
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRadical defects modulate the photocatalytic response in 2D-graphitic carbon nitride†
Edoardo
Raciti
 ab,
Sai Manoj
Gali
a,
Michele
Melchionna
b,
Giacomo
Filippini
b,
Arianna
Actis
c,
Mario
Chiesa
c,
Manuela
Bevilacqua
de,
Paolo
Fornasiero
be,
Maurizio
Prato
bfg,
David
Beljonne
*a and
Roberto
Lazzaroni
*a
ab,
Sai Manoj
Gali
a,
Michele
Melchionna
b,
Giacomo
Filippini
b,
Arianna
Actis
c,
Mario
Chiesa
c,
Manuela
Bevilacqua
de,
Paolo
Fornasiero
be,
Maurizio
Prato
bfg,
David
Beljonne
*a and
Roberto
Lazzaroni
*a
aLaboratory for Chemistry of Novel Materials, Materials Research Institute, University of Mons, Place du Parc 20, Mons 7000, Belgium. E-mail: david.beljonne@umons.ac.be; roberto.lazzaroni@umons.ac.be
bDepartment of Chemical and Pharmaceutical Sciences, INSTM, University of Trieste, Via L. Giorgieri 1, Trieste 34127, Italy
cDepartment of Chemistry, University of Torino, NIS Centre of Excellence, Via Giuria 9, Torino 10125, Italy
dInstitute of Chemistry of OrganoMetallic Compounds (ICCOM-CNR), via Madonna del Piano 10, Sesto Fiorentino 50019, Italy
eCenter for Energy, Environment and Transport Giacomo Ciamician and ICCOM-CNR Trieste Research Unit, University of Trieste, via L. Giorgieri 1, I-34127 Trieste, Italy
fCenter for Cooperative Research in Biomaterials (CIC biomaGUNE), Basque Research and Technology Alliance (BRTA), Paseo de Miramón 182, Donostia San Sebastián 20014, Spain
gBasque Foundation for Science, Ikerbasque, Bilbao 48013, Spain
First published on 12th August 2022
Abstract
Graphitic carbon nitride (gCN) is an important heterogeneous metal-free catalytic material. Thermally induced post-synthetic modifications, such as amorphization and/or reduction, were recently used to enhance the photocatalytic response of these materials for certain classes of organic transformations, with structural defects possibly playing an important role. The knowledge of how these surface modifications modulate the photocatalytic response of gCN is therefore not only interesting from a fundamental point of view, but also necessary for the development and/or tuning of metal-free gCN systems with superior photo-catalytic properties. Herein, employing density functional theory calculations and combining both the periodic and molecular approaches, in conjunction with experimental EPR measurements, we demonstrate that different structural defects on the gCN surface generate distinctive radical defect states localized within the electronic bandgap, with only those correlated with amorphous and reduced gCN structures being photo-active. To this end, we (i) model defective gCN surfaces containing radical defect states; (ii) assess the interactions of these defects with the radical precursors involved in the photo-driven alkylation of electron-rich aromatic compounds (namely perfluoroalkyl iodides); and (iii) describe the photo-chemical processes triggering the initial step of that reaction at the gCN surface. We provide a coherent structure/photo-catalytic property relationship on defective gCN surfaces, elaborating how only specific defect types act as binding sites for the perfluoroalkyl iodide reagent and can favor a photo-induced charge transfer from the gCN surface to the molecule, thus triggering the perfluoroalkylation reaction.
Introduction
Photocatalysis with metal-free catalysts is gaining scientific and industrial attention in view of the growing demand for new sustainable synthetic schemes to access chemicals of extensive use.1 In comparison to homogeneous systems, heterogeneous metal-free catalysts offer an edge due to their recyclability, low cost, good compatibility with specific reaction environments and sustained reactivity over time. Graphitic carbon nitride (hitherto abbreviated as gCN), a relatively wide band-gap semiconductor, with easy availability, low cost, good stability and excellent electronic properties2,3 is being extensively used in photocatalytic applications such as H2 production, water oxidation and CO2 reduction.4–6 Recent studies have extended the use of gCN to a wider range of industry-relevant chemical reactions which span from photocatalytic oxidation of small molecules7,8 to C–C and C-heteroatom coupling reactions,9–12 including functionalization of arenes and heteroarenes with fluoroalkylated groups.12–14 The heterogeneous forms of gCN such as mesoporous carbon nitride, polymeric carbon nitride, potassium poly(heptazine imides) were also successfully employed for a plethora of applications such as the photocatalytic/photoredox sulfonylation of alkenes, ketalization of different ketones, aerobic oxidative coupling of resveratrol, and aerobic oxidation of benzylamines.15Structural modifications by simple and scalable approaches were also proposed16 to fine-tune the properties and reactivity of gCN surfaces. For instance, protonation and doping of gCN can be employed to fine-tune the electronic properties and also modulate the available surface area,6,16 whereas inclusion of metallic ions via intercalation can decrease the band gap, enhance charge carrier mobility and provide additional catalytic sites.17,18 More recently, carbon nitride surfaces, modified to include electron-rich phenyl moieties, were employed for the metal-free, visible light-responsive photocatalytic synthesis of methyl esters from benzylic alcohols, photocatalytic oxidative esterification by the inclusion of surface acidic sites19 and for the photocatalytic aerobic oxidation of alcohols, under irradiation using an extended visible-light wavelength of 620 nm .20 Some of us have recently demonstrated that the visible-light photocatalytic activity of gCN for the perfluoroalkylation of electron-rich organic substrates can be substantially enhanced by diverse surface modifications.21 Briefly, this strategy includes post-synthetic modifications of melamine-derived gCN (Fig. 1a) to introduce structural and chemical variations into pristine gCN by adopting specific protocols for reduction (red-gCN), oxidation (oxy-gCN) and thermal amorphization (am-gCN). The photocatalytic response of these materials was then investigated for the grafting of perfluorinated groups on aromatic cores, which is a reaction of major interest in medicinal chemistry, agro-chemistry and materials science.14,22–24 The test case that has been described in ref. 21 is the reaction between 1,3,5-trimethoxy-benzene (1a), a model electron-rich aromatic molecule, and perfluorobutyl iodide (2a, C4F9–I) as the radical precursor to give the fluorinated product 3a (Fig. 1b). It has been proposed that the key step in the reaction between C4F9–I and 1,3,5-trimethoxy-benzene is a photo-induced electron transfer from the gCN catalyst surface to the C4F9–I molecule.
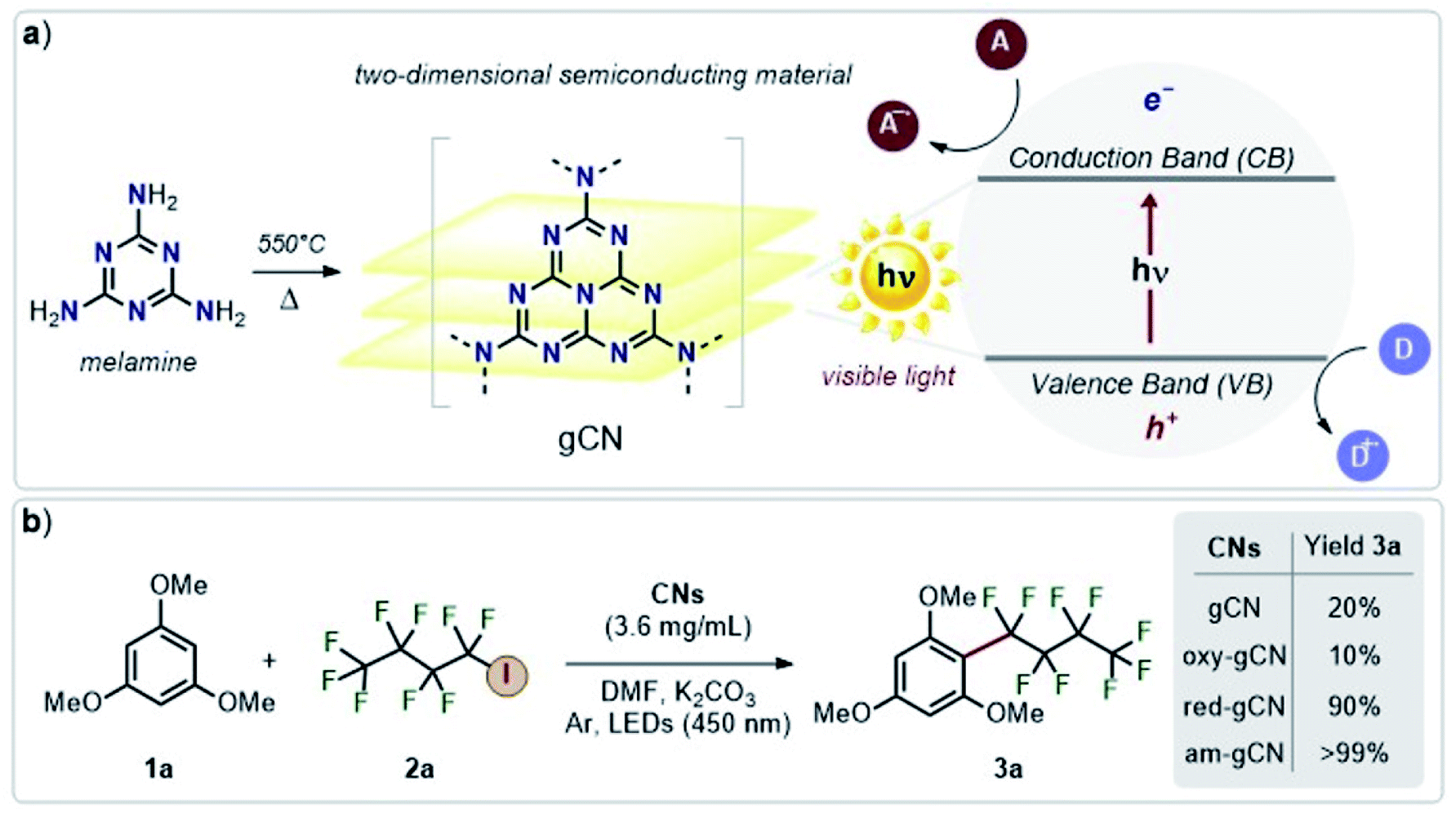 | ||
| Fig. 1 Schematic representation of (a) graphitic carbon nitride obtained by the pyrolysis of melamine and its conduction and valence bands (CB and VB); (b) the reaction under study and its yield, highlighting the difference in the catalytic efficiency of the various types of gCN. A: electron acceptor; D: electron donor. | ||
Interestingly, it was found that the photo-catalytic activity of the reduced and amorphous gCN forms is much higher (≥90%) compared to that of the gCN surface in its pristine (20%) and oxidized (10%) forms. However, all these post-synthetically modified gCN structures show: (i) a similar C/N ratio, as indicated by the XPS data, with the exception of am-gCN and red-gCN forms that show a comparatively lower N-content attributed to the presence of nitrogen vacancies, consistent with previous observations;15,25–28 (ii) a similar optical bandgap, as obtained by diffuse reflectance spectroscopy (DRS); and (iii) the presence of native and photo-induced radical species in all samples, as observed by electron paramagnetic resonance (EPR) measurements.29
Therefore, despite the structural and spectroscopic data at hand, we are still missing a detailed fundamental understanding of: (i) the nature of the interaction between the perfluoroalkyl iodide and the (modified) gCN surface(s), (ii) the excited-state dynamics triggering the reaction and (iii) the influence of the gCN surface composition on the photocatalytic activity. Such an understanding is crucial to establish a solid structure/property/activity relationship for gCN photocatalysis and to further broaden the spectrum of the reactions that could be catalyzed by gCN, aiding the process of rational function-driven catalyst design for future applications.
