
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEndowing matrix-free carbon dots with color-tunable ultralong phosphorescence by self-doping†
Huixian
Shi‡
*a,
Zuoji
Niu‡
a,
He
Wang
b,
Wenpeng
Ye
b,
Kai
Xi
 c,
Xiao
Huang
b,
Hongliang
Wang
d,
Yanfeng
Liu
c,
Hengwei
Lin
e,
Huifang
Shi
*b and
Zhongfu
An
*b
c,
Xiao
Huang
b,
Hongliang
Wang
d,
Yanfeng
Liu
c,
Hengwei
Lin
e,
Huifang
Shi
*b and
Zhongfu
An
*b
aCollege of Materials Science and Engineering, Taiyuan University of Technology, Taiyuan 030024, China. E-mail: shihuixian@tyut.edu.cn
bKey Laboratory of Flexible Electronics (KLoFE), Institute of Advanced Materials (IAM), Nanjing Tech University, 30 South Puzhu Road, Nanjing 211800, China. E-mail: iamhfshi@njtech.edu.cn; iamzfan@njtech.edu.cn
cSchool of Chemistry and Chemical Engineering, Nanjing University, Nanjing, 210023, China
dCollaborative Innovation Center for Molecular Imaging of Precision Medicine, Shanxi Medical University, Taiyuan, 030001, China
eInternational Joint Research Center for Photo-responsive Molecules and Materials, School of Chemical and Material Engineering, Jiangnan University, Wuxi, 214122, China
First published on 30th March 2022
Abstract
Color-tunable ultralong phosphorescence is urgently desired in optoelectronic applications. Herein, we report a new type of full-color-tunable ultralong phosphorescence carbon dots (CDs) without matrix-assistance by a self-doping method under ambient conditions. The phosphorescence color can be rationally tuned from blue to red by changing the excitation wavelength from 310 to 440 nm. The CDs exhibit an ultralong lifetime of up to 1052.23 ms at 484 nm. From the experimental data, we speculate that the excitation-dependent multi-color phosphorescence is attributed to the presence of multiple emitting centers related to carbonyl units. Given the unique color-tunability of CDs, we demonstrate their potential applications in information encryption, light detection ranging from UV to visible light and LED devices. This finding not only takes a step towards the fundamental design of full-color emissive materials, but also provides a broader scope for the applications of phosphorescent materials.
Introduction
Room temperature phosphorescence (RTP) has attracted considerable attention in lighting, sensing, and anti-counterfeiting owing to its long-lived emission lifetime and large Stokes shift characteristics.1–5 However, it is very challenging to realize phosphorescence in metal-free organic phosphors because of the spin-forbidden radiative and fast nonradioactive transitions at room temperature. During the past few years, great efforts have been devoted to achieving RTP following two ways: (1) accelerating intersystem crossing (ISC) between singlet and triplet excited states by introducing heavy atoms, heteroatoms, carbonyl groups, or other substituents,6–10 and (2) suppressing the nonradiative transitions for phosphorescence enhancement by constructing a rigid molecular environment.6,11,12 So far, diverse metal-free phosphorescence materials with colourful emission, including small molecular crystals, amorphous polymers, metal–organic frameworks, etc., have been developed via a series of feasible approaches, such as crystal engineering,13,14 host–guest doping,15 polymerization16,17 and so forth. Under the stimulation of ultraviolet light, most phosphorescent materials normally show single emission. Compared with traditional mono-color materials, materials with multi-color emission have superior optical performance to meet the growing demand for advanced optoelectronic applications, such as high-capacity data storage,18 three-dimensional display,19 multiplexed bioimaging,20 and so on. Therefore, it is of great significance to develop phosphorescence materials with multi-color emission.Carbon dots (CDs), a type of zero-dimensional nano-material with excellent optical properties, biocompatibility and low toxicity, have received increasing attention in many fields.21–23 Specifically, due to their unique property of excitation-dependent photoluminescence (PL), CDs easily achieve color-tunable emission in the visible region by changing the excitation wavelength.24 However, this excitation-dependent multi-color emission is hardly realized for phosphorescence. In small molecule and polymer systems, we found that the construction of multi-emission centers is essential to realize color-tunable organic phosphorescence.13,14 Besides, it is worth noting that doping is a concise and effective method to modulate the photophysical and photoelectronic properties of materials. For instance, the electrical conductivity and work function values can be greatly improved by electrical p-doping in organic semiconductors.25 The efficiency of RTP can be significantly boosted by host–guest doping.26 N-doping endows carbon materials with novel catalytic performance.27 Thus, we speculate that the introduction of the self-doping method is beneficial to control and improve the phosphorescence properties of CDs.
