DOI:
10.1039/D2SC00377E
(Edge Article)
Chem. Sci., 2022,
13, 4397-4405
An efficient factor for fast screening of high-performance two-dimensional metal–organic frameworks towards catalyzing the oxygen evolution reaction†
Received
20th January 2022
, Accepted 7th March 2022
First published on 9th March 2022
Abstract
Two-dimensional (2D) metal–organic frameworks (MOFs) are promising materials for catalyzing the oxygen evolution reaction (OER) due to their abundant exposed active sites and high specific surface area. However, how to rapidly screen out highly-active 2D MOFs from numerous candidates is still a great challenge. Herein, based on the high-throughput density functional theory (DFT) calculations for 20 kinds of different transition metal-based MOFs, we propose a factor for fast screening of 2D MOFs for the OER under alkaline conditions (pH = 14.0), that is, when the Gibbs free energy change of the O–O bond formation (defined as ΔG1) is located at ∼1.15 eV, the peak OER performance would be achieved. Based on the high-throughput calculation results, the prediction factor can be further simplified by replacing the Gibbs free energy with the sum of the associated single point energy (SPE) and a binding energy-dependent term. Guided by this factor, we successfully predicted and then obtained the high-performance Ni-based 2D MOFs. This factor would be a practical approach for fast screening of 2D MOF candidates for the OER, and also provide a meaningful reference for the study of other materials.
Introduction
Developing clean energy materials is of great importance since the environmental issues caused by fossil fuels have been increasingly emerging.1–5 Hydrogen, which is a kind of environmentally-friendly energy carrier with a high energy density,6 has drawn increasing attention.7,8 Water splitting has been widely considered to be a promising strategy to generate hydrogen.9,10 However, the electrocatalyzed oxygen evolution reaction (OER) is the efficiency-determining step of water splitting because the OER is a multistep, four-electron transfer process, which is a Gibbs free energy uphill process and then suffers from slow kinetics and requires a high overpotential to drive the reaction.11–13 Hence, designing highly efficient OER materials is a key subject to further improving the application of hydrogen energy technology.14–17
Metal–organic frameworks (MOFs) are a kind of porous material formed by the coordination bonds between inorganic metal atoms and organic ligands.18,19 Due to their unique structural features, MOFs could deliver a series of inherent advantages in heterogeneous catalysis, such as a well-defined chemical structure, large specific surface area and high porosity.20–22 Unfortunately, MOFs are usually assembled in the form of a three-dimensional (3D) network structure, which hinders the metal atoms in the body phase from being accessed by the reactants, and thus limits the enhancement of catalytic activity.23 To address this issue, ultra-thinning MOFs into two-dimensional (2D) nanosheets is an efficient strategy to obtain MOF-based high-performance catalysts due to the ultrathin thickness for rapid mass and electron transfer, and high percentages of exposed active metal sites.24 Thus, 2D MOFs have been widely studied and have become one of the hottest subjects in the catalysis field.25,26 However, a noteworthy fact is that more than 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 kinds of MOFs have been reported so far, and this value is still growing rapidly.27 Screening out desired high-performance 2D MOFs from numerous candidates efficiently remains a challenge. Therefore, a convenient screening factor is urgently needed to improve screening efficiency. However, there has been no correlative report for a MOF screening factor in the OER catalysis system so far. Exploring a general factor that can be used to accelerate the screening process would be greatly significant.28–30
000 kinds of MOFs have been reported so far, and this value is still growing rapidly.27 Screening out desired high-performance 2D MOFs from numerous candidates efficiently remains a challenge. Therefore, a convenient screening factor is urgently needed to improve screening efficiency. However, there has been no correlative report for a MOF screening factor in the OER catalysis system so far. Exploring a general factor that can be used to accelerate the screening process would be greatly significant.28–30
Since the formation of the O–O bond and the deprotonation step in the OER process have a great influence on the reaction thermodynamics, the binding strength of the related intermediates may be highly correlated with the overpotential. This view was verified by our high-throughput calculations and experimental results. Based on these, we propose a factor for constructing high-performance 2D MOFs for the OER in this work, and it points out that when the Gibbs free energy change of the O–O bond formation step (defined as ΔG1) is around 1.15 eV, the peak OER performance would be obtained. With the ΔG1 further deviating from this value, a higher overpotential for the OER would be needed to drive the reaction. We further predicted that Ni-based MOFs would offer better activity than other counterparts based on this factor, which was validated by the experimental results and so as to the predictive power of the factor. This successful practice indicates that the factor would offer credible guidance for constructing highly active MOFs for the OER, and would significantly reduce the research cost and shorten the development period thanks to its lower amount of computation compared with direct high-throughput calculation screening.