Here, we present a detailed theoretical study of model perfluoroalkyl iodide/gCN systems with the aim of providing a comprehensive understanding of the recently reported NMR, XPS and EPR spectroscopic data and photocatalytic activities.21,29 More specifically, we focus on the specific interaction between different gCN surfaces and the C4F9–I molecule, which is considered to be the crucial first step governing the experimental photo-catalytic efficiency, quantified only by the global yield of 2-nonafluorobutyl-1,3,5-trimetoxybenzene (product 3a in Fig. 1).21 The subsequent reaction steps leading to the formation of product 3a correspond to a classical case of homolytic aromatic substitution.
For that purpose, we apply a two-fold modelling strategy to: (i) evaluate the ground-state interaction between the pristine, red-gCN, oxy-gCN and am-gCN surfaces and the C4F9–I molecule to quantify the adsorption profiles and the electronic structure of the combined gCN/C4F9–I systems and (ii) to determine, with time-dependent DFT (TD-DFT) calculations on the gCN/C4F9–I systems, the excited-state electronic structure, in order to shed light on the photo-chemical response of these gCN surfaces in the presence of the C4F9–I molecule. A detailed description of the model gCN structures, the computational protocol employed, and the results pertaining to the ground-state and excited-state gCN/C4F9–I interactions are presented in the following sections. We find a clear correlation between the nature of the defects on the gCN surface, their electronic structure, and the viability of a photo-induced electron transfer from gCN to the C4F9–I molecule, which is most probably the key step that triggers its coupling with the electron-rich aromatic molecule.
Modelling methodology
Modelling protocol
The following modelling protocol was employed to evaluate the ground-state and excited-state interactions between defective gCN surfaces and the C4F9–I molecule:First, starting from pristine gCN, we select model systems including structural point defects to mimic the reduced, amorphous and oxidized forms and we assess how these defects perturb the gCN electronic structure.
We then compute the ground-state interaction between the different defective gCN surfaces and the C4F9–I molecule to: (i) extract the electronic structure of the defective gCN surfaces and the C4F9–I molecule adsorbed on those gCN surfaces (hitherto referred as gCN/C4F9–I for simplicity), (ii) evaluate the adsorption energies, (iii) quantify the ground state (partial) charge transfer between the gCN surface and the molecule, and (iv) evaluate the bond order parameters in relation to the 19F-NMR experimental results21,29 for a qualitative assessment of halogen bonding.
Finally, we perform Time-Dependent DFT (TD-DFT) calculations on the gCN/C4F9–I systems to determine the excited-state electronic structure, in order to shed light on the photo-chemical response of different gCN surfaces in the presence of the C4F9–I molecule.
These steps, which form the core of the gCN/C4F9–I interaction, represent the ground-state and excited-state interactions between a given defective form of the gCN surface and the C4F9–I molecule. More specifically, we investigate the ground-state electronic interactions between gCN surfaces and the C4F9–I molecule using both the 2D periodic approach and a molecular approach. For the latter, we use a model triangular heptazine trimer representing a fragment of the (defective) gCN surface (see Fig. SI-1†) that is made to interact with C4F9–I, as this offers computationally easy access to excited-state calculations. The choice of using a molecular (heptazine trimer) model for excited-state studies is mainly motivated by the fact that: (i) the molecular representation of gCN, as often considered in previous studies,30–32 opens up a wide range of hybrid functionals to evaluate the optical properties, which would be computationally prohibitive within the periodic approaches, and (ii) as the gCN systems are characterized by limited band dispersion, the interaction of the C4F9–I molecule with a molecular fragment considered to represent the periodic equivalent, turns out to be computationally appropriate, as detailed in the subsequent sections.
Computational details
All periodic DFT calculations were performed using the VASP code and the projector-augmented wave (PAW) basis set.33,34 Exchange and correlation effects are treated at the Perdew–Burke–Ernzerhof (PBE) level with the dispersion forces treated by using Grimme correction (PBE + D2),35,36 with a kinetic energy cut-off of 500 eV and using a Monkhorst–Pack sampling of 3 × 3 × 1 for the Brillouin zone (BZ) integration on all the gCN unit cells replicated 4 × 4 × 1 times, with the vacuum spacing set to be 30 Å to avoid the interaction with periodic images. Dipole moment correction was employed along the ‘c’ axis (Z direction & perpendicular to the gCN surface). The geometries of pristine and defective gCN surfaces along with C4F9–I adsorbed were fully optimized at the PBE/GGA level of theory prior to the calculation of the electronic properties at the hybrid (HSE06)37 level of theory. Bond orders and ground state partial charge transfers were computed using the DDEC06 method.38 DFT and TD-DFT calculations of the molecular equivalent gCN units with and without C4F9–I adsorption were performed using the Gaussian16 program39 at the B3LYP/6-311G(d,p) level of theory for molecular optimizations. The subsequent electronic structure and UV-vis absorption spectra calculations, along with the corresponding NTO40 and attachment-detachment density41,42 analysis were performed on the molecular systems at the hybrid level of theory (HSE1HSE), to maintain consistency with the electronic structure obtained with the periodic DFT calculations. Prior to TD-DFT calculations on the molecular systems, the consistency between the periodic and molecular gCN/C4F9–I descriptions was ensured by comparing the electronic structure at the hybrid level (HSE06) of theory.gCN structural models
