
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpatiotemporal dynamics of supramolecular polymers by in situ quantitative catalyst-free hydroamination†
Minghan
Tan
 ab,
Masayuki
Takeuchi
*ab and
Atsuro
Takai
*a
ab,
Masayuki
Takeuchi
*ab and
Atsuro
Takai
*a
aMolecular Design and Function Group, National Institute for Materials Science (NIMS), 1-2-1 Sengen, Tsukuba, Ibaraki 305-0047, Japan. E-mail: TAKEUCHI.Masayuki@nims.go.jp; TAKAI.Atsuro@nims.go.jp
bDepartment of Materials Science and Engineering, Faculty of Pure and Applied Sciences, University of Tsukuba, 1-1-1 Tennodai, Tsukuba, Ibaraki 305-8577, Japan
First published on 23rd March 2022
Abstract
Implementing chemical reactivity into synthetic supramolecular polymers based on π-conjugated molecules has been of great interest to create functional materials with spatiotemporal dynamic properties. However, the development of an in situ chemical reaction within supramolecular polymers is still in its infancy, because one needs to design optimal π-conjugated monomers having excellent reactivity under mild conditions possibly without byproducts or a catalyst. Herein we report the synthesis of a supramolecular polymer based on ethynyl core-substituted naphthalenediimide (S-NDI2) molecules that react with various amines quantitatively in a nonpolar solvent, without a catalyst, at 298 K. Most interestingly, the in situ reaction of the S-NDI2 supramolecular polymer with a linear aliphatic diamine proceeded much faster than the homogeneous reaction of a monomeric naphthalenediimide with the same diamine, affording diamine-linked S-NDI2 oligomers and polymers. The acceleration of in situ hydroamination was presumably due to rapid intra-supramolecular cross-linking between ethynyl and amino groups fixed in close proximity within the supramolecular polymer. Such intra-supramolecular cross-linking did not occur efficiently with an incompatible diamine. The systematic kinetic studies of in situ catalyst-free hydroamination within supramolecular polymers provide us with a useful, facile and versatile tool kit for designing dynamic supramolecular polymeric materials based on electron-deficient π-conjugated monomers.
Introduction
Supramolecular polymers composed of π-conjugated molecules have been attracting increasing attention as soft materials that could play pivotal roles in organic electronics, energy conversion and life-like systems in a sustainable society.1–5 Toward these goals, in addition to conventional static or thermodynamically controlled supramolecular polymers, the development of dynamic supramolecular materials that are responsive to external stimuli such as chemical reactions has been extensively studied.6–18 To design supramolecular polymers based on π-conjugated monomers that can implement chemical reactivity, one needs to introduce a reaction site where the chemical reaction proceeds under mild conditions without byproducts or a catalyst that could have unintended effects on the supramolecular polymers. These stringent requirements for the design of π-conjugated molecules hinder the further development of dynamic supramolecular polymers, although elaborate dynamic supramolecular assemblies that incorporate efficient chemical reactions have been reported.9,19–38 Besides, there are only a handful of in situ chemical reactions within the supramolecular polymers, such as the topochemical photoreaction and redox reaction, that can lead to significant changes in the optical and electronic properties of the π-systems.33–38Meanwhile, we recently reported quantitative, catalyst-free reactions between an ethynyl group directly attached to electron-accepting π-conjugated molecules and amines in various media.39–41 An ethynyl core-substituted naphthalenediimide (NDI) is a representative electron-accepting π-conjugated molecule that reacts with an amine rapidly under mild conditions without byproducts or a catalyst (Scheme 1). This atom-economical hydroamination of NDIs possesses unique characteristics that are distinctively different from other existing chemical reactions for π-systems: (1) various amines, many of which are commercially available, are applicable to functionalize the π-systems, and thus (2) the π-conjugated structure is changed by the reaction to exhibit remarkable changes in the optical and electronic properties. In this context, considering that NDIs have been widely known as building blocks of supramolecular polymers,42–45 we conceived that our catalyst-free hydroamination of NDIs could be used as a new family of in situ reactions to trigger macroscopic changes of supramolecular polymers based on NDI molecules using various amines. The in situ reaction of π-conjugated molecules within supramolecular polymers is also expected to exhibit characteristic reactivity different from that in homogeneous monodisperse solutions, because the molecules are pre-organized and locally condensed. Such a proximity effect on chemical reactivity has been studied in intramolecular and enzymatic reactions,46–50 but little is known in supramolecular polymers.
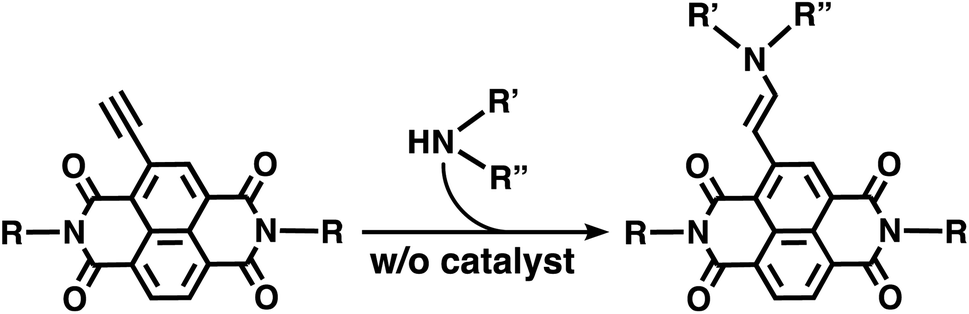 | ||
| Scheme 1 Quantitative catalyst-free hydroamination of an ethynyl core-substituted NDI without a catalyst. | ||
We report herein the formation of a supramolecular polymer based on ethynyl core-substituted NDI monomers in nonpolar solvents, its unique reactivity with amines, and the spatiotemporal dynamics through in situ catalyst-free hydroamination. As shown in Fig. 1, we designed S-NDI2 having two ethynyl groups at the NDI π-core as reaction sites. The imide side chains of S-NDI2 and a reference compound (ref-S-NDI) include the amide and trialkoxyphenyl groups, which reinforce the π-stacking of the NDIs to enhance the formation of supramolecular polymers.51–57 The butano –(CH2)4– spacer58 of the imide side chain was introduced because it could facilitate supramolecular structural changes upon external stimuli.53 The reactivity and the reaction kinetics of the supramolecular polymers of S-NDI2 with various monoamines and diamines were investigated by UV-vis absorption spectroscopy, mass spectrometry and chromatography techniques. The morphological and structural changes of the supramolecular polymers of S-NDI2 during the course of hydroamination were studied by atomic force microscopy (AFM), Fourier-transform infrared (FT-IR) spectroscopy and the X-ray diffraction (XRD) method. Hydroamination of a reference compound (NDI2) that does not form any supramolecular polymers was also studied for comparison. The present study shows for the first time that in situ reactions within supramolecular polymers based on π-conjugated monomers gave completely different reaction kinetics and products from the homogeneous reactions in monodisperse solutions of π-conjugated molecules.