Results and discussion
To verify our hypothesis, biuret with rich carbonyl groups was selected as an ideal phosphorescent carbon dot precursor because it is easily decomposed at high temperatures to form various by-products such as urea and 1,3,5-triazinane-2,4,6-trione (CYAD),28 making the construction of multiple emission centers for color-tunable phosphorescence possible by self-doping. Besides, the carbonyl groups can facilitate the spin–orbit coupling to populate the triplet excitons.6,29 Here we synthesized a new type of CDs, namely FP-CDs, via a one-pot hydrothermal reaction of biuret and phosphoric acid aqueous solution (Scheme 1). As anticipated, FP-CDs show color-tunable phosphorescence without any treatment in the solid state, ranging from blue to red with the excitation wavelength changed from 310 to 440 nm. Further research showed that during the hydrothermal reaction, biuret was decomposed to form urea and CYAD together with biuret to form various chromophores. Phosphoric acid served as both a catalyst and a crosslinking agent to boost dehydration, carbonization and polymerization of biuret for the construction of a rigid chromophore environment by cross-linking.30,31 Finally, full-color-tunable ultralong phosphorescence is realized. Given the unique optical performance, FP-CDs were successfully applied in optical potential applications of anti-counterfeiting, UV-visible light detection and LED devices.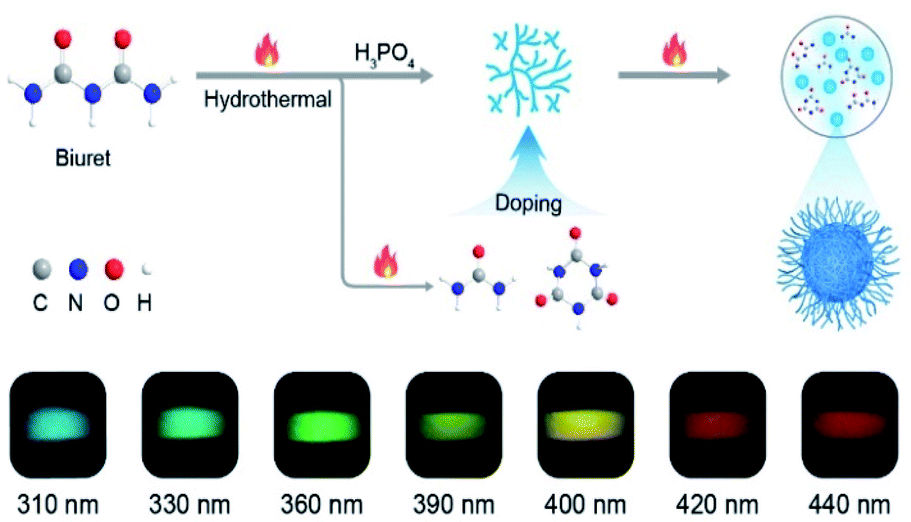 | ||
| Scheme 1 Schematic illustration of the formation of CDs and the process of self-doping for full-color-tunable phosphorescence in CDs. | ||
First, the morphology and chemical compositions of the as-prepared CDs were characterized by transmission electron microscopy (TEM) and Fourier transform infrared spectroscopy (FT-IR). As shown in Fig. 1a and b, the CDs exhibit well-dispersed spherical shapes with an average size of about 2.4 nm. Moreover, a high-resolution TEM image shows that the CDs had well-resolved lattice fringes with a lattice spacing of 0.21 nm (Fig. 1 inset), which is ascribed to the (100) plane of graphite.32 Therefore, we concluded that the core of the FP-CDs was the graphitic structure. As shown in the IR spectrum in Fig. 1c, the FP-CDs display distinguishable peaks at 3212 and 3051 cm−1, resulting from the stretching vibrations of the –NH2 and –OH groups.30 The strong absorption peaks at 1754 and 1718 cm−1 are assigned to the stretching vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and CO, respectively.33,34 The absorption peaks at 1460, 1396, 1053 and 846 cm−1 could be attributed to the stretching vibrations of PO, C–N, C–O/P–O, and P–N, respectively. Finally, the peak at 536 cm−1 implies the presence of phosphate salts.33,34
N and CO, respectively.33,34 The absorption peaks at 1460, 1396, 1053 and 846 cm−1 could be attributed to the stretching vibrations of PO, C–N, C–O/P–O, and P–N, respectively. Finally, the peak at 536 cm−1 implies the presence of phosphate salts.33,34
 | ||
| Fig. 1 Structural characterization of carbon dots. (a) TEM image. (b) Size distribution. (c) FT-IR spectrum. (d) XPS spectrum. The high-resolution XPS fitting results for the C 1s (e) and O 1s spectra (f) of the CDs, respectively. | ||
To further confirm the above FT-IR analyses, X-ray photoelectron spectroscopy (XPS) of FP-CDs was conducted. The FP-CDs mainly contain C, N, O and P elements with compositions of 40.84, 26.53, 30.90 and 1.73%, respectively (Fig. 1d). As shown in Fig. 1e, the high-resolution XPS spectrum of C 1s indicates the presence of CO (289.5 eV), C–N/C–O (286.4 eV) and C–C (284.6 eV) bonds in the FP-CDs.35 Notably, it was found that the ratio of carbonyl was much higher than that in previous studies.36–38 The O 1s spectrum also shows a main peak at 531.8 eV for CO, further demonstrating the abundance of CO on the FP-CDs surface. The other two peaks are attributed to PO