For the periodic approach, we started from a monolayer of pristine gCN surface (P.0 in Fig. 2), and incorporated a large variety of structural defects on the gCN surface, classified based on the post-synthetic experimental treatment,21 chemical intuition and literature data, and we calculated their electronic structure and stability. The most representative gCN systems in their pristine (gCN), red-gCN, oxy-gCN and red-gCN forms are shown in Fig. 2. To simulate the formation of amorphous gCN (am-gCN), for which some nitrogen loss has been observed experimentally,21,29 defects were created by removing one nitrogen atom from the surface of the gCN unit cell. The nitrogen atom is removed from either the core of a heptazine unit or from the edge of a heptazine unit (Fig. 2(1.a and 1.b), respectively). Both these forms of nitrogen vacancies are thermodynamically stable. Here we find that the defect formation energy is 4.71 eV (108.6 kcal mol−1) and 4.80 eV (110.7 kcal mol−1) respectively, consistent with previous results.43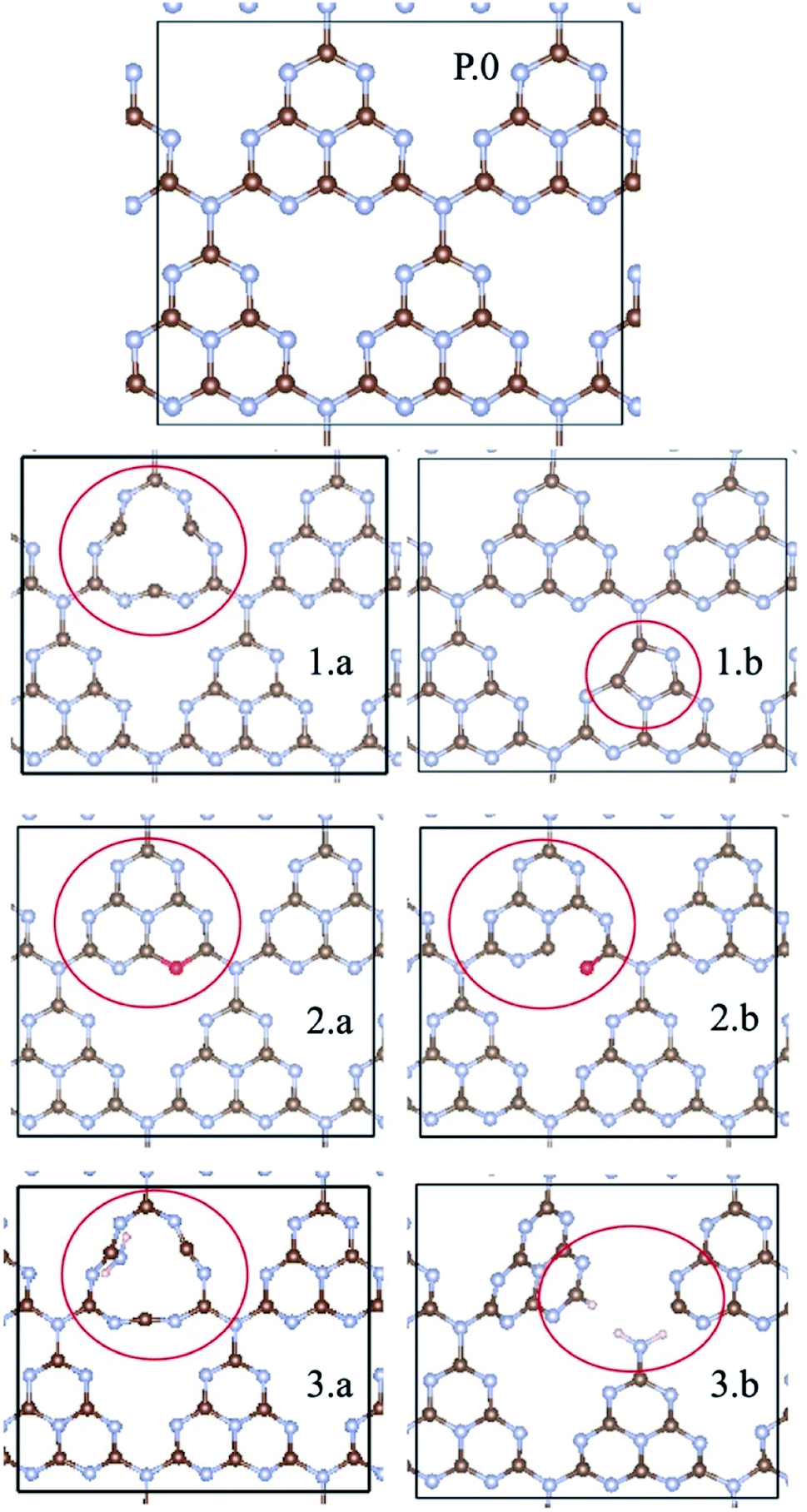 | ||
| Fig. 2 gCN surfaces in their pristine (P.0) and defective forms (1.a/b, 2.a/b, and 3.a/b) considered in this work (carbon: brown; nitrogen: blue; oxygen: red; hydrogen: pink). am-gCN surfaces are represented to contain nitrogen vacancies either at the centre or at the edge of a heptazine unit, as shown in 1.a to 1.b; oxy-gCN surfaces in their pyran-like (2.a) and carbonyl-like (2.b) forms were created by substituting the heptazine edge nitrogen vacancy. Structural defects with disconnected bonds between nitrogen and carbon atoms, either at the heptazine centre or the edge (3.a and 3.b, respectively) represent the red-gCN forms. | ||
To represent gCN treated under oxidative conditions, oxygenated gCN models (oxy-gCN), considered with respect to recent investigations,32 were obtained by substituting the nitrogen atoms at the edge of a heptazine unit, to obtain either a pyran-like unit (see Fig. 2(2.a)) or a carbonyl-containing gCN form (see Fig. 2(2.b)).
Since the reductive treatment is carried out in the presence of H2, we hypothesize that structural defects appearing upon bond cleavage during reductive treatment are likely to be at least partly saturated by hydrogen atoms. To model the situation of reduced gCN forms (red-gCN), two disconnected gCN forms were considered wherein the bonds between the nitrogen atom and the surrounding carbon atoms were cleaved, either at the center of the heptazine unit (see Fig. 2(3.a)) or at the edge of the heptazine unit (see Fig. 2(3.b)) and were partly saturated with hydrogen atoms.
It must be noted that all these structures, as shown in Fig. 2(1.a/b–2.3.a/b), are open-shell systems with at least one unpaired electron per unit cell, consistent with the experimental EPR results that indicate the presence of radicals on all the gCN surfaces, irrespective of their treatment (oxidation, reduction, and amorphization). The defect formation energies of the oxy-gCN forms were found to be in the 0.6 eV to 1.0 eV range (1.08 eV and 0.66 eV for structures 2.a and 2.b, respectively) whereas that of red-gCN (structure 3.a) is ≈4.1 eV. See ESI, Fig. SI-3,† for the defect formation energies of these defective gCN forms, along with the corresponding schematics of the reaction mechanism considered.
The molecular equivalents to the periodic structures for pristine and defective gCN forms were built by suitably modifying the pristine heptazine trimer; those model defective molecules are shown in Fig. SI-2.† The specific nomenclature of the different gCN structures pertaining to periodic and molecular gCN forms (related to Fig. 2 and SI-2,† respectively) along with their reference numbers are listed in Table 1. Hitherto, to ease the description, the reference number from Table 1 is used to refer to any given gCN surface/form that is discussed in this work.
| Ref. | Type | Description of the gCN surface |
|---|---|---|
| P.0 | Pristine | gCN-pristine |
| 1.a | am-gCN | gCN-N vacancy at the center of a heptazine unit |
| Nature of the defect: radical with one unpaired electron | ||
| 1.b | gCN-N vacancy at the edge of a heptazine unit | |
| Nature of the defect: radical with one unpaired electron | ||
| 2.a | oxy-gCN | Oxygen substitution at the edge of a heptazine unit: pyran-like form |
| Nature of the defect: radical with one unpaired electron | ||
| 2.b | Oxygen substitution at the edge of a heptazine unit: carbonyl form | |
| Nature of the defect: radical with one unpaired electron | ||
| 3.a | red-gCN | Disconnected gCN network, with N–C bonds cleaved at the center of a heptazine unit |
| Nature of the defect: radical with two unpaired electrons (bi-radical form) | ||
| 3.b | Disconnected gCN network, with N–C cleaved at the edge of a melamine unit | |
| Nature of the defect: radical with one unpaired electron |
Results and discussion
Electronic structure of the defective gCN forms
The electronic structure of the different gCN derivatives from Fig. 2, obtained at the HSE06 level of theory, is shown in Fig. 3. While pristine gCN shows an electronic band gap of ≈2.8 eV, the radical gCN forms show additional defect levels within the electronic band gap. More specifically, for the radical am-gCN form with a nitrogen vacancy at the center of the heptazine unit (see structure 1.a and Fig. 3(1.a)), a localized occupied defect level (viz. the radical state with one unpaired ‘α’ electron) appears at around mid-gap, i.e. ≈1.2 eV from the conduction band (CB) edge and its corresponding unoccupied electronic level (viz. the ‘β’ state pertaining to the occupied ‘α’ state at ≈1.2 eV) merges with the CB edge. When the nitrogen vacancy is present at the edge of the heptazine unit in am-gCN (see structure 1.b and Fig. 3(1.b)), an occupied defect level (‘α’ state) is positioned at ≈1.8 eV from the CB edge, while a new, corresponding unoccupied (empty ‘β’ state) electronic level, is positioned within the band gap at ≈0.6 eV from the CB edge.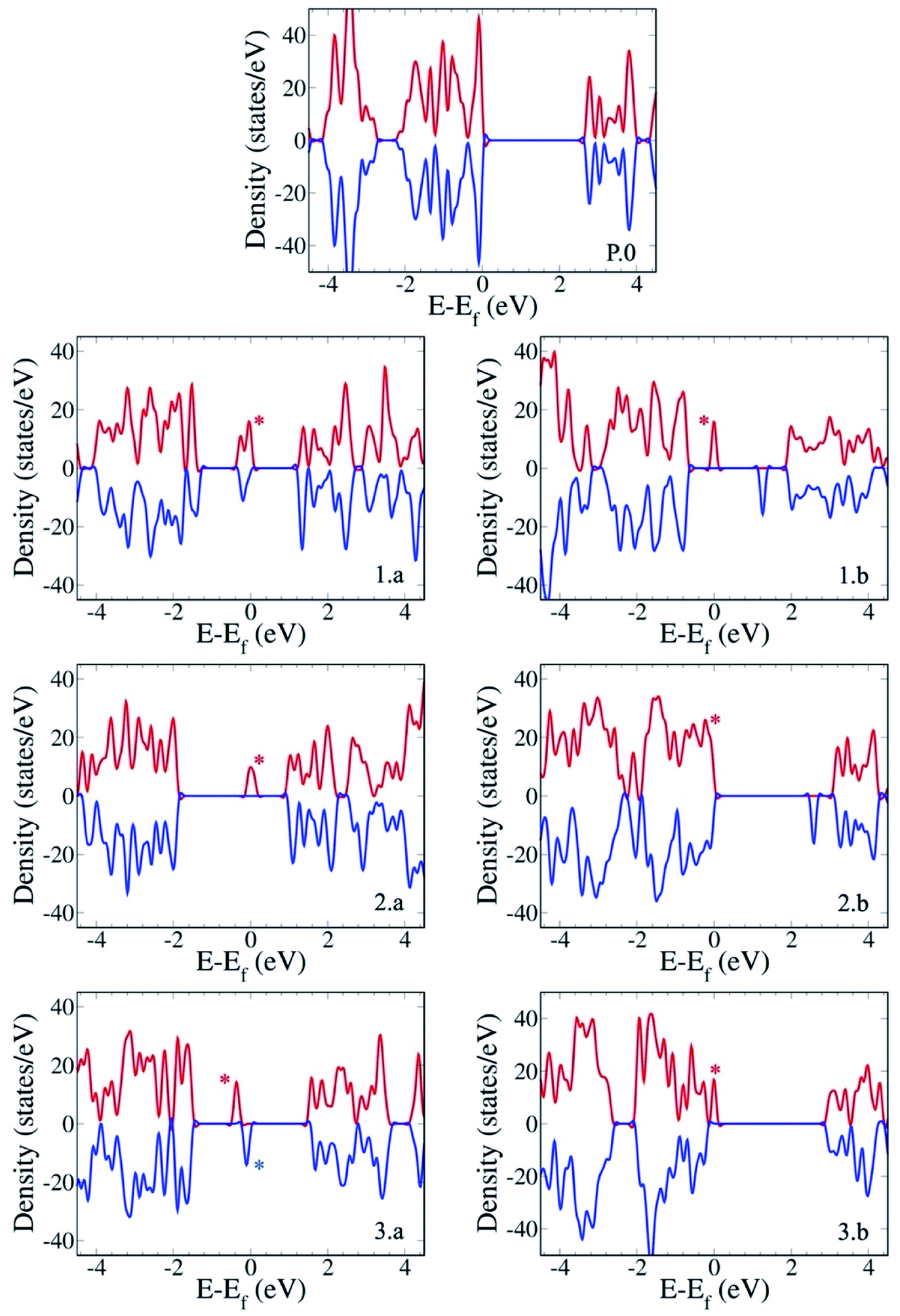 | ||
| Fig. 3 Electronic structure of the gCN surfaces in their pristine (P.0) and defective forms considered in this study (with am-gCN represented by 1.a/b, oxy-gCN represented by 2.a/b and red-gCN represented by 3.a-b). The positive (red) and negative (blue) density-of-states curves correspond to the α and β electron densities, respectively. The zero of the energy scale, i.e., the Fermi level, corresponds to the highest-occupied electronic state. Electronic energy levels corresponding to the localized occupied defect states at the Fermi level are indicated by an asterisk. | ||
For the oxygenated gCN surface with a pyran-like defect (see structure 2.a and Fig. 3(2.a)), the occupied radical defect level is positioned within the bandgap at ≈0.9 eV from the CB edge. For the carbonyl-containing oxy-gCN form (see structure 2.b and Fig. 3(2.b)), the occupied radical defect level basically merges with the top of the valence band (VB) edge while its corresponding empty level is positioned within the band gap at ≈0.5 eV from the CB edge.