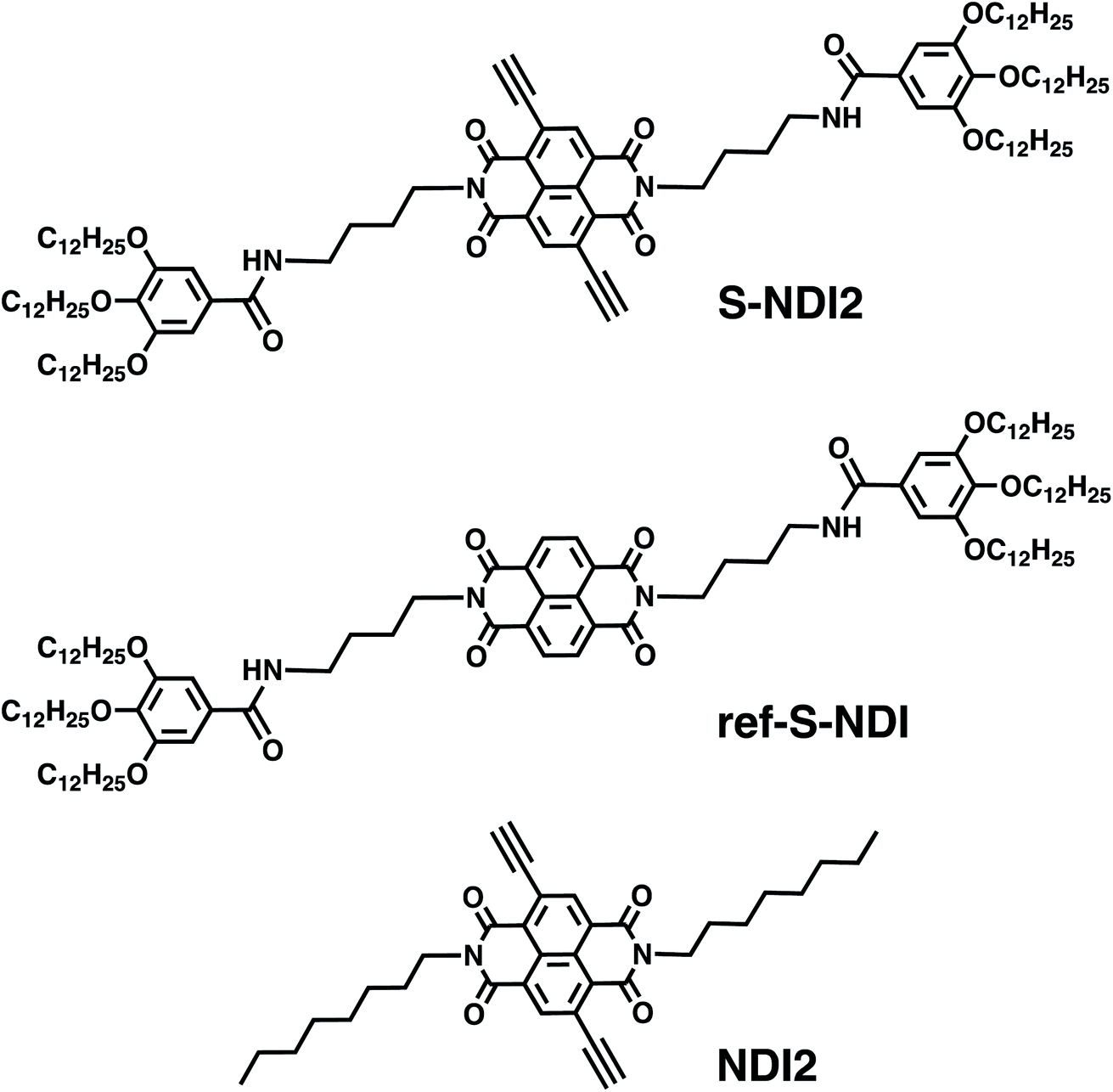 | ||
| Fig. 1 Structures of S-NDI2, ref-S-NDI and NDI2. | ||
Results and discussion
Supramolecular polymerization of NDIs
The syntheses of S-NDI2 and ref-S-NDI and their full characterization data are given in the ESI (S2. Synthesis and characterization†). The synthesis of NDI2 was reported previously.39 Supramolecular polymerization of S-NDI2 was examined in several solvents such as methylcyclohexane (MCH) and toluene. Among them, we chose a MCH/toluene (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) mixed solvent, owing to the optimal supramolecular polymerization behavior of S-NDI2 and its stability; no precipitation was formed during the analyses. When S-NDI2 (25 μM) in MCH/toluene (4:1 v/v) was cooled from 363 K to 278 K at a rate of 1 K min−1, the UV-vis absorption spectrum of S-NDI2 changed with a typical Davydov splitting; a new red-shifted absorption band at 446 nm and a blue-shifted absorption band at 418 nm appeared, suggesting the formation of supramolecular J-aggregates (Fig. 2a).59,60 The degree of polymerization (αagg) was calculated by using eqn (1);
:1 v/v) mixed solvent, owing to the optimal supramolecular polymerization behavior of S-NDI2 and its stability; no precipitation was formed during the analyses. When S-NDI2 (25 μM) in MCH/toluene (4:1 v/v) was cooled from 363 K to 278 K at a rate of 1 K min−1, the UV-vis absorption spectrum of S-NDI2 changed with a typical Davydov splitting; a new red-shifted absorption band at 446 nm and a blue-shifted absorption band at 418 nm appeared, suggesting the formation of supramolecular J-aggregates (Fig. 2a).59,60 The degree of polymerization (αagg) was calculated by using eqn (1);| αagg ≈ (A − AMonomer)/(APolymer − AMonomer) | (1) |
| αagg = αSAT[1 − exp(−ΔHe(T − Te)/RTe2)] | (2) |
:1 v/v) without the formation of precipitate up to 2 hours.