at 530.8 eV and C–O at 532.8 eV, respectively (Fig. 1f).39 The N 1s spectrum contains three components that can be assigned to C–NC (399.8 eV), N–(C)3 (400.5 eV) and N–H (401.3 eV), respectively (Fig. S1a†).35 And the P 2p spectrum is divided into two peaks at 133.5 and 134.1 eV, attributed to P–O and P–N bonds, respectively (Fig. S1b†).30 Taking the FT-IR and XPS results together, we concluded that there might exist a large number of CO and heteroatom-containing functional groups on the surface of CDs, which could populate triplet excitons by promotion of the ISC process and suppressing non-radiative transitions with the assistance of multiple hydrogen bonds for boosting phosphorescence simultaneously.30,31
Subsequently, the photophysical properties of FP-CDs in the solid state were systematically investigated. As shown in Fig. S3,† the absorption spectrum of FP-CDs shows two bands at around 220 and 365 nm, which could be attributed to the π–π* transition of the CC units and the n–π* transition of the CO groups, respectively.38,40 The FP-CD powders exhibit similar blue emission under 310 and 365 nm ultraviolet (UV)-light excitation. Accordingly, the photoluminescence (PL) spectra revealed that the emission peaks shifted slightly with the excitation wavelength change (Fig. S4a†). Compared with the excitation-dependent PL emission in solution (Fig. S4b†), the related emission of FP-CDs shows some degree of redshift in the solid state, indicating that there might exist aggregation induced self-quenching caused by excessive resonance energy transfer.33
Astonishingly, after removing the excitation sources of 310 nm and 365 nm UV lamps, the FP-CD powders show a distinguishing blue and green afterglow under ambient conditions, respectively. To explore this unique emission behavior, we measured the phosphorescence spectra of FP-CDs at different excitation wavelengths (Fig. 2a–c). As shown in Fig. 2c, the excitation-phosphorescence spectra show the strongest phosphorescence emission of FP-CDs with a peak at 484 nm under 310 nm excitation. With the change of the excitation wavelengths from 310 to 440 nm, the ultralong phosphorescence shows a significant bathochromic shift from blue to red with the main peaks changing from 484 to 633 nm. From Fig. S5,† it is easily found that the phosphorescent colors gradually changed from blue to red spanning the entire visible region. And the corresponding Commission International de l'Eclairage (CIE) coordinates of the spectra can visualize the color-tunable ultralong phosphorescence more directly (Fig. 2b). To the best of our knowledge, such full-color-tunable phosphorescence has rarely been reported in CD materials previously.41–43 From the analysis of the time-resolved emission spectra (Fig. 2d), it was found that FP-CDs showed the longest lifetime of 1229.39 ms under the excitation at 360 nm under ambient conditions. And the emission peak at 633 nm exhibits red ultralong phosphorescence with an ultralong lifetime of up to 256.24 ms. To further confirm the nature of phosphorescence, the effects of temperature and oxygen on phosphorescence were studied (Fig. 2e, f and S6†). With an increase in the temperature from 183 to 423 K, the emission intensity showed a significant decrease at different excitation wavelengths, owing to the enhancement of nonradioactive transitions caused by thermal activation. It is worth noting that the FP-CDs have excellent thermal stability with a decomposition temperature of 568 K (Fig. S2†). Similarly, after being exposed to oxygen, the intensity of phosphorescence spectra of FP-CDs has a certain decrease (Fig. 2f), indicating its phosphorescence nature.44 Additionally, we found that the phosphorescence showed a slight decrease under a high humidity atmosphere over a period of time (Fig. S7†), indicating that moisture had little influence on the phosphorescence of FP-CDs in the solid state.