For the reduced gCN system with a disconnection in the C–N bonding at the center of the heptazine unit (see structure 3.a and Fig. 3(3.a)), the occupied radical defect level is positioned within the bandgap at ≈1.7 eV from the CB edge. This gCN form (structure 3.a) is a bi-radical, as confirmed by the ground-state broken symmetry calculations, with an energy difference of ≈0.5 eV between the partly occupied α and β defect states (see the ESI† for details). For the red-gCN form with a C–N disconnection at the heptazine edge (see structure 3.b and Fig. 3(3.b)), the occupied defect level is positioned within the band gap but very close (0.2 eV) to the VB edge.
The electronic structure of the pristine and defective molecular gCN models, obtained with pristine and modified heptazine units, shows a very good quantitative agreement with the periodic gCN forms in terms of the electronic structure, energy gap and orbital symmetry of the respective gCN forms, as shown in the ESI (Fig. SI-4 and SI-5†), implying the transferability between the periodic and the molecular approaches. In addition, all the intra-gap levels in the radical gCN forms are localized in the vicinity of the defect for both periodic and molecular equivalents, as can be seen in the crystalline/molecular orbitals shown in the ESI (Fig. SI-5†). Indeed, it is the nature of the localized radical defect states present on the gCN surface that generate a significant and specific interaction with the perfluoroalkyl iodide reagent, which subsequently modulate the photo-induced charge transfer from the gCN surface to the C4F9–I molecule (vide infra).
It is to be noted that gCN samples prepared by direct thermo-condensation of precursors could form a melon-based structure, as reported previously.44 A melon-based carbon nitride principally represents a one-dimensional (1D) class wherein some of the heptazine sub-units form 1D polymer strands and the other heptazine sub-units align in a zigzag manner due to a hydrogen bonding network with the nitrogen atoms in the 1D strands, resulting in an interconnected network of heptazine units over a flat basal plane. The periodic and the molecular models considered in this work represent the two limit structures of a CN surface, namely, the periodic monolayer represents the result of the complete crosslinking of a melon model into a two-dimensional (2D) structure whereas the molecular model is the zero-dimensional (0D) representation of independent heptazine units, which are also present in the melon-like 1D structure. As both the defective periodic (2D) and molecular (0D) models show a similar electronic structure (see Fig. SI-4 and SI-5† for details), we believe that the conclusions drawn from this work could be extended to other gCN forms, in particular the melon-based 1D carbon nitride structures.
It is also of interest to analyze the computed spin-density distributions on various defective gCN forms. These systematically show larger spin densities on carbon atoms, along with smaller contributions on the nitrogen atoms (see the case of the am-gCN models in Fig. 4(1.a/b)). This result concurs with EPR experiments shown in Fig. 4c, which indicate that all the gCN structures (including pristine gCN) contain radical carbon species, with the radical concentration being the largest in the am-gCN form (4.5 ×1016 spins/g vs. 2.5 ×1016 in pristine gCN). Importantly, 14N 2D-EPR experiments (see ESI, section S1†) performed on am-gCN (Fig. 4d) provide evidence for a minute 14N hyperfine coupling (Amax ≈ 1 MHz), consistent with the delocalization of the unpaired electron wave function over a number of nitrogen atoms, which further confirms the relevance of the model systems chosen to represent the actual surface-active sites.
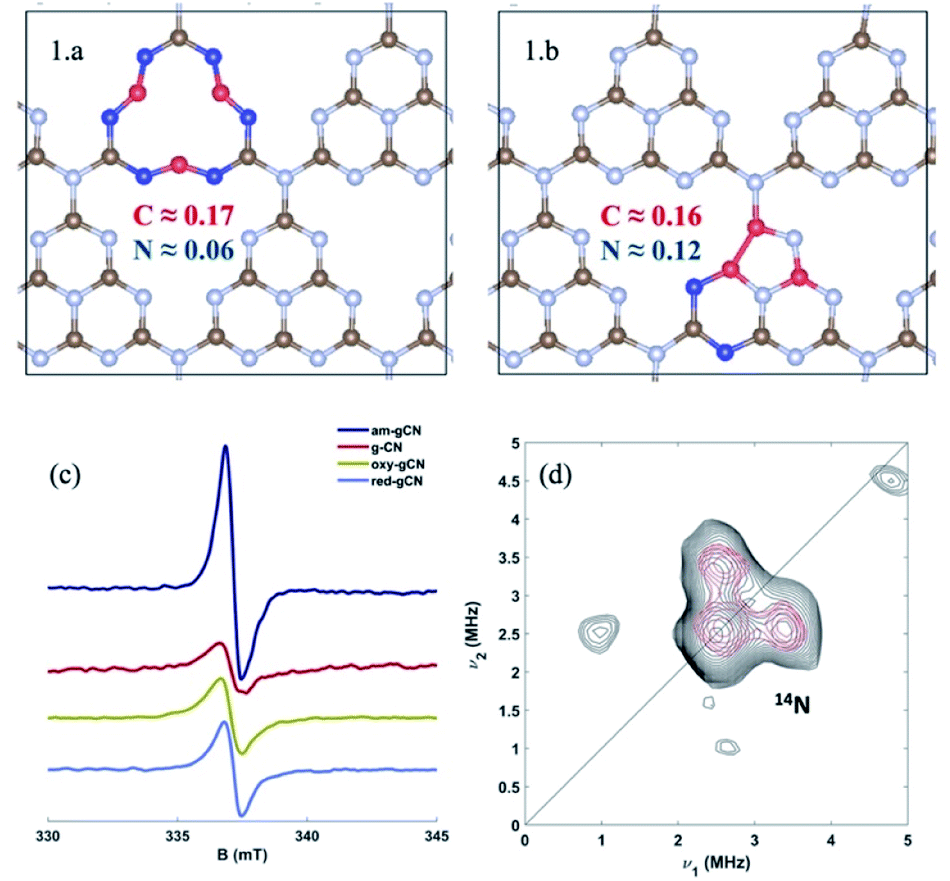 | ||
| Fig. 4 Atomic spin density distribution in the models for amorphous gCN (structures 1.a and 1.b), extracted using the DDEC6 methodology, and bottom: left (c) continuous wave X-band EPR spectra of the four gCN forms considered in this work recorded in the dark at 77 K; bottom: right (d) Q-band 14N HYSCORE spectrum of the amorphous gCN system (black experiment, red simulation). The spectrum was recorded at 110 K. Experimental details are shown in the ESI (Section S1†), along with the simulation parameters. | ||
In addition, the H2 evolution reaction by photo-reformation of ethanol in aqueous solution was performed using all four gCN derivatives, employing Pt as a co-catalyst to decrease the recombination rates of the photo-generated electrons and holes. Both red-CN and ox-CN forms achieve higher maximum H2 formation rates when compared to the g-CN or am-CN forms, confirming that specific tuning of the CN structure is instrumental for the specific catalytic application (see ESI, section S2† for details). Furthermore, electrochemical impedance spectroscopy (EIS) measurements were performed on the pristine and am-gCN forms, both in the dark and with a light source, to investigate the effect on the charge transfer mechanism at the electrode–electrolyte interface (see ESI, section S3† for details). According to the data summarized in Table SI-1,† a decrease in the charge transfer resistance (Rct) in the presence of a light source was found with respect to the dark condition for both am-CN and g-CN materials. The differences in the absolute Rct values can be attributed to differences in the homogeneity of the catalyst deposition on the GC support for the different materials. Nevertheless, the evolution of the Rct values clearly shows that the charge transfer at the electrode–electrolyte interface, already occurring under the dark condition, has been further improved in the presence of light, clearly confirming the photo-induced nature of the processes taking place at the CN-solution interface.