 | ||
| Fig. 2 (a) Temperature-dependent UV-vis absorption spectral changes of S-NDI2 (25 μM) in MCH/toluene (4:1 v/v) upon cooling from 363 K (red line) to 278 K (blue line) at a rate of 1 K min−1. (b) Plot of the degree of polymerization (αagg) calculated from the absorbance at 446 nm against temperature (blue circle). Blue solid line denotes a theoretical curve fitted by using a cooperative model with an R2 value of 0.996. (c) AFM image of a spin-coated sample from the MCH/toluene (4:1 v/v) solution used for the UV-vis absorption measurement. Scale bar: 200 nm. (d) The height profile of the cross-section (cyan dashed line) in (c). (e) Optimized structure of S-NDI2 calculated at the B3LYP/6-31G* level. The alkoxy groups (–OC12H25) were substituted by methoxy groups for simplicity. Atom colour code: grey, C; red, O; blue, N; white, H. | ||
Similarly, when a MCH/toluene (4:1 v/v) solution of ref-S-NDI (25 μM) was cooled from 363 K to 298 K,62 the UV-vis absorption spectrum of ref-S-NDI changed with an abrupt point at 323 K, indicating the cooperative formation of a supramolecular assembly, as shown in Fig. S2.† Note that the degree of polymerization of ref-S-NDI at 298 K (76%) was smaller than that of S-NDI2 (96%). The ΔHe and the Te values of ref-S-NDI at CT of 25 μM are −47.3 kJ mol−1 and 323 K, respectively.
AFM images for the spin-coated samples of the S-NDI2 supramolecular assembly exhibited well-developed nanofibers with over micrometers in length (Fig. 2c). The average height of the nanofibers (Fig. 2d) was comparable with the distance between the two ethynyl protons of S-NDI2 (1.2 nm) obtained from the DFT calculations (Fig. 2e), indicating the formation of nanofibers with a unimolecular height. On the other hand, the AFM image of the ref-S-NDI supramolecular assemblies showed the formation of nanoparticles (Fig. S2d†). NDI2 did not form any supramolecular assembly. These results indicate that the ethynyl-extended π-core of S-NDI2 facilitates the formation of the supramolecular nanofibers with the help of the imide side chains.
In situ hydroamination of the S-NDI2 supramolecular polymer with monoamines
We then decided to study the reactivity of the S-NDI2 supramolecular polymer with an amine and its reaction kinetics. As a first step, we confirmed that hydroamination occurred between NDI2 and diethylamine (DEA), as a typical case, in a nonpolar solvent. Upon addition of DEA (1.24 mM) to a MCH/toluene (4:1 v/v) solution of NDI2 (25 μM) at 298 K, hydroamination started to occur and the UV-vis absorption spectrum of NDI2 remarkably changed with isosbestic points (Fig. S3†). The appearance and saturation of a characteristic charge-transfer band at 607 nm indicates the quantitative formation of an amine monoadduct NDI2–DEA.39–41,63,64 The rate of formation of NDI2–DEA in the presence of excess DEA obeyed pseudo-first order kinetics (eqn S1–S3†). The time profile of the change in the absorbance at 607 nm was fitted well by using eqn S3† with an R2 value of 0.999 to give a second-order rate constant (k) as 2.9 × 10−1 M−1 s−1 (Fig. S3b† and Table 1).
:1 v/v) at 298 K
| Amine | NDI | k (M−1 s−1) |
|---|---|---|
| Diethylamine (DEA) | S-NDI2 | 6.3(±0.3) × 10−2 |
| NDI2 | 2.9(±0.2) × 10−1 | |
| Diisopropylamine | S-NDI2 | 1.2(±0.06) × 10−4 |
| NDI2 | 3.1(±0.2) × 10−3 | |
| 1,4-Diaminobutane | S-NDI2 | 3.1(±0.2) × 10−1 |
| NDI2 | 6.5(±0.3) × 10−2 | |
| cis-1,4-Cyclohexanediamine | S-NDI2 | 8.7(±0.4) × 10−2 |
| NDI2 | 1.7(±0.09) × 10−2 | |
| trans-1,4-Cyclohexanediamine | S-NDI2 | 4.1(±0.2) × 10−2 |
| NDI2 | 2.8(±0.1) × 10−2 |
When DEA (1.24 mM) was added to a MCH/toluene (4:1 v/v) solution of S-NDI2 supramolecular polymers (CT = 25 μM) at 298 K, the UV-vis absorption spectrum was significantly changed as shown in Fig. 3a. The decrease in the absorption band at 446 nm and the increase in the new absorption band at 615 nm indicated that the reaction between the ethynyl group of S-NDI2 and DEA proceeded. Judging from the UV-vis absorption spectrum (see the latter discussion) and the MALDI TOF-MS chart (Fig. S4†) after the reaction completed, the resultant product is a supramolecular assembly of the corresponding amine monoadduct (S-NDI2–DEA). The formation of an amine bisadduct, S-NDI2–(DEA)2 was much less than that of S-NDI2–DEA under the measurement conditions (Fig. S5†).65 It must be noted that, in the initial stage of the reaction, we observed distinct peak shifts of the charge-transfer band at 615 nm to a longer wavelength (ca. 645 nm) and that of the π–π* transition band at 446 nm toa shorter wavelength (ca. 440 nm) as shown in Fig. 3a. This result strongly indicates the existence of an intermediate during the transition from the supramolecular assembly of S-NDI2 to that of S-NDI2–DEA. Despite the existence of such an intermediate (Fig. 3b), the time profiles of the absorbance changes at 446 nm and 615 nm obeyed pseudo-first order kinetics with a high R2 value of 0.995, and the second-order rate constant (k) could be obtained as 6.3 × 10−2 M−1 s−1. The rate constant for the S-NDI2 supramolecular polymer was 4.7-fold smaller than that for NDI2 (Table 1), probably because the steric hindrance around the ethynyl groups within the S-NDI2 supramolecular polymer was larger than that in the monodisperse state. The slower reaction rate for the S-NDI2 supramolecular polymer than that for NDI2 eliminates the possibility that hydroamination took place at the terminal of the S-NDI2 supramolecular polymer or at the free S-NDI2 monomer, considering that the S-NDI2 monomer has slightly higher reactivity than the NDI2 monomer in pure toluene at 298 K (see Fig. S6† for details). On the other hand, the reaction of the S-NDI2 supramolecular polymer with diisopropylamine was 525-fold slower than the case of DEA (Fig. S7†), although the basicity of diisopropylamine (pKa = 11.05) and DEA (pKa = 10.98) are almost the same.66 This result indicates that the steric hindrance of the nucleophile (amine) also critically affects the reaction rate.