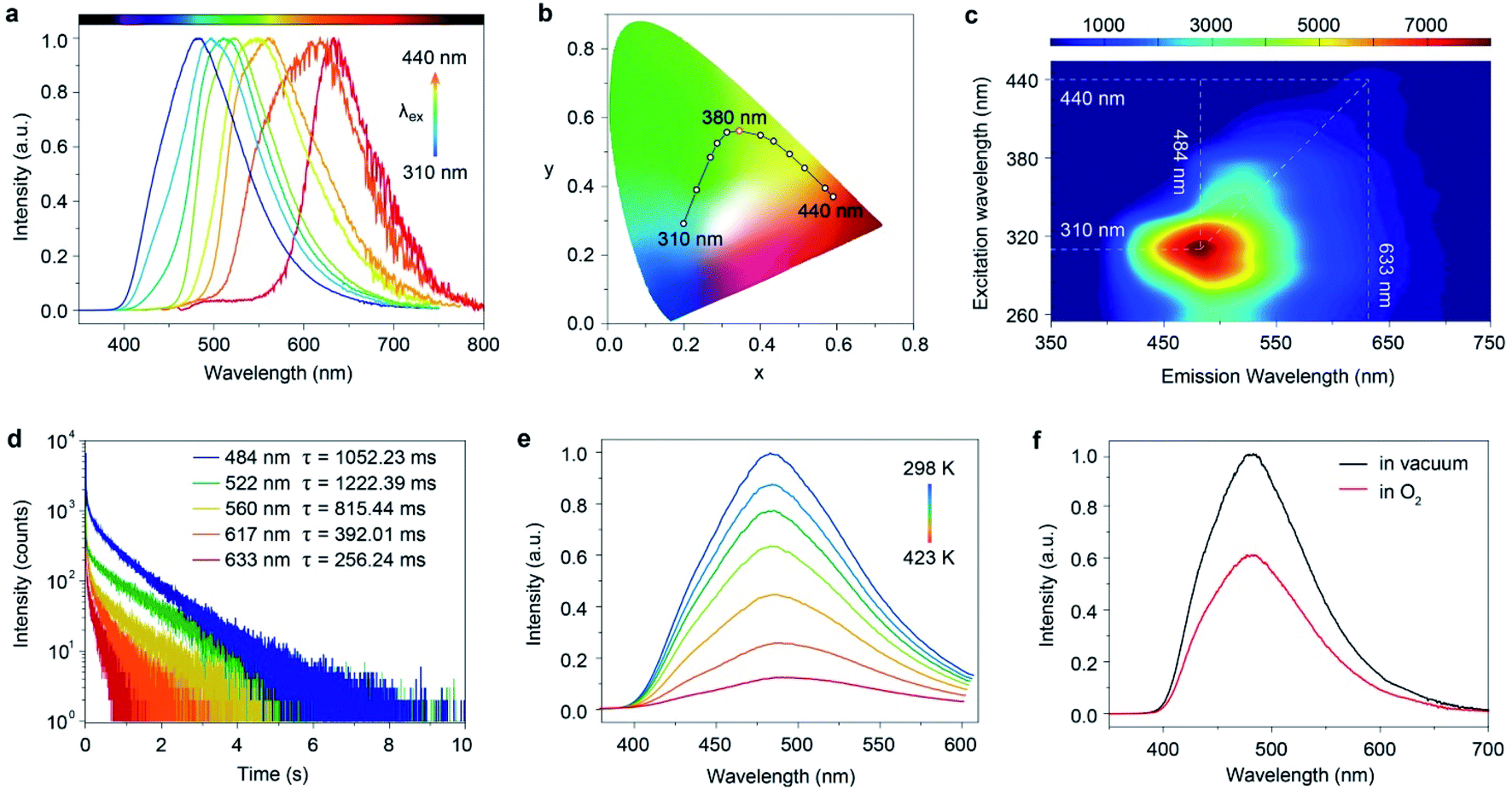 | ||
| Fig. 2 Photophysical properties of the FP-CDs. (a) Excitation-dependent phosphorescence spectra and the (b) corresponding CIE coordinates. (c) Excitation-phosphorescence mapping of FP-CD powders under ambient conditions. (d) Time-resolved decay curves excited at 310, 360, 400, 420 and 440 nm under ambient conditions. (e) Phosphorescence spectra under 310 nm excitation at different temperatures from 298 to 423 K. (f) Phosphorescence spectra in O2 (red) and vacuum (black) atmospheres under 310 nm excitation at room temperature. | ||
To gain insights into the mechanism of the ultralong phosphorescence for FP-CDs, the phosphorescence properties of FP-CDs in aqueous solution were investigated at room temperature and 77 K (Fig. 3a, b, S4b and S8†). Under irradiation by 365 nm UV-light, the FP-CDs aqueous solution displayed blue emission. However, no afterglow emission was observed after the stoppage of the UV-light, and even no phosphorescence signal was detected by a spectrophotometer. At 77 K, however, intense excitation-dependent ultralong phosphorescence was observed (Fig. S8†), indicating that confinement of the chromophores on the surface of CDs plays a critical role in boosting ultralong phosphorescence. Because the chromophores on the surface of FP-CDs show intense molecular motions, leading to the phosphorescence quenching in aqueous solution, while being fixed at 77 K, enhanced phosphorescence was obtained owing to suppressing non-radiative transitions of the chromophores.
