
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemically-promoted synthesis of benzo[b]thiophene-1,1-dioxides via strained quaternary spirocyclization†
Ruitao
Li
a,
Dafu
Yuan
a,
Mengqi
Ping
a,
Yuyi
Zhu
b,
Shaofei
Ni
b,
Ming
Li
a,
Lirong
Wen
 a and
Lin-Bao
Zhang
*a
a and
Lin-Bao
Zhang
*a
aState Key Laboratory Base of Eco-Chemical Engineering, College of Chemistry and Molecular Engineering, Qingdao University of Science & Technology, Qingdao, 266042, P. R. China. E-mail: zhang_linbao@126.com
bDepartment of Chemistry, Shantou University, Shantou, Guangdong, 515063 P. R. China
First published on 28th July 2022
Abstract
We report an approach for the synthesis of benzothiophene motifs under electrochemical conditions by the reaction of sulfonhydrazides with internal alkynes. Upon the formation of a quaternary spirocyclization intermediate by the selective ipso-addition instead of an ortho-attack, the S-migration process was rationalized to lead to the products. Computational studies revealed the selectivity and the compatibility of drug molecules showcased the potential application of the protocols.
Introduction
Benzothiophenes have diverse applications in medicinal chemistry and materials science, furthermore, they arepresent in many natural products.1 Consequently, the development of efficient methods for the construction of benzothiophene and its derivatives is of great importance.2 Among these strategies, the intermolecular coupling of aromatic substrates with alkynes presents a straightforward route that benefits from step- and atom-economy.3 However, the site selectivity of the abovementioned protocol can be problematic for meta-substituted substrates with a mixture of products formed. In addition, the construction of benzothiophene motifs demands metal catalysts, elaborate starting materials, and harsh reaction conditions, reducing the efficiency of the existing protocols.4 Thus, developing novel and green methods for such functional benzothiophene derivatives will be highly desirable.The development of radical chemistry has drawn wide attention as it offers diverse and unique reaction pathways in synthetic chemistry.5 Recently, intramolecular radical involved transformations toward the ortho- or ipso-carbon of the benzene ring have been extensively investigated, thus producing valuable molecular scaffolds by convenient synthetic means (Scheme 1a).6 The ipso-counterpart provides unusual means for achieving the attainable products via a highly versatile spirocyclic intermediate compared to the ortho-addition strategy (Avs.B). In fact, the synthetic diversity of the spirocyclic pathway has successfully been demonstrated by the oxidative or reductive dearomative process,7 as well as by various migratory reactions.8 In this context, the Cho group developed spirocyclization reactions by applying iminyl radicals, respectively via a photocatalytic ipso-attack process (Scheme 1b).9 Nevado and co-workers revealed the ipso-reactivity of sulfamidyl radicals with subsequent aryl migration.10 The group of Chang showcased an approach of visible-light-induced selective C(sp2)–H amidative cyclization to furnish δ-benzolactams via selective ipso-addition of amidyl radicals (Scheme 1c).11
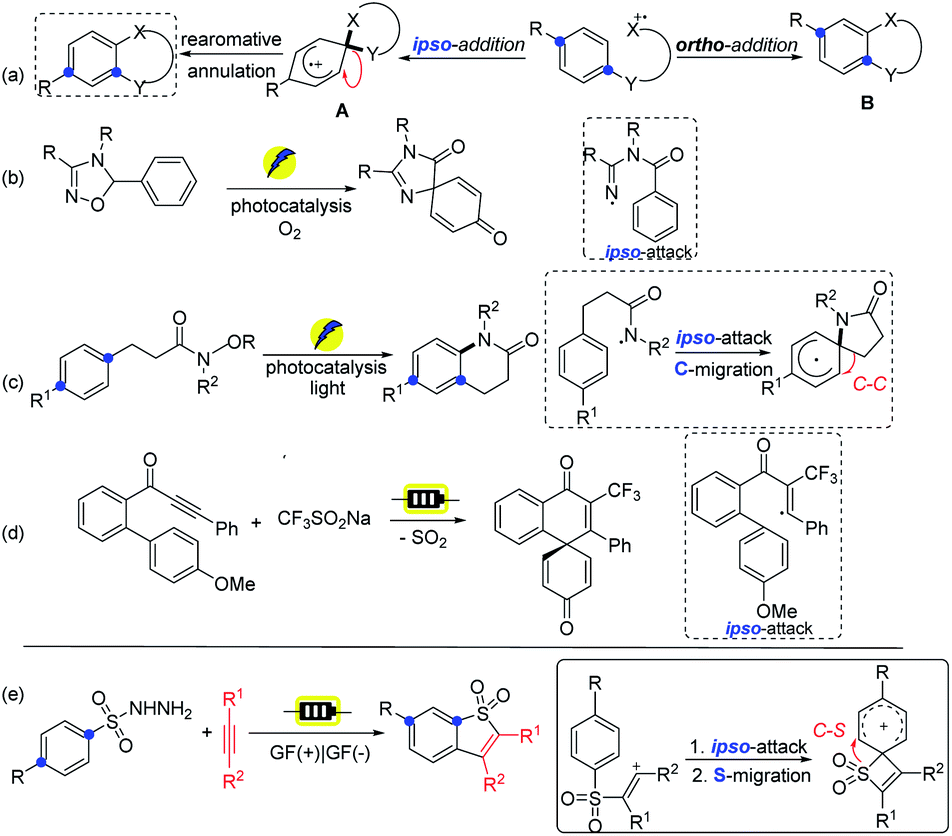 | ||
| Scheme 1 The developed strategy involving an ipso-attack pathway (a–e). | ||
On the other hand, organic electrochemistry has attracted broad interest as an enabling synthetic tool by the advantages of traceless electrons as a green reagent.12 It can also promote dehydrogenative processes through H2 release without the requirement of metal catalysts or chemical oxidants.13 Moreover, the convenient tunability of the potential of the reaction system provides a fine handle to precisely harness redox transformations.14 Recently, the group of Ackermann revealed an efficient approach for electrooxidative dearomatization of biaryls through radical spirocyclization via the ipso-attack route (Scheme 1d).15 In our previous work, we have reported regio- and stereoselective electrochemical syntheses of sulfonylated enethers, which lead to benzo[b]thiophene-1,1-dioxides via the ortho-attack process.16 Hence, our interest in electrochemical transformation and green chemistry prompts us to develop a new strategy for the direct formation of such benzothiophene motifs via an alkenyl radical intermediate followed by the selective ipso-addition instead of the ortho-attack pathway, which underwent S-migration via more strained quaternary spirocyclization (Scheme 1e).17
Results and discussion