 | ||
| Fig. 3 (a) UV-vis absorption spectral changes of the S-NDI2 supramolecular polymer (CT = 25 μM) observed upon addition of DEA (1.24 mM) in MCH/toluene (4:1 v/v) at 298 K. (b) Time profiles of absorbance changes at 446 nm (black circle) and 615 nm (blue circle) during the reaction, fitted by pseudo-first-order kinetic curves with R2 values of 0.995. The inset shows the structure of S-NDI2–DEA. (c) AFM images observed during the reaction. The samples were prepared by spin-coating from the MCH/toluene (4:1 v/v) solution at 13 min, 180 min, 414 min and 1200 min after the addition of DEA. Scale bar: 1 μm. | ||
In conjunction with the UV-vis absorption spectral changes, the morphologies of the S-NDI2 supramolecular polymer were also changed as shown in Fig. 3c. The pristine nanofibers became gradually shorter until the unreacted S-NDI2 units were consumed. The short nanofibers at 414 min when 89% of the reaction completed were then bundled to change into larger assemblies at 1200 min, although there was no significant UV-vis absorption spectral change between 414 min and 1200 min. This result merely indicates that the number of polymerized monomers remains the same.27
In order to clarify the reaction product and the kinetics of the in situ reaction of the S-NDI2 supramolecular polymer with DEA, we first confirmed supramolecular polymerization behavior of S-NDI2–DEA by means of UV-vis absorption spectroscopy and AFM (Fig. S8†). The amine monoadduct (S-NDI2–DEA) was prepared by the reaction of S-NDI2 with DEA in CHCl3 at room temperature in 93% isolated yield (see the ESI† for details). When a MCH/toluene (4:1 v/v) solution of S-NDI2–DEA (25 μM) was cooled from 363 K to 298 K,67 the supramolecular polymer of S-NDI2–DEA was formed, showing a sigmoidal absorbance change at its charge-transfer band. The plot of αaggversus temperature (Fig. S8b†) was fitted by using an isodesmic model expressed in eqn S6,†61 giving an aggregation enthalpy (ΔH = −61.0 kJ mol−1) and a melting temperature (Tm = 312 K) where αagg is 0.5 at CT of 25 μM. The degree of polymerization of S-NDI2–DEA at 298 K was calculated to be 74%. A thin film spin-coated from a MCH/toluene (4:1 v/v) solution after cooling to 298 K exhibited a morphology of short nanofibers with an average height of 4 nm (Fig. S8e and f†). The height is comparable with twice the distance between the ethynyl proton and the amine moiety of S-NDI2–DEA, which could be because S-NDI2–DEA tends to form a slipped-stack dimer to cancel out the large dipole moment in the π-plane (7.96 Debye; Fig. S8g†).68,69 The UV-vis absorption spectrum and the morphology of the supramolecular polymer of S-NDI2–DEA were fairly similar to the one obtained from the in situ reaction between the S-NDI2 supramolecular polymer and DEA (Fig. 3c). Based on these results, it is reasonable to conclude that the final product of in situ hydroamination was the supramolecular assembly of S-NDI2–DEA.
We next conducted two control experiments: (i) mixing two different supramolecular polymers of S-NDI2 and S-NDI2–DEA in a 1:1 ratio at 298 K and (ii) mixing two different monomers of S-NDI2 and S-NDI2–DEA in a 1:1 ratio at 363 K and cooling to 298 K. In both cases, the UV-vis absorption spectra were consistent with the sum of those of each supramolecular assembly (Fig. S9 and S10†), indicating the formation of self-sorted assemblies. This self-sorting behavior is reasonable, given that the elongation mechanisms of S-NDI2 and S-NDI2–DEA are different between cooperative and isodesmic, respectively. An AFM image of a spin-coated film from the MCH/toluene (4:1 v/v) solution used for the UV-vis absorption measurements also indicated the existence of two different supramolecular assemblies (Fig. S10c†). In contrast, the UV-vis absorption spectrum of the intermediate during in situ hydroamination was different from that of the sum of the self-sorted supramolecular assemblies as shown in Fig. S9.† The UV-vis absorption spectrum of the intermediate did not match with that of the sum of monomeric S-NDI2 and S-NDI2–DEA, either. Thus, hydroamination should take place within the supramolecular polymer, not at the terminal of the S-NDI2 supramolecular polymer or the S-NDI2 monomer, and the intermediate supramolecular assembly was only observed during in situ hydroamination of the S-NDI2 supramolecular polymer, as shown in Fig. 4.70
 | ||
| Fig. 4 Schematic illustration of self-assembled behaviors of S-NDI2 and S-NDI2–DEA and in situ hydroamination of the S-NDI2 supramolecular polymer with DEA. | ||
S-NDI2 followed a cooperative mechanism of self-assembly, whereas S-NDI2–DEA followed an isodesmic mechanism. To understand this difference in the supramolecular polymerization processes, we measured the FT-IR spectra of the supramolecular assemblies of S-NDI2, ref-S-NDI and S-NDI2–DEA and compared the strength of the intermolecular hydrogen-bonding between the amide moieties. FT-IR samples were prepared by drying MCH/toluene (4:1 v/v) solutions of S-NDI2, ref-S-NDI and S-NDI2–DEA that were cooled from 363 K to 298 K at a rate of 1 K min−1. We confirmed that the UV-vis absorption spectrum of each sample was consistent with that in MCH/toluene (4:1 v/v) at 298 K, indicating that the dried sample retained its supramolecular structure (Fig. S11†). As shown in Fig. 5, no peaks at energies higher than 3400 cm−1 were observed, suggesting the absence of non-hydrogen-bonded amide moieties.53,54 The N–H stretching vibration of the supramolecular polymer of S-NDI2 was found at 3210 cm−1, which was a significantly lower frequency compared to those of ref-S-NDI and S-NDI2–DEA. The peak shift of the N–H stretching vibration to lower energy is indicative of stronger hydrogen-bonding interactions among the amide moieties. Such a strong intermolecular hydrogen-bonding interaction allows S-NDI2 to form its supramolecular polymers through a cooperative mechanism, unlike S-NDI2–DEA, which forms its supramolecular polymers through an isodesmic mechanism.