Notably, the polyvinyl alcohol (PVA) matrix is not only a good oxygen barrier but also a hydrophilic polymer with plenty of hydroxyl groups. It has a strong capability to form hydrogen bonds for suppressing non-radiative transitions, which is similar to the confinement of the chromophores by lowering the temperature. After being doped into the PVA film at room temperature, as expected, FP-CDs demonstrate obvious excitation-dependent ultralong phosphorescence (Fig. 3a), further confirming the importance of the surface chromophore confinement (SCC) for ultralong phosphorescence generation.
 | ||
| Fig. 3 Mechanistic investigation of color-tunable phosphorescence for FP-CDs. (a) Excitation-dependent phosphorescence spectra of the FP-CD doped PVA film excited from 310 to 440 nm. (b) The phosphorescence excitation spectra of FP-CDs by monitoring at different emission wavelengths. (c) Phosphorescent photographs of U-CDs, U/C-CDs-1, U/C-CDs-2 and B/C-CDs at different excitation wavelengths. (d) The corresponding photographs of phosphorescence with different delay times at different excitation wavelengths. (e) A plausible mechanism for excitation-dependent ultralong phosphorescence. Note that S0 represents the ground state. S1 and T1 are the lowest excited singlet and triplet states. ISC is the abbreviation of intersystem crossing. | ||
According to previous reports on excitation-dependent emission, we speculated that excitation-dependent full-color phosphorescence might stem from multiple triplet excited states of the emitting centers formed by space electronic delocalization between carbonyl and different groups such as C–O, C–N, PO, etc.45 To prove the existence of multiple triplet excited states, the phosphorescence excitation spectra of FP-CDs were collected by monitoring different phosphorescence emission wavelengths. As shown in Fig. 3b, the excitation spectra exhibited distinct excitation bands at around 310, 360, and 440 nm, respectively, which exactly suggested the existence of multiple triplet emission centers.
To further verify our speculation, urea was selected as a carbon source due to its similar structure to biuret. Through a similar method, we synthesized U-CDs. TEM shows that the U-CDs exhibit well-dispersed spherical shapes (Fig. S9a†). As per our expectation, U-CDs show excitation-dependent emission (Fig. S10†) and XPS indicates that U-CDs also demonstrate a large number of carbonyl groups like FP-CDs (Fig. S13†). However, as the excitation wavelength changes, the phosphorescent colors can only change from blue to yellow (Fig. 3c, d and S15†), where the phosphorescence color span is not nearly as wide as FP-CDs. It is worth noting that urea cannot be converted to the CYAD dopant. For biuret, however, it can decompose into CYAD at 466 K.27 Therefore, we reasoned that the doping of CYAD not only resulted in phosphorescence redshift but also made the phosphorescence cover the entire visible range.46 In order to verify our guess, we incorporated a certain proportion of CYAD to synthesize three more kinds of carbon dots, namely U/C-CDs-1, U/C-CDs-2 and B/C-CDs (Scheme S2†). The TEM images show that the three newly synthesized CDs all exhibit well-dispersed spherical shapes (Fig. S9†). As shown in Fig. 3c, d, S11, S12, S15 and S16,† it is found that low concentrations of CYAD could result in blue to orange emission (U/C-CDs-1), and high concentrations of CYAD could result in green to red emission (U/C-CDs-2). Similarly, B/C-CDs can only change from green to red with the excitation wavelength change (Fig. 3d and S16†). Moreover, these four CDs synthesized by the doping method all show ultralong phosphorescence lifetimes (Fig. S17†). Therefore, we reasoned that the self-doping of CYAD played a vital role in color-tunable ultralong phosphorescence, which might be a reasonable way to expand the phosphorescence colors of CD materials. Specifically, the centers of phosphorescence in short-wavelength bands are the clusters that are composed of carbonyl groups with some heteroatom-containing functional groups, while the phosphorescence emission centers in long-wavelength bands are related to the CYAD dopant.
Taking the above results together, we proposed a plausible mechanism for full-color-tunable ultralong phosphorescence of CDs by self-doping (Fig. 3d and S18†). First, biuret and phosphoric acid are dehydrated to form longer polymer chains. At the same time, part of the biuret is pyrolyzed to form urea and CYAD. As the heating time increases, the preformed polymer chains are entangled with each other. Then, free urea, biuret, CYAD and phosphoric acid served as linking units between polymer chains to form a covalently cross-linked framework. Finally, the aggregation of carbonyl groups with other groups forms carbonyl assistant clusters and the presence of urea, biuret and CYAD may promote the formation of carbonyl clusters of different through-space conjugations, enabling the formation of multiple emitting centers with distinguished energy gaps. With excitation wavelength variation, the different emitting centers endow the CDs with color-tunable phosphorescence in the solid state.