gCN/C4F9–I systems: adsorption energy and bond order analysis
To better quantify the interaction between pristine/defective gCN surfaces and the C4F9–I molecule and to get further insights, we calculated the adsorption energies between the model gCN surfaces and the C4F9–I molecule, along with estimations of the bond order (focusing on the iodine atom and the neighboring fluorine atoms) and the ground-state partial charge transfer between the partners, as shown in Table 2. Herein, we describe only the lowest-energy conformation of the C4F9–I molecule adsorbed on pristine/defective gCN surfaces, which corresponds to the long axis of the molecule being parallel to the basal plane of the gCN surface, as presented in Fig. 5 and SI-7.† The alternate conformation, wherein the long axis of the molecule is perpendicular to the gCN surface, is ≈2 kcal mol−1 higher in energy, irrespective of the gCN form.| Ref. | E Ads (kcal mol−1) | Bond order | Carbon–Iodine | Partial charge transfer (∂q/A) | |
|---|---|---|---|---|---|
| System | I | Bond length (Δd, Å) | |||
| P.0 | 6.1 | 1.28 | -na- | -na- | +0.03 |
| 1.a | 17.4 | 1.57 | 0.20 | 0.061 | −0.10 |
| 1.b | 11.2 | 1.38 | 0.05 | 0.051 | −0.07 |
| 2.a | 8.1 | 1.35 | 0.04 | 0.006 | −0.01 |
| 2.b | 7.0 | 1.21 | 0.01 | 0.004 | −0.01 |
| 3.a | 12.5 | 1.51 | 0.15 | 0.051 | −0.10 |
| 3.b | 11.4 | 1.35 | 0.05 | 0.008 | −0.03 |
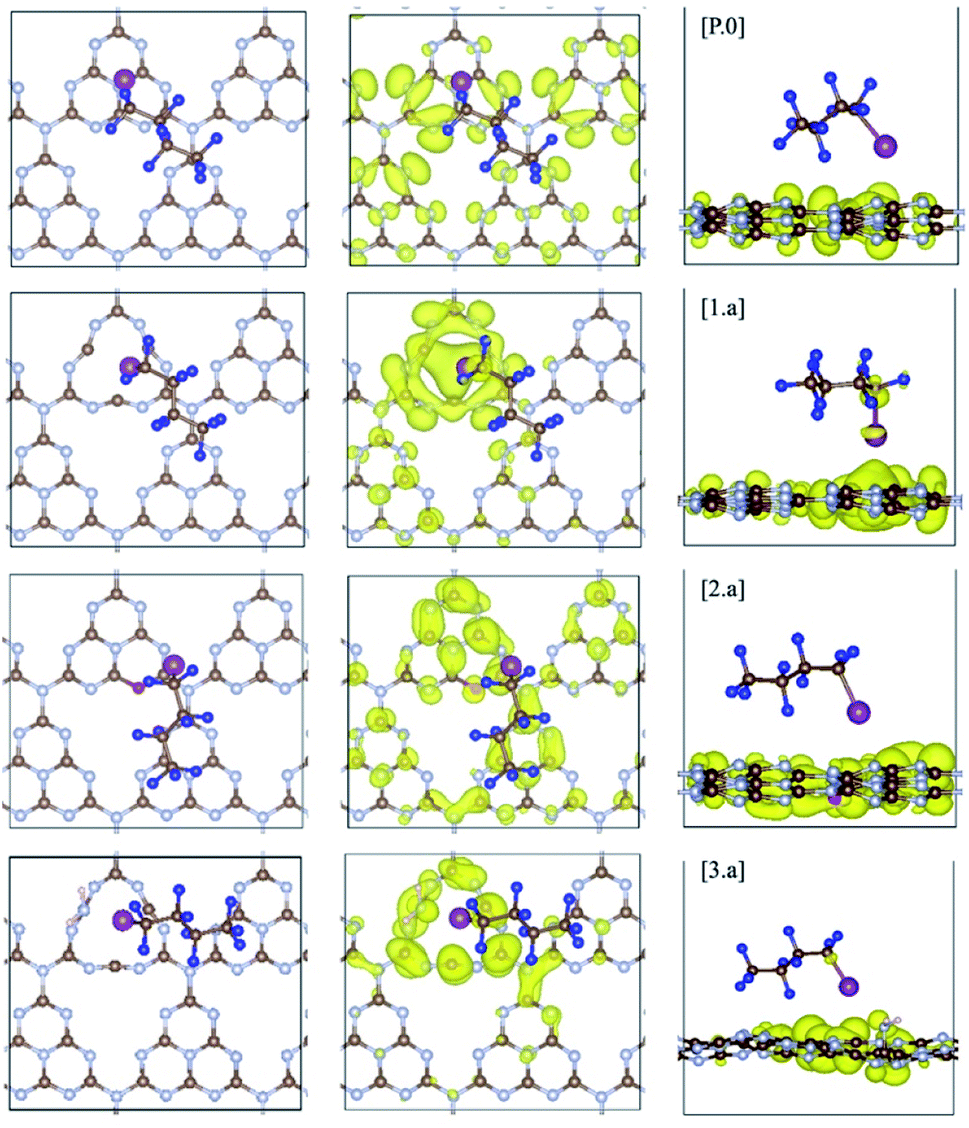 | ||
| Fig. 5 C4F9–I adsorbed on pristine (P.0) and defective gCN surfaces (am-gCN: 1.a, oxy-gCN: 2.a and red-gCN: 3.a), in their lowest-energy conformations wherein the long axis of the molecule is parallel to the basal plane of the given gCN surface (carbon: brown; nitrogen: blue; oxygen: red; hydrogen: pink, fluorine: navy and iodine: magenta). Wavefunction plots at the fermi level (highest-occupied electronic state) for gCN/C4F9–I systems are also reported for gCN surfaces in its (P.0) pristine and (1.a, 2.a and 3.a) defective forms. The wavefunction is found to be partially localized on the C4F9–I molecule for structures 1.a and 3.a. Corresponding data for gCN structures 1.b, 2.b and 3.b are shown in Fig. SI-6.† | ||
The adsorption energy of C4F9–I is ≈6 kcal mol−1 on the pristine gCN surface (structure P.0) but it has significantly higher values when radical defects are present. More specifically, for the am-gCN forms (structures 1.a and 1.b), the adsorption energy of C4F9–I ranges from ≈11 kcal mol−1 to ≈17 kcal mol−1, the highest value being found when a nitrogen vacancy is present in the middle of a heptazine unit (structure 1.a). In contrast, when oxygenated defects (pyran/carbonyl-like) are present on the gCN surface, the adsorption energies are reduced to ≈8 kcal mol−1. The adsorption energies of C4F9–I on the molecular gCN models are in good quantitative agreement with the periodic models (see Table SI-2†). This decrease in adsorption energy for oxygenated defects, despite the radical nature of these defects, could be attributed to the electrostatic repulsion between the oxygen and iodine atoms. In addition, the strong adsorption of C4F9–I on most red/am-gCN surfaces (structures 1.a/b and 3.a) is also associated with a: (i) non-negligible ground-state partial charge transfer from the gCN surface to the C4F9–I molecule (≈0.1q) and (ii) a small elongation of the carbon-iodine (C–I) bond of the C4F9–I molecule, of ≈0.06 Å.
Interestingly, the adsorption energies also reflect a variation in the net bond order of the iodine atom: the highest bond order on iodine is observed when the C4F9–I molecule is adsorbed on gCN surfaces containing nitrogen vacancies at the center of the heptazine unit (structures 1a and 3a, and Table 2). This is an indication of the stronger interaction of the iodine atom with the surface defects in these systems. In addition, a variation of net bond order is also observed on the two fluorine atoms that are in proximity to the iodine atom. The relative change in the bond order of these fluorine atoms for different defect types compared to pristine gCN is also the highest for the gCN surfaces containing a nitrogen vacancy at the center of the heptazine unit (see Table 2).
The matching between the increased adsorption energy of the C4F9–I molecule on defective surfaces representing the amorphous and reduced gCN and the relative variation in the bond orders of iodine and fluorine atoms correlates well with the variation in the experimental 19F-NMR T1/T2 ratios,21,29 with the highest T1/T2 ratios observed for the amorphous/reduced gCN forms. More specifically, these earlier studies,21,29 using perfluorohexyl iodide as a probe molecule, reported higher 19F-NMR T1/T2 ratios for am-gCN and red-gCN forms compared to pristine-/oxy-gCN forms, pointing to stronger molecule/surface interactions in the former systems. These strong interactions between the defective/modified gCN surface and the perfluorohexyl iodide molecule, were hypothetically ascribed to the formation of halogen bonding (N⋯I) between the freely accessible nitrogen (N) atoms from the defective amorphized gCN surfaces and the iodine(I) atom of the probe molecule.
To validate this hypothesis, we considered C4F9–I, C4F10 and C4H10 as probe molecules and computed their interactions on the pristine gCN surface (structure P.0) and on the am-gCN surface containing a nitrogen vacancy at the center of the heptazine unit (structure 1.a), as shown in the ESI (Fig. SI-8†). On the pristine gCN surface, all the three probe molecules show a similar adsorption energy in the order of ≈4.5 to 6 kcal mol−1, but on the am-gCN surface, their adsorption energies are larger, ranging from ≈11.5 kcal mol−1 to ≈17.5 kcal mol−1. Of the three probe molecules adsorbed on the am-gCN surface, C4F9–I has the largest adsorption energy (17.4 kcal mol−1) while both C4F10 and C4H10 molecules show comparatively smaller adsorption energies of 12.7 and 11.4 kcal mol−1, respectively. In addition, both C4F10 and C4H10 molecules adsorbed on am-gCN surfaces are characterized by a negligible partial charge transfer between the surface and molecule (≈0.01 ∂q/A) and no variations are observed: (i) in the bond orders of fluorine or hydrogen atoms; (ii) in the C–F/C–H bond lengths. In contrast, C4F9–I adsorbed on the am-gCN surface shows larger variations in terms of partial ground-state charge transfer (∂q/A), a relative change in the bond order of fluorine atoms (ΔF) and a relative change in the C–I bond length (Δd), as shown in Table 2, and consequently shows the highest adsorption energy, which is indicative of the strong interaction between the C4F9–I molecule and the defective gCN surface.