 | ||
Fig. 5 FT-IR spectra (ATR mode) of the supramolecular polymers of (a) S-NDI2, (b) ref-S-NDI and (c) S-NDI2–DEA. The peaks at 2100 cm−1 and 3233 cm−1 of S-NDI2 and S-NDI2–DEA are assigned to the stretching vibrations of C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C and C–H of the terminal ethynyl group, respectively.39 The FT-IR spectrum of the S-NDI2–DEA supramolecular assembly in the range of 1500 cm−1 and 1750 cm−1 exhibited more peaks than the others owing to its asymmetric structure. C and C–H of the terminal ethynyl group, respectively.39 The FT-IR spectrum of the S-NDI2–DEA supramolecular assembly in the range of 1500 cm−1 and 1750 cm−1 exhibited more peaks than the others owing to its asymmetric structure. | ||
XRD analyses also support the different packing structures between the supramolecular polymers of S-NDI2 and S-NDI2–DEA. The XRD pattern of the S-NDI2 supramolecular polymer exhibited peaks attributed to the lamellar and π-stacking structure of S-NDI2 molecules, whereas that of the S-NDI2–DEA supramolecular polymer exhibited a peak matching with the length of S-NDI2–DEA along the short axis (ca. 1.5 nm) and a broad peak attributed to the alkyl chain halo and π-stacking distances, as shown in Fig. S12.† We infer that the difference in the packing structures of the supramolecular polymers of S-NDI2 and S-NDI2–DEA may result from the steric effects of the DEA moiety and the large dipole moment of S-NDI2–DEA in the π-plane (7.96 Debye), which diminishes long-range interactions in the growth direction of the supramolecular polymer.39,71,72 Indeed, the supramolecular assemblies of other amine adducts such as S-NDI2–(DEA)2 showed different AFM morphologies (Fig. S5c†), suggesting different packing structures. Note that the influence of excess DEA on the stability and the packing structure of supramolecular assemblies during in situ hydroamination was considered negligible; upon addition of DEA (1.24 mM) to a MCH/toluene (4:1 v/v) solution of the ref-S-NDI supramolecular polymer (CT = 25 μM), there were no changes in both the UV-vis absorption spectra and AFM images before and after the addition of DEA (Fig. S13†).
In situ hydroamination of the S-NDI2 supramolecular polymer with diamines
The S-NDI2 supramolecular nanofibers also exhibited unique reactivity toward a diamine for cross-linkage. When 1,4-diaminobutane (0.62 mM)73 was added to the supramolecular nanofibers of S-NDI2 (CT = 25 μM) in MCH/toluene (4:1 v/v) at 298 K, the UV-vis absorption spectrum was changed (Fig. 6a and b), as was observed for the reaction with DEA. The increase in the new absorption band at around 615 nm indicated that the reaction of the ethynyl groups of S-NDI2 with an amino group in 1,4-diaminobutane proceeded to show a charge-transfer band. The resulting charge-transfer band was broader than that observed in the reaction with DEA, reaching the near-IR (NIR) region above 1000 nm. From the time course of the absorbance at 615 nm, the second-order rate constant was determined to be 3.1 × 10−1 M−1 s−1.
 | ||
| Fig. 6 (a) UV-vis-NIR absorption spectral changes of the S-NDI2 supramolecular polymer (CT = 25 μM) observed upon addition of 1,4-diaminobutane (0.62 mM) in MCH/toluene (4:1 v/v) at 298 K. (b) Time profile of the absorbance change at 615 nm (blue circle) during the reaction, fitted by pseudo-first-order kinetic curves with an R2 value of 0.992. (c) MALDI TOF-MS (positive ion, linear mode) chart obtained 470 min after the reaction. Some oligomers' peaks were split into several peaks, which were assigned to the oligomers with different terminal groups, i.e. ethynyl and amino groups. (d) GPC charts obtained before (black line) and after (red line) the reaction of S-NDI2 (1.2 mM) with 1,4-diaminobutane (1.2 mM) in MCH/toluene (4:1 v/v) for 18.5 h at 298 K. The chromatograms were normalized to the highest peaks. (e) AFM images observed before and after the reaction of S-NDI2 (1.2 mM) with 1,4-diaminobutane (1.2 mM). The samples were prepared by spin-coating on silicon wafers from a MCH/toluene (4:1 v/v) solution. Scale bar: 2 μm. | ||
The MALDI TOF-MS chart after the reaction showed the formation of oligomers and polymers up to 12-mer (Fig. 6c). This result strongly indicates that the cross-linking reaction took place even in a diluted solution of S-NDI2 (CT = 25 μM). The formation of the covalently linked oligomers and polymers were also confirmed by gel permeation chromatography (GPC) after the reaction of S-NDI2 (1.2 mM) with 1,4-diaminobutane (1.2 mM), as shown in Fig. 6d.74 There were two peaks with average molecular weights of 2.8 kDa and 8.1 kDa; the shorter one may be assigned to the S-NDI2 dimer linked by 1,4-diaminobutane, while the longer one is assigned to the S-NDI2 oligomers and polymers. The maximum molecular weight reached around 23 kDa, which corresponds to 12-mer.
AFM studies before and after the cross-linking reaction between S-NDI2 (1.2 mM) and 1,4-diaminobutane (1.2 mM) indicated that nano-fibrous structures of the S-NDI2 assemblies changed two-dimensionally extended structures made up of short fibers upon addition of 1,4-diaminobutane (Fig. 6e). The inter-supramolecular cross-linking reaction should also occur at high concentrations as well as intra-supramolecular cross-linking. Such two-dimensionally extended morphologies were not observed for the thin films prepared from isolated S-NDI2 oligomers and polymers (Fig. S14†), indicating that the net-like assemblies are only formed through the in situ reaction between the S-NDI2 supramolecular polymer and 1,4-diaminobutane.