Results and discussion
According to a previous report,31 the potential determining step (PDS) of the OER under alkaline conditions (pH = 14.0) is the formation of O–O bonding (eqn (1)) or the deprotonation process (eqn (2)).| | | M–O + OH− → M–O–OH + e− | (1) |
| | | M–O–OH + OH− → M–O–O + H2O + e− | (2) |
Here, M is the active metal site of catalysts, e− is the electron and OH− is hydroxyl, and the Gibbs free energy change of eqn (1) (that is the ΔG1), and the Gibbs free energy change of the second deprotonation step (the reaction in eqn (2), defined as ΔG2) could be derived as follows:| | | ΔG1 = G(M–O–OH) − G(OH−) − G(M–O) | (3) |
| | | ΔG2 = G(M–O–O) + G(H2O) − G(OH−) − G(M–O–OH) | (4) |
Here, G is the Gibbs free energy of the corresponding species, and similarly hereinafter. The overall equation is as follows:| | | ΔG1 + ΔG2 = G(M–O–O) − G(M–O) − 2G(OH−) + G(H2O) | (5) |
The high-throughput DFT calculations (see computational details in the ESI†) of the MOFs based on the first twenty kinds of transition metals, which cover a wide range of the commonly used transition metals to prepare MOFs, indicate that ΔG1 + ΔG2 remain unchanged as the type of metal ions varies due to the constant difference between G(M–O–O) and G(M–O). Here, we defined the difference between G(M–O–O) and G(M–O) as ΔGO:
| | | ΔGO = G(M–O–O) − G(M–O) | (6) |
As shown in Fig. 1a, twenty kinds of different transition metal-based MOFs possess an almost constant value of ΔGO (the fluctuation is about 0.01 eV per cell, 10−3 eV per atom), which agrees with the tendency on oxide surfaces that was reported in the previous literature,32 indicating that OER processes on MOF surfaces follow the same scaling relationship as that on metal oxide surfaces. This could be explained as follows: in this equation, the G(OH−) and G(H2O) are constants no matter what the metal is. Besides, the ΔGO was also observed to be a constant due to their similar structure derived nearly equal zero point energy (ZPE) and entropy correction TS term (Fig. S1†), that is, M–O–O is just an additional oxygen atom bonded to the adsorbed oxygen atoms of M–O, so the ΔGO should be approximately the sum of the atomic energy of the oxygen atom and the bonding energy of the O–O bond.
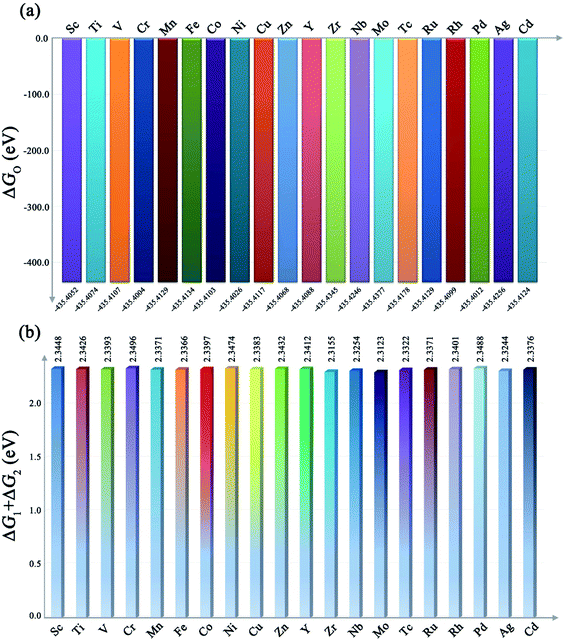 |
| | Fig. 1 (a) The value of ΔGO of different metal-based MOFs. (b) The values of ΔG1 + ΔG2 of different metal-based MOFs. | |
Taking the OH− and H2O molecules into consideration, the value of ΔG1 + ΔG2 would be obtained. In this work, the G(OH−) was derived from the definition of the standard hydrogen electrode (SHE) because the periodic DFT calculations can't predict the thermodynamic properties of OH− since it would cause the artificial electrostatic interaction between the periodic images of the charged ions.33 Hence, the G(OH−) is derived as follows:
| | | G(OH−) = G(H2O) − 1/2G(H2) + 0.0592pH | (7) |
Here, the pH is the pH value of the system. Hence,
eqn (5) could be transformed to be the following equation:
| | | ΔG1 + ΔG2 = G(M–O–O) − G(M–O) − G(H2O) + G(H2) + 0.1184pH | (8) |
When the calculated Gibbs free energies of H2O (G(H2O) = −466.98 eV) and H2 (G(H2) = −30.89 eV) were substituted in eqn (8) with pH = 14.0, we can conclude the following equation:
| | | ΔG1 + ΔG2 = G(M–O–O) − G(M–O) + 437.75 eV | (9) |
Combined with the result of ΔGO shown in Fig. 1a, we can obtain the value of ΔG1 + ΔG2. As shown in Fig. 1b, the value of ΔG1 + ΔG2 is also a constant value of ∼2.3 eV for different metal-based MOFs since the ΔGO is almost constant as mentioned above. Based on this, it is reasonable to conclude that when ΔG1 = ΔG2 = 1.15 eV, the lowest overpotential, namely, the peak OER performance would be achieved since the overall overpotential is dependent on the max[ΔG1, ΔG2] = max[ΔG1, 2.3 − ΔG1]. This implies that the following condition must be met to achieve the peak OER performance:
| | | ΔG1 = G(M–O–OH) − G(OH−) − G(M–O) = 1.15 eV | (10) |
Here, we defined the difference between
G(M–O–OH) −
G(M–O) and
G(OH
−) as Δ
GOH:
| | | ΔGOH = G(M–O–O–H) − G(M–O) | (11) |
Considering that ΔG1 is the difference between ΔGOH and G(OH−), we can replace the G(OH−) with eqn (7) and obtain the below factor to achieve the peak OER performance:
| | | ΔGOH = G(M–O–OH) − G(M–O) = −449.56 eV | (12) |
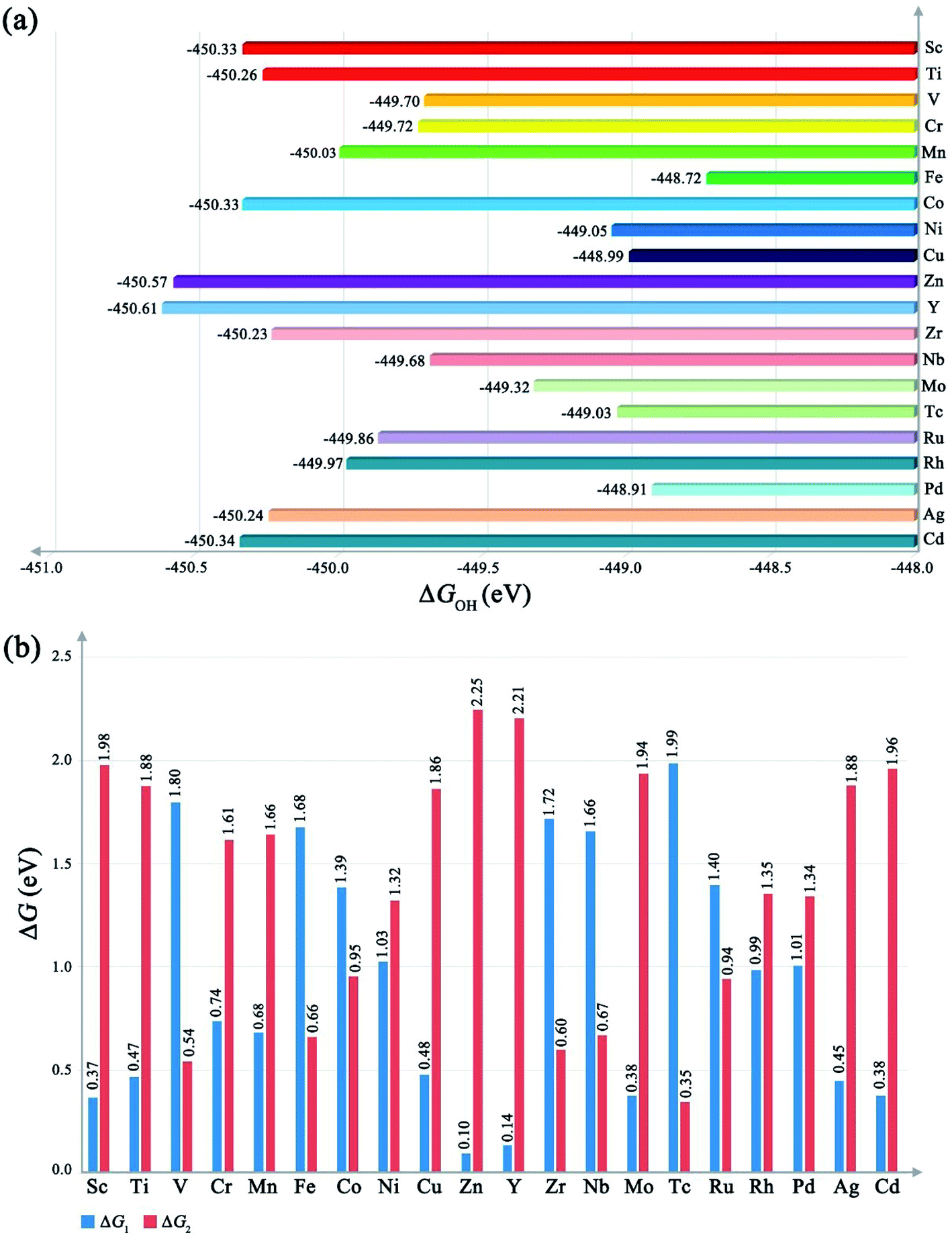
As shown in Fig. 2a, our results indicate that the ΔGOH of different metal-based MOFs is not a constant like ΔGO. This should be attributed to the more complex potential surface (PES) induced by the more structural freedom of M–OOH, especially for the H atom, which would significantly contribute to ZPE due to the light mass. The different ΔGOH values lead to the different performances of the OER. A too high ΔGOH value will lead to a high overpotential for the formation of O–O bonding, while a too small ΔGOH value will cause a high overpotential for the deprotonation process. From the ΔGOH, we could obtain the ΔG1 and ΔG2 respectively. As shown in Fig. 2b, different metal-based MOFs have different ΔG1 and ΔG2, implying that the PDS and overall overpotential are different as the metal ions change. Meanwhile, from the ΔG1 and ΔG2, we could also rank the OER performance for different MOFs theoretically according to eqn (10), that is, the closer to 1.15 eV of ΔG1, the lower the overpotential would be delivered.
