1-chloropropyl 4-methylbenzoate 1 and methyl 4-bromobenzoate (Table 1). After an extensive screening of reaction parameters,19 reaction under the optimized reaction conditions gave the chiral product 2 in 92% isolated yield and 94% ee, using NiBr2(DME) as precatalyst, L1 as ligand and Mn as reductant (entries 1 and 2). Other chiral bisoxazolines or BiIm ligands (L2–L8) were also examined (entries 3–9). A trace amount of product was detected without TBAI (tetrabutylammonium iodide) and LiI (entry 10). In the absence of TBAI or LiI, equivalent enantioselectivity with 70% and 20% yields were detected, respectively (entries 11 and 12). This is likely due to a necessary Cl/I exchange to generate more reactive alkyl iodides at low concentration. While LiI is possibly more effective for halide exchange, TBAI may also promote the homogeneity of the reaction. A gram-scale reaction gave the product in high yield without causing the enantioselectivity to deteriorate (entry 13). Finally, 2 was facilely converted into free alcohol 3 in 77% yield with retained enantiomeric excess upon treatment with MeONa in MeOH (Scheme S1†).19 The absolute configuration of alcohol 3 was determined by comparison of the optical rotation with literature data.20|
|
|||
|---|---|---|---|
| Entrya | Deviation from the standard conditions | Yield (%) | ee (%) |
a Reactions conducted on a 0.15 mmol scale for 40 h, DME/THF = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, NMR yield using 2,5-dimethyl furan as the internal standard, ee was determined by chiral HPLC analysis, DME = 1,2-dimethoxyethane.
b Isolated yield. :2, NMR yield using 2,5-dimethyl furan as the internal standard, ee was determined by chiral HPLC analysis, DME = 1,2-dimethoxyethane.
b Isolated yield.
|
|||
| 1 | No changes | 95 (92)b | 94 |
| 2 | (S,S)-L1 instead of (R,R)-L1 | 93 | −94 |
| 3 | L2 instead of (R,R)-L1 | 94 | −90 |
| 4 | L3 instead of (R,R)-L1 | 95 | −89 |
| 5 | L4 instead of (R,R)-L1 | 90 | −87 |
| 6 | L5 instead of (R,R)-L1 | 82 | −62 |
| 7 | L6 instead of (R,R)-L1 | 36 | −54 |
| 8 | L7 instead of (R,R)-L1 | 9 | −17 |
| 9 | L8 instead of (R,R)-L1 | 50 | −56 |
| 10 | Without TBAI and LiI | Trace | - |
| 11 | Without TBAI | 70 | 93 |
| 12 | Without LiI | 20 | 92 |
| 13 | Gram-scale with 5 mmol of methyl 4-bromobenzoate | 85 | 93 |
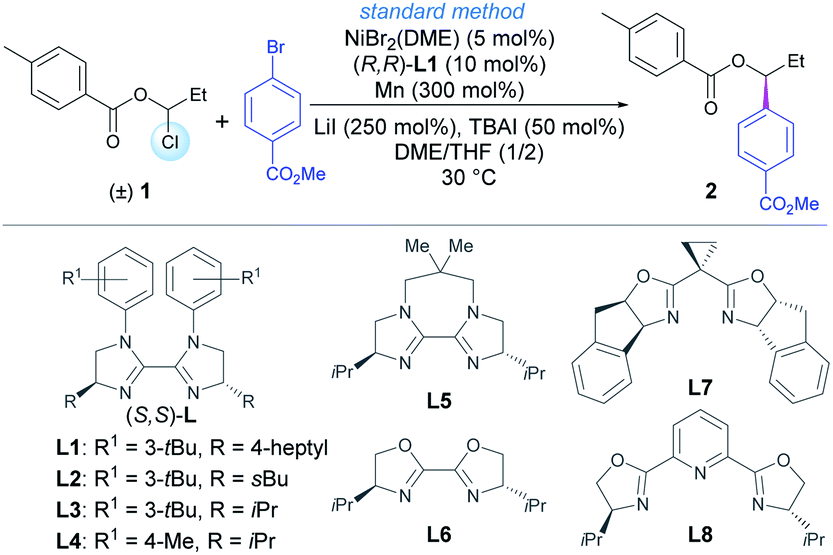
With the optimized conditions (Table 1, entry 1) in hand, a set of 1-chloropropyl benzoates decorated with methyl, F and tBu on the phenyl moieties or 2-naphthoate were tested to examine their coupling with methyl 4-bromobenzoate. The resultant chiral benzylic esters 4–8 were obtained in high yields and ees (Scheme 2). Varying the alkyl chain length of the esters did not affect the coupling yields and enantioselectivity as demonstrated by the examples of 9–13. Of note was the competency of 1-chloropropyl isobutyrate that delivered 14 in high yield and ee, indicating that an alkanoyl ester is also suitable under the reaction conditions. The alkyl chains containing different terminal groups such as iPr (15), Ph (16), OBz (18, 19), an ester group (20), MeS (21), Ts (22) and a cholic acid derivative (23) were all compatible in this method, but a large β group such as tBu significantly affects the yield (17).