In contrast, the reaction of NDI2 (25 μM) with 1,4-diaminobutane (0.62 mM) under the same experimental conditions mainly afforded its amine monoadduct as shown in Fig. S15.† The UV-vis absorption spectrum after the reaction showed a charge-transfer band at 582 nm that was blue-shifted and much narrower than the case of the S-NDI2 supramolecular polymer (Fig. 6a). MALDI TOF-MS and GPC results also indicated the formation of the corresponding amine monoadduct as a main product and a trace amount of the NDI2 dimer. These results indicated that the intermolecular cross-linking reaction scarcely occurred under the conditions where NDI2 was monodispersed in solution. In other words, it is indicated that the cross-linking reaction of the S-NDI2 supramolecular polymer at CT of 25 μM was mainly the intra-supramolecular reaction between the pre-organized NDI units and diamines.
The second-order rate constants of the series of hydroamination are summarized in Table 1. Interestingly, the second-order rate constant of the S-NDI2 supramolecular polymer with 1,4-diaminobutane (3.1 × 10−1 M−1 s−1) was 4.8-fold larger than the case of NDI2 (6.5 × 10−2 M−1 s−1). This tendency is totally opposite to the reaction with monoamines such as DEA and diisopropylamine. The acceleration of the reaction of the S-NDI2 supramolecular polymer with a diamine can be explained by the proximity effect in a cross-linking reaction; once an amine moiety of 1,4-diaminobutane reacted with an ethynyl group of S-NDI2, the other amine moiety simultaneously reacted with another ethynyl group of an adjacent S-NDI2 within the supramolecular polymer, as shown in Fig. 7. Accordingly, despite the crowded circumstances of the ethynyl groups in the supramolecular polymer, the in situ reaction of the S-NDI2 supramolecular polymer with 1,4-diaminobutane was accelerated compared to the reaction of NDI2 with 1,4-diaminobutane in monodisperse solution.
 | ||
| Fig. 7 Schematic illustration of the in situ cross-linking reaction of the S-NDI2 supramolecular assembly with diamines. | ||
The reactivity of the S-NDI2 supramolecular polymer for cross-linking may depend on the shape of the diamines. To verify this point, we compared the in situ reaction of the S-NDI2 supramolecular polymer with different diamines; we chose cis- and trans-1,4-cyclohexanediamine. As in the case of 1,4-diaminobutane, the reaction between the S-NDI2 supramolecular polymer and cis-1,4-cyclohexanediamine afforded the corresponding oligomers and polymers through intra-supramolecular cross-linking (Fig. S16†). The reaction rate constant for the S-NDI2 supramolecular polymer was 5.1-fold faster than that for NDI2, as shown in Table 1 and Fig. S17.†
In stark contrast, the reaction between the S-NDI2 supramolecular polymer and trans-1,4-cyclohexanediamine only afforded an amine monoadduct and a dimer linked by the diamine (Fig. S18†). The reaction rate for the S-NDI2 supramolecular polymer was comparable with that for NDI2, as shown in Table 1 and Fig. S19.† These results indicated that the reaction of the S-NDI2 supramolecular polymer with trans-1,4-cyclohexanediamine did not form cross-linked structures efficiently, but rather the reaction occurred similar to that of a monodisperse solution. The reluctance of the cross-linking reaction by trans-1,4-cyclohexanediamine was also supported by the DFT optimized structure of the amine monoadduct where the residual amine moiety faced away from the NDI units (Fig. S20†). On the basis of these results, the intra-supramolecular polymer cross-linking reaction of S-NDI2 proceeded efficiently only when the orientation of the amine moieties was matched with the configuration of the NDI units in the supramolecular polymers (Fig. 7).
Conclusions
In summary, we have demonstrated dynamic structural changes of a supramolecular polymer based on ethynyl core-substituted naphthalenediimide (S-NDI2) molecules by quantitative hydroamination at 298 K without a catalyst. The in situ reaction of the S-NDI2 supramolecular polymer with a monoamine afforded another supramolecular assembly of the corresponding amine-adduct through transient intermediate supramolecular assemblies. The quantitative reaction of the S-NDI2 supramolecular polymer with a remarkable change in its optical properties allowed us to determine the second-order reaction rate constant using UV-vis absorption spectroscopy. The rate of the reaction within the S-NDI2 supramolecular polymer was slower than that in a monodisperse solution of a reference monomer (NDI2), probably because of the large steric hindrance around the ethynyl groups within the supramolecular polymer. On the other hand, the reaction of the S-NDI2 supramolecular polymer with a flexible diamine such as 1,4-diaminobutane occurred faster than the homogeneous reaction of NDI2 in a monodisperse solution, affording unique diamine cross-linked oligomers and polymers of S-NDI2. The acceleration of this reaction should be due to intra-supramolecular cross-linking between preorganized S-NDI2 units and flexible diamines in close proximity. Intra-supramolecular cross-linking did not occur efficiently when an incompatible diamine was used. The present study provides an excellent opportunity to systematically compare the reaction kinetics of an in situ reaction within supramolecular polymers and a homogeneous reaction in monodisperse solutions. The molecular design that can implement chemical reactivity in supramolecular polymers and the proximity effect of in situ reactions shown here will be useful for further development of supramolecular polymers based on π-conjugated molecules with unique dynamic properties and reactivity.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
Atsuro Takai conceived and co-supervised the project. Minghan Tan mainly performed the experiments, analyzed the data and co-wrote the original draft with Atsuro Takai. Masayuki Takeuchi co-supervised the project. All the authors reviewed and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Ms Izumi Matsunaga, Ms Nozomi Kishida and Dr Takanobu Hiroto (NIMS) for their help in the syntheses of precursor compounds, UV-vis absorption spectral measurements and XRD measurements, respectively. We are also grateful to Ms Debbie Le and Dr Mark MacLachlan (The University of British Columbia) for their contribution in the early stage of this work. This research was supported by a Grant-in-Aid for Scientific Research (C) from JSPS 19K05640 to A. T., a Grant-in-Aid from the Murata Science Foundation to A. T., a Grant-in-Aid for Transformative Research Areas (A) “Condensed Conjugation” from MEXT (20H05868 to M. T.), and a MEXT “NIMS Molecule and Material Synthesis Platform” program and the Materials Analysis Station of NIMS. The quantum chemical calculations in this study were partially performed on the Numerical Materials Simulator at NIMS.References
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491 CrossRef CAS PubMed.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687 CrossRef CAS PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813 CrossRef CAS PubMed.
- D. B. Amabilino, D. K. Smith and J. W. Steed, Chem. Soc. Rev., 2017, 46, 2404 RSC.