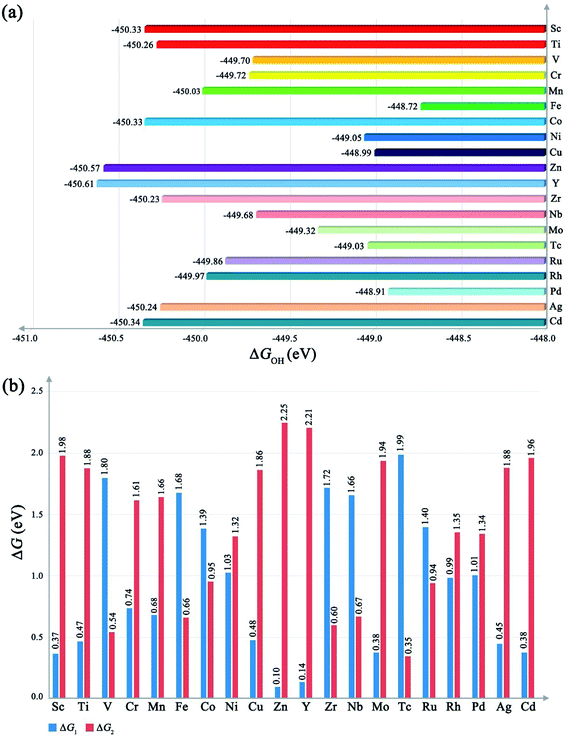 |
| | Fig. 2 (a) The value of ΔGOH of different metal-based MOFs. (b) The value of ΔG1 and ΔG2 of different metal-based MOFs. | |
Considering the requirement of the frequency, the calculation of Gibbs free energy is usually computationally intensive, leading to a high calculation cost.34 Hence, an ideal way to obtain the Gibbs free energy is by deriving from a specific quantity that is easy to obtain and strongly correlated with the Gibbs free energy. Based on this purpose, we seek to explore the desired specific quantity. Quite sensibly, the SPE, which is easily acquired, may be a promising candidate. Note that the difference between SPE and the Gibbs free energy is the contribution from the vibration at the limited temperature, and the vibration frequency is strongly dependent on the bond strength.35 Since the bond strength is usually described by the bond order, the difference between SPE and Gibbs free energy should have a certain correlation with the associated bond order, that is, the O–O bond and O–H bond. However, the O–O bond orders and O–H bond orders in different metal-based MOFs should not vary significantly, which is not enough to accurately distinguish different metal-based MOFs. Hence, the binding energy, derived from the formula below, is employed.36
| | | Ebinding(A–B) = E(A–B) − E(A) − E(B) | (13) |
Here, the
Ebinding(A–B) is the binding energy of A and B.
E(A–B) is the total energy of the binding configuration of A and B.
E(A) and
E(B) is the total energy of component A and component B, respectively. This inference was confirmed by the phonon calculations of M–OOH and M–O. As shown in
Fig. 3, the difference between Δ
GOH and Δ
EOH is a function of scaled O–O binding energy and scaled O–H binding energy. More specifically, the equation derived from machine learning based multiple linear regression (MLR)
37 is shown below with
R2 = 0.98:
| | | ΔGOH − ΔEOH = 0.78Ebinding(O–H) + 0.52Ebinding(O–O) | (14) |
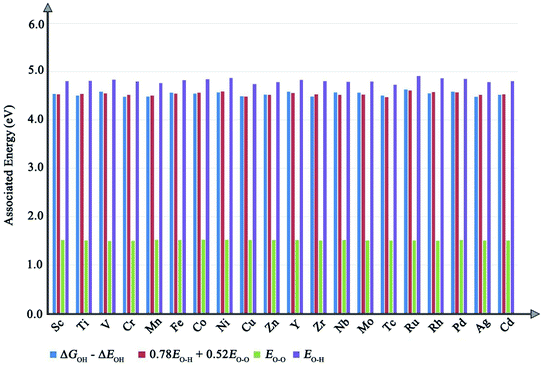 |
| | Fig. 3 The value of ΔGOH–ΔEOH, scaled and unscaled binding energies of O–O bonding and O–H bonding in different columns. | |
Hence, the ΔGOH could be replaced by the ΔEOH plus an additional binding energy-dependent term. As a result, this substitution would significantly reduce the amount of computation, lowering the calculation cost and shortening the calculation time as the computationally intensive frequency calculation was rationally substituted. Collectively, eqn (10) could be transformed as follows:
| | | ΔG1 = ΔEOH − G(OH−) + 0.78Ebinding(O–H) + 0.52Ebinding(O–O) = 1.15 eV | (15) |
Based on the factor proposed by eqn (15), we could predict the OER performance for different metal-based 2D MOFs rapidly, which is significant for catalyst design and screening.
Based on the prediction from the factor, the Ni, Co, Mn and Fe based 2D MOFs should deliver incremental performance in the order of Ni > Co > Mn > Fe. For validation purpose, we seek to verify the factor experimentally since the electrochemical measurements would offer a validation of the predictive power of the factor. Hence, four kinds of different metal-based 2D MOFs were prepared for further validation. The morphology of these four kinds of 2D MOF nanosheets is shown in Fig. 4 and S2,† in which the 2D ultrathin feature of the as-prepared MOFs could be identified clearly from the scanning electron microscope (SEM) and transmission electron microscopy (TEM) images, and this agrees with the simulated morphology (Fig. S3†) by means of the Bravais–Friedel–Donnay–Harker (BFDH) method.38 The high education ring dark field scanning transmission electron microscope (HAADF-STEM) images and the corresponding energy disperse spectroscopy (EDS) mapping images are shown in Fig. S4–S7,† indicating that the metal elements and N and O are uniformly distributed on the entire surface of these 2D MOFs, and the carbon element that dominates the field of view arises from the carbon membrane that was used to prepare the TEM sample. The size of the nanosheets was estimated at hundreds of nanometers and the thickness was measured at ∼10 nm through the side view SEM images of Ni-MOFs (Fig. S8†), which agree well with the atomic force microscopy (AFM) measurement shown in Fig. 4e–h. Besides, the powder X-ray diffraction (PXRD) results (Fig. S9†) showed that all these as-prepared MOFs are isostructural, and no obvious peak shift was detected, indicating that the interplanar spacing and lattice parameters almost remained unchanged, and this should be attributed to the similar atomic radius for Ni, Co, Fe and Mn. Moreover, the PXRD patterns are in accordance with the simulated pattern of the optimized model structure, which further confirmed the successful preparation of the 2D-MOFs. The high-resolution X-ray photoelectron spectroscopy (XPS) spectra of the metal elements are shown in Fig. S10.† According to previous reports,23,24 the location of these main peaks indicates that the metal nodes are coordinated octahedrally by six oxygen atoms. Based on the above results, the atomic arrangements in 2D MOFs could be well defined as shown in Fig. 4i and j. In addition, the N2 isotherm (Fig. S11†) of these four kinds of 2D MOFs at 77 K was supposed to be type IV with a H4-type hysteresis loop, which should be attributed to the formation of narrow slits. Note that these 2D MOFs have nearly the same Brunauer–Emmett–Teller (BET) specific surface area (25.91 m2 g−1, 26.32 m2 g−1, 25.47 m2 g−1, and 23.92 m2 g−1 for Ni-MOFs, Co-MOFs, Fe-MOFs and Mn-MOFs, respectively), which should be assigned to their similar lattice structure. Collectively, these results confirmed the successful preparation of 2D MOF nanosheets and were able to be used for subsequent evaluation of the OER performance experimentally.
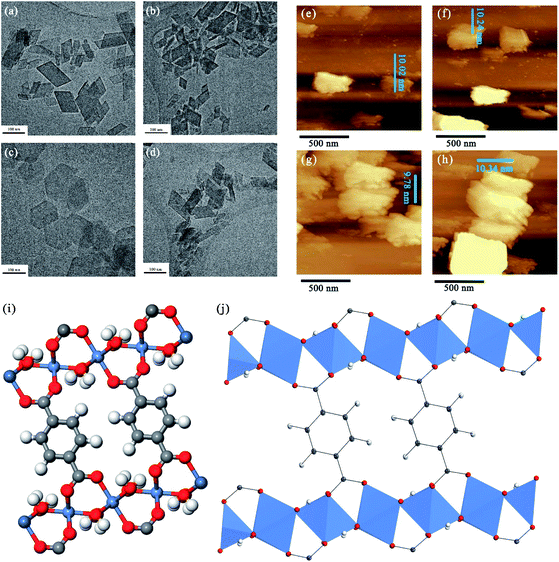 |
| | Fig. 4 TEM images of different metal-based 2D MOFs: (a) Ni-MOFs, (b) Co-MOFs, (c) Fe-MOFs and (d) Mn-MOFs; AFM images of different metal-based 2D MOFs: (e) Ni-MOFs, (f) Co-MOFs, (g) Fe-MOFs and (h) Mn-MOFs; (i) atomic arrangements of 2D MOFs and (j) coordination mode of 2D MOFs. | |
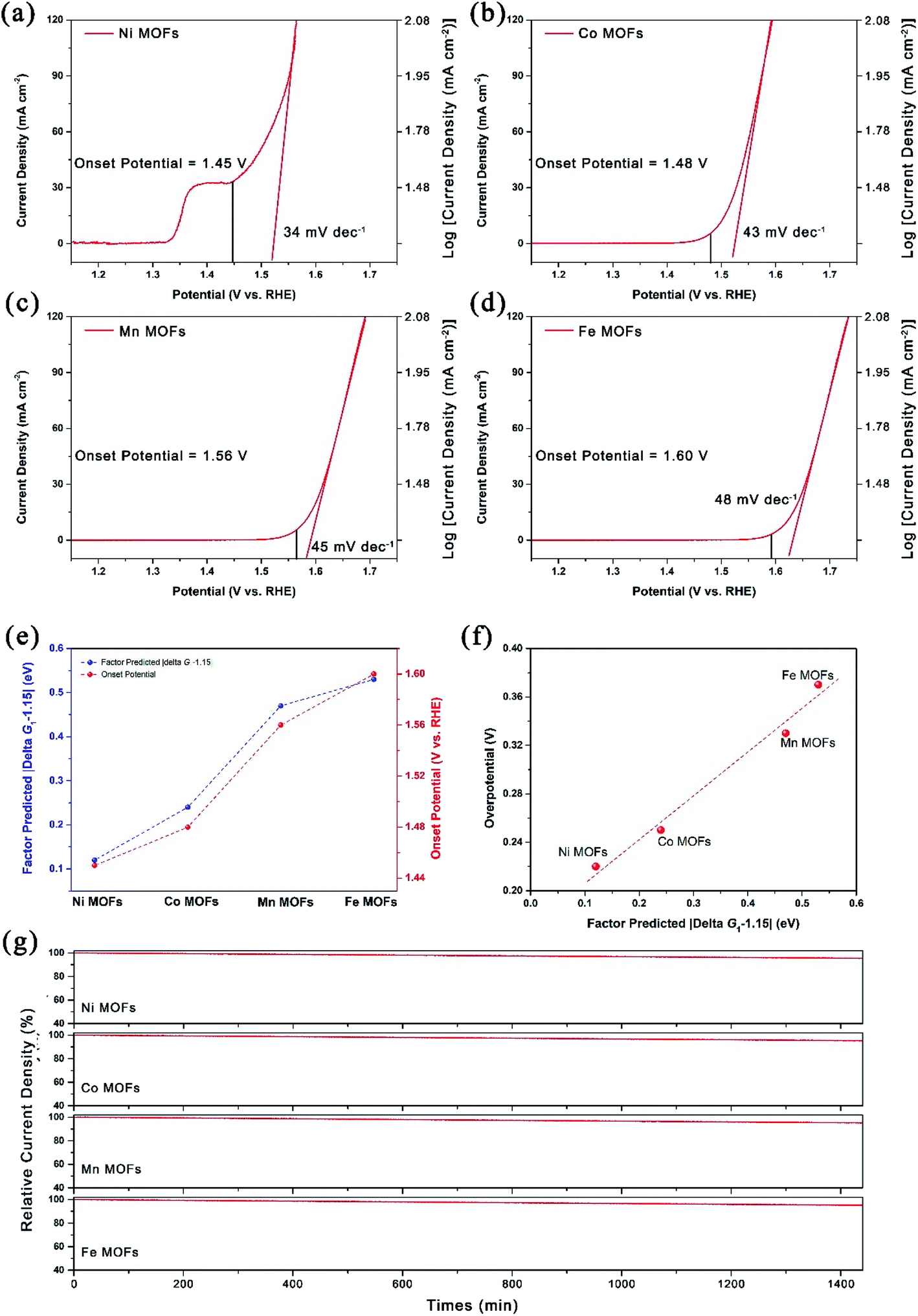
The OER performance was measured with a typical three-electrode configuration. For comparison purpose, OER performance was evaluated via the onset potential (defined as the potential to achieve a current density of 1 mA cm−2 for the OER) of these four kinds of MOFs. Note that the peak of Ni-MOFs around 1.37 V should be attributed to the oxidation of Ni.24 As shown in Fig. 5a–d, the OER performance was ranked in the order of Ni-MOFs > Co-MOFs > Mn-MOFs > Fe-MOFs with an onset potential of 1.45 V, 1.48 V, 1.56 V and 1.60 V versus the reversible hydrogen electrode (RHE) respectively, which agrees well with the calculated results and the factor proposed by eqn (10), that is, the ΔG1 of Ni-MOFs is the closest to 1.15 eV and thus delivered the best OER performance. As ΔG1 goes away from 1.15 eV, more positive potential was needed to drive the reaction (Fig. 5e). Note that the value of |ΔG1 − 1.15| in Fig. 5e is derived from eqn (15), rather than from the Gibbs free energy calculations, and it obviously indicates that the factor also has the predictive power after the ΔGOH in eqn (10) is substituted by the ΔEOH, and no significant loss of accuracy was detected. As a result, a good compromise is achieved between accuracy and computational cost. Besides, the factor in eqn (10) indicated that the onset potential should be linearly correlated to the |ΔG1 − 1.15|, and this was also observed in Fig. 5f, which further confirmed the credibility of the proposed factor.
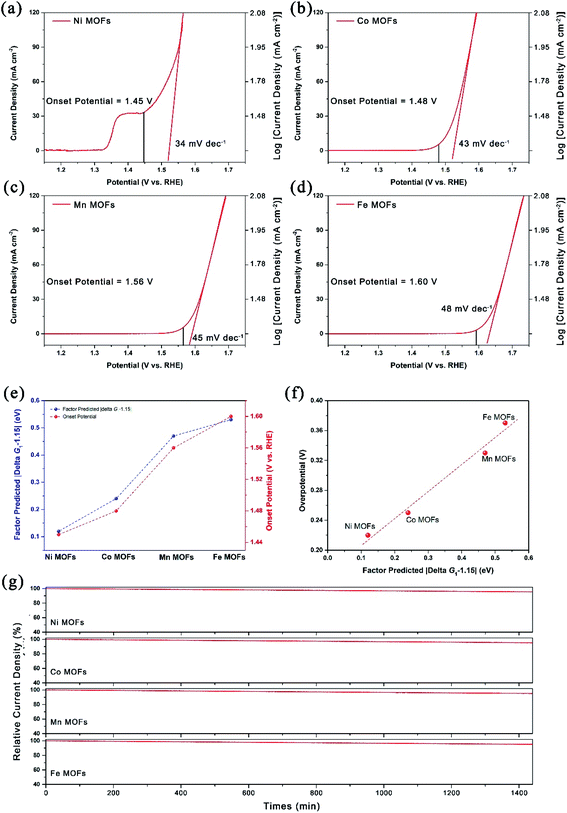 |