It therefore follows that the increased interaction between the defective gCN surface and C4F9–I does not appear to be related to a conventional halogen bonding between the freely available nitrogen atoms from the defective gCN surface and the iodine atom of the molecule, as hypothesized previously based on the increased experimental 19F-NMR T1/T2 ratios. Instead, the calculations indicate that it results from a more complex interaction mechanism involving a reorganization of the electronic density on the terminal iodine, which is characterized by: (i) a partial ground state charge transfer from the defective gCN surface to the molecule; (ii) an increase in the ground state C–I bond length; and (iii) a consequential increase (decrease) in the bond order (length) between carbon and fluorine atoms neighboring the iodine atom. These specific interactions between the C4F9–I molecule and the defects present on the gCN surface could indeed be manifested into an increase in 19F-NMR T1/T2 ratios, observed experimentally.
Electronic structure of the gCN/C4F9–I systems
The ground-state electronic structure of the gCN/C4F9–I systems is analyzed here with a specific focus on the energy separation between the highest-occupied crystalline states of gCN surfaces and the lowest unoccupied molecular states of C4F9–I, to get insights into the possibility of electron transfer between the partners upon photoexcitation. The electronic structure shown in Fig. 6 is extracted from the partial density of states (pDOS) obtained at the HSE06 level of theory, as shown in the ESI (Fig. SI-6 and SI-7†).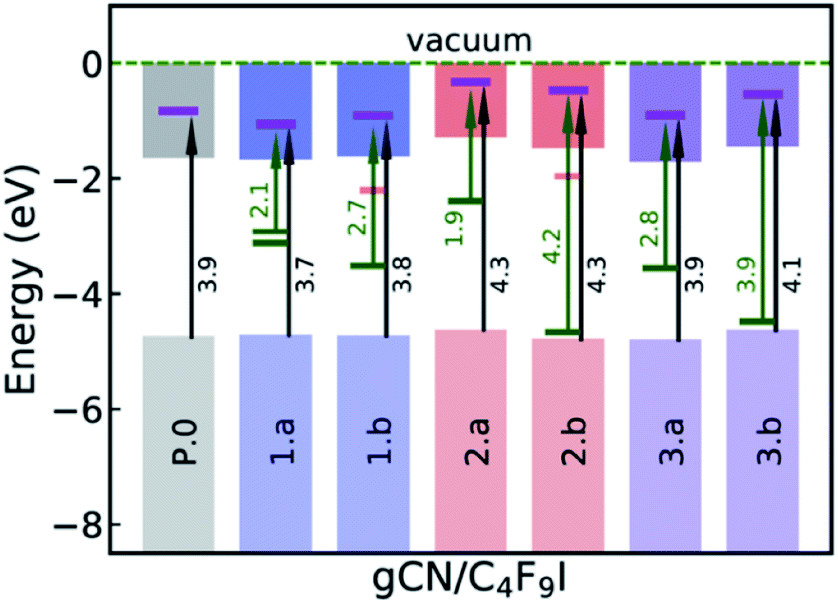 | ||
| Fig. 6 Summarized electronic structure of the gCN/C4F9–I systems. The occupied (unoccupied) localized mid-gap defect states within the electronic band gap are indicated with green (red) bars while the iodine-centered electronic energy levels of the C4F9–I molecule within the conduction band are indicated with a magenta bar. Green (black) arrows correspond to the energy difference between the occupied localized mid-gap state (valence band edge) and the iodine-centered electronic level. | ||
On the pristine gCN surface (P.0), the iodine-localized electronic level of the C4F9–I molecule lies deep in the conduction band of the system, at ≈3.9 eV from the VB edge (see Fig. 6). However, when radical defects are present on the gCN surface, gCN/C4F9–I systems show localized defect levels within the electronic band gap, similar to the description in Fig. 3. In the model systems for the amorphous gCN, structure 1.a, two mid-gap levels are positioned at ≈1.5 eV from the CB edge. The iodine centered C4F9–I electronic energy level within the conduction band is positioned at ≈2.1 eV (≈2.3 eV) from the singly (doubly) occupied defect state and at ≈3.7 eV from the VB edge. For structure 1.b, the singly occupied mid-gap level is positioned at ≈1.9 eV from the CB edge, while the iodine-centered C4F9–I electronic energy level within the conduction band is positioned at ≈2.7 eV from this defect state and ≈3.8 eV from the VB edge.
For defective surfaces modeling oxidized gCN, the pyran-like structure (2.a) shows a singly occupied defect level at ≈0.9 eV from the CB edge, which is ≈1.9 eV from the iodine-centered C4F9–I electronic energy level within the conduction band. In contrast, when a carbonyl-like defect (2.b) is present on the gCN surface, the singly occupied defect level is very close to the VB edge positioned at ≈4.2 eV from the iodine-centered C4F9–I electronic energy level. For both the oxy-gCN structures (2.a/b) the iodine centered C4F9–I electronic energy level within the conduction band is positioned at ≈4.3 eV from the VB edge. In the model systems representing reduced gCN, the localized defect state is ≈1.8 eV from the CB edge and is positioned at ≈2.8 eV from the iodine-centered electronic energy level in system 3a, whereas the corresponding values are ≈3.0 eV and ≈3.9 eV in system 3b.
Interestingly, in the amorphous and reduced systems showing mid-gap states (structures 1.a and 3.a), the wavefunction at the mid-gap defect level is partially localized on C4F9–I, as shown in Fig. 5. This indicates strong hybridization between the molecule and the defective surface (as also suggested by the ground-state partial electron transfer in Table 2), which is expected to ease a photoinduced electron transfer reaction from the substrate to the molecule. It is also to be noted that the adsorption energies of the C4F9–I molecule atop am-gCN (1.a) and red-gCN (3.a) surfaces, as well as the electronic structures of the gCN/C4F9–I systems at the localized mid-gap defect state, remain unaltered irrespective of the size of the replicated gCN surface unit cell (see ESI Fig. SI-9† for details). This further indicates that it is the localized radical nature of the defects on the gCN surface, rather than the density of defects or the closeness of the molecules, that governs the interaction of the gCN surface with the C4F9–I molecule.
Photo-physical properties of the gCN/C4F9–I systems
The excited-state electronic structure of the gCN/C4F9–I systems is analyzed by considering the representative heptazine trimers (see Fig. SI-2†). The theoretical UV-vis absorption spectra for the gCN/C4F9–I systems involving the pristine, am-gCN, and oxy-gCN model surfaces are shown in Fig. 7, while those involving the red-gCN models are shown in Fig. SI-14.† The calculated absorption profile of the pristine gCN/C4F9–I system shows a very intense peak with a maximum at ≈300 nm and a second band centered at ≈360 nm, along with a very weak tail extending into the visible domain. This second band can be related to the broad absorption peak in the 350–400 nm region in the experimental spectrum of pristine gCN (see Fig. 7, left panel).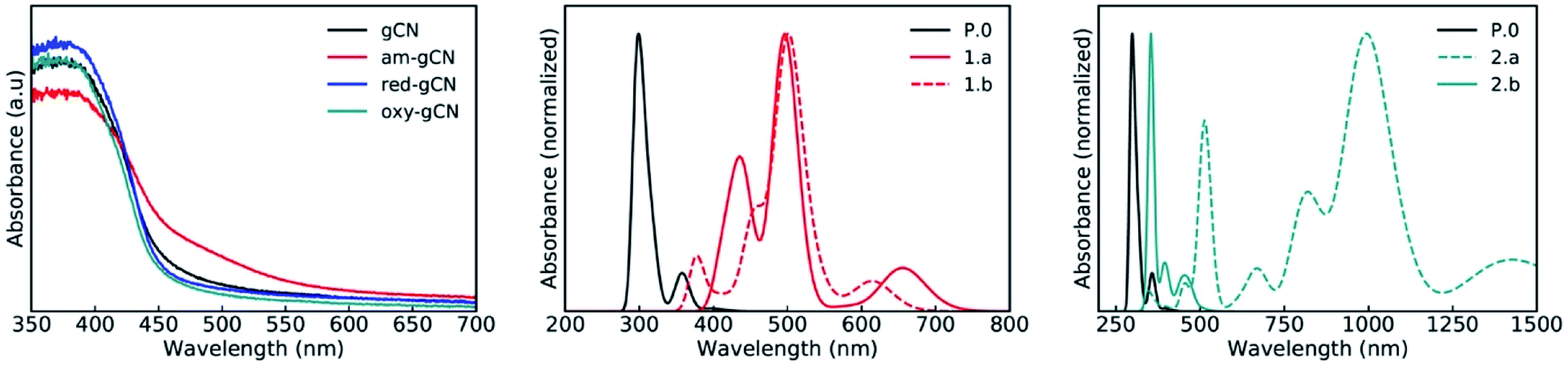 | ||
| Fig. 7 Experimental (left panel) and theoretical absorption spectra (normalized) of gCN/C4F9–I systems for the gCN surface in its pristine (black curves) and defective forms; with (middle panel) C4F9–I on top of the 1.a (full red line) and 1.b (dashed red line) gCN forms and (right) C4F9–I on top of the 2.a (dashed green line) and 2.b (full green line) gCN forms. | ||
A major change in the calculated optical properties occurs upon the formation of defects in amorphous gCN (structures 1.a and 1.b; Fig. 7, middle panel). Namely, new absorption features appear in the visible domain (an absorption band peak at ≈500 nm, with additional transitions at higher wavelengths). These changes in the optical absorption predicted by the modelling are in line with the appearance of a shoulder near 500 nm in the experimental absorption spectrum of amorphous gCN. However, it is to be noted that a direct comparison between the calculated and measured intensities should be considered with care, as the defect density considered in the calculations could be different from those present experimentally.