- P. K. Hashim, J. Bergueiro, E. W. Meijer and T. Aida, Prog. Polym. Sci., 2020, 105, 101250 CrossRef CAS.
- J.-M. Lehn, Angew. Chem., Int. Ed., 2015, 54, 3276 CrossRef CAS PubMed.
- E. Mattia and S. Otto, Nat. Nanotechnol., 2015, 10, 111 CrossRef CAS PubMed.
- J.-F. Lutz, J.-M. Lehn, E. W. Meijer and K. Matyjaszewski, Nat. Rev. Mater., 2016, 1, 16024 CrossRef CAS.
- S. A. P. van Rossum, M. Tena-Solsona, J. H. van Esch, R. Eelkema and J. Boekhoven, Chem. Soc. Rev., 2017, 46, 5519 RSC.
- B. A. Grzybowski, K. Fitzner, J. Paczesny and S. Granick, Chem. Soc. Rev., 2017, 46, 5647 RSC.
- A. Sorrenti, J. Leira-Iglesias, A. J. Markvoort, T. F. A. de Greef and T. M. Hermans, Chem. Soc. Rev., 2017, 46, 5476 RSC.
- J. W. Chen, F. K. C. Leung, M. C. A. Stuart, T. Kajitani, T. Fukushima, E. van der Giessen and B. Feringa, Nat. Chem., 2018, 10, 132 CrossRef CAS PubMed.
- G. Ragazzon and L. J. Prins, Nat. Nanotechnol., 2018, 13, 882 CrossRef CAS PubMed.
- S. H. Jung, M. Takeuchi and K. Sugiyasu, in Kinetic Control in Synthesis and Self-Assembly, ed. M. Numata, S. Yagai and T. Hamura, Academic Press, 2019, p. 205 Search PubMed.
- J. Matern, Y. Dorca, L. Sánchez and G. Fernández, Angew. Chem., Int. Ed., 2019, 58, 16730 CrossRef CAS PubMed.
- Out-of-Equilibrium (Supra)molecular Systems and Materials, ed. N. Giuseppone and A. Walther, Wiley-VCH, Weinheim, Germany, 2021 Search PubMed.
- M. Weißenfels, J. Gemen and R. Klajn, Chem, 2021, 7, 23 Search PubMed.
- S. Amano, S. Borsley, D. A. Leigh and Z. Sun, Nat. Nanotechnol., 2021, 16, 1057 CrossRef CAS PubMed.
- J. Boekhoven, A. M. Brizard, K. N. K. Kowlgi, G. J. M. Koper, R. Eelkema and J. H. van Esch, Angew. Chem., Int. Ed., 2010, 49, 4825 CrossRef CAS PubMed.
- H. Shigemitsu and I. Hamachi, Acc. Chem. Res., 2017, 50, 740 CrossRef CAS PubMed.
- M. Kumar, N. L. Ing, V. Narang, N. K. Wijerathne, A. I. Hochbaum and R. V. Ulijn, Nat. Chem., 2018, 10, 696 CrossRef CAS PubMed.
- S. Dhiman and S. J. George, Bull. Chem. Soc. Jpn., 2018, 91, 687 CrossRef CAS.
- A. Mishra, S. Dhiman and S. J. George, Angew. Chem., Int. Ed., 2021, 60, 2740 CrossRef CAS PubMed.
- M. Masuda, P. Jonkheijm, R. P. Sijbesma and E. W. Meijer, J. Am. Chem. Soc., 2003, 125, 15935 CrossRef CAS PubMed.
- M. F. C. Romera, X. W. Lou, J. Schill, G. ter Huurne, P. P. K. H. Fransen, I. K. Voets, C. Storm and R. P. Sijbesma, J. Am. Chem. Soc., 2018, 140, 17547 CrossRef PubMed.
- T. Fukui, T. Uchihashi, N. Sasaki, H. Watanabe, M. Takeuchi and K. Sugiyasu, Angew. Chem., Int. Ed., 2018, 57, 15465 CrossRef CAS PubMed.
- M. F. J. Mabesoone, G. M. ter Huurne, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2020, 142, 12400 CrossRef CAS PubMed.
- K. Gao, Z. Zhang, L. Ma, L. Chen, X. Chen, Y. Zhang and M. Zhang, Giant, 2020, 4, 100034 CrossRef.
- K. M. Vonk, E. W. Meijer and G. Vantomme, Chem. Sci., 2021, 12, 13572 RSC.
- Z. Chen, Y. Suzuki, A. Imayoshi, X. Ji, K. V. Rao, Y. Omata, D. Miyajima, E. Sato, A. Nihonyanagi and T. Aida, Nat. Mater., 2022, 21, 253 CrossRef CAS PubMed.
- K. Sato, W. Ji, L. C. Palmer, B. Weber, M. Barz and S. I. Stupp, J. Am. Chem. Soc., 2017, 139, 8995 CrossRef CAS PubMed.
- S. M. Chin, C. V. Synatschke, S. P. Liu, R. J. Nap, N. A. Sather, Q. F. Wang, Z. Álvarez, A. N. Edelbrock, T. Fyrner, L. C. Palmer, I. Szleifer, M. O. de la Cruz and S. I. Stupp, Nat. Commun., 2018, 9, 2395 CrossRef PubMed.
- K. Sada, M. Takeuchi, N. Fujita, M. Numata and S. Shinkai, Chem. Soc. Rev., 2007, 36, 415 RSC.
- Y. Kitamoto, Z. Pan, D. D. Prabhu, A. Isobe, T. Ohba, N. Shimizu, H. Takagi, R. Haruki, S. Adachi and S. Yagai, Nat. Commun., 2019, 10, 4578 CrossRef PubMed.
- E. Krieg, E. Shirman, H. Weissman, E. Shimoni, S. G. Wolf, I. Pinkas and B. Rybtchinski, J. Am. Chem. Soc., 2009, 131, 14365 CrossRef CAS PubMed.
- J. Leira-Iglesias, A. Sorrenti, A. Sato, P. A. Dunne and T. M. Hermans, Chem. Commun., 2016, 52, 9009 RSC.
- E. Moulin, J. J. Armao and N. Giuseppone, Acc. Chem. Res., 2019, 52, 975 CrossRef CAS PubMed.
- W. A. Ogden and Z. Guan, ChemSystemsChem, 2020, 2, e1900030 CrossRef CAS.
- A. Takai and M. Takeuchi, Bull. Chem. Soc. Jpn., 2018, 91, 44 CrossRef CAS.