| | Fig. 5 (a) LSV curves and Tafel plots of (a) Ni-MOFs, (b) Co-MOFs, (c) Mn-MOFs and (d) Fe-MOFs; (e) correlation between onset potential and associated factor predicted values of |ΔG1 − 1.15|; (f) the linear correlation of overpotential and associated factor predicted values of |ΔG1 − 1.15|; (g) chronoamperometry plots of different 2D MOFs at 1.65 V vs. RHE for 24 h (1440 min). | |
Apart from the onset potential, the Tafel slope, which reflects the kinetics of the reaction, was also calculated based on the corresponding linear sweep voltammetry (LSV) curves. As shown in Fig. 5a, the Ni-MOFs could deliver the lowest Tafel slope of 34 mV dec−1 compared with other samples (43 mV dec−1 for Co-MOFs, 45 mV dec−1 for Mn-MOFs and 48 mV dec−1 for Fe-MOFs) in this work, confirming the outstanding reaction kinetics for the OER. Compared to state-of-the-art RuO2, IrO2 and Pt/C (Fig. S12†), the as prepared Ni-MOFs could deliver much better OER performance, which opened up fine prospects for practical applications. Moreover, the correlation between OER performance and the d-band center was also investigated. As shown in Fig. S13,† when the d-band center increased, the adsorption energy of *OH also increased, which agrees well with the d-band center proposed by Hammer and Nørskov.39–41 Meanwhile, a too high d-band center would lead to decreased OER activity, which could be explained by Sabatier's principle, which means that the best catalytic activity can be achieved with moderate binding strength, and too strong or too weak binding is not conducive to the catalytic process. Besides, the long-term stability of the catalysts is another key parameter in evaluating the OER performance. As shown in Fig. 5g, the chronoamperometry measurements at 1.65 V vs. RHE were also conducted and only <5% current density decay was observed after the 24 h test for all the samples, indicating the excellent stability of these prepared 2D MOFs, which possess the potential for production run.
Conclusions
In conclusion, this work proposes a factor for fast screening of 2D-MOFs for catalysis of the OER under alkaline conditions aided by the high-throughput DFT calculations. This factor is straightforward and feasible for practical application. When the ΔG1 is located at 1.15 eV, the peak OER performance will be delivered. Further deviation from this value would lead to a higher overpotential. Further simplification was achieved by replacing the Gibbs free energy with the sum of the associated SPE and a binding energy-dependent term. The predictive power of this factor was validated by our successful prediction and preparation of the high-performance Ni-based 2D MOFs. This factor would be a practical approach for fast screening of 2D MOF candidates for the OER, and also provides a meaningful reference for the study of other materials.
Data availability
All the calculation data and experimental data reported in this paper could be accessed from the authors.
Author contributions
Guangtong Hai: conceptualization, data curation, formal analysis, investigation, computation, writing – original manuscript, writing – review & editing manuscript. Hongyi Gao: data curation, funding acquisition. Xiubing Huang: data curation. Li Tan: data curation. Xiangdong Xue: data curation. Shihao Feng: data curation. Ge Wang: supervision, project administration, funding acquisition.
Conflicts of interest
No conflicts of interest are declared for any of the authors.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 51972024) and the Fundamental Research Funds for the Central Universities (FRF-IDRY-20-004).
References
- H. Furukawa and O. M. Yaghi, Storage of hydrogen, methane, and carbon dioxide in highly porous covalent organic frameworks for clean energy applications, J. Am. Chem. Soc., 2009, 131(25), 8875–8883 CrossRef CAS PubMed.
- Z. Wang, J. Xu and J. Yang,
et al., Ultraviolet/ozone treatment for boosting OER activity of MOF nanoneedle arrays, Chem. Eng. J., 2022, 427, 131498 CrossRef CAS.
- F. Li, Y. Tian and S. Su,
et al., Theoretical and experimental exploration of tri-metallic organic frameworks (t-MOFs) for efficient electrocatalytic oxygen evolution reaction, Appl. Catal., B, 2021, 299, 120665 CrossRef CAS.
- J. Du, F. Li and L. Sun, Metal-organic frameworks and their derivatives as electrocatalysts for the oxygen evolution reaction, Chem. Soc. Rev., 2021, 50(4), 2663–2695 RSC.
- C. P. Wang, Y. Feng and H. Sun,
et al., Self-optimized metal-organic framework electrocatalysts with structural stability and high current tolerance for water oxidation, ACS Catal., 2021, 11, 7132–7143 CrossRef CAS.
- G. Korotcenkov, S. D. Han and J. R. Stetter, Review of electrochemical hydrogen sensors, Chem. Rev., 2009, 109(3), 1402–1433 CrossRef CAS PubMed.
- G. Li, J. Yu and Z. Zhou,
et al., N-doped Mo2C nanobelts/graphene nanosheets bonded with hydroxy nanocellulose as flexible and editable electrode for hydrogen evolution reaction, iScience, 2019, 19, 1090–1100 CrossRef CAS PubMed.
- T. Liu, P. Li and N. Yao,
et al., CoP-doped MOF-based electrocatalyst for pH-universal hydrogen evolution reaction, Angew. Chem., Int. Ed., 2019, 58(14), 4679–4684 CrossRef CAS PubMed.
- C. S. Gopinath and N. Nalajala, A scalable and thin film approach for solar hydrogen generation: a review on enhanced photocatalytic water splitting, J. Mater. Chem. A, 2021, 9(3), 1353–1371 RSC.
- L. Jin, H. Zhao and Z. M. Wang,
et al., Quantum dots-based photoelectrochemical hydrogen evolution from water splitting, Adv. Energy Mater., 2021, 11(12), 2003233 CrossRef CAS.
- W. Cheng, X. Zhao and H. Su,
et al., Lattice-strained metal-organic-framework arrays for bifunctional oxygen electrocatalysis, Nat. Energy, 2019, 4(2), 115–122 CrossRef CAS.
- F. Jaouen, Predicting electrochemical activity, Nat. Catal., 2018, 1(5), 314–315 CrossRef.
- S. W. Boettcher, Oxygen stays put during water oxidation, Nat. Catal., 2018, 1(11), 814–815 CrossRef CAS.
- Z. Y. Yu, Y. Duan and X. Y. Feng,
et al., Clean and affordable hydrogen fuel from alkaline water splitting: past, recent progress, and future prospects, Adv. Mater., 2021, 33(31), 2007100 CrossRef CAS PubMed.
- Y. Li, X. Wei and S. Han,
et al., MnO2 electrocatalysts coordinating alcohol oxidation for ultra-durable hydrogen and chemical productions in acidic solutions, Angew. Chem., Int. Ed., 2021, 60(39), 21464–21472 CrossRef CAS PubMed.
- W. Li, J. Liu and P. Guo,
et al., Co/CoP heterojunction on hierarchically ordered porous carbon as a highly efficient electrocatalyst for hydrogen and oxygen evolution, Adv. Energy Mater., 2021, 11(42), 2102134 CrossRef CAS.
- B. Zhao, J. Liu and C. Xu,
et al., Hollow NiSe nanocrystals heterogenized with carbon nanotubes for efficient electrocatalytic methanol upgrading to boost hydrogen Co-production, Adv. Funct. Mater., 2021, 31(8), 2008812 CrossRef CAS.
- J. R. Long and O. M. Yaghi, The pervasive chemistry of metal-organic frameworks, Chem. Soc. Rev., 2009, 38(5), 1213–1214 RSC.