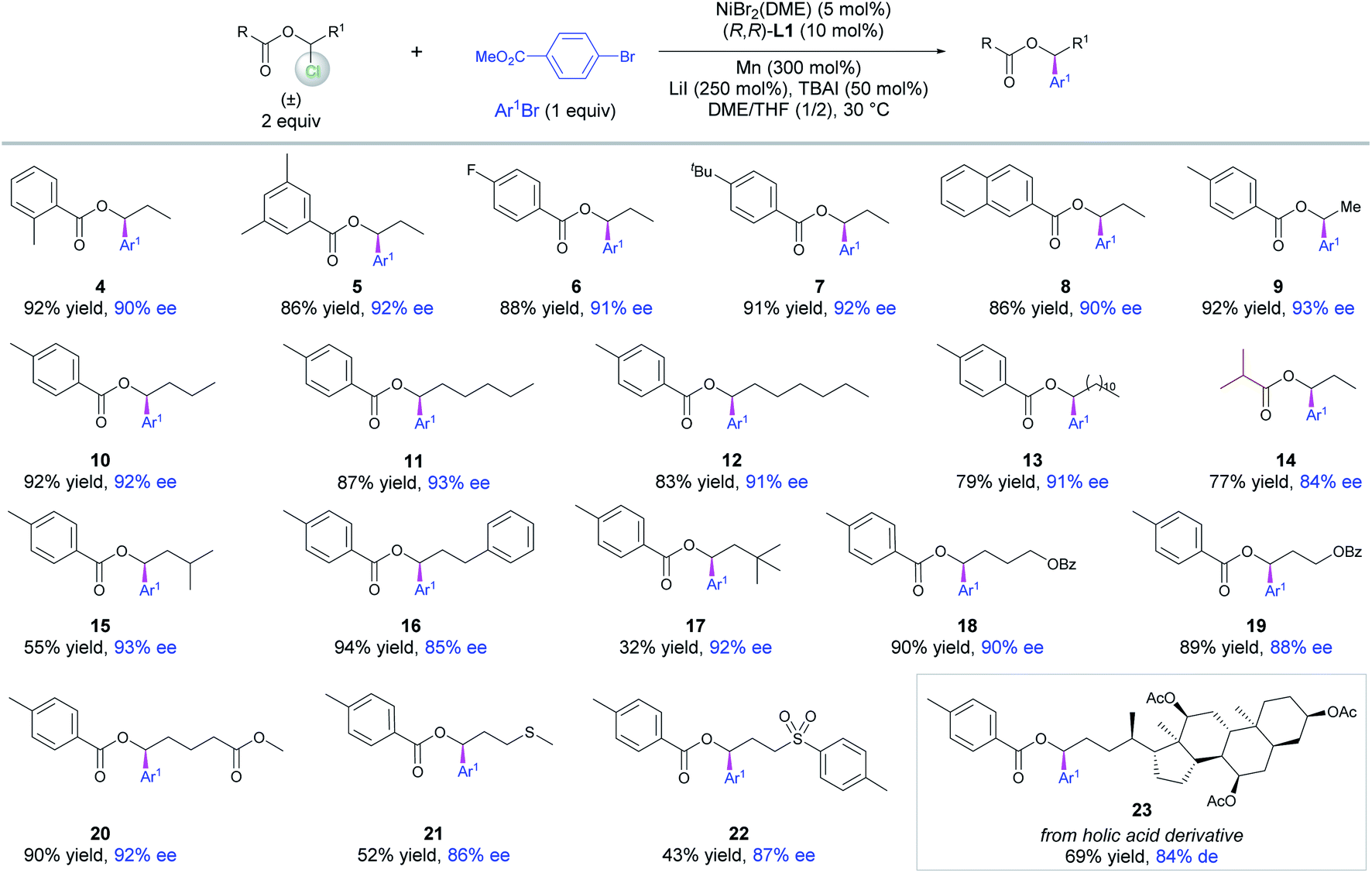 | ||
| Scheme 2 Scope of 1-chloro-1-alkanol esters. Reactions were conducted on a 0.15 mmol scale for 40 h, and ee was determined by chiral HPLC analysis. | ||
The scope of aryl halides was examined next for the coupling with 1 (Scheme 3, Part a). A diverse array of electron-poor aryl bromides were highly compatible with the method as exemplified by 24–34. The functional groups that survived in these transformations included esters, alkene, amides, aldehyde, acetones, sulfone, phosphonate and chloride, which provided opportunities for further synthetic elaboration. Moreover, heteroaryl bromides such as chloro- and trifluormethyl-substituted pyridinyl and pyrimidinyl were successfully incorporated into the products 35–39, wherein moderate coupling yields and high ees were detected. The applicability of this reaction was showcased by the introduction of a chiral ester motif to drug frameworks, e.g., fenofibrate (40), procanine (41) and indomethacine (42), affording the coupling products in good yields and high ees.
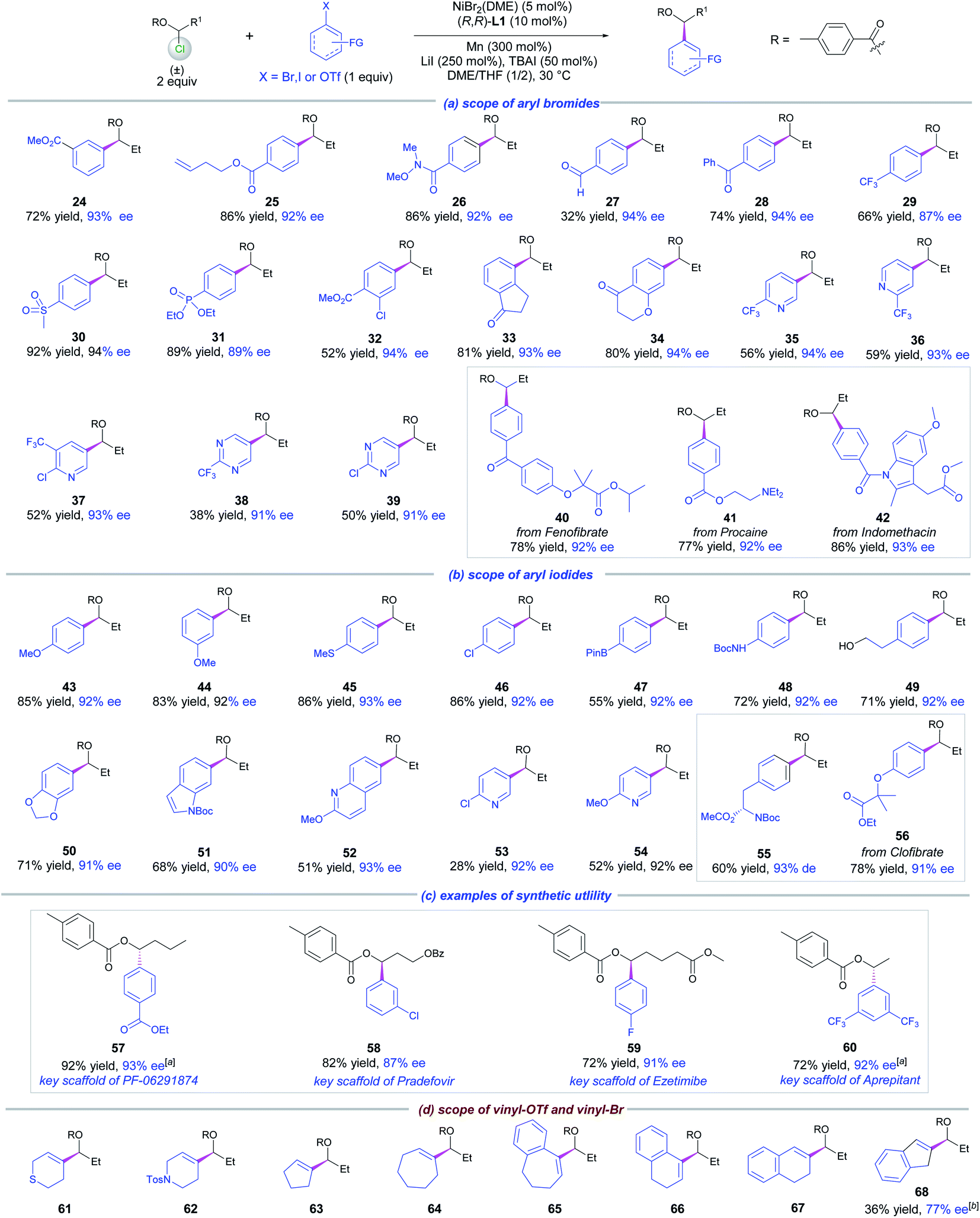 | ||
| Scheme 3 Scope of aryl/vinyl electrophiles. Reactions conducted on a 0.15 mmol scale for 40 h, ee was determined by chiral HPLC analysis. [a] (S,S)-L1 used as the chiral ligand. [b] With vinyl–OTf. [c] With vinyl–Br. | ||
For electron-neutral and electron-rich arenes, aryl iodides were more appropriate coupling partners for 1 under the standard conditions (Scheme 3, Part b). A wide spectrum of functional groups was tolerated as evidenced by the results of 43–50. The notable functional groups included ether, thioether, Bpin, chloride, BocNH and free hydroxyl groups. A number of heteroaryl iodides were also examined. While the electron-rich indole iodide also gave a good result (51), the electron-deficient 6-iodo-2-methoxyquinoline and 2-chloro- and 2-methoxy 5-iodo-pyridines produced 52–54 in low to moderate yields. In all these cases, high levels of enantioselectivity were constantly obtained. Likewise, the present method was also suitable for the diversification of a natural product, L-phenylalanine, and a drug molecule, clofibrate, furnishing the desired chiral products 55 and 56 in good yields and excellent ees respectively.
To further demonstrate the synthetic utility of this method, we prepared the key structural scaffolds of four biologically active molecules shown in Scheme 3 (Part c). Chiral products 57–60 were obtained in high yields and ees.
We next studied the reaction of 1 with vinyl electrophiles. In this case, vinyl triflates are usually more effective (Scheme 3, Part d). To our delight, 61 bearing 3,6-dihydro-2H-thiopyran-4-yl was afforded in 60% yield and 90% ee, using 3,6-dihydro-2H-thiopyran-4-yl triflate as a substrate. The vinylation protocol was also suitable for other five to seven membered rings giving chiral products 62–67 in moderate to good yields and high enantioselectivities. However, for a 1H-inden-2-yl substrate, vinyl bromide gave a better result (68). In contrast, a terminal alkenyl bromide such as (E)-(4-bromobut-3-en-1-yl)benzene gave a trace amount of product, due to severe homodimerization of the vinyl bromide.
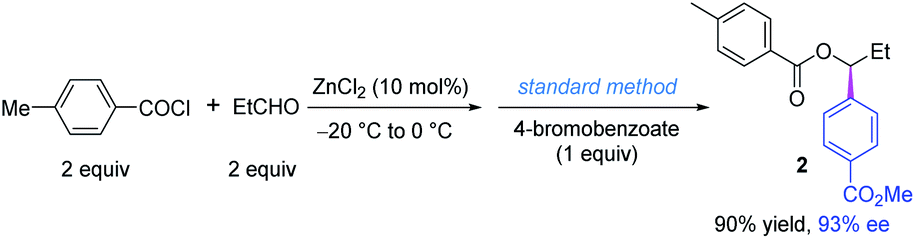
Finally, we found that the reaction conditions of the in situ substrate preparation and the coupling reaction are compatible. To our delight, a one-pot procedure afforded the desired product in excellent yield and ee as well, and only two equivalents of acid chloride and aldehyde were used (Scheme 4).21 The ZnCl2 catalyst of the former step has almost no influence on the coupling step. Actually, ZnCl2 is a byproduct of some Ni-catalyzed cross-electrophile couplings using organochlorides and zinc.22
 | ||
| Scheme 4 One-pot reductive arylation. | ||
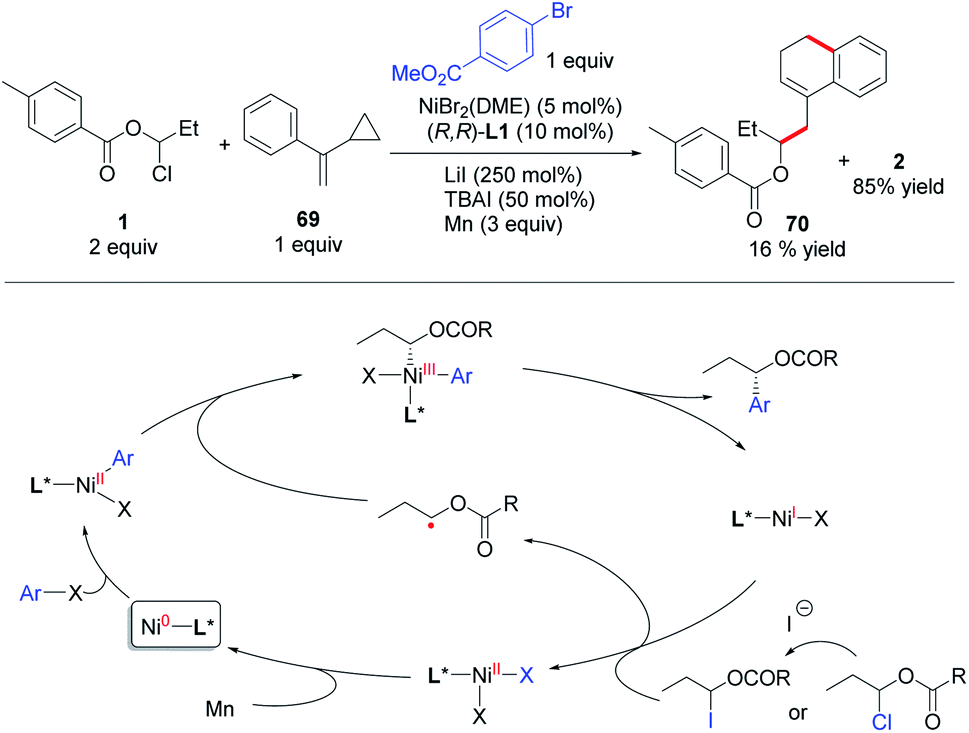
Based on Weix's study on the Ni-catalyzed arylation of secondary alkyl halides with aryl halides,23 we propose a radical chain mechanism for this coupling reaction (Scheme 5). The oxidative addition of aryl halide to Ni(0) forms an Ar–Ni(II)–X species, which intercepts an α-oxa carbon radical derived from a 1-Cl/I-1-alkanol ester and then gives an Ar–Ni(III)X–alkyl species. Upon reductive elimination, the chiral product is produced along with a Ni(I)X species. The Ni(I)X is responsible for reduction or halide abstraction of the 1-Cl/I-1-alkanol ester to afford the alkyl radical and Ni(II)X2. Reduction of this Ni(II) to Ni(0) closes the catalytic cycle. The radical nature of the reaction was evidenced by a radical clock experiment, wherein the addition of an alkyl radical derived from 1 to 69 followed by cyclopropyl ring-opening, intramolecular cyclization and hydrogen abstraction affords 70. Moreover, the reaction of a (dtbbpy)BrNi(II)Ar complex and 1 gave the coupling product (rac-2) in 36% yield in the absence of reductant (Mn), while the reaction with addition of 3 equiv. of Mn cannot provide a higher yield of the product (see ESI part 5†). This result is consistent with a radical chain mechanism.24 However, due to the small energy barrier difference between the possible mechanisms, a subtle change of the reaction parameters (e.g., concentration of the catalyst, ligands, and type of substrate) may lead the reaction to proceed via a different main pathway. So a sequential reduction (or double oxidative addition, see ESI part 5†) mechanism24d,25 also cannot be ruled out in these coupling reactions.