- K. Nakano, H. Sanematsu, Y. Kaji, A. Takai and K. Tajima, Chem.–Eur. J., 2020, 26, 15931 CrossRef CAS PubMed.
- M. Tan, R. Chrostowski, H. Sanematsu, M. Takeuchi and A. Takai, Asian J. Org. Chem., 2021, 10, 918 CrossRef CAS.
- N. Sakai, J. Mareda, E. Vauthey and S. Matile, Chem. Commun., 2010, 46, 4225 RSC.
- G. M. Prentice, L. Emmett, V. Luxami and G. D. Pantoş, in Naphthalenediimide and its Congeners: From Molecules to Materials, ed. G. D. Pantoş, The Royal Society of Chemistry, Croydon, UK, 2017, ch. 1, p. 1 Search PubMed.
- S. V. Bhosale, M. Al Kobaisi, R. W. Jadhav, P. P. Morajkar, L. A. Jones and S. George, Chem. Soc. Rev., 2021, 50, 9845 RSC.
- A. Mukherjee and S. Ghosh, Org. Mater., 2021, 3, 281 CrossRef CAS.
- H. D. Martin and B. Mayer, Angew. Chem., Int. Ed., 1983, 22, 283 CrossRef.
- A. H. Hoveyda, D. A. Evans and G. C. Fu, Chem. Rev., 1993, 93, 1307 CrossRef CAS.
- M. C. Whisler, S. MacNeil, V. Snieckus and P. Beak, Angew. Chem., Int. Ed., 2004, 43, 2206 CrossRef CAS PubMed.
- S. Santra and P. R. Andreana, Angew. Chem., Int. Ed., 2011, 50, 9418 CrossRef CAS PubMed.
- J. F. Shi, Y. Z. Wu, S. H. Zhang, Y. Tian, D. Yang and Z. Y. Jiang, Chem. Soc. Rev., 2018, 47, 4295 RSC.
- P. Mukhopadhyay, Y. Iwashita, M. Shirakawa, S. Kawano, N. Fujita and S. Shinkai, Angew. Chem., Int. Ed., 2006, 45, 1592 CrossRef CAS PubMed.
- X.-Q. Li, V. Stepanenko, Z. J. Chen, P. Prins, L. D. A. Siebbeles and F. Würthner, Chem. Commun., 2006, 3871 RSC.
- M. R. Molla and S. Ghosh, Chem. Mater., 2011, 23, 95 CrossRef CAS.
- S. Ogi, V. Stepanenko, J. Theirs and F. Würthner, J. Am. Chem. Soc., 2016, 138, 670 CrossRef CAS PubMed.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962 CrossRef PubMed.
- J. Fan, X. Chang, M. He, C. Shang, G. Wang, S. Yin, H. Peng and Y. Fang, ACS Appl. Mater. Interfaces, 2016, 8, 18584 CrossRef CAS PubMed.
- E. E. Greciano, J. Calbo, E. Ortí and L. Sánchez, Angew. Chem., Int. Ed., 2020, 59, 17517 CrossRef CAS PubMed.
- IUPAC Nomenclature of Organic Chemistry, https://www.acdlabs.com/iupac/nomenclature/79/r79_166.htm, accessed March 2022 Search PubMed.
- S. Ghosh, X. Q. Li, V. Stepanenko and F. Würthner, Chem.–Eur. J., 2008, 14, 11343 CrossRef CAS PubMed.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376 CrossRef PubMed.
- M. M. J. Smulders, M. M. L. Nieuwenhuizen, T. F. A. de Greef, P. van der Schoot, A. P. H. J. Schenning and E. W. Meijer, Chem.–Eur. J., 2010, 16, 362 CrossRef CAS PubMed.
- It should be mentioned that the formation of precipitates of ref-S-NDI aggregates was observed below 298 K.
- F. Würthner, S. Ahmed, C. Thalacker and T. Debaerdemaeker, Chem.–Eur. J., 2002, 8, 4742 CrossRef.
- H. Kar, D. W. Gehrig, N. K. Allampally, G. Fernández, F. Laquai and S. Ghosh, Chem. Sci., 2016, 7, 1115 RSC.
- This is because the reaction between the amine monoadduct (S-NDI2–DEA) and DEA is much slower than that between S-NDI2 and DEA. In the presence of a large excess amount of DEA (159 mM), we observed a two step reaction of the S-NDI2 supramolecular polymer, indicating the formation of a supramolecular assembly based on S-NDI2–(DEA)2. In contrast, in the presence of DEA (1.24 mM), the concentration of S-NDI2–(DEA)2 is less than 3 μM even 1200 min after the start of the reaction. See Fig. S5† for details.
- H. K. Hall, J. Am. Chem. Soc., 1957, 79, 5441 CrossRef CAS.
- It should be mentioned that the formation of precipitates of S-NDI2–DEA aggregates was observed below 297 K.
- A. Arjona-Esteban, J. Krumrain, A. Liess, M. Stolte, L. Z. Huang, D. Schmidt, V. Stepanenko, M. Gsänger, D. Hertel, K. Meerholz and F. Würthner, J. Am. Chem. Soc., 2015, 137, 13524 CrossRef CAS PubMed.
- F. Würthner, Acc. Chem. Res., 2016, 49, 868 CrossRef PubMed.
- Some reviewers pointed out the terminology of the supramolecular assemblies observed in the middle of the reaction. We agree with the reviewers that these species are not the same as the transient assemblies of single monomers formed over time. On the other hand, we should note that these species are also different from the conventional reaction intermediate that has a local potential energy minimum.
- C. Kulkarni, S. Balasubramanian and S. J. George, ChemPhysChem, 2013, 14, 661 CrossRef CAS PubMed.
- C. Kulkarni, K. K. Bejagam, S. P. Senanayak, K. S. Narayan, S. Balasubramanian and S. J. George, J. Am. Chem. Soc., 2015, 137, 3924 CrossRef CAS PubMed.
- We unified the concentration of the amine to be 1.24 mM, which is the same as in the experiments using DEA.
- A. Ashcraft, K. X. Liu, A. Mukhopadhyay, V. Paulino, C. Liu, B. Bernard, D. Husainy, T. Phan and J. H. Olivier, Angew. Chem., Int. Ed., 2020, 59, 7487 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2sc00035k |
| This journal is © The Royal Society of Chemistry 2022 |