- O. M. Yaghi, Metal-organic frameworks: a tale of two entanglements, Nat. Mater., 2007, 6(2), 92–93 CrossRef CAS PubMed.
- D. Britt, D. Tranchemontagne and O. M. Yaghi, Metal-organic frameworks with high capacity and selectivity for harmful gases, Proc. Natl. Acad. Sci., 2008, 105(33), 11623–11627 CrossRef CAS PubMed.
- H. Furukawa, N. Ko and Y. B. Go,
et al., Ultrahigh porosity in metal-organic frameworks, Science, 2010, 329(5990), 424–428 CrossRef CAS PubMed.
- Q. Li, W. Zhang and O. Š. Miljanić,
et al., Docking in metal-organic frameworks, Science, 2009, 325(5942), 855–859 CrossRef CAS PubMed.
- G. Hai, X. Jia and K. Zhang,
et al., High-performance oxygen evolution catalyst using two-dimensional ultrathin metal-organic frameworks nanosheets, Nano Energy, 2018, 44, 345–352 CrossRef CAS.
- S. Zhao, Y. Wang and J. Dong,
et al., Ultrathin metal-organic framework nanosheets for electrocatalytic oxygen evolution, Nat. Energy, 2016, 1(12), 16184 CrossRef CAS.
- R. Dong and X. Feng, Making large single crystals of 2D MOFs, Nat. Mater., 2021, 20(2), 122–123 CrossRef CAS PubMed.
- M. Wang, R. Dong and X. Feng, Two-dimensional conjugated metal-organic frameworks (2D c-MOFs): chemistry and function for MOFtronics, Chem. Soc. Rev., 2021, 50(4), 2764–2793 RSC.
- H. L. Nguyen, Perspectives on titanium-based metal-organic frameworks, J. Phys.: Energy, 2021, 3(2), 021003 CAS.
- M. Pardakhti, E. Moharreri and D. Wanik,
et al., Machine learning using combined structural and chemical descriptors for prediction of methane adsorption performance of metal organic frameworks (MOFs), ACS Comb. Sci., 2017, 19(10), 640–645 CrossRef CAS PubMed.
- J. Zhang, Z. Zhou and F. Wang,
et al., Two-dimensional metal hexahydroxybenzene frameworks as promising electrocatalysts for an oxygen reduction reaction, ACS Sustainable Chem. Eng., 2020, 8(19), 7472–7479 CrossRef CAS.
- T. Li, M. Li and X. Zhu,
et al., Conductive two-dimensional M3(C6S3O3)2 monolayers as effective electrocatalysts for the oxygen reduction reaction, J. Mater. Chem. A, 2021, 9, 24887–24894 RSC.
- J. Suntivich, K. J. May and H. A. Gasteiger,
et al., A perovskite oxide optimized for oxygen evolution catalysis from molecular orbital principles, Science, 2011, 334(6061), 1383–1385 CrossRef CAS PubMed.
- I. C. Man, H. Y. Su and F. Calle-Vallejo,
et al., Universality in oxygen evolution electrocatalysis on oxide surfaces, ChemCatChem, 2011, 3(7), 1159–1165 CrossRef CAS.
- Y. Yao, S. Hu and W. Chen,
et al., Engineering the electronic structure of single atom Ru sites via compressive strain boosts acidic water oxidation electrocatalysis, Nat. Catal., 2019, 2(4), 304–313 CrossRef CAS.
- B. Cheng and M. Ceriotti, Computing the absolute Gibbs free energy in atomistic simulations: applications to defects in solids, Phys. Rev. B, 2018, 97(5), 054102 CrossRef CAS.
- E. Voyiatzis and M. C. Böhm, An open-source implementation of a quasi harmonic approach to compute the intramolecular entropy of particle systems, Comput. Phys. Commun., 2019, 234, 286–293 CrossRef CAS.
- G. Hai, H. Gao and G. Zhao,
et al., Difference between metal-S and metal-O bond orders: a descriptor of oxygen evolution activity for isolated metal atom-doped MoS2 nanosheets, iScience, 2019, 20, 481–488 CrossRef CAS PubMed.
- Z. Li, B. J. Bucior and H. Chen,
et al., Machine learning using host/guest energy histograms to predict adsorption in metal-organic frameworks: application to short alkanes and Xe/Kr mixtures, J. Chem. Phys., 2021, 155(1), 014701 CrossRef CAS PubMed.
- J. D. H. Donnay and D. Harker, A new law of crystal morphology extending the law of Bravais, Am. Mineral., 1937, 22(5), 446–467 CAS.
- V. Pallassana, M. Neurock and L. B. Hansen,
et al., Theoretical analysis of hydrogen chemisorption on Pd(111), Re(0001) and PdML/Re(0001), ReML/Pd(111) pseudomorphic overlayers, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60(8), 6146 CrossRef CAS.
- B. Hammer, Y. Morikawa and J. K. Nørskov, CO chemisorption at metal surfaces and overlayers, Phys. Rev. Lett., 1996, 76(12), 2141 CrossRef CAS PubMed.
- A. Ruban, B. Hammer and P. Stoltze,
et al., Surface electronic structure and reactivity of transition and noble metals, J. Mol. Catal. A: Chem., 1997, 115(3), 421–429 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2sc00377e |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Li
Tan
c,
Xiangdong
Xue
a,
Shihao
Feng
a and
Ge
Wang
a,
Li
Tan
c,
Xiangdong
Xue
a,
Shihao
Feng
a and
Ge
Wang