 | ||
| Scheme 5 Radical clock experiment and a working hypothesis of the reaction mechanism. | ||
Conclusions
In summary, we have developed a method for preparation of enantioenriched aryl/vinyl alkyl carbinols via Ni-catalyzed asymmetric reductive cross-coupling between 1-chloro-1-alkanol esters and aryl/vinyl electrophiles. This transformation exhibited a broad substrate scope and tolerated a variety of functional groups, generally furnishing the coupling products in moderate to high yields with high enantioselectivities. The present work provides a convenient handle for the functionalization and preparation of bioactive and drug molecules and their intermediates. This method adds a new entry to accessing and diversifying chiral secondary benzylic and allylic alcohol derivatives using readily accessible electrophiles.Data availability
All experimental and characterization data is available in the ESI.†Author contributions
Conceptualization: D. S., G. M. and Y. C.; investigation: D. S. and X. T.; formal analysis, D. S. and Y. C.; writing – original draft: D. S., J. W. and Y. C.; writing – review & editing: J. W. and Y. C.; supervision: G. M. and J. W.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support of the National Natural Science Foundation of China (21871173, 21901162).Notes and references
- (a) G. Wang and M. J. Krische, J. Am. Chem. Soc., 2016, 138, 8088–8091 CrossRef CAS PubMed; (b) Y.-M. Siu, J. Roane and M. J. Krische, J. Am. Chem. Soc., 2021, 143, 10590–10595 CrossRef CAS PubMed.
- (a) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921–2944 CrossRef CAS PubMed; (b) E. Skucas, M.-Y. Ngai, V. Komanduri and M. J. Krische, Acc. Chem. Res., 2007, 40, 1394–1401 CrossRef CAS PubMed; (c) A. Lumbroso, M. L. Cooke and B. Breit, Angew. Chem., Int. Ed., 2013, 52, 1890–1932 CrossRef CAS PubMed; (d) A. Gil, F. Albericio and M. Álvarez, Chem. Rev., 2017, 117, 8420–8446 CrossRef CAS PubMed.
- (a) Z. Li, W. Zhang and H. Yamamoto, Angew. Chem., Int. Ed., 2008, 47, 7520–7522 CrossRef CAS PubMed; (b) B. Liu, P. Xie, J. Zhao, J. Wang, M. Wang, Y. Jiang, J. Chang and X. Li, Angew. Chem., Int. Ed., 2021, 60, 8396–8400 CrossRef CAS PubMed.