Given the excitation-dependent colorful ultralong phosphorescent feature of the FP-CDs, we further demonstrate potential applications in light detection and information encryption. After exposure to different light excitations with an interval of 10 nm, colorful ultralong phosphorescence was observed by the naked eye, from blue to red, covering the entire visible range. By comparing with the standard color chart (Fig. 4a), the wavelength of the excitation source can be determined by the naked eye quickly and easily, making the UV-blue light detection visible. Furthermore, from Fig. 4b, it can be found that there exists a one-to-one correspondence between the CIE coordinates and excitation wavelengths. The wavelength of the excitation source can be accurately determined using the corresponding functional relationship once the CIE coordinates are identified. Considering the long-lived emission lifetimes, FP-CDs are successfully used for information encryption. As shown in Fig. 4c, a digit pattern “8888” was painted by marking the black parts with FP-CDs and the grey parts with a fluorescent luminogen (Scheme S3 and Fig. S18†). Under irradiation by a 365 nm UV lamp, the pattern displayed a blue “8888” while a green digit “2020” was captured by the naked eye when the UV lamp was switched off. In view of the excitation-dependent full-color afterglow emission, FP-CDs have great potential in multicolor afterglow display and anti-counterfeiting. As shown in Fig. 4d, a beautiful rose was fabricated by using FP-CDs and PACDs-3/PAA (PACD-3 doped polyacrylic acid) with cyan afterglow.47 By changing the excitation wavelength from 310 to 440 nm, the afterglow color of the flower turned from blue to green, yellow and red, while the color of the branches and leaves remained almost unchanged. Additionally, the integrated LED device displayed visual afterglow emission after the stoppage of electricity (Fig. 4e). As the excitation wavelength of the LED changes, the afterglow color also changes. In view of the eco-friendly, low cost and facile synthesis, FP-CDs will be promising in a variety of advanced optical applications.
 | ||
| Fig. 4 Demonstration of color-tunable phosphorescence for light detection, encryption and LED devices under ambient conditions. (a) A standard color chart showing the ability of using FP-CD powder to detect specific wavelengths from 310 nm to 440 nm. (b) The fitting function based on FP-CD emission upon the change of the excitation wavelength from 310 to 440 nm. (c) Demonstration of ultralong phosphorescence for security application using FP-CDs in combination with a fluorescent luminogen. (d) Demonstration of full-color phosphorescence for multicolour display by varying the excitation wavelengths from 310 to 440 nm. (e) Photographs of the afterglow emission from the integrated LED devices after the stoppage of electricity. | ||
Conclusions
In summary, we have developed a self-doping method to synthesize a new type of matrix-free carbon dots with full-color-tunable ultralong phosphorescence. Impressively, by changing the excitation wavelength, the carbon dots can exhibit controlled emission color changes from blue (484 nm) to red (633 nm) under ambient conditions. The ultralong phosphorescence shows an ultralong lifetime of up to 1229.39 ms. Taking the experimental results together, we reasoned that the carbonyl-related clusters on the surface of the CDs are responsible for the excitation-dependent ultralong phosphorescence and the addition of high-proportion carbonyl molecules contributes to the emission of redshift. Regarding the color-tunability and ultralong lifetime features, the CDs were successfully applied to UV-visible light detection, information encryption and LED devices. Our finding not only provides a design concept for multicolor room temperature phosphorescent materials, but also extends the scope of potential applications for carbon dot materials.Data availability
The experimental data associated with this article have been provided in ESI.†Author contributions
H. X. Shi, H. F. Shi and Z. An conceived the experiments. H. Shi, Z. Niu and Z. An prepared the manuscript. Z. Niu, H. Wang and W. Ye were primarily responsible for the experiments. K. Xi, Y. Liu and X. Wang conducted the TEM measurements. H. Wang and H. Lin gave suggestions for the manuscript. All authors contributed to the data analyses.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21975120 and 21875104) and Open Research Fund Program of Collaborative Innovation Center for Molecular Imaging of Precision Medicine (2020-ZD01 and 2020-MS03).Notes and references
- D. Chaudhuri, E. Sigmund, A. Meyer, L. Röck, P. Klemm, S. Lautenschlager, A. Schmid, S. R. Yost, T. V. Voorhis, S. Bange, S. Höger and J. M. Lupton, Angew. Chem., Int. Ed., 2013, 52, 13449–13452 CrossRef CAS PubMed.
- T. Wang, X. Su, X. Zhang, X. Nie, L. Huang, X. Zhang, X. Sun, Y. Luo and G. Zhang, Adv. Mater., 2019, 31, 1904273 CrossRef CAS PubMed.
- D. Lee, O. Bolton, B. C. Kim, J. H. Youk, S. Takayama and J. Kim, J. Am. Chem. Soc., 2013, 135, 6325–6329 CrossRef CAS PubMed.