Initial studies to determine the optimal reaction conditions were performed using 1-propynylbenzene (1a) to react with 4-methylbenzenesulfonhydrazide (2a) in HFIP under electrochemical anodic oxidation conditions (Table 1). Gratifyingly, the benzo[b]thiophene-1,1-dioxide (3aa) was obtained with a yield of 75%, using graphite felt (GF) electrodes under 5.0 mA constant current electrolysis in an undivided electrolytic cell at room temperature with Et4NPF6 as the electrolyte and HFIP/CH3NO2 as the co-solvent (Table 1, entry 1). Various reaction conditions were explored and it was found that both increasing and decreasing the constant current would lead to decreased reaction yields (Table 1, entries 2 and 3). The electrode material had an important impact on this reaction. Lower yields were obtained when the GF was replaced by a platinum cathode or a nickel foam cathode, giving 3aa in 51% and 47% yields, respectively (Table 1, entries 4 and 5). As for the choice of electrolytes, the results revealed that other electrolytes such as nBu4NPF6 or Me4NPF6 did not give superior yields (Table 1, entries 6 and 7). In addition, diminished reaction yields were achieved when HFIP was replaced with other solvents, such as DCM, DCE, THF and TFE (Table 1, entries 8–11), indicating that HFIP was crucial for the transformation, and this might be due to the fact that HFIP increased the reactivity of the carbocation intermediates involved in this process.18 A control experiment showed that 3aa was not generated without electricity (Table 1, entry 12). When the reaction was conducted under an air atmosphere (Table 1, entry 13), the yield was only 30%, indicating that the O2 in air might quench the sulfonyl radical in the reaction process.|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yieldb (%) |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.6 mmol), Et4NPF6 (0.2 mmol), HFIP (4.7 mL), CH3NO2 (0.3 mL), graphite felt anode (10 mm × 10 mm × 5 mm), constant current = 5.0 mA, RT, 8 h, undivided cell (7.46 F, faradaic efficiency: 42%). b Isolated yield. | ||
| 1 | None | 75 |
| 2 | 3 mA, 13 h | 70 |
| 3 | 10 mA, 4 h | 65 |
| 4 | Pt plate cathode | 51 |
| 5 | Nickel foam cathode | 47 |
| 6 | n Bu4NPF6 instead of Et4NPF6 | 64 |
| 7 | Me4NPF6 instead of Et4NPF6 | 69 |
| 8 | DCM instead of HFIP | 62 |
| 9 | DCE instead of HFIP | 60 |
| 10 | THF instead of HFIP | 38 |
| 11 | TFE instead of HFIP | Trace |
| 12 | No electric current | n.r |
| 13 | Under air | 30 |
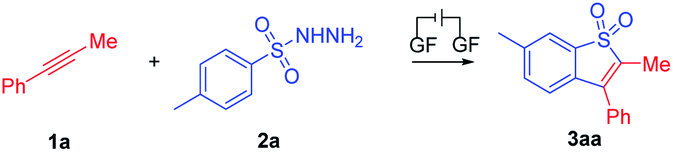
With the optimal reaction conditions confirmed, a series of alkynes were first used to systematically examine the general protocol. The results are presented in Scheme 2. The reaction showed a broad compatibility with various alkyl groups of the internal alkynes (3aa–3qa). In addition, the structure of 3aa was further confirmed by X-ray crystallography analysis (see the ESI for details; CCDC: 2044891†). The unsymmetrical alkynes bearing a benzene ring and long chain alkyl groups resulted in 3aa–3ka with yields of 75–39%. In addition, we speculate that the products were mixed with regioisomers and the ratio was determined through 1H NMR analysis (3ca′–3ka′, see the ESI† for details). For substrates bearing isopropyl (1la), tert-butyl (1ma), sec-butyl (1na) and cyclopropyl (1oa), the corresponding products 3la–3oa were smoothly delivered with yields of 69–42%, which indicated that the steric hindrance factor did not influence the efficiency of the electrochemical transformations. The alkane terminal OTs group was also compatible with the reaction system, providing corresponding products 3pa and 3qa in 36% and 55% yields, respectively. Disappointingly, we did not detect the products 3ra and 3sa when employing internal alkynes containing the methyl or chlorine group, possibly due to their relatively lower oxidation potential (see ESI, Fig. S3†). Protected amine (3ta) and ester (3ua) were smoothly delivered with yields of 44% and 30%, respectively. It should be noted that trace byproducts consistent with reduction of the benzothiophene core to the corresponding dihydrobenzothiophene were detected (see the ESI† for details).
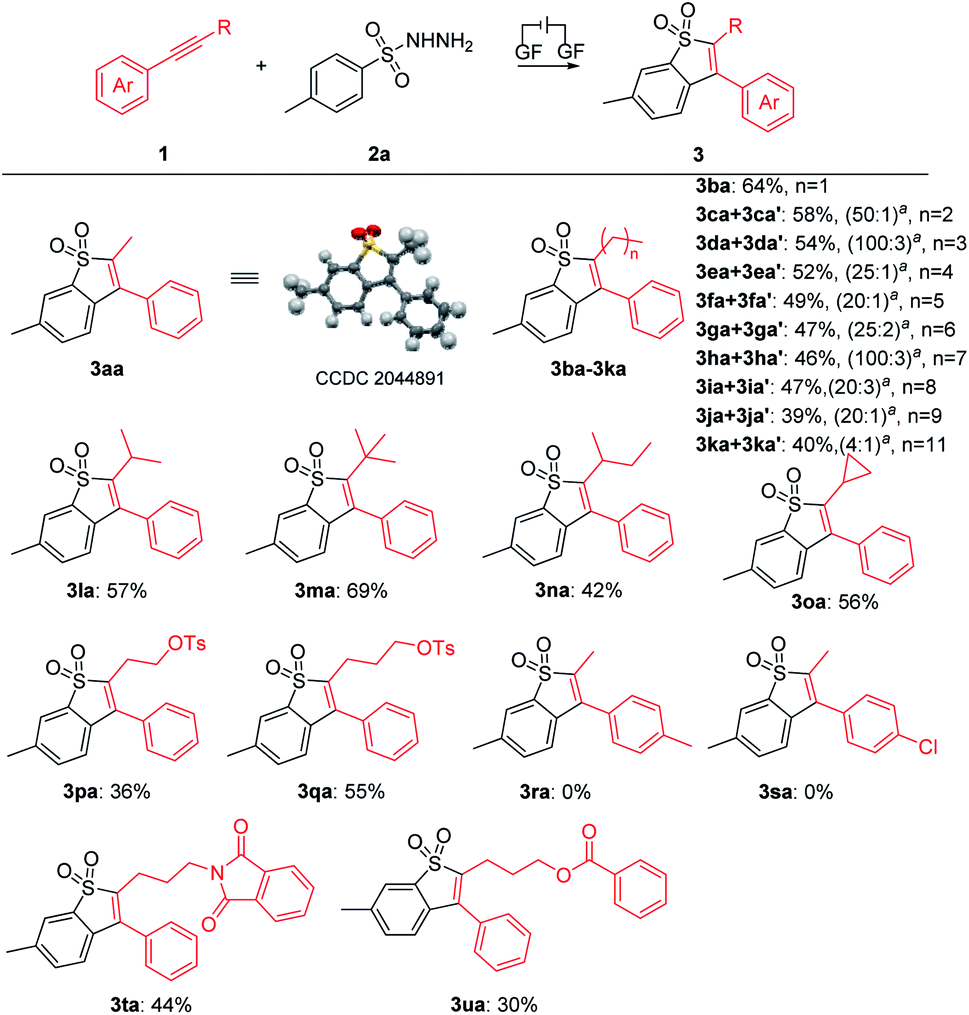 | ||
| Scheme 2 Substrate scope of 1. General reaction conditions: 1 (0.2 mmol), 2a (0.6 mmol), Et4NPF6 (0.2 mmol), HFIP (4.7 mL), CH3NO2 (0.3 mL), graphite felt anode (10 mm × 10 mm × 5 mm), constant current = 5.0 mA, RT, 8 h, undivided cell. aThe ratio was determined by 1H NMR analysis. | ||
Furthermore, the scope of aryl internal alkynes was further investigated, as summarized in Scheme 3. Similarly, the structure of 3Aa was also confirmed by X-ray crystallography analysis (see the ESI† for details; CCDC: 2044867). We found that functional groups including methyl, ethyl and tert-butyl were well tolerated, generating the products 3Ba–3Da in 43–64% yields. Moreover, halide substituents, including F, Cl and Br at the para-position of the benzene ring did not inhibit the reaction, affording 3Ea, 3Fa and 3Ga in yields of 58%, 49% and 44%, respectively. The unsymmetric aryl alkyne (1I) underwent the electrochemical annulation to give 3Ia and 3Ia′ with yields of 26% and 24%, respectively. In addition, the structure of 3la′ was further confirmed by X-ray crystallography analysis (see the ESI for details; CCDC: 2171791†). Unfortunately, the trial of the (hetero)cyclic alkynes under the conditions was unsuccessful.
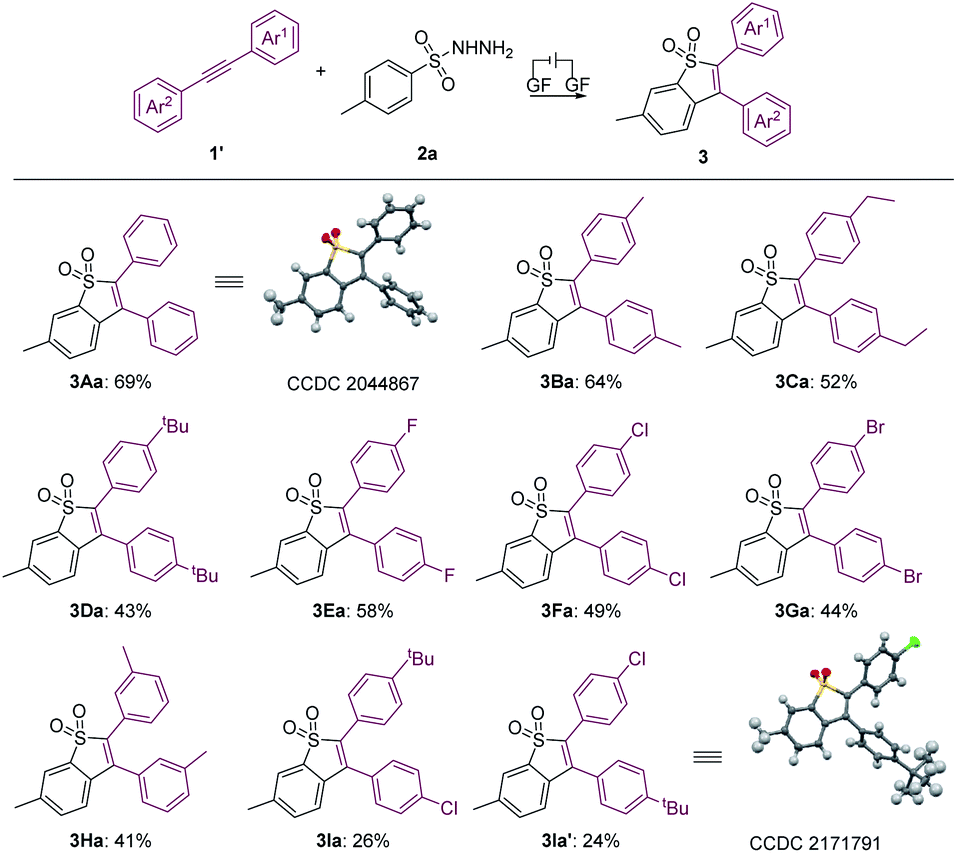 | ||
| Scheme 3 Substrate scope of 1′. General reaction conditions: 1′ (0.2 mmol), 2a (0.6 mmol), Et4NPF6 (0.2 mmol), HFIP (4.7 mL), CH3NO2 (0.3 mL), graphite felt anode (10 mm × 10 mm × 5 mm), constant current = 5.0 mA, RT, 8 h, undivided cell. | ||
Subsequently, the scope of arylsulfonyl hydrazides was explored as summarized in Scheme 4. We found that for benzenesulfonyl hydrazides bearing electron-donating groups such as ethyl (2b), the electron effect played a significant role in this transformation, as propyl (2c), cyclohexyl (2d) and tert-butyl (2e) at the para-position of the benzene ring underwent the transformation smoothly with yields of 51–69%. 3-Methylbenzenesulfonohydrazide 2f reacted well with 1a, producing corresponding rearrangement product 3af in a yield of 56%. Moreover, the sulfonyl hydrazine 2g decorated with 2-naphthalene finally provided naphtho[1,2-b]thiophene derivative 3ag in moderate yield. We found that electron-donating groups such as methoxy (2h) could enable the reaction, while the electron-withdrawing substituent Cl (2i) at the same position of the benzene ring totally inhibited the electrolysis reaction.
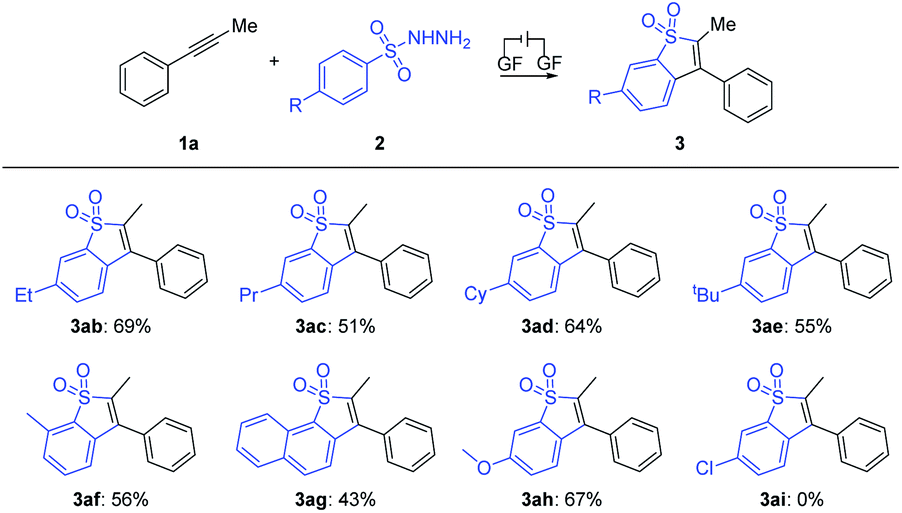 | ||
| Scheme 4 Substrate scope of 2. General reaction conditions: 1a (0.2 mmol), 2 (0.6 mmol), Et4NPF6 (0.2 mmol), HFIP (4.7 mL), CH3NO2 (0.3 mL), graphite felt anode (10 mm × 10 mm × 5 mm), constant current = 5.0 mA, RT, 8 h, undivided cell. | ||
To demonstrate the practicability of the reaction, a 5.0 mmol scale reaction of 1a with 2a was conducted, affording 3aa in 82% yield under the conditions (Scheme 5a). Moreover, the triazo group was easily introduced under the reaction of 3qa with sodium azide, which could be further functionalized via the click reaction (Scheme 5b).19 It is particularly noteworthy that a series of natural products and pharmaceutical motifs such as indometacin, ibuprofen, gemfibrozil, loxoprofen, lambdacyhalothric acid, naproxen and isoxepac could undergo nucleophilic attack by 3qa, generating benzo[b]thiophene-1,1-dioxide linked drug derivatives 6–12 in good yields (see ESI, Scheme S2†).
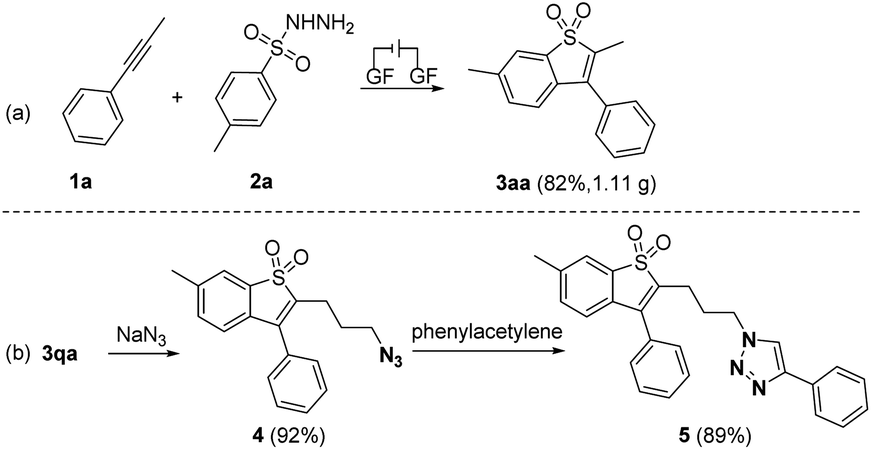 | ||
| Scheme 5 (a) Gram-scale reaction. Reaction conditions: 1a (5 mmol), 2a (15 mmol), Et4NPF6 (5 mmol), HFIP (117.5 mL), CH3NO2 (7.5 mL), graphite felt anode (40 mm × 10 mm × 15 mm), constant current = 5.0 mA, RT, 67 h, undivided cell. (b) Functionalization of 3qa. Reaction conditions: step 1, 3qa (0.2 mmol), NaN3 (0.24 mmol), DMF (1.1 mL), RT; step 2, 4 (0.2 mmol), phenylacetylene (0.26 mmol), CuSO4·5H2O (0.02 mmol), sodium ascorbate (0.05 mmol), THF (2.5 mL), H2O (2.5 mL), RT, overnight. | ||
To gain some insights into the reaction mechanism, control experiments were performed. First of all, cyclic voltammetry (CV) experiments were performed on 1a and 2a and the results revealed that the oxidation peak of 1a emerged at 2.67 V, which is consistent with the observation of the radical cation.20 However the oxidation peaks of 2a emerged at 1.80 V and 2.48 V, respectively, indicating that 2a was oxidized preferentially at the anode (Fig. 1).
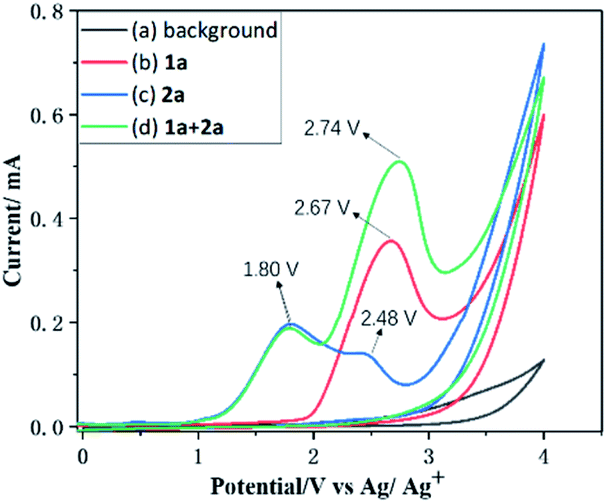 | ||
| Fig. 1 Results of the cyclic voltammetry studies. Conditions: a glassy carbon working electrode, aqueous kit-Ag/Ag+ reference electrode, and a platinum wire counter electrode, Et4NPF6 (0.01 M in CH3CN), 100 mV s−1 scan rate with 1a (0.02 M); 2a (0.01 M); 1a (0.02 M) + 2a (0.01 M); background. | ||
Control experiments were performed to probe the mechanism (Scheme 6), and the addition of radical scavengers, butylated hydroxytoluene (BHT, 3.0 equiv. Scheme 6a) and 2,2,6,6-tetramethylpiperidinooxy (TEMPO, 3.0 equiv. Scheme 6b) totally inhibited the reaction, indicating that the radical pathway was involved in the process. In addition, compound 13 was isolated in 55% yield and 14 was detected by HPLC-MS analysis (see ESI, Fig. S4†). Moreover, complex 15 was isolated in 79% yield, when 1,1-diphenylethylene (3.0 equiv. Scheme 6c) was subjected to the reaction, further demonstrating the existence of the sulfonyl radical. We found that sulfonylated enether 16 was produced in 51% yield by adding EtOH as the co-solvent under the electrochemical conditions (Scheme 6d), which might act as indirect evidence of the alkenyl cation in the process.16
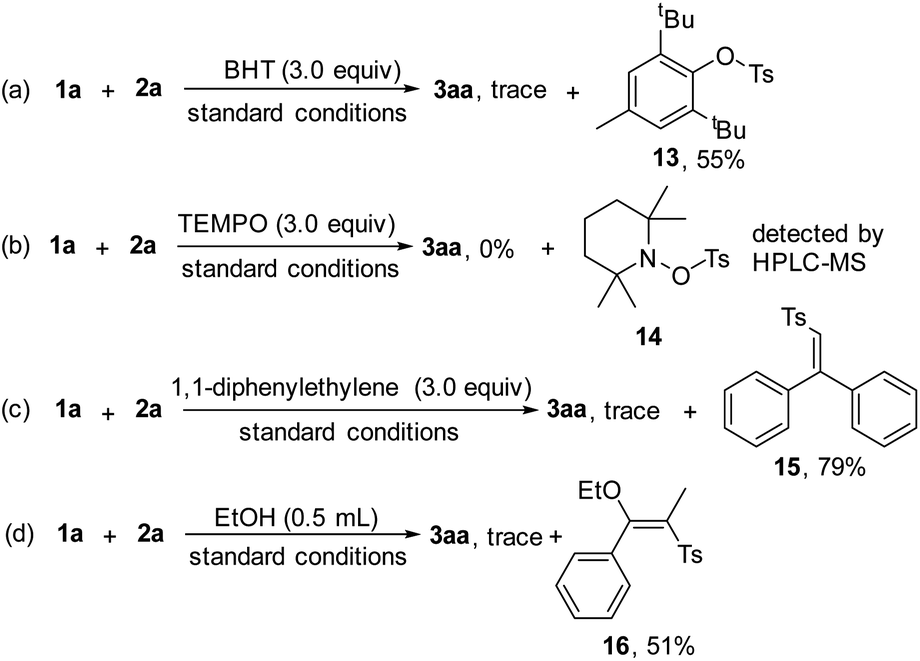 | ||
| Scheme 6 Control experiments (a–d). | ||
On the basis of our experiments, a plausible route is presented and the theoretically calculated results are presented in Fig. 2 and Scheme 7. The sulfonyl radical II formation involved deprotonation, anodic oxidation and nitrogen liberation. Then the sulfonyl radical II reacted with 1a to form the alkenyl radical III, which underwent further anodic oxidation to produce the alkenyl cation intermediate IV and HFIP in the solvent can play the role of stabilizing positive ions. As shown in Fig. 2, the free energy barrier for this radical addition process viaTS1 is calculated to be 6.7 kcal mol−1, about 8 kcal mol−1 lower than that of the regioselective transition state TS1′, agreeing well with the experimentally observed regioselectivity. Intermediate IV undergoes intra-molecular cyclization via transition state TS2 (Fig. 2) to form quaternary spirocyclization species V, which then rapidly undergoes ring expansion involving 1,2-S-migration to give VI. The calculated reaction barriers for the cyclization and 1,2-S-migration processes are 6.4 and 1.3 kcal mol−1, respectively, which are affordable under the current reaction conditions. Further oxidation and deprotonation processes gave the target product 3aa. The ring expansion transition state TS4 involving 1,2-C-migration is also located with the reaction barrier calculated to be 18.1 kcal mol−1, which is rather higher than that of S-migration.
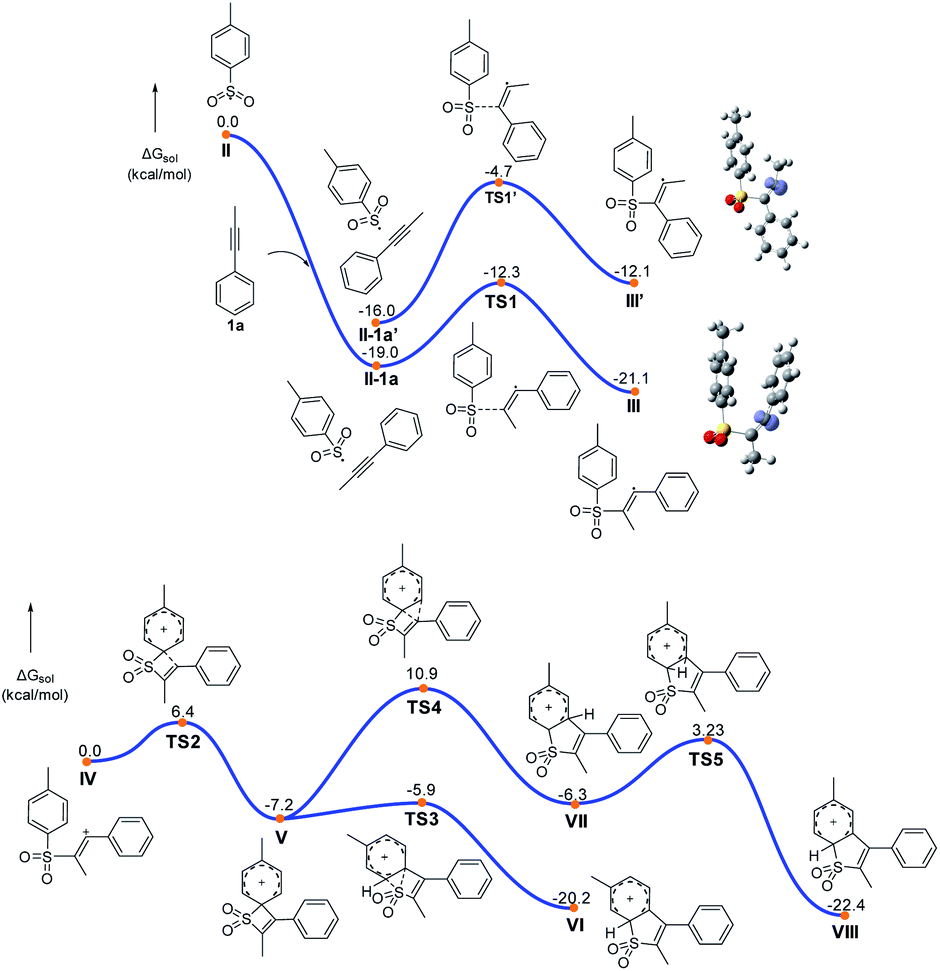 | ||
| Fig. 2 DFT calculations. | ||
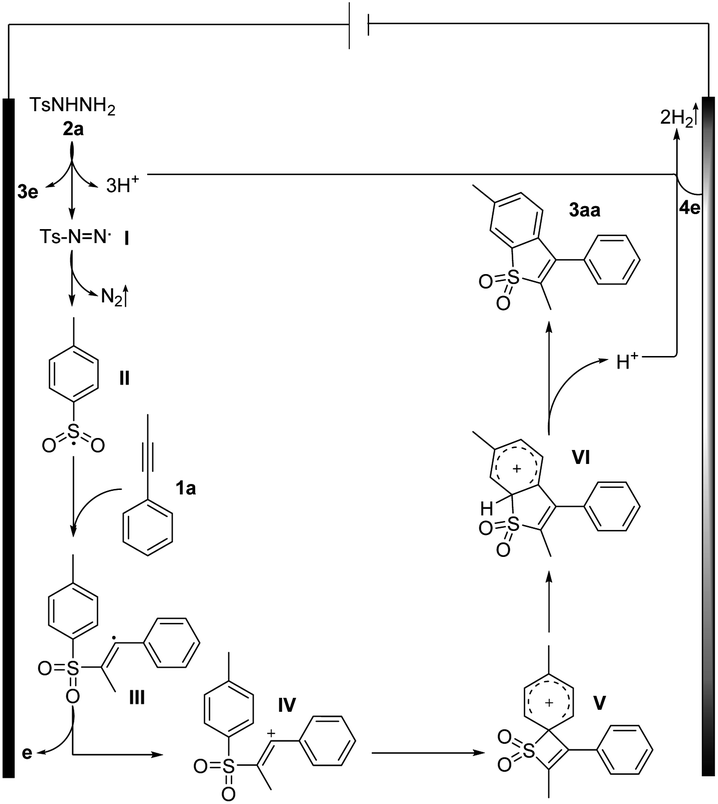 | ||
| Scheme 7 Proposed mechanism. | ||
The fluorescent properties of 3Aa were further studied in a solvent mixture of DMSO and water (Fig. 3a). These fluorophores showed relatively weak blue fluorescence in DMSO, and the emission peaks are slightly red-shifted with the increase of water fraction. Initially, addition of water had little effect on the emission intensity. When the water fraction was greater than 80%, the fluorescence intensity increased dramatically (Fig. 3b), which indicated that 3Aa exhibits good aggregation-induced emission.21
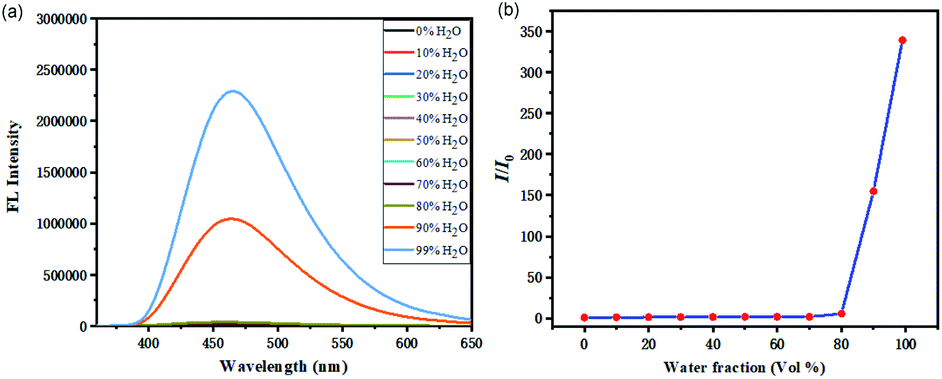 | ||
| Fig. 3 Fluorescence intensities and dependence of I/I0 ratios of 3Aa (a and b) in DMSO/water mixed solvent with different water fractions. Concentration: 10−5 M. | ||
The fluorescent emission of these compounds (3Aa–3Ia′) is obviously blue-shifted or red-shifted in solution (Fig. 4a). They exhibit strong blue emission with maximum emission wavelength around 334–442 nm. Further investigation reveals that these benzothiophene derivatives are good emitters with CIE coordinates as follows: 3Aa (0.16, 0.13), 3Ba (0.18, 0.10), 3Ca (0.18, 0.12), 3Da (0.14, 0.10), 3Ea (0.15, 0.07), 3Fa (0.17, 0.10), 3Ga (0.16, 0.14), 3Ha (0.15, 0.08), 3Ia (0.16, 0.16), and 3Ia′ (0.16, 0.13) (Fig. 4b).
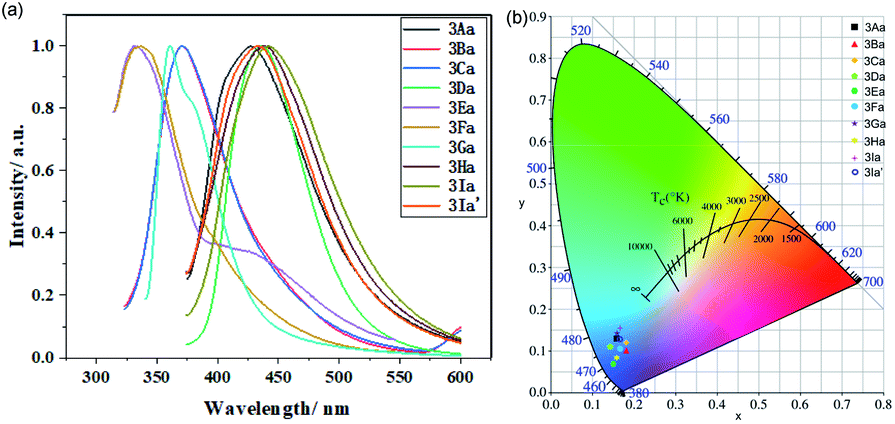 | ||
| Fig. 4 (a) Normalized liquid-state fluorescence spectra of 3Aa–3Ia′. (b) The CIE coordinates of 3Aa–3Ia′. | ||