- (a) D. C. Ebner, J. T. Bagdanoff, E. M. Ferreira, R. M. McFadden, D. D. Caspi, R. M. Trend and B. M. Stoltz, Chem.–Eur. J., 2009, 15, 12978–12992 CrossRef CAS PubMed; (b) O. Verho and J.-E. Backvall, J. Am. Chem. Soc., 2015, 137, 3996–4009 CrossRef CAS PubMed.
- (a) G. R. A. Adair and J. M. J. Williams, Chem. Commun., 2007, 2608–2609 RSC; (b) M. M. Musa, F. Hollmann and F. G. Mutti, Catal. Sci. Technol., 2019, 9, 5487–5503 RSC.
- (a) R. Noyori and T. Ohkuma, Angew. Chem., Int. Ed., 2001, 40, 40–73 CrossRef CAS; (b) H. Wang, J. Wen and X. Zhang, Chem. Rev., 2021, 121, 7530–7567 CrossRef CAS PubMed; (c) J.-H. Xie and Q.-L. Zhou, in Asymmetric Hydrogenation and Transfer Hydrogenation, ed. V. Ratovelomanana-Vidal and P. Phansavath, WILEY-VCH GmbH, 2021, pp. 87–128 Search PubMed.
- S. Suga and M. Kitamura, in Asymmetric 1,2-addition of organometallics to carbonyl and imine groups; synthetic methods III-catalytic methods: C–C bond formation, comprehensive chirality, ed. H. Yamamoto and E. M. Carreira, Elsevier Science, Oxford, 2012, pp. 328–342 Search PubMed.
- G. Li, X. Huo, X. Jiang and W. Zhang, Chem. Soc. Rev., 2020, 49, 2060–2118 RSC.
- F.-H. Zhang, F.-J. Zhang, M.-L. Li, J.-H. Xie and Q.-L. Zhou, Nat. Catal., 2020, 3, 621–627 CrossRef CAS.
- X. M.-H. Zhu Dong-Xing, Chin. J. Org. Chem., 2020, 40, 255–275 CrossRef.
- (a) H.-T. Chang, M. Jeganmohan and C.-H. Cheng, Chem.–Eur. J., 2007, 13, 4356–4363 CrossRef CAS PubMed; (b) K. Yabushita, A. Yuasa, K. Nagao and H. Ohmiya, J. Am. Chem. Soc., 2019, 141, 113–117 CrossRef CAS PubMed; (c) S. Zhang, S. Perveen, Y. Ouyang, L. Xu, T. Yu, M. Zhao, L. Wang, P. Song and P. Li, Angew. Chem., Int. Ed., 2022, 61, e202117843 CAS; (d) Z. Zhu, J. Xiao, M. Li and Z. Shi, Angew. Chem., Int. Ed., 2022, 61, e202201370 CAS.
- X. Jiang, H. Jiang, Q. Yang, Y. Cheng, L.-Q. Lu, J. A. Tunge and W.-J. Xiao, J. Am. Chem. Soc., 2022, 144, 8347–8354 CrossRef CAS PubMed.
- Z.-P. Yang and G. C. Fu, J. Am. Chem. Soc., 2020, 142, 5870–5875 CrossRef CAS PubMed.
- S. Wang, J.-X. Zhang, T.-Y. Zhang, H. Meng, B.-H. Chen and W. Shu, Nat. Commun., 2021, 12, 2771 CrossRef CAS PubMed.
- X.-X. Wang, L. Yu, X. Lu, Z.-L. Zhang, D.-G. Liu, C. Tian and Y. Fu, CCS Chem., 2022, 4, 605–615 CrossRef CAS.
- (a) A. H. Cherney, N. T. Kadunce and S. E. Reisman, J. Am. Chem. Soc., 2013, 135, 7442–7445 CrossRef CAS; (b) A. H. Cherney and S. E. Reisman, J. Am. Chem. Soc., 2014, 136, 14365–14368 CrossRef CAS PubMed; (c) J. L. Hofstra, A. H. Cherney, C. M. Ordner and S. E. Reisman, J. Am. Chem. Soc., 2018, 140, 139–142 CrossRef CAS PubMed; (d) H. Guan, Q. Zhang, P. J. Walsh and J. Mao, Angew. Chem., Int. Ed., 2020, 59, 5172–5177 CrossRef CAS PubMed; (e) H.-Y. Tu, F. Wang, L. Huo, Y. Li, S. Zhu, X. Zhao, H. Li, F.-L. Qing and L. Chu, J. Am. Chem. Soc., 2020, 142, 9604–9611 CrossRef CAS PubMed; (f) D. Sun, G. Ma, X. Zhao, C. Lei and H. Gong, Chem. Sci., 2021, 12, 5253–5258 RSC; (g) P. Zheng, P. Zhou, D. Wang, W. Xu, H. Wang and T. Xu, Nat. Commun., 2021, 12, 1646 CrossRef CAS PubMed.
- (a) Y. Jin and C. Wang, Synlett, 2020, 31, 1843–1850 CrossRef CAS; (b) K. E. Poremba, S. E. Dibrell and S. E. Reisman, ACS Catal., 2020, 10, 8237–8246 CrossRef CAS PubMed; (c) A. Lipp, S. O. Badir and G. A. Molander, Angew. Chem., Int. Ed., 2021, 60, 1714–1726 CrossRef CAS; (d) S. Zhu, X. Zhao, H. Li and L. Chu, Chem. Soc. Rev., 2021, 50, 10836–10856 RSC.
- H.-M. Huang, P. Bellotti, J. E. Erchinger, T. O. Paulisch and F. Glorius, J. Am. Chem. Soc., 2022, 144, 1899–1909 CrossRef CAS PubMed.
- See the ESI† for details.
- (a) M. Rouen, P. Chaumont, G. Barozzino-Consiglio, J. Maddaluno and A. Harrison-Marchand, Chem.–Eur. J., 2018, 24, 9238–9242 CrossRef CAS PubMed; (b) Q. Wang, S. Li, C.-J. Hou, T.-T. Chu and X.-P. Hu, Appl. Organomet. Chem., 2019, 33, e5108 Search PubMed.
- The sequential operation is more efficient, avoiding the loss of 1-chloro-1-alkanol esters (about 50% yield after isolation) in the quench and silica gel chromatography process.
- (a) Y. Ye, H. Chen, J. L. Sessler and H. Gong, J. Am. Chem. Soc., 2019, 141, 820–824 CrossRef CAS PubMed; (b) S. Kim, M. J. Goldfogel, M. M. Gilbert and D. J. Weix, J. Am. Chem. Soc., 2020, 142, 9902–9907 CrossRef CAS PubMed.
- S. Biswas and D. J. Weix, J. Am. Chem. Soc., 2013, 135, 16192–16197 CrossRef CAS PubMed.
- (a) N. D. Schley and G. C. Fu, J. Am. Chem. Soc., 2014, 136, 16588–16593 CrossRef CAS PubMed; (b) O. Gutierrez, J. C. Tellis, D. N. Primer, G. A. Molander and M. C. Kozlowski, J. Am. Chem. Soc., 2015, 137, 4896–4899 CrossRef CAS PubMed; (c) S. A. Green, S. Vásquez-Céspedes and R. A. Shenvi, J. Am. Chem. Soc., 2018, 140, 11317–11324 CrossRef CAS PubMed; (d) X. Wang, G. Ma, Y. Peng, C. E. Pitsch, B. J. Moll, T. D. Ly, X. Wang and H. Gong, J. Am. Chem. Soc., 2018, 140, 14490–14497 CrossRef CAS PubMed.
- (a) A. H. Cherney, N. T. Kadunce and S. E. Reisman, J. Am. Chem. Soc., 2013, 135, 7442–7445 CrossRef CAS PubMed; (b) Q. Lin and T. Diao, J. Am. Chem. Soc., 2019, 141, 17937–17948 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc02806a |
| This journal is © The Royal Society of Chemistry 2022 |