The optical absorption spectra of the oxygen-containing defects are quite contrasting (Fig. 7, right panel): on one hand, the presence of pyran-like oxygenated defects (structure 2.a) on the gCN surface is expected to red-shift the absorption very strongly, up to ≈1000 nm. On the other hand, the absorption of carbonyl-like oxygenated defects (structure 2.b) shows a peak centered at ≈350 nm and only a weak response in the visible domain, around 500 nm. The fact that the experimental spectrum of the oxidized gCN shows no intensity at low energy clearly indicates that pyran-like defects are not likely to be present in real samples.Also, the carbonyl-like oxygenated gCN structure is lower in energy by ≈0.6 eV, indicating that the carbonyl-like form is more stable in comparison. Probably, the low concentration of carbonyl defects and their low expected absorption explain the absence of any new features in the experimental spectrum of oxidized gCN.
To get further insight into the nature of the excited states, we performed the attachment-detachment density (ADD) analysis41,42 on all the pristine/defective gCN/C4F9–I systems, wherein the detachment density corresponds to the electron density associated with single electronic levels vacated in an electronic transition and the attachment density corresponds to the electron density associated with the single electron levels occupied in the transition. The difference between these densities is called the difference density matrix; it is shown in Fig. 8, wherein the cyan color represents the electronic density of the vacated transition electronic level (detachment density) and the yellow color represents the electronic density of the occupied transition electronic level (attachment density). We also performed the complementary natural transition orbital (NTO) analysis on all the pristine/defective gCN/C4F9–I systems and the representative lowest unoccupied transition orbitals (LUTOs) are shown in the ESI (see Fig. SI-12 to SI-14† for details).
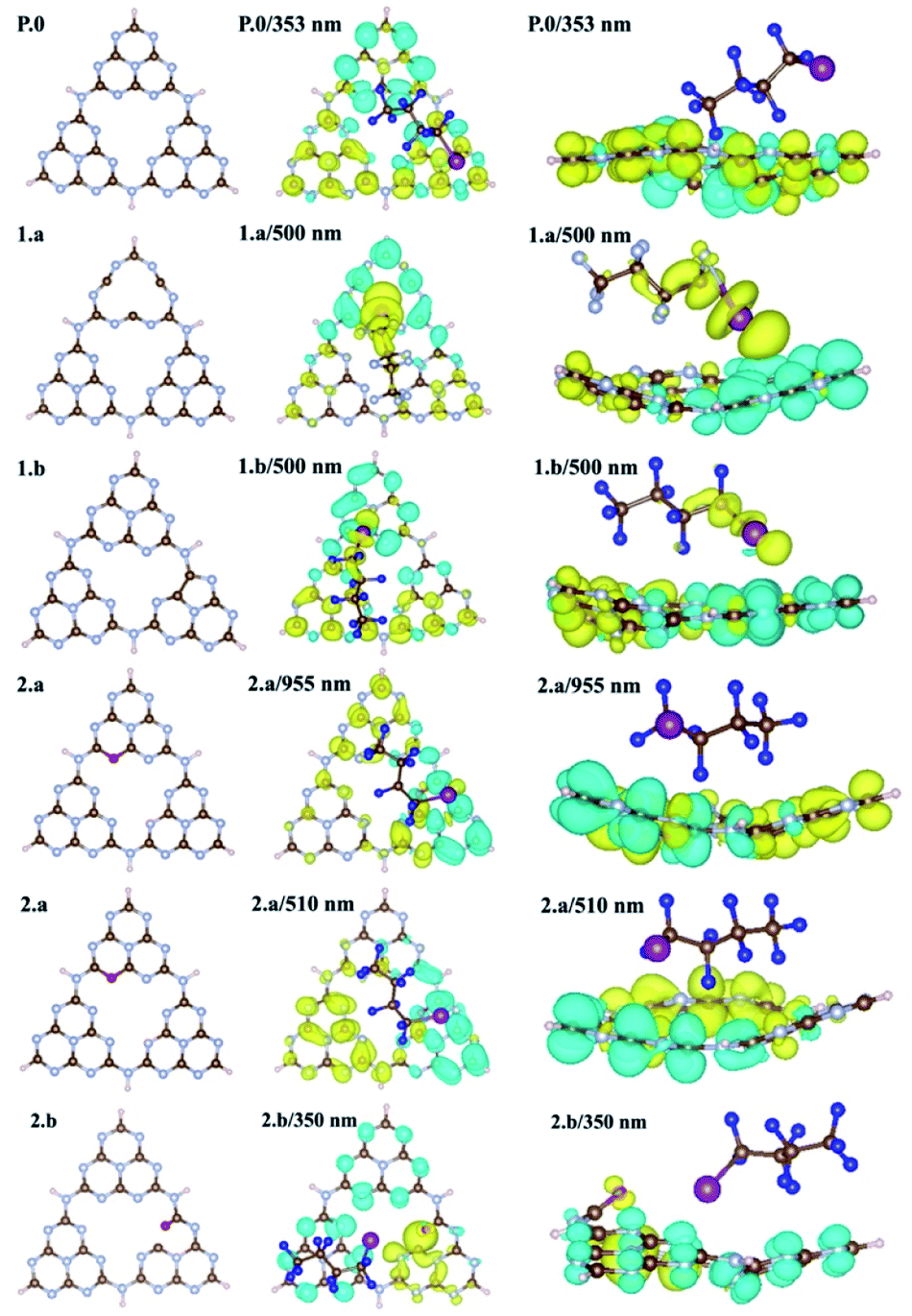 | ||
| Fig. 8 Top (center) and side (right) views of the attachment-detachment density (ADD) differences of gCN/C4F9–I systems for the gCN surface in its pristine (P.0) and amorphous forms (1.a/b) and pyran-like and carbonyl-like oxygenated forms (2.a/b). Iodine and fluorine atoms of the C4F9–I molecule adsorbed on pristine/defective gCN units are represented with magenta and blue colors, for clarity. The attachment density is represented in yellow and the detachment density in cyan. | ||
In the case of pristine gCN, the ADD difference corresponding to low-energy excitations (≈350 nm) is confined on the semiconductor with both the attachment (in yellow) and detachment (in cyan) transition densities observed only on the surface. Only with excitation at high energy (≈300 nm) the transition density partly spreads over the C4F9–I molecule (see Fig. SI-13†). Interestingly, for the models representing the amorphous gCN surface (structures 1.a/b), the ADD difference calculated at 500 nm now shows major contributions from both the gCN surface and the C4F9–I molecule. While the detachment density is on the am-gCN surface, the attachment density is delocalized on the C4F9–I molecule, in particular on the iodine atom (with a dz2-like character). Such transitions are prompted by the molecule-gCN hybridization described above and involve a direct electron excitation from the localized defect state of gCN to the C4F9–I molecule.
In contrast, the oxygenated gCN forms do not show any electronic transition involving the unoccupied C4F9–I molecular orbital as landing levels (Fig. 8, structures 2.a/b) but feature electron transitions occurring exclusively within the gCN surface even for the high-energy excitations (≈350 nm), irrespective of the pyran-like or carbonyl-like oxygenated system. In comparison, for models representing the reduced gCN surface (structures 3.a and 3.b), a small delocalization of transition density is observed on the C4F9–I molecule adsorbed on structure 3.a at the lowest energy excitation wavelength of ≈500 nm, whereas for structure 3.b, the transition density is fully localized only on the gCN surface (see Fig. SI-14† for details).
These results convey the following message: (i) band-edge optical absorption in pristine gCN is not expected to induce an electron transfer to the C4F9–I molecule; (ii) oxygenated defects on gCN surfaces tend to suppress any photo-induced electron transfer from the gCN surface to C4F9–I; and (iii) optical absorption in the visible domain for amorphous and reduced gCN forms promotes a photo-induced electron transfer reaction from the semiconductor to the C4F9–I molecule, thus likely prompting the initial reaction step in the addition of perfluoroalkyl groups to aromatic compounds.
To further corroborate the possibility of electron transfer from the pristine and defective gCN surfaces to the C4F9–I molecule, we calculated the reaction enthalpies (ΔH) considering a schematic ground-state electron transfer from the pristine/defective gCN surface to the C4F9–I molecule: C4F9–I + gCN → C4F9–I− + gCN+ (see Fig. SI-15† for details). Electron transfer to the molecule from the amorphous and reduced gCN forms (structures 1.a and 3.a) is associated with a much lower ΔH value of ≈25 kcal mol−1, compared to that of the pristine gCN form (ΔH of ≈90 kcal mol−1, ESI Table SI-3† for details). Implicitly, it follows that a photo-induced electron transfer from the am-/red-gCN surface to the C4F9–I molecule is favorable on excitations in the visible domain, consistent with the electronic and excited energy levels shown in Fig. 6 and 8, whereas the electron transfer would happen at much higher energies when the pristine gCN surface is considered.
Very interestingly, upon electron transfer from the gCN surface, the C4F9–I molecule undergoes an elongation of the C–I bond length by ≈1 Å, with ≈80% of the charge localized on the iodine atom, while the unpaired electron is strongly localized (≈65%) on the neighboring carbon atom (see Fig. SI-16†). This is fully consistent with the mechanism proposed for gCN-assisted perfluoroalkylation of electron-rich aromatic compounds, in which the photoexcitation is thought to generate a perfluoroalkyl radical along with the release of iodide.17 Importantly, the modelling results suggest that the photo-induced electron transfer and C–I bond breaking reactions occur in a concerted way. We further note that the amorphous gCN forms considered in this work show an atomic spin density that is centered on the carbon atoms along with some delocalization on nitrogen atoms, consistent with the EPR results shown in Fig. 4.
These computational results, when compared to the experimental observations in terms of the enhanced photo-catalytic response of am-gCN and red-gCN surfaces, combined with their lower nitrogen contents recorded from the XPS data and higher radical densities observed from the EPR measurements, consistently and strongly imply that specific amorphous/reductive treatments of gCN promotes the formation of more nitrogen vacancies on the gCN surface. These nitrogen vacancies not only favor the adsorption of the C4F9–I molecule at the defective sites but also promote the photo-induced electron transfer from the radical defect states present on the defective gCN surfaces to the C4F9–I molecule, which happens at lower excitation energies (at ≈500 nm), and consequently fully rationalize the superior photo-catalytic response of amorphous and reduced gCN surfaces.
Conclusions
In conclusion, we employ comprehensive density functional theory (DFT) calculations, using both periodic and molecular approaches, to investigate the nature of radical defects present on the gCN surfaces, in order to provide a detailed structure/photo-physical property relationship between the nature of these defects, their electronic structures, their interactions with a perfluoroalkyl iodide compound in the ground- and excited states and the resulting photo-catalytic efficiencies of the corresponding materials measured experimentally for the perfluoroalkylation of electron-rich organic substrates. It was found that, when radical defect states are created on gCN samples, either by amorphization or reductive or oxidative treatments, electronic defect states appear within the electronic band gap. But only amorphous/reduced gCN forms containing nitrogen vacancies act as strong binding sites for the perfluoroalkyl iodide molecule, via partial ground-state charge transfer from these defect states to the substrate molecule and the associated change in bond orders. These defect states are also photo-active in the visible domain: upon photo-excitation electron transfer occurs from these defect states to the C4F9–I molecule, which is the crucial, initial step in the addition of perfluoroalkyl groups to aromatic compounds. The presence of photo-active defect states within the electronic band gap can therefore directly enhance the resulting photo-catalytic efficiencies of defective gCN samples. This study, therefore, shows the potential of determining the nature and properties of electronic defects in gCN materials for the rational design of gCN-based photocatalysts for other organic substrates.Data availability
Data for this paper, including the input and output files of the calculations, are available at the UMONS Institutional Repository (https://orbi.umons.ac.be).Author contributions
E. R. and S. M. G. performed all the quantum chemical calculations. G. F synthesized and characterized the experimental gCN samples, under the supervision of M. M and P. F. A. A performed the EPR measurements under the supervision of M. C. M. B. performed the EIS measurements. M. P., D. B., and R. L., conceived and designed the study. All authors discussed the results and contributed to the interpretation of the data as well as to the writing and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Energy Transition Fund of the Belgian Federal Government (FPS Economy) within the T-REX project, the Belgian National Fund for Scientific Research (FRS-FNRS) within the Consortium des Équipements de Calcul Intensif – CÉCI, under Grant 2.5020.11, and by the Walloon Region (ZENOBE Tier-1 supercomputer, under grant 1117545). E. R. is grateful to the University of Trieste and the University of Mons for a joint doctoral fellowship. M. M. and G. F. kindly acknowledge FRA2021 funded by the University of Trieste. G. F. also acknowledges Microgrants 2021 funded by Region FVG (LR 2/2011, ART. 4). This work was supported by the University of Trieste, INSTM, and the Italian Ministry of Education MIUR (cofin Prot. 2017PBXPN4). Part of this work was performed under the Maria de Maeztu Units of Excellence Program from the Spanish State Research Agency Grant No. MDM-2017-0720. We acknowledge Prof. E. Salvadori for useful discussions on the EPR results and Dr G. Londi for his assistance in the attachment/detachment density analysis. D. B. is Research Director of FNRS. M. P. is the AXA Chair for Bionanotechnology (2016–2023).Notes and references
- C. Rosso, G. Filippini, A. Criado, M. Melchionna, P. Fornasiero and M. Prato, ACS Nano, 2021, 15, 3621–3630 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS PubMed.
- J. Zhang, Y. Chen and X. Wang, Energy Environ. Sci., 2015, 8, 3092–3108 RSC.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS.
- J. Liu, H. Wang and M. Antonietti, Chem. Soc. Rev., 2016, 45, 2308–2326 RSC.
- X. Ye, Y. Cui, X. Qiu and X. Wang, Appl. Catal., B, 2014, 152–153, 383–389 CrossRef CAS.
- X. Chen, J. Zhang, X. Fu, M. Antonietti and X. Wang, J. Am. Chem. Soc., 2009, 131, 11658–11659 CrossRef CAS PubMed.
- L. Möhlmann, M. Baar, J. Rieß, M. Antonietti, X. Wang and S. Blechert, Adv. Synth. Catal., 2012, 354, 1909–1913 CrossRef.
- C. Cavedon, A. Madani, P. H. Seeberger and B. Pieber, Org. Lett., 2019, 21, 5331–5334 CrossRef CAS PubMed.
- B. Pieber, J. A. Malik, C. Cavedon, S. Gisbertz, A. Savateev, D. Cruz, T. Heil, G. Zhang and P. H. Seeberger, Angew. Chem., Int. Ed., 2019, 58, 9575–9580 CrossRef CAS.
- I. Ghosh, J. Khamrai, A. Savateev, N. Shlapakov, M. Antonietti and B. König, Science, 2019, 365, 360–366 CrossRef CAS PubMed.
- K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS PubMed.
- A. Savateev, I. Ghosh, B. König and M. Antonietti, Angew. Chem., Int. Ed., 2018, 57, 15936–15947 CrossRef CAS PubMed.
- Y. Zhang, A. Thomas, M. Antonietti and X. Wang, J. Am. Chem. Soc., 2009, 131, 50–51 CrossRef CAS.
- H. Pan, Y.-W. Zhang, V. B. Shenoy and H. Gao, ACS Catal., 2011, 1, 99–104 CrossRef CAS.
- X. Wang, X. Chen, A. Thomas, X. Fu and M. Antonietti, Adv. Mater., 2009, 21, 1609–1612 CrossRef CAS.
- C. Wang, Q. Wan, J. Cheng, S. Lin, A. Savateev, M. Antonietti and X. Wang, J. Catal., 2021, 393, 116–125 CrossRef CAS.
- C. Wang, Y. Hou, J. Cheng, M.-J. Lin and X. Wang, Appl. Catal., B, 2021, 294, 120259 CrossRef CAS.
- G. Filippini, F. Longobardo, L. Forster, A. Criado, G. Di Carmine, L. Nasi, C. D'Agostino, M. Melchionna, P. Fornasiero and M. Prato, Sci. Adv., 2020, 6, eabc9923 CrossRef CAS PubMed.
- R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496 RSC.
- C.-J. Wallentin, J. D. Nguyen, P. Finkbeiner and C. R. J. Stephenson, J. Am. Chem. Soc., 2012, 134, 8875–8884 CrossRef CAS PubMed.
- S. Barata-Vallejo, M. V. Cooke and A. Postigo, ACS Catal., 2018, 8, 7287–7307 CrossRef CAS.
- N. Robinson, C. Robertson, L. F. Gladden, S. J. Jenkins and C. D'Agostino, ChemPhysChem, 2018, 19, 2472–2479 CrossRef CAS PubMed.
- P. Politzer, P. Lane, M. C. Concha, Y. Ma and J. S. Murray, J. Mol. Model., 2007, 13, 305–311 CrossRef CAS PubMed.
- P. Niu, L.-C. Yin, Y.-Q. Yang, G. Liu and H.-M. Cheng, Adv. Mater., 2014, 26, 8046–8052 CrossRef CAS PubMed.
- Y. Kang, Y. Yang, L.-C. Yin, X. Kang, G. Liu and H.-M. Cheng, Adv. Mater., 2015, 27, 4572–4577 CrossRef CAS PubMed.
- F. Longobardo, G. Gentile, A. Criado, A. Actis, S. Colussi, V. Dal Santo, M. Chiesa, G. Filippini, P. Fornasiero, M. Prato and M. Melchionna, Mater. Chem. Front., 2021, 5, 7267–7275 RSC.
- T. Suter, V. Brázdová, K. McColl, T. S. Miller, H. Nagashima, E. Salvadori, A. Sella, C. A. Howard, C. W. M. Kay, F. Corà and P. F. McMillan, J. Phys. Chem. C, 2018, 122, 25183–25194 CrossRef CAS.
- A. Ugolotti and C. Di Valentin, Nanomaterials, 2021, 11, 1863 CrossRef CAS PubMed.
- X. Song, X. Li, X. Zhang, Y. Wu, C. Ma, P. Huo and Y. Yan, Appl. Catal., B, 2020, 268, 118736 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 224106 CrossRef PubMed.
- N. G. Limas and T. A. Manz, RSC Adv., 2018, 8, 2678–2707 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. a. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. a. Petersson, H. Nakatsuji, X. Li, M. Caricato, a. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, a. F. Izmaylov, J. L. Sonnenberg, D.-Y. Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. a. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. a. Keith, R. Kobayashi, J. Normand, K. Raghavachari, a. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, 2016, Gaussian 16, Revision C.01, Gaussian, Inc., Wallin Search PubMed.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
- T. Etienne, X. Assfeld and A. Monari, J. Chem. Theory Comput., 2014, 10, 3906–3914 CrossRef CAS.
- A. J. Gillett, C. Tonnelé, G. Londi, G. Ricci, M. Catherin, D. M. L. Unson, D. Casanova, F. Castet, Y. Olivier, W. M. Chen, E. Zaborova, E. W. Evans, B. H. Drummond, P. J. Conaghan, L.-S. Cui, N. C. Greenham, Y. Puttisong, F. Fages, D. Beljonne and R. H. Friend, Nat. Commun., 2021, 12, 6640 CrossRef CAS.
- O. Faye, U. Eduok and J. A. Szpunar, J. Phys. Chem. C, 2019, 123, 29513–29523 CrossRef CAS.
- F. K. Kessler, Y. Zheng, D. Schwarz, C. Merschjann, W. Schnick, X. Wang and M. J. Bojdys, Nat. Rev. Mater., 2017, 2, 17030 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Protocols for the EPR, EIS and H2 evolution measurements; molecular models for defective gCNs; electronic structure of defective gCNs; electronic structure and adsorption energies of C4F9–I on defective gCNs; NTO analysis of C4F9–I/gCN systems; CIF files for the defective gCN structures. See https://doi.org/10.1039/d2sc03964h |
| This journal is © The Royal Society of Chemistry 2022 |