- Y. Zhou, W. Qin, C. Du, H. Gao, F. Zhu and G. Liang, Angew. Chem., Int. Ed., 2019, 58, 12102–12106 CrossRef CAS PubMed.
- Z. Tian, D. Li, E. V. Ushakova, V. G. Maslov, D. Zhou, P. Jing, D. Shen, S. Qu and A. L. Rogach, Adv. Sci., 2018, 5, 1800795 CrossRef PubMed.
- S. Hirata, Adv. Opt. Mater., 2017, 5, 1700116 CrossRef.
- H. Wu, W. Chi, G. Baryshnikov, B. Wu, Y. Gong, D. Zheng, X. Li, Y. Zhao, X. Liu, H. Ågren and L. Zhu, Angew. Chem., Int. Ed., 2019, 58, 4328–4333 CrossRef CAS PubMed.
- Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685–690 CrossRef CAS PubMed.
- H. Shi, L. Song, H. Ma, C. Sun, K. Huang, A. Lv, W. Ye, H. Wang, S. Cai, W. Yao, Y. Zhang, R. Zheng, Z. An and W. Huang, J. Phys. Chem. Lett., 2019, 10, 595–600 CrossRef PubMed.
- Y. Zhan, C. Xu, W. Li, Z. Mao, X. Ge, Q. Huang, H. Deng, J. Zhao, F. Gu, Y. Zhang and Z. Chi, Angew. Chem., Int. Ed., 2020, 59, 17451–17455 CrossRef PubMed.
- M. Baroncini, G. Bergamini and P. Ceroni, Chem. Commun., 2017, 53, 2081–2093 RSC.
- S. Kuila and S. J. George, Angew. Chem., Int. Ed., 2020, 59, 9393–9397 CrossRef CAS PubMed.
- L. Gu, H. Shi, L. Bian, M. Gu, K. Ling, X. Wang, H. Ma, S. Cai, W. Ning, L. Fu, H. Wang, S. Wang, Y. Gao, W. Yao, F. Huo, Y. Tao, Z. An, X. Liu and W. Huang, Nat. Photonics, 2019, 13, 406–411 CrossRef CAS.
- H. Wu, W. Chi, G. Baryshnikov, B. Wu, Y. Gong, D. Zheng, X. Li, Y. Zhao, X. Liu, H. Ågren and L. Zhu, Angew. Chem., Int. Ed., 2019, 58, 4328–4333 CrossRef CAS PubMed.
- Y. Lei, W. Dai, J. Guan, S. Guo, F. Ren, Y. Zhou, J. Shi, B. Tong, Z. Cai, J. Zheng and Y. Dong, Angew. Chem., Int. Ed., 2020, 59, 16054 CrossRef CAS PubMed.
- L. Gu, H. Wu, H. Ma, W. Ye, W. Jia, H. Wang, H. Chen, N. Zhang, D. Wang, C. Qian, Z. An, W. Huang and Y. Zhao, Nat. Commun., 2020, 11, 944–951 CrossRef CAS PubMed.
- X. Lin, J. Wang, B. Ding, X. Ma and H. Tian, Angew. Chem., Int. Ed., 2020, 60, 3459–3463 CrossRef PubMed.
- J. Lee, P. W. Bisso, R. L. Srinivas, J. J. Kim, A. J. Swiston and P. S. Doyle, Nat. Mater., 2014, 13, 524–529 CrossRef CAS PubMed.
- R. Deng, F. Qin, R. Chen, W. Huang, M. Hong and X. Liu, Nat. Nanotechnol., 2015, 10, 237–242 CrossRef CAS PubMed.
- L. Pan, S. Sun, A. Zhang, K. Jiang, L. Zhang, C. Dong, Q. Huang, A. Wu and H. Lin, Adv. Mater., 2015, 27, 7782–7787 CrossRef CAS PubMed.
- F. Yuan, Z. Wang, X. Li, Y. Li, Z. Tan, L. Fan and S. Yang, Adv. Mater., 2017, 29, 1604436 CrossRef PubMed.
- J. Li, S. Yang, Y. Deng, P. Chai, Y. Yang, X. He, X. Xie, Z. Kang, G. Ding, H. Zhou and X. Fan, Adv. Funct. Mater., 2018, 28, 1800881 CrossRef.
- X. Miao, D. Qu, D. Yang, B. Nie, Y. Zhao, H. Fan and Z. Sun, Adv. Mater., 2018, 30, 1704740 CrossRef PubMed.
- H. Ding, S. B. Yu, J. S. Wei and H. M. Xiong, ACS Nano, 2016, 10, 484–491 CrossRef CAS PubMed.
- V. A. Kolesov, C. F. Hernandez, W. F. Chou, N. Aizawa, F. A. Larrain, M. Wang, A. Perrotta, S. Choi, S. Graham, G. C. Bazan, T. Q. Nguyen, S. R. Marder and B. Kippelen, Nat. Mater., 2017, 16, 474–480 CrossRef CAS PubMed.