Inspired by the potential biological applications of benzothiophene,22 we performed fluorescence imaging of HeLa cells incubated with 3Ha for 10 min, with excitation at 405 nm and the collection wavelength range was 420–460 nm. The result showed that green fluorescence was observed throughout the whole cytoplasm (see ESI, Fig. S5†).23
Conclusions
In summary, we have developed an efficient electrochemical method for the synthesis of benzothiophene dioxides by the reaction of sulfonhydrazides with internal alkynes at room temperature in undivided cells by constant current electrolysis.24 We observed that selective ipso-addition instead of the ortho-attack pathway led to strained quaternary spirocyclization, which produced the products via the S-migration process. The protocol avoided the requirement of transition metal catalysts or stoichiometric amounts of oxidants, and a variety of functional groups were tolerated. Moreover, gram-scale experiments and the transformation involving drug molecules might provide an effective route for the synthesis of value-added compounds.Data availability
All experimental data and detailed experimental procedures are available in the ESI.†Author contributions
R. L. and D. Y. conceived and performed the majority of experiments. M. P. performed a part of the research work. S. N. and Y. Z. performed DFT calculations. M. L. and L.-R. W. polished the manuscript. L.-B. Z. conceived and directed the project and wrote the paper. All the authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Natural Science Foundation of China (21801152 and 21572110) and the Natural Science Foundation of Shandong Province (ZR2019BB005 and ZR2019MB010) for financial support. We thank the Youth Innovation Science and Technology Plan of Colleges and Universities in Shandong Province (2021KJ076). L. B. Zhang acknowledges the Hong Kong Scholars Program (XJ2020013).Notes and references
- (a) R. S. Keri, K. Chand, S. Budagumpi, S. B. Somappa, S. A. Patil and B. M. Nagaraja, Eur. J. Med. Chem., 2017, 138, 1002–1033 CrossRef CAS; (b) A. J. Seed, K. J. Toyne, J. W. Goodby and M. Hird, J. Mater. Chem., 2000, 10, 2069 RSC; (c) I. Fouad, Z. Mecbal, K. I. Chane-Ching, A. Adenier, F. Maurel, J.-J. Aaron, P. Vodicka, K. Cernovska, V. Kozmik and J. Svoboda, J. Mater. Chem., 2004, 14, 1711–1721 RSC; (d) J. W. Ellingboe, T. R. Alessi, T. M. Dolak, T. T. Nguyen, J. D. Tomar, F. Guzzo, J. F. Bagli and M. L. Mccaleb, J. Med. Chem., 1992, 35, 1176–1183 CrossRef CAS; (e) M. S. Malamas, J. Sredy, C. Moxham, A. Katz, W. Xu, R. Mcdevitt, F. O. Adebayo, D. R. Sawicki, L. Seestaller, D. Sullivan and J. R. Taylor, J. Med. Chem., 2000, 43, 1293–1310 CrossRef CAS PubMed.
- (a) Y. Ma, K. Wang, D. Zhang and P. Sun, Adv. Synth. Catal., 2019, 361, 597–602 CrossRef CAS; (b) I. Nakamura, T. Sato and Y. Yamamoto, Angew. Chem., Int. Ed., 2006, 45, 4473–4475 CrossRef CAS PubMed; (c) E. Ramesh, T. Guntreddi and A. K. Sahoo, Eur. J. Org. Chem., 2017, 2017, 4405–4413 CrossRef CAS; (d) R. Che, Z. Wu, Z. Li, H. Xiang and X. Zhou, Chem.–Eur. J., 2014, 20, 7258–7261 CrossRef CAS; (e) H. Ebata, E. Miyazaki, T. Yamamoto and K. Takimiya, Org. Lett., 2007, 9, 4499–4502 CrossRef CAS; (f) B. L. Flynn, P. Verdier-Pinard and E. Hamel, Org. Lett., 2001, 3, 651–654 CrossRef CAS.
- (a) E. Ramesh, M. Shankar, S. Dana and A. K. Sahoo, Org. Chem. Front., 2016, 3, 1126–1130 RSC; (b) R. Zhu, J. Wei and Z. Shi, Chem. Sci., 2013, 4, 3706–3711 RSC; (c) R. C. Larock, E. K. Yum, M. J. Doty and K. K. C. Sham, J. Org. Chem., 1995, 60, 3270–3271 CrossRef CAS.
- S. Ejaz, M. Zubair, K. Rizwan, I. Karakaya, T. Rasheed and N. Rasool, Curr. Org. Chem., 2021, 25, 40–67 CrossRef CAS.
- (a) T. Pintauer and K. Matyjaszewski, Chem. Soc. Rev., 2008, 37, 1087–1097 RSC; (b) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58–102 CrossRef CAS; (c) K. J. Romero, M. S. Galliher, D. A. Pratt and C. R. J. Stephenson, Chem. Soc. Rev., 2018, 47, 7851–7866 RSC; (d) H. Wang, W. Shi, Y. Li, M. Yu, Y. Gao and A. Lei, CCS Chem., 2020, 2, 1710–1717 Search PubMed.
- (a) C. L. Perrin, J. Org. Chem., 2021, 86, 14245–14249 CrossRef CAS; (b) C. L. Perrin and G. A. Skinner, J. Am. Chem. Soc., 1971, 93, 3389–3394 CrossRef CAS; (c) C. L. Perrin, J. Org. Chem., 1971, 36, 420–425 CrossRef CAS; (d) X. Liu, F. Xiong, X. Huang, L. Xu, P. Li and X. Wu, Angew. Chem., Int. Ed., 2013, 52, 6962–6966 CrossRef CAS PubMed; (e) Z.-M. Chen, W. Bai, S.-H. Wang, B.-M. Yang, Y.-Q. Tu and F.-M. Zhang, Angew. Chem., Int. Ed., 2013, 52, 9781–9785 CrossRef CAS; (f) A. Bunescu, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2015, 54, 3132–3135 CrossRef CAS.
- (a) J. Boivin, M. Yousfi and S. Z. Zard, Tetrahedron Lett., 1997, 38, 5985–5988 CrossRef CAS; (b) H. Ohno, S.-i. Maeda, M. Okumura, R. Wakayama and T. Tanaka, Chem. Commun., 2002, 316–317 RSC; (c) H. Ohno, M. Okumura, S.-i. Maeda, H. Iwasaki, R. Wakayama and T. Tanaka, J. Org. Chem., 2003, 68, 7722–7732 CrossRef CAS; (d) H. Iwasaki, T. Eguchi, N. Tsutsui, H. Ohno and T. Tanaka, J. Org. Chem., 2008, 73, 7145–7152 CrossRef CAS; (e) T. Yamada, Y. Ozaki, M. Yamawaki, Y. Sugiura, K. Nishino, T. Morita and Y. Yoshimi, Tetrahedron Lett., 2017, 58, 835–838 CrossRef CAS; (f) A. R. Flynn, K. A. McDaniel, M. E. Hughes, D. B. Vogt and N. T. Jui, J. Am. Chem. Soc., 2020, 142, 9163–9168 CrossRef CAS; (g) H. Takeuchi, S. Inuki, K. Nakagawa, T. Kawabe, A. Ichimura, S. Oishi and H. Ohno, Angew. Chem., Int. Ed., 2020, 59, 21210–21215 CrossRef CAS; (h) L. Wu, Y. Hao, Y. Liu, H. Song and Q. Wang, Chem. Commun., 2020, 56, 8436–8439 RSC.
- (a) N. Fuentes, W. Kong, L. Fernández-Sánchez, E. Merino and C. Nevado, J. Am. Chem. Soc., 2015, 137, 964–973 CrossRef CAS PubMed; (b) W. Kong, M. Casimiro, E. Merino and C. Nevado, J. Am. Chem. Soc., 2013, 135, 14480–14483 CrossRef CAS; (c) H. Jiang and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 10707–10711 CrossRef CAS; (d) N. Radhoff and A. Studer, Angew. Chem., Int. Ed., 2021, 60, 3561–3565 CrossRef CAS; (e) Z. Wu, D. Wang, Y. Liu, L. Huan and C. Zhu, J. Am. Chem. Soc., 2017, 139, 1388–1391 CrossRef CAS; (f) X. Wu, M. Wang, L. Huan, D. Wang, J. Wang and C. Zhu, Angew. Chem., Int. Ed., 2018, 57, 1640–1644 CrossRef CAS; (g) X. Wu and C. Zhu, Acc. Chem. Res., 2020, 53, 1620–1636 CrossRef CAS PubMed; (h) Z.-M. Chen, X.-M. Zhang and Y.-Q. Tu, Chem. Soc. Rev., 2015, 44, 5220–5245 RSC; (i) M. Huynh, M. D. Abreu, P. Belmont and E. Brachet, Chem.–Eur. J., 2021, 27, 3581–3607 CrossRef CAS; (j) Y.-R. Chen and W.-L. Duan, J. Am. Chem. Soc., 2013, 135, 16754–16757 CrossRef CAS.
- V. K. Soni, H. S. Hwang, Y. K. Moon, S. W. Park, Y. You and E. J. Cho, J. Am. Chem. Soc., 2019, 141, 10538–10545 CrossRef CAS.
- W. Kong, E. Merino and C. Nevado, Angew. Chem., Int. Ed., 2014, 53, 5078–5082 CrossRef CAS.
- H. Keum, H. Jung, J. Jeong, D. Kim and S. Chang, Angew. Chem., Int. Ed., 2021, 60, 25235–25240 CrossRef CAS PubMed.
- (a) C. Ma, P. Fang and T.-S. Mei, ACS Catal., 2018, 8, 7179–7189 CrossRef CAS; (b) C. Ma, P. Fang, D. Liu, K.-J. Jiao, P.-S. Gao, H. Qiu and T.-S. Mei, Chem. Sci., 2021, 12, 12866–12873 RSC; (c) K.-J. Jiao, Y.-K. Xing, Q.-L. Yang, H. Qiu and T.-S. Mei, Acc. Chem. Res., 2020, 53, 300–310 CrossRef CAS; (d) H. Wang, X. Gao, Z. Lv, T. Abdelilah and A. Lei, Chem. Rev., 2019, 119, 6769–6787 CrossRef CAS PubMed; (e) Y. Yuan, J. Yang and A. Lei, Chem. Soc. Rev., 2021, 50, 10058–10086 RSC; (f) T. H. Meyer, I. Choi, C. Tian and L. Ackermann, Chem, 2020, 6, 2484–2496 CrossRef CAS; (g) R. C. Samanta, T. H. Meyer, I. Siewert and L. Ackermann, Chem. Sci., 2020, 11, 8657–8670 RSC.
- (a) C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415–431 CrossRef CAS PubMed; (b) S.-H. Shi, Y. Liang and N. Jiao, Chem. Rev., 2021, 121, 485–505 CrossRef CAS PubMed; (c) L. Ackermann, Acc. Chem. Res., 2020, 53, 84–104 CrossRef CAS; (d) S. D. Minteer and P. Baran, Acc. Chem. Res., 2020, 53, 545–546 CrossRef CAS; (e) P. Xiong and H.-C. Xu, Acc. Chem. Res., 2019, 52, 3339–3350 CrossRef CAS PubMed; (f) W. Zhang and S. Lin, J. Am. Chem. Soc., 2020, 142, 20661–20670 CrossRef CAS PubMed; (g) J. C. Siu, N. Fu and S. Lin, Acc. Chem. Res., 2020, 53, 547–560 CrossRef CAS PubMed; (h) Y. Kawamata and P. S. Baran, Joule, 2020, 4, 701–704 CrossRef.
- (a) C. Kingston, M. D. Palkowitz, Y. Takahira, J. C. Vantourout, B. K. Peters, Y. Kawamata and P. S. Baran, Acc. Chem. Res., 2020, 53, 72–83 CrossRef CAS PubMed; (b) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS; (c) C. Huang, Z.-Y. Li, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2021, 60, 11237–11241 CrossRef CAS; (d) L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC; (e) N. Fu, G. S. Sauer, A. Saha, A. Loo and S. Lin, Science, 2017, 357, 575–579 CrossRef CAS; (f) Z. Zhang, L. Zhang, X. Zhang, J. Yang, Y. Yin, Y. Jiang, C. Zeng, G. Lu, Y. Yang and F. Mo, Chem. Sci., 2020, 11, 12021–12028 RSC; (g) Y. Liang, S.-H. Shi, R. Jin, X. Qiu, J. Wei, H. Tan, X. Jiang, X. Shi, S. Song and N. Jiao, Nat. Catal., 2021, 4, 116–123 CrossRef CAS; (h) Y. Yuan, Y. Cao, Y. Lin, Y. Li, Z. Huang and A. Lei, ACS Catal., 2018, 8, 10871–10875 CrossRef CAS.
- Y. Zhang, C. Ma, J. Struwe, J. Feng, G. Zhu and L. Ackermann, Chem. Sci., 2021, 12, 10092–10096 RSC.
- W.-B. Du, N.-N. Wang, C. Pan, S.-F. Ni, L.-R. Wen, M. Li and L.-B. Zhang, Green Chem., 2021, 23, 2420–2426 RSC.
- (a) Z. Kong, C. Pan, M. Li, L. Wen and W. Guo, Green Chem., 2021, 23, 2773–2777 RSC; (b) L.-B. Zhang, R.-S. Geng, Z.-C. Wang, G.-Y. Ren, L.-R. Wen and M. Li, Green Chem., 2020, 22, 16–21 RSC; (c) Z.-C. Wang, R.-T. Li, Q. Ma, J.-Y. Chen, S.-F. Ni, M. Li, L.-R. Wen and L.-B. Zhang, Green Chem., 2021, 23, 9515–9522 RSC; (d) L.-R. Wen, N.-N. Wang, W.-B. Du, M.-Z. Zhu, C. Pan, L.-B. Zhang and M. Li, Chin. J. Chem., 2021, 39, 1831–1837 CrossRef CAS; (e) L.-R. Wen, Y. Rao, M. Zhu, R. Li, J. Zhan, L. Zhang, L. Wang, M. Li, S. Pang and Z. Zhou, Angew. Chem., Int. Ed., 2021, 60, 17356–17361 CrossRef CAS; (f) D.-F. Yuan, Z.-C. Wang, R.-S. Geng, G.-Y. Ren, J. S. Wright, S.-F. Ni, M. Li, L.-R. Wen and L.-B. Zhang, Chem. Sci., 2022, 13, 478–485 RSC; (g) L.-B. Zhang, Z.-C. Wang, S.-Z. Sun, S.-F. Ni, L.-R. Wen and M. Li, Chin. J. Chem., 2018, 36, 587–593 CrossRef.
- (a) A. Berkessel, J. A. Adrio, D. Hüttenhain and J. M. Neudörfl, J. Am. Chem. Soc., 2006, 128, 8421–8426 CrossRef CAS; (b) J. M. Ramos-Villaseñor, E. Rodríguez-Cárdenas, C. E. B. Díaz and B. A. Frontana-Uribe, J. Electrochem. Soc., 2020, 167, 155509 CrossRef.
- P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905–4979 CrossRef CAS PubMed.
- Z. Yang, F. Lu, H. Li, Y. Zhang, W. Lin, P. Guo, J. Wan, R. Shi, T. Wang and A. Lei, Org. Chem. Front., 2020, 7, 4064–4068 RSC.
- Y. Fang, Y. Meng, C. Yuan, C. Du, K.-P. Wang, S. Chen and Z.-Q. Hu, Spectrochimica Acta, Part A: Molecular and Biomolecular Spectroscopy, 2022, 267, 120575 CrossRef CAS PubMed.
- Y.-C. Jeong, S. I. Yang, E. Kim and K.-H. Ahn, Tetrahedron, 2006, 62, 5855–5861 CrossRef CAS.
- (a) X. Cui, W. Cheng and X. Han, J. Mater. Chem. B, 2018, 6, 8078–8084 RSC; (b) Q. Zhang, C. Fu, X. Guo, J. Gao, P. Zhang and C. Ding, ACS Sens., 2021, 6, 1138–1146 CrossRef CAS PubMed.
- During the submission stage of this paper, a similar electrochemical migratory cyclization was reported. Z. Shi, Y. Li, N. Li, W.-Z. Wang, H.-K. Lu, H. Yan, Y. Yuan, J. Zhu and K. Ye, Angew. Chem., Int. Ed., 2022, 61 DOI:10.1002/anie.202206058.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2044891, 2044867 and 2171791. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc01175a |
| This journal is © The Royal Society of Chemistry 2022 |