- O. Bolton, K. Lee, H. J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205–210 CrossRef CAS PubMed.
- L. He, F. Weniger, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 12582–12594 CrossRef CAS PubMed.
- P. M. Schaber, J. Colson, S. Higgins, D. Thielen, B. Anspach and J. Brauer, Thermochim. Acta, 2004, 424, 131–142 CrossRef CAS.
- Z. Yang, Z. Mao, X. Zhang, D. Ou, Y. Mu, Y. Zhang, C. Zhao, S. Liu, Z. Chi, J. Xu, Y. C. Wu, P. Y. Lu, A. Lien and M. R. Bryce, Angew. Chem., Int. Ed., 2016, 55, 2181–2185 CrossRef CAS PubMed.
- K. Jiang, Y. Wang, X. Gao, C. Cai and H. Lin, Angew. Chem., Int. Ed., 2018, 57, 6216–6220 CrossRef CAS PubMed.
- S. Tao, S. Lu, Y. Geng, S. Zhu, S. A. T. Redfern, Y. Song, T. Feng, W. Xu and B. Yang, Angew. Chem., Int. Ed., 2018, 57, 2393–2398 CrossRef CAS PubMed.
- Y. Chen, M. Zheng, Y. Xiao, H. Dong, H. Zhang, J. Zhuang, H. Hu, B. Lei and Y. Liu, Adv. Mater., 2016, 28, 312–318 CrossRef CAS PubMed.
- T. Yuan, F. Yuan, X. Li, Y. Li, L. Fan and S. Yang, Chem. Sci., 2019, 10, 9801–9806 RSC.
- J. Zhu, X. Bai, X. Chen, H. Shao, Y. Zhai, G. Pan, H. Zhang, E. V. Ushakova, Y. Zhang, H. Song and A. L. Rogach, Adv. Opt. Mater., 2019, 7, 1801599 CrossRef.
- K. Jiang, Y. Wang, C. Cai and H. Lin, Adv. Mater., 2018, 30, 1800783 CrossRef PubMed.
- Z. Wang, Y. Liu, S. Zhen, X. Li, W. Zhang, X. Sun, B. Xu, X. Wang, Z. Gao and X. Meng, Adv. Sci., 2020, 7, 1902688 CrossRef CAS PubMed.
- Y. Deng, D. Zhao, X. Chen, F. Wang, H. Song and D. Shen, Chem. Commun., 2013, 49, 5751–5753 RSC.
- W. Li, W. Zhou, Z. Zhou, H. Zhang, X. Zhang, J. Zhuang, Y. Liu, B. Lei and C. Hu, Angew. Chem., Int. Ed., 2019, 58, 7278–7283 CrossRef CAS PubMed.
- C. Lu, Q. Su and X. Yang, Nanoscale, 2019, 11, 16036–16042 RSC.
- H. Yang, Y. Liu, Z. Guo, B. Lei, J. Zhuang, X. Zhang, Z. Liu and C. Hu, Nat. Commun., 2019, 10, 1798–1799 CrossRef PubMed.
- C. Wang, Y. Chen, T. Hu, Y. Chang, G. Ran, M. Wang and Q. Song, Nanoscale, 2019, 11, 11967–11974 RSC.
- K. Jiang, S. Hu, Y. Wang, Z. Li and H. Lin, Small, 2020, 16, 2001909 CrossRef CAS PubMed.
- C. Xia, S. Zhu, S. T. Tang, Q. Zeng, S. Tao, X. Tao, X. Tian, Y. Li and B. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 38593–38601 CrossRef CAS PubMed.
- H. Wang, H. Shi, W. Ye, X. Yao, Q. Wang, C. Dong, W. Jia, H. Ma, S. Cai, K. Huang, L. Fu, Y. Zhang, J. Zhi, L. Gu, Y. Zhao, Z. An and W. Huang, Angew. Chem., Int. Ed., 2019, 131, 18952–18958 CrossRef.
- S. Zheng, T. Zhu, Y. Wang, T. Yang and W. Z. Yuan, Angew. Chem., Int. Ed., 2020, 59, 10018–10022 CrossRef CAS PubMed.
- Q. Wang, X. Dou, X. Chen, Z. Zhao, S. Wang, Y. Wang, K. Sui, Y. Tan, Y. Gong, Y. Zhang and W. Z. Yuan, Angew. Chem., Int. Ed., 2019, 58, 12667–12673 CrossRef CAS PubMed.
- H. Gou, Y. Liu, G. Zhang, Q. Liao, X. Huang, F. Ning, C. Ke, Z. Meng and K. Xi, Nanoscale, 2019, 11, 18311–18319 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, additional figures and tables. See DOI: 10.1039/d2sc01167k |
| ‡ Co-first author, they contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |