
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controllable access to trifluoromethyl-containing indoles and indolines: palladium-catalyzed regioselective functionalization of unactivated alkenes with trifluoroacetimidoyl chlorides†
Hefei
Yang‡
a,
Le-Cheng
Wang‡
a,
Yu
Zhang
a,
Dongling
Zheng
a,
Zhengkai
Chen
 *a and
Xiao-Feng
Wu
*bc
*a and
Xiao-Feng
Wu
*bc
aDepartment of Chemistry, Key Laboratory of Surface & Interface Science of Polymer Materials of Zhejiang Province, Zhejiang Sci-Tech University, Hangzhou 310018, People's Republic of China. E-mail: zkchen@zstu.edu.cn
bDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, Liaoning, China. E-mail: xwu2020@dicp.ac.cn
cLeibniz-Institut für Katalyse e. V., Albert-Einstein-Straβe 29a, 18059 Rostock, Germany. E-mail: xiao-feng.wu@catalysis.de
First published on 3rd March 2022
Abstract
The synthesis of diverse products from the same starting materials is always attractive in organic chemistry. Here, a palladium-catalyzed substrate-controlled regioselective functionalization of unactivated alkenes with trifluoroacetimidoyl chlorides has been developed, which provides a direct but controllable access to a variety of structurally diverse trifluoromethyl-containing indoles and indolines. In more detail, with respect to γ,δ-alkenes, 1,1-geminal difunctionalization of unactivated alkenes with trifluoroacetimidoyl chloride enables the [4 + 1] annulation to produce indoles; as for β,γ-alkenes, a [3 + 2] heteroannulation with the hydrolysis product of trifluoroacetimidoyl chloride through 1,2-vicinal difunctionalization of alkenes occurs to deliver indoline products. The structure of alkene substrates differentiates the regioselectivity of the reaction.
Introduction
Transition-metal-catalyzed difunctionalization of alkenes has emerged as a powerful synthetic strategy for the assembly of structurally complex molecules.1 The transformation can forge two new C–C or C–X bonds for installing two different components into the C–C double bonds, which has aroused tremendous interest from many research groups. For the more difficult difunctionalization of unactivated alkenes, in recent years, the Engle group and others have developed a series of transition-metal-catalyzed alkene 1,2-difunctionalization reactions with the assistance of the 8-aminoquinoline (AQ) auxiliary as a strongly coordinating bidentate directing group, including hydrofunctionalization,2 dicarbofunctionalization,3 carboamination,4 carboboration and aminoboration,5 and so on (Scheme 1a). In most cases of the above transformations, the nucleopalladated alkylpalladium(II) species stabilized by a bidentate directing group were initially generated, and then protodepalladation or oxidative addition with electrophiles occurred to afford the difunctionalizated products.2a,b Meanwhile, a competitive β-hydride elimination process was usually suppressed by the conformational rigidity of the directing group.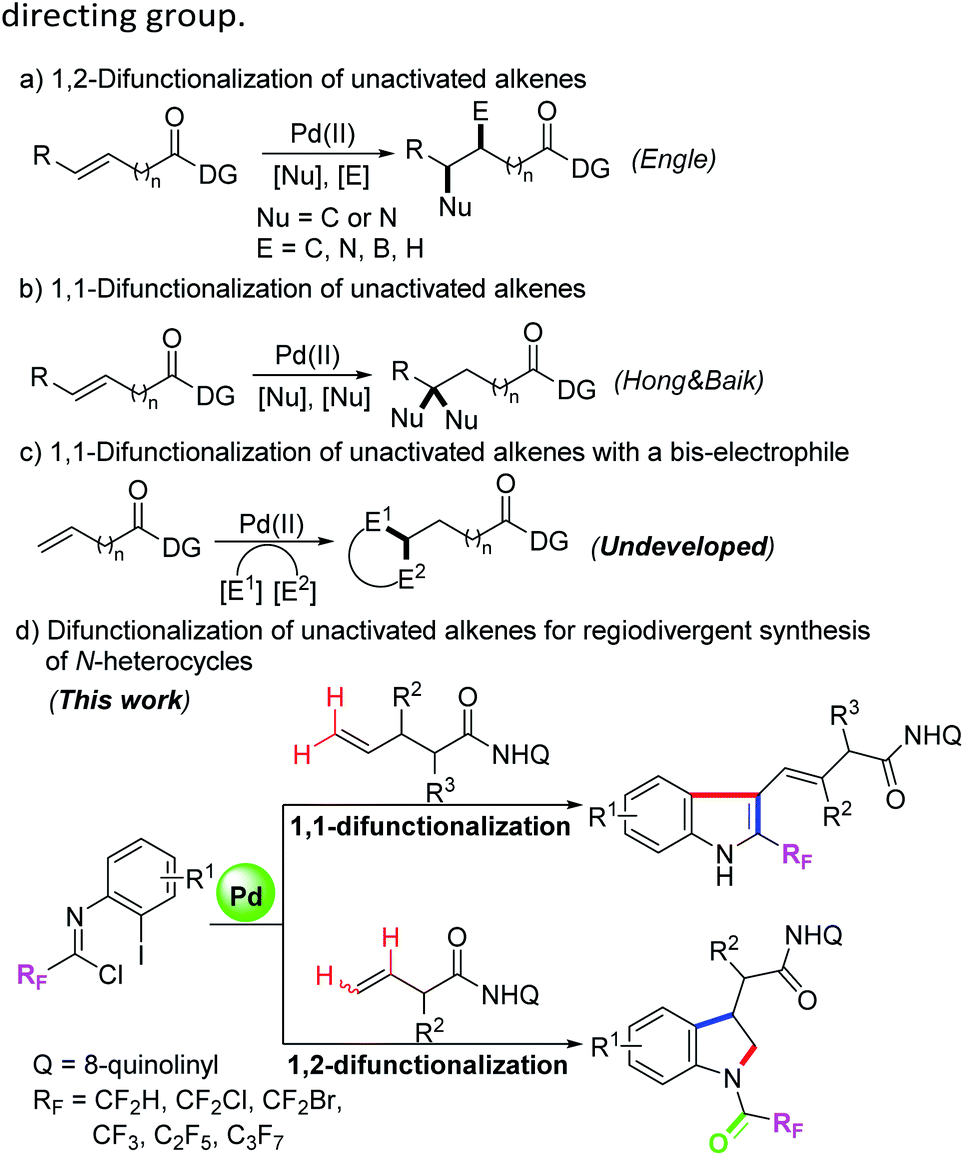 | ||
| Scheme 1 Palladium-catalyzed difunctionalization of unactivated alkenes. | ||
Compared with the rapid development of 1,2-vicinal difunctionalization protocols, reactions involving 1,1-geminal difunctionalization of unactivated alkenes have been rarely reported. To date, palladium-catalyzed 1,1-arylhalogenation, 1,1-diarylation and 1,1-arylborylation of alkenes have been explored by the use of prefunctionalized aryl sources.6 In 2019, Hong, Baik and their co-workers demonstrated a palladium(II)-catalyzed site-selective 1,1-difunctionalization of unactivated alkenes with two nucleophiles, wherein a regioselective β-H elimination of the cationic palladacycle and subsequent migratory insertion were involved (Scheme 1b).7 In the 1,1-difunctionalization reactions, the key step lies in the β-H elimination from a less stable six-membered palladacycle to regenerate an olefin moiety, thereby enabling the following olefin insertion.2b We surmised that the structural flexibility of the in situ formed palladacycle results in the regioselective difference of the 1,1-difunctionalization and 1,2-difunctionalization reactions. Pertinent to the present research, the utilization of difunctionalization of unactivated alkenes with two different electrophiles to produce structurally diverse heterocycles remains undeveloped and is of great significance (Scheme 1c).
Trifluoromethyl-substituted nitrogen-containing heterocycles as core skeletons widely exist in numerous bioactive and pharmaceutical molecules.8 Due to the unique properties of fluorine atoms, the physicochemical and pharmacological properties of the parent heterocyclic molecules could be obviously improved.9 Trifluoroacetimidoyl chlorides have been applied as a powerful and versatile fluorinated synthon for the assembly of trifluoromethyl-substituted N-heterocycles.10 Our group and others have developed a variety of synthetic methods for preparing various trifluoromethyl-containing N-heterocycles by the employment of trifluoroacetimidoyl chlorides as reactive partners.11 When the halogen atom was located at the ortho position of the aryl moiety of trifluoroacetimidoyl chloride, it will serve as a 4-atom reaction precursor with two electrophilic reactive sites, which can be adopted as useful building blocks to construct trifluoromethyl-substituted N-heterocycles. For instance, Zhu, Chen and co-workers demonstrated a palladium-catalyzed directed C-2 and C-3 dual C–H functionalization of N-(2-pyrimidyl)-indoles with trifluoroacetimidoyl chlorides for accessing fluorinated isocryptolepine analogues.12 Nevertheless, the more challenging task involving the combination of dual C–H functionalization of unactivated alkenes with trifluoroacetimidoyl chlorides is still elusive. Herein, we report our research finding of a palladium-catalyzed bidentate-directed 1,1-geminal and 1,2-vicinal difunctionalization of unactivated alkenes with trifluoroacetimidoyl chlorides for the regioselective synthesis of biologically important trifluoromethyl-containing indoles and indolines13 (Scheme 1d).
Results and discussion
We commenced our studies by using trifluoroacetimidoyl chloride 1a and 4-pentenoic acid derivative 2a with an 8-aminoquinoline directing group as the model substrate (Table 1). The reaction was performed at 80 °C under N2 atmosphere in different solvents in the presence of Pd(OAc)2, PPh3 and Na3PO4. The results indicated that only THF could afford the 1,1-geminal difunctionalization product 3a with an alkenyl moiety at 3-position of indole in 37% yield (Table 1, entries 1–5). The exact structure of indole 3a was unambiguously confirmed by single X-ray diffraction analysis (CCDC: 2144265†).14 Other bidentate directing groups were also examined, including 2-methyl-8-quinolinyl, pyridyl, picolyl and 2-methylmercaptophenyl, and only trace of product 3a could be detected. The replacement of 8-quinolinyl with 1-naphthyl totally inhibited the reaction, highlighting the necessity of the 8-aminoquinoline directing group for the success of this reaction. Then, various bases were surveyed under the reaction system, which suggested the product 3a was obtained in 47% yield applying Na2CO3 as the base (Table 1, entries 6–9). The reaction conditions were further optimized by the use of a series of palladium catalysts, and Pd(hfac)2 could afford the best outcome (Table 1, entries 10–14). Furthermore, changing PPh3 with other phosphine ligands failed to give a better result (Table 1, entries 15–18). Elevating or lowering the reaction temperature exerted a negative effect on the reaction (Table 1, entries 19 and 20). Gratifyingly, the employment of a mixed solvent of THF and PhCF3 (v/v = 4/1) promoted the reaction to deliver 3a in 58% yield (Table 1, entry 21). Further optimization of the reaction conditions was implemented by the addition of diverse additives, such as TBAI, BQ, AgOAc or TEMPO, but the desired product 3a was not detected. The relatively lower yield of this reaction came from the inevitable residue of alkene 2a and the susceptibility of trifluoroacetimidoyl chloride 1a under the current reaction conditions.|
|
|||||
|---|---|---|---|---|---|
| Entry | [Pd] (mol%) | Ligand (mol%) | Base (equiv.) | Solvent (mL) | Yieldb (%) |
| a Reaction conditions: 1a (0.4 mmol), 2a (0.2 mmol), [Pd] (10 mol%), ligand (20 mol%), base (2.0 equiv.) in solvent (2.0 mL) at 80 °C under N2 atmosphere for 48 h. b Isolated yields. c 110 °C. d 60 °C. e The reaction was conducted with the addition of 2.0 equiv. of additive (TBAI, BQ, AgOAc or TEMPO). ND = no detection of the product. Pd(hfac)2 = palladium(II) hexafluoroacetylacetonate. | |||||
| 1 | Pd(OAc)2 | PPh3 | Na3PO4 | MeCN | ND |
| 2 | Pd(OAc)2 | PPh3 | Na3PO4 | 1,4-Dioxane | Trace |
| 3 | Pd(OAc)2 | PPh3 | Na3PO4 | Toluene | Trace |
| 4 | Pd(OAc)2 | PPh3 | Na3PO4 | THF | 37 |
| 5 | Pd(OAc)2 | PPh3 | Na3PO4 | HFIP | ND |
| 6 | Pd(OAc)2 | PPh3 | K3PO4 | THF | 38 |
| 7 | Pd(OAc)2 | PPh3 | K2CO3 | THF | Trace |
| 8 | Pd(OAc)2 | PPh3 | Na2CO3 | THF | 47 |
| 9 | Pd(OAc)2 | PPh3 | Et3N | THF | 13 |
| 10 | Pd(dba)2 | PPh3 | Na2CO3 | THF | 40 |
| 11 | Pd(PPh3)4 | PPh3 | Na2CO3 | THF | 35 |
| 12 | Pd(PPh3)2Cl2 | PPh3 | Na2CO3 | THF | 40 |
| 13 | Pd(TFA)2 | PPh3 | Na2CO3 | THF | 38 |
| 14 | Pd(hfac)2 | PPh3 | Na2CO3 | THF | 51 |
| 15 | Pd(hfac)2 | P(4-F-Ph)3 | Na2CO3 | THF | 48 |
| 16 | Pd(hfac)2 | P(4-OMe-Ph)3 | Na2CO3 | THF | 23 |
| 17 | Pd(hfac)2 | dppf | Na2CO3 | THF | 32 |
| 18 | Pd(hfac)2 | Xantphos | Na2CO3 | THF | ND |
| 19 | Pd(hfac)2 | PPh3 | Na2CO3 | THF | 42c |
| 20 | Pd(hfac)2 | PPh3 | Na2CO3 | THF | 23d |
| 21 | Pd(hfac) 2 | PPh 3 | Na 2 CO 3 | THF/PhCF 3 (4/1) | 58 |
| 22 | Pd(hfac)2 | PPh3 | Na2CO3 | THF/PhCF3 (4/1) | NDe |
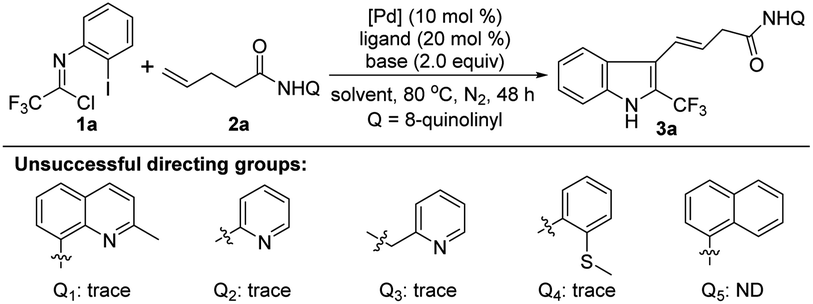
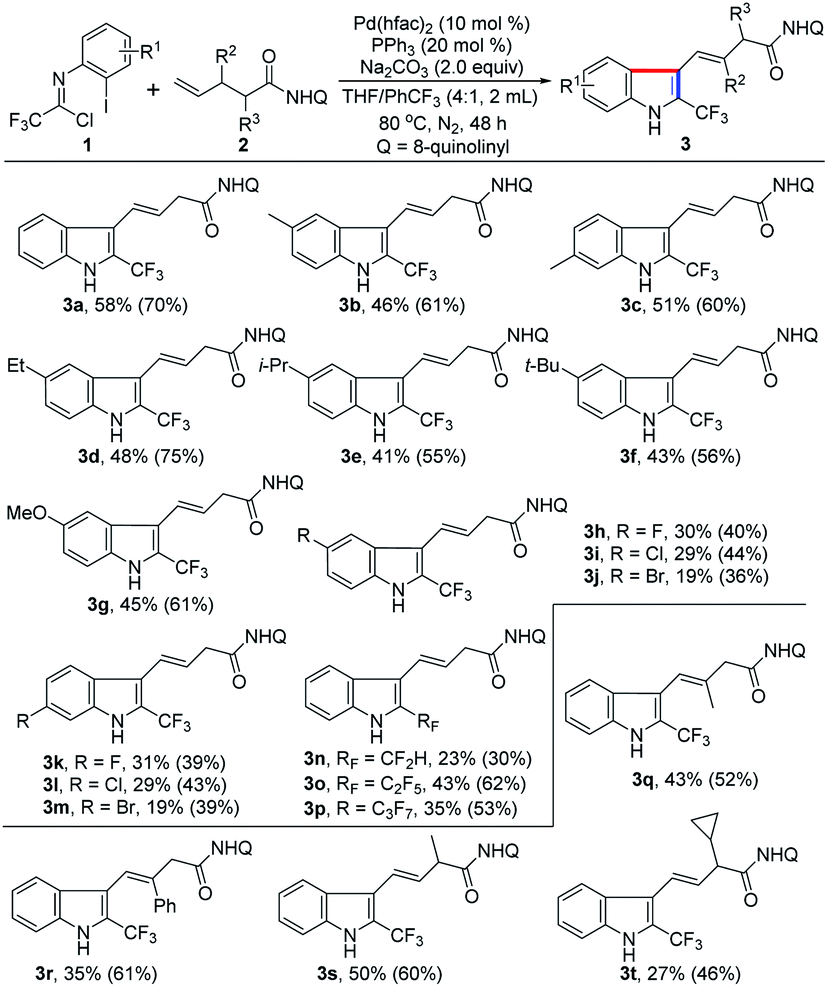
With the establishment of the optimal reaction conditions, the scope and limitation of this 1,1-geminal difunctionalization reaction was explored by adopting a range of trifluoroacetimidoyl chlorides (Table 2). In general, the trifluoroacetimidoyl chlorides with electron-donating or electron-withdrawing groups in aryl moiety all could participate in this transformation to give rise to the corresponding 2-CF3-indole products 3b–m in low to moderate yields. In all cases, it was found that the γ,δ-alkene 2 remained and the excess trifluoroacetimidoyl chloride 1 was decomposed under the current reaction conditions. Extensive effort towards the screening of the reaction conditions was devoted to fully consume the alkene substrate 2 but without success. Therefore, another group of yield data was provided in the parentheses calculated based on the recovery of alkene 2. Trifluoroacetimidoyl chlorides bearing electron-donating groups 3b–g showed higher reactivity than that of substrates with electron-withdrawing groups 3h–m. The halogen substituents (F, Cl and Br) were also tolerated in this reaction (3h–m), but only lower efficiency was observed. Noteworthy is that other fluoroalkyl groups, including CF2H, C2F5 and C3F7, were also smoothly incorporated into the indole products 3n–p with moderate reactivity. Then, several substituted unactivated alkenes were evaluated to further explore the generality of the protocol. For instance, β-methyl- and phenyl-substituted 4-pentenoic amides reacted with 1a to deliver the indole products 3q–r in acceptable yields. α-Methyl- and cyclopropyl-substituted 4-pentenoic amides also acted as viable coupling partners (3s–t). Unfortunately, 1,2- and 1,1-disubstituted alkenes were not compatible with the reaction, presumably due to the remarkable steric factors. According to the structure of the obtained indole product 3, the presence of substituent at δ-position of 4-pentenoic amides will impede the formation of indole product.
Interestingly, a unexpected N-trifluoroacetyl-substituted indoline product 4a was afforded in moderate yield when we used β,γ-alkenes as substrates to further evaluate the generality of this transformation (Table 3). We speculated that the hydrolysis of trifluoroacetimidoyl chlorides 1 occurs to give trifluoroacetamides, which undergo [3 + 2] heteroannulation with 3-butenoic acid derivatives in a 1,2-vicinal difunctionalization manner. An analogous process involving palladium-catalyzed [3 + 2] heteroannulation of non-conjugated alkenyl amides and ortho-iodoanilines/phenols for preparing 2,3-dihydrobenzofurans and indolines was disclosed by Engle and co-workers.15
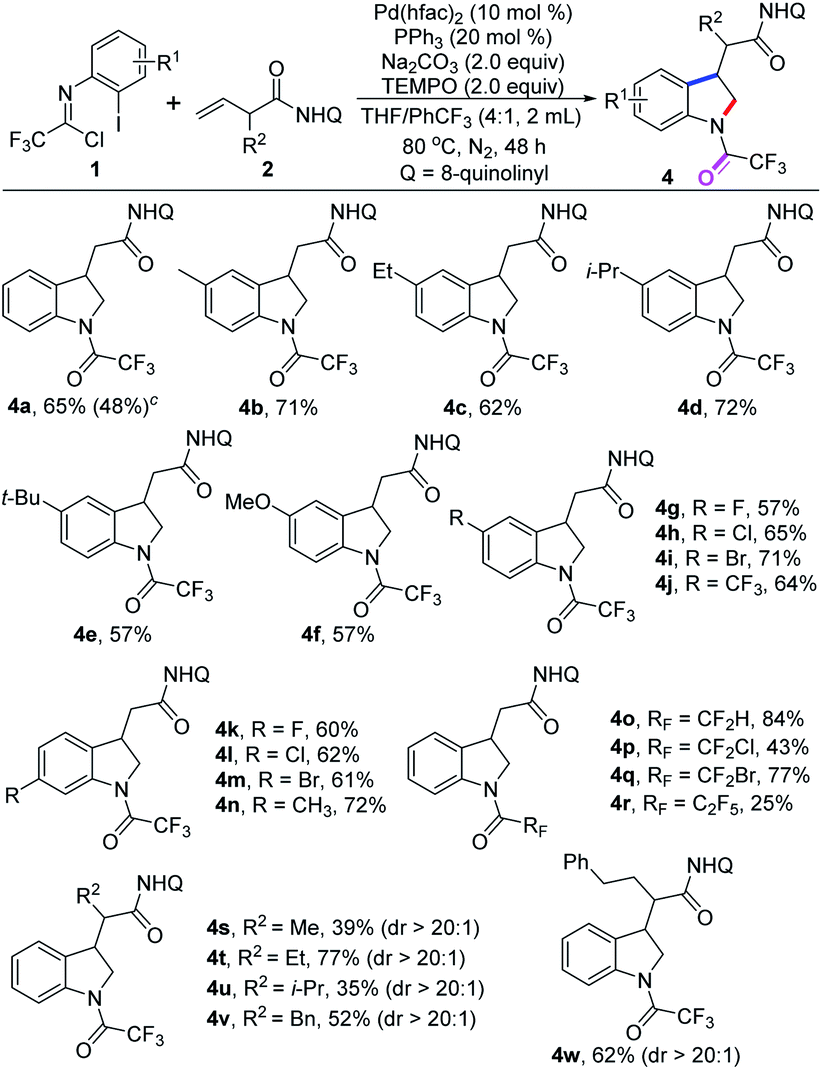
We next explored this protocol through slight modification of the reaction conditions by the addition of 2.0 equiv. of TEMPO, and the yield of product 4a was significantly increased to 65%. The scope of this 1,2-difunctionalization reaction was examined with diverse trifluoroacetimidoyl chlorides, and to our delight, the N-trifluoroacetyl-substituted indoline products 4a–n was delivered in moderate to good yields (Table 3).
A wide variety of trifluoroacetimidoyl chlorides were applicable to the reaction system, regardless of the electron properties of the aryl moiety. The halogen groups, even strong electron-withdrawing –CF3 group, could survive well in the reaction (4g–m). Apart from trifluoromethyl, other fluoroalkyl groups were also compatible with the reaction for producing the corresponding indoline products 4o–r in 25–84% yields. The structure of product 4l was also determined by single X-ray diffraction analysis (CCDC: 2144260†).14 In addition, β,γ-alkene substrates bearing different substituents at the α-position were suitable substrates under the optimal conditions to provide the desired products 4s–w in reasonable yields with excellent stereoselectivity. However, when internal alkenes were tested under the standard conditions, no desired reaction occurred. The yields of the 1,2-vicinal difunctionalization reaction are generally higher than that of 1,1-geminal difunctionalization reaction. It is well-known that chelation-stabilized five-membered palladacycle species which either generated from nucleopalladation of β,γ-alkenes or from C–H activation of the corresponding aliphatic chains could favorably react with carbon electrophiles, which was pioneered by Daugulis and co-workers.16
In order to gain some deeper insight into the reaction pathway, a series of control experiments were performed as shown in Scheme 2. First, the reaction of alkene 2a with trifluoroacetimidoyl chloride 1′ without iodine substituent was carried out under standard conditions, but the coupling product 5 was not observed (Scheme 2a). The coupling reaction of 2a with iodobenzene proceeded smoothly to give the Heck-coupling product 6 in 36% yield (Scheme 2b). The above results indicated that the coupling reaction occurred initially between the alkene 2a and the C–I bond of trifluoroacetimidoyl chloride. When deuterated water as an additive was subjected into the reaction, a Heck-coupling product 7 with an amide moiety was obtained in 14% yield with the concomitant formation of the product 3a in 48% yield (Scheme 2c). The N–H bonds of amide moiety and the C(sp2)–H bonds of alkene moiety were both partially deuterated. The replacement of trifluoroacetimidoyl chloride 1a with its hydrolysis product 8 to react with alkene 2a under the standard conditions could afford the coupling product 7 in 31% yield without generation of the [3 + 2] heteroannulation indoline product (Scheme 2d). For the indoline synthesis using β,γ-alkenes, the addition of water into the reaction could give improved yields, whereas the reaction yield decreased in the absence of TEMPO or with higher loading of water (Scheme 2e). Combined with the yield data of standard conditions (65%) or without TEMPO (42%), it is evident that TEMPO greatly promotes the reaction, but the exact role of TEMPO in the reaction is still unclear. We also utilized the hydrolysis product 8 as the coupling partner in the optimal conditions or in the absence of TEMPO, the comparable poor yields were obtained (Scheme 2f), which revealed that trifluoroacetimidoyl chloride was possibly not rapidly transformed into amide 8 to participate in the reaction. Notably, the alkene 2l with the longer chain was unreactive in the reaction system, presumably due to the very unstable palladacycle intermediate (Scheme 2g). Only trace of the desired product could be detected when bromide or chloride analogues of trifluoroacetimidoyl chlorides were tested (Scheme 2h). Finally, reactions between trifluoroacetimidoyl chloride and TEMPO were also carried out to exclude the possible reaction pathway (Scheme 2i).
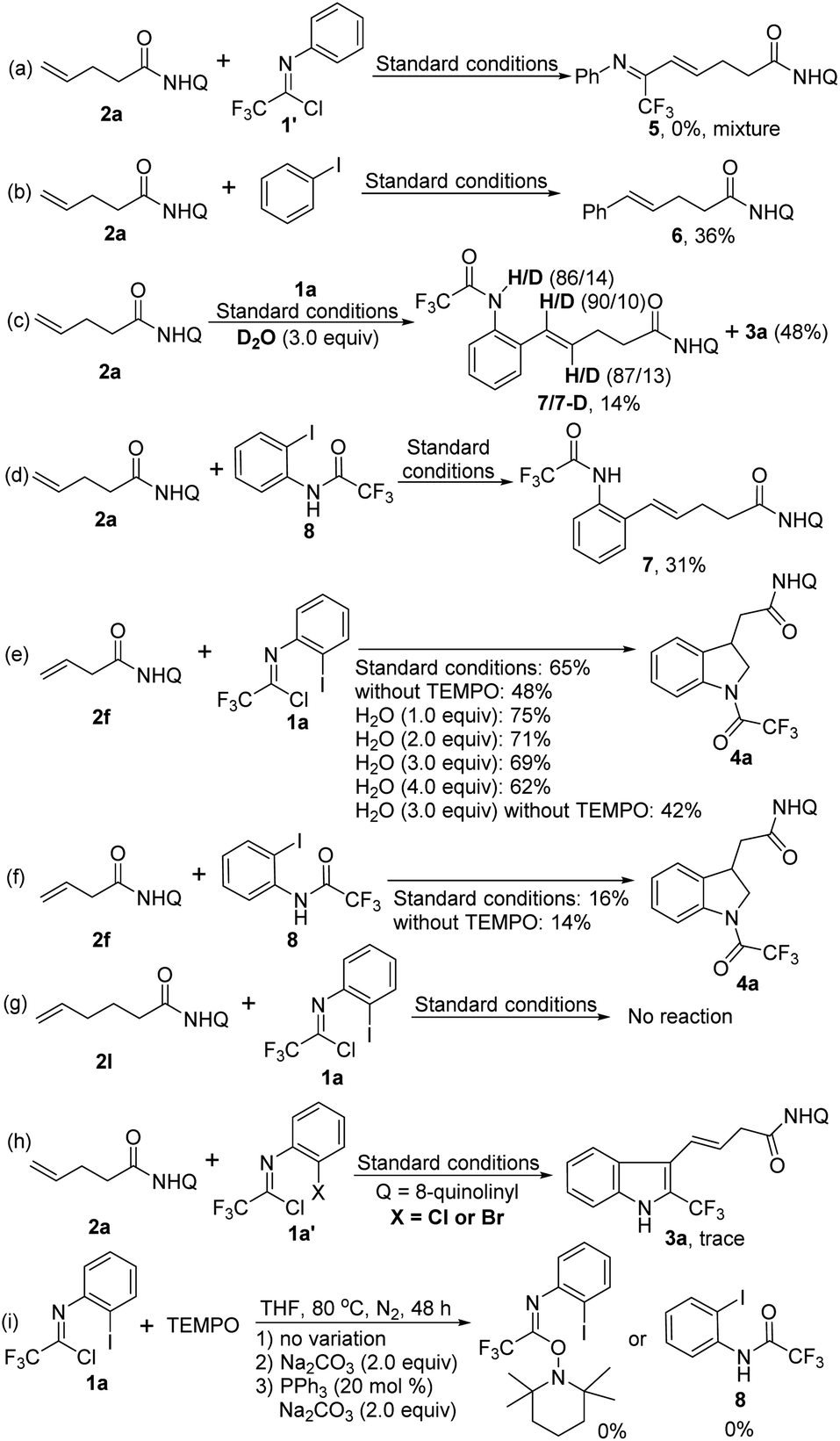 | ||
| Scheme 2 Control experiments. | ||
Based on the mechanistic investigation and the precedent literature,2g,12,15–17 the plausible reaction mechanisms of two different difunctionalizations of unactivated alkenes were proposed as depicted in Scheme 3. For the 1,1-difunctionalization reaction, the oxidative addition of C–I bond of trifluoroacetimidoyl chloride 1a to Pd(0) generated imidoyl Pd(II) intermediate A, which coordinated with the directing group in the alkene 2a to form complex B. Then, the 1,2-migratory insertion of B afforded the 6-membered palladacycle intermediate C, followed by the β-H elimination to deliver the alkene-tethered trifluoroacetimidoyl chloride D. Subsequently, the second sequence of oxidative addition of C–Cl bond of D, migratory insertion of E and β-H elimination of F occurred to give the 3H-indole G, which underwent the double bond isomerization process to furnish the desired 2-CF3-indole product 3a. For the 1,2-difunctionalization reaction, a similar Pd(II)/Pd(IV) catalytic pathway based on the Engle's work was reasonable.15 Initially, the hydrolysis of trifluoroacetimidoyl chloride in the presence of trace amount of water in reaction system occurred to produce amide 8. The alkene coordination with Pd(II) catalyst generated Pd(II) species H, which underwent nucleopalladation with 8 to afford Pd(II) complex I. Then, the oxidative addition of C–I bond of aryl moiety to Pd(II) center delivered Pd(IV) intermediate J, followed by the reductive elimination step to give rise to the final indoline product 4a. Although the exact role of TEMPO in the 1,2-difunctionalization reaction is still unclear, we propose that TEMPO reoxidize Pd(0) that was formed from during the reaction to the catalytically active Pd(II) form.
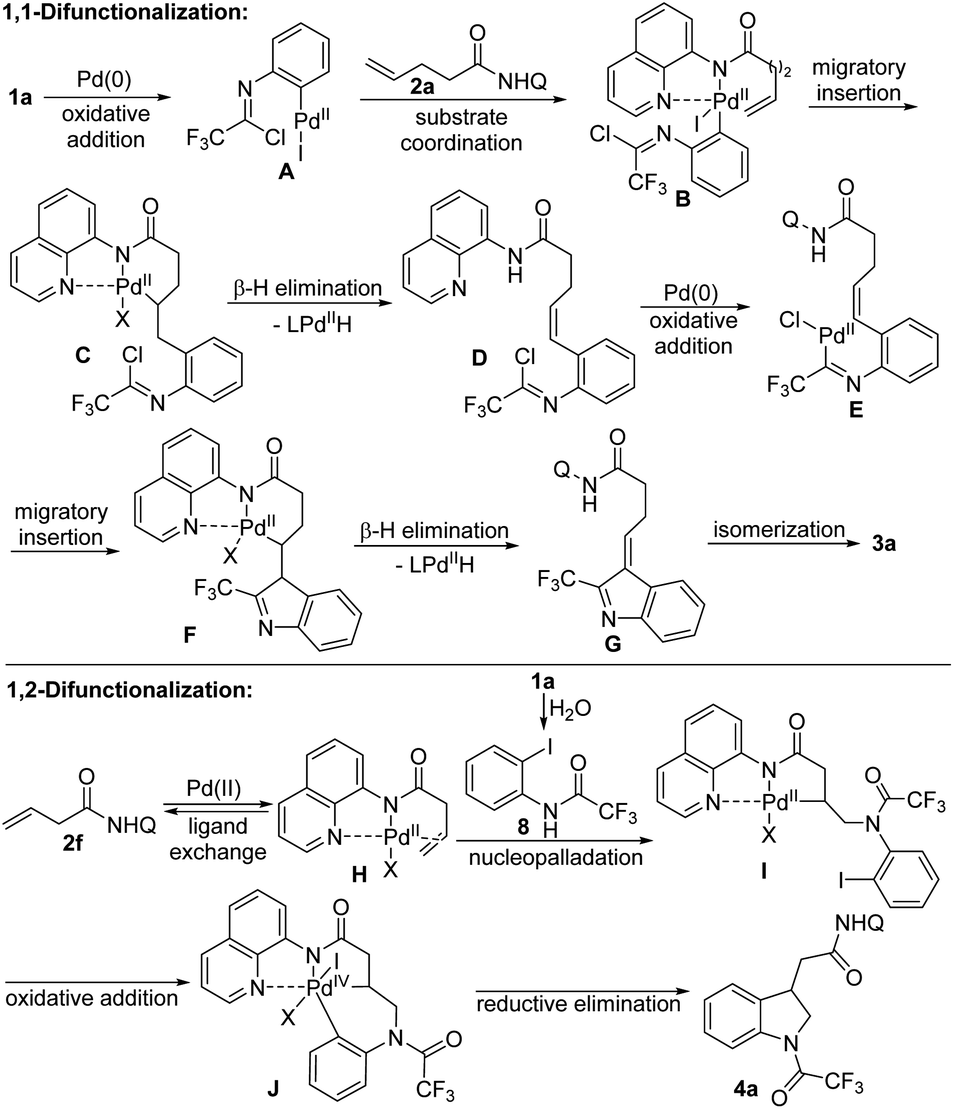 | ||
| Scheme 3 Plausible reaction mechanism. | ||
We also explored the practicability of the reaction by performing a scale-up reaction. The reaction could be easily reproducible at 1.0 mmol scale for product 4n with slightly decreased efficiency (66%) and the trifluoroacetyl group of 4n could be readily removed in the presence of NaBH4 (Scheme 4a). With regard to product 4q with a CF2Br moiety, a palladium-catalyzed radical annulation reaction with isocyanide18 was conducted for incorporating a phenanthridine scaffold into the indoline 4q to afford compound 10, albeit with low yield (Scheme 4b).
 | ||
| Scheme 4 Scale-up reaction and synthetic transformations. | ||
Conclusions
In conclusion, we have developed a straightforward approach for the regioselective synthesis of structurally diverse trifluoromethyl-containing indoles and indolines via a palladium-catalyzed dual functionalization of unactivated alkenes with trifluoroacetimidoyl chlorides. The structure of alkene substrates controls the regioselectivity of the annulation reaction, which enables the divergence of 1,1-geminal and 1,2-vicinal difunctionalization of unactivated alkenes. Several control experiments have been conducted to elucidate the reaction mechanism. Further efforts towards the additional application of trifluoroacetimidoyl chlorides as versatile coupling partners in the C–H functionalization field should be pursued in future.Author contributions
XFW and ZC directed this project and prepared this manuscript. HY, LCW, YZ, and DZ performed all the experiments and prepared ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the Natural Science Foundation of Zhejiang Province (LY19B020016) and K. C. Wong Education Foundation (GJTD-2020-08).Notes and references
- (a) K. H. Jensen and M. S. Sigman, Org. Biomol. Chem., 2008, 6, 4083 RSC; (b) R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981 CrossRef CAS PubMed; (c) G. Yin, X. Mu and G. Liu, Acc. Chem. Res., 2016, 49, 2413 CrossRef CAS PubMed.
- (a) J. A. Gurak, K. S. Yang, Z. Liu and K. M. Engle, J. Am. Chem. Soc., 2016, 138, 5805 CrossRef CAS PubMed; (b) K. S. Yang, J. A. Gurak, Z. Liu and K. M. Engle, J. Am. Chem. Soc., 2016, 138, 14705 CrossRef CAS PubMed; (c) R. Matsuura, T. C. Jankins, D. E. Hill, K. S. Yang, G. M. Gallego, S. Yang, M. He, F. Wang, R. P. Marsters, I. McAlpine and K. M. Engle, Chem. Sci., 2018, 9, 8363 RSC; (d) S. K. Nimmagadda, M. Liu, M. K. Karunananda, D. W. Gao, O. Apolinar, J. S. Chen, P. Liu and K. M. Engle, Angew. Chem., Int. Ed., 2019, 58, 3923 CrossRef CAS PubMed; (e) H. Wang, Z. Bai, T. Jiao, Z. Deng, H. Tong, G. He, Q. Peng and G. Chen, J. Am. Chem. Soc., 2018, 140, 3542 CrossRef CAS PubMed; (f) C. Wang, G. Xiao, T. Guo, Y. Ding, X. Wu and T.-P. Loh, J. Am. Chem. Soc., 2018, 140, 9332 CrossRef CAS PubMed; (g) K. Zheng, G. Xiao, T. Guo, Y. Ding, C. Wang, T. P. Loh and X. Wu, Org. Lett., 2020, 22, 694 CrossRef CAS PubMed; (h) X. Chen, W. Rao, T. Yang and M. J. Koh, Nat. Commun., 2020, 11, 5857 CrossRef CAS PubMed.
- (a) Z. Liu, T. Zeng, K. S. Yang and K. M. Engle, J. Am. Chem. Soc., 2016, 138, 15122 CrossRef CAS PubMed; (b) J. Derosa, V. T. Tran, M. N. Boulous, J. S. Chen and K. M. Engle, J. Am. Chem. Soc., 2017, 139, 10657 CrossRef CAS PubMed; (c) J. Derosa, V. A. van der Puyl, V. T. Tran, M. Liu and K. M. Engle, Chem. Sci., 2018, 9, 5278 RSC; (d) Y. Zhang, G. Chen and D. Zhao, Chem. Sci., 2019, 10, 7952 RSC.
- (a) Z. Liu, Y. Wang, Z. Wang, T. Zeng, P. Liu and K. M. Engle, J. Am. Chem. Soc., 2017, 139, 11261 CrossRef CAS PubMed; (b) V. A. van der Puyl, J. Derosa and K. M. Engle, ACS Catal., 2019, 9, 224 CrossRef CAS.
- (a) Z. Liu, H.-Q. Ni, T. Zeng and K. M. Engle, J. Am. Chem. Soc., 2018, 140, 3223 CrossRef CAS PubMed; (b) Z. Liu, X. Li, T. Zeng and K. M. Engle, ACS Catal., 2019, 9, 3260 CrossRef CAS PubMed; (c) Z. Liu, J. Chen, H.-X. Lu, X. Li, Y. Gao, J. R. Coombs, M. J. Goldfogel and K. M. Engle, Angew. Chem., Int. Ed., 2019, 58, 17068 CrossRef CAS PubMed; (d) Z. Bai, S. Zheng, Z. Bai, F. Song, H. Wang, Q. Peng, G. Chen and G. He, ACS Catal., 2019, 9, 6502 CrossRef CAS.
- (a) D. Kalyani and M. S. Sanford, J. Am. Chem. Soc., 2008, 130, 2150 CrossRef CAS PubMed; (b) A. D. Satterfield, A. Kubota and M. S. Sanford, Org. Lett., 2011, 13, 1076 CrossRef CAS PubMed; (c) Y. He, Z. Yang, R. T. Thornbury and F. D. Toste, J. Am. Chem. Soc., 2015, 137, 12207 CrossRef CAS PubMed; (d) H. M. Nelson, B. D. Williams, J. Miró and F. D. Toste, J. Am. Chem. Soc., 2015, 137, 3213 CrossRef CAS PubMed; (e) E. Yamamoto, M. J. Hilton, M. Orlandi, V. Saini, F. D. Toste and M. S. Sigman, J. Am. Chem. Soc., 2016, 138, 15877 CrossRef CAS PubMed; (f) A. M. Bergmann, S. K. Dorn, K. B. Smith, K. M. Logan and M. K. Brown, Angew. Chem., Int. Ed., 2019, 58, 1719 CrossRef CAS PubMed.
- J. Jeon, H. Ryu, C. Lee, D. Cho, M. H. Baik and S. Hong, J. Am. Chem. Soc., 2019, 141, 10048 CrossRef CAS PubMed.
- (a) F. Meyer, Chem. Commun., 2016, 52, 3077 RSC; (b) F. Zhang, X. Peng and J.-A. Ma, Chin. J. Org. Chem., 2019, 39, 109 CrossRef CAS.
- (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (b) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed; (c) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS PubMed; (d) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315 CrossRef CAS PubMed; (e) N. A. Meanwell, J. Med. Chem., 2018, 61, 5822 CrossRef CAS PubMed.
- (a) K. Tamura, H. Mizukami, K. Maeda, H. Watanabe and K. Uneyama, J. Org. Chem., 1993, 58, 32 CrossRef CAS; (b) Z. Chen, S. Hu and X.-F. Wu, Org. Chem. Front., 2020, 7, 223 RSC.
- (a) Z. Chen, W.-F. Wang, H. Yang and X.-F. Wu, Org. Lett., 2020, 22, 1980 CrossRef CAS PubMed; (b) L.-C. Wang, S. Du, Z. Chen and X.-F. Wu, Org. Lett., 2020, 22, 5567 CrossRef CAS PubMed; (c) Z. Chen, L.-C. Wang, J. Zhang and X.-F. Wu, Org. Chem. Front., 2020, 7, 2499 RSC; (d) S. Du, L.-C. Wang, Z. Yang, Z. Chen and X.-F. Wu, Adv. Synth. Catal., 2020, 362, 5130 CrossRef CAS; (e) L.-C. Wang, Y. Zhang, Z. Chen and X.-F. Wu, Adv. Synth. Catal., 2021, 363, 1417 CrossRef CAS; (f) H. Yang, S.-N. Lu, Z. Chen and X.-F. Wu, J. Org. Chem., 2021, 86, 4361 CrossRef CAS PubMed; (g) H. Yang, T.-H. Xu, S.-N. Lu, Z. Chen and X.-F. Wu, Org. Chem. Front., 2021, 8, 3440 RSC; (h) H. Yang, S.-N. Lu, Y. Song, Z. Chen and X.-F. Wu, Org. Chem. Front., 2021, 8, 5040 RSC; (i) J. F. Rodríguez, A. Zhang, R. Arora and M. Lautens, Org. Lett., 2021, 23, 7540 CrossRef PubMed; (j) A.-A. Zhang, C. Chen, Y. Gao, M. Mo, R.-Z. Shen, Y.-H. Zhang, N. Ishida, M. Murakami and L. Liu, Green Synth. Catal., 2021, 2, 311 CrossRef.
- C. Chen, Y. Wang, X. Shi, W. Sun, J. Zhao, Y. P. Zhu, L. Liu and B. Zhu, Org. Lett., 2020, 22, 4097 CrossRef CAS PubMed.
- (a) Y. Fukuda, H. Furuta, F. Shiga, Y. Oomori, Y. Kusama, H. Ebisu and S. Terashima, Bioorg. Med. Chem. Lett., 1997, 7, 1683 CrossRef CAS; (b) Y. Fukuda, H. Furuta, Y. Kusama, H. Ebisu, Y. Oomori and S. Terashima, J. Med. Chem., 1999, 42, 1448 CrossRef CAS PubMed; (c) M. A. Akanmu, C. Songkram, H. Kagechika and K. Honda, Neurosci. Lett., 2004, 364, 199 CrossRef CAS PubMed; (d) J. E. Wilson, R. Kurukulasuriya, M. Reibarkh, M. Reiter, A. Zwicker, K. Zhao, F. Zhang, R. Anand, V. J. Colandrea, A.-M. Cumiskey, A. Crespo, R. A. Duffy, B. A. Murphy, K. Mitra, D. G. Johns, J. L. Duffy and P. Vachal, ACS Med. Chem. Lett., 2016, 7, 261 CrossRef CAS PubMed.
- Compounds 3a and 4l were analyzed by X-ray crystallography. See the ESI for full details. CCDC 2144265 and 2144260 contain the ESI crystallographic data for this paper.†.
- H. Q. Ni, I. Kevlishvili, P. G. Bedekar, J. S. Barber, S. Yang, M. Tran-Dube, A. M. Romine, H. X. Lu, I. J. McAlpine, P. Liu and K. M. Engle, Nat. Commun., 2020, 11, 6432 CrossRef CAS PubMed.
- (a) D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2010, 132, 3965 CrossRef CAS PubMed; (b) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053 CrossRef CAS PubMed.
- (a) J.-B. Peng, F.-P. Wu, D. Li, H.-Q. Geng, X. Qi, J. Ying and X.-F. Wu, ACS Catal., 2019, 9, 2977 CrossRef CAS; (b) Y. Li, J.-F. Gong and M.-P. Song, Org. Chem. Front., 2020, 7, 2216 RSC.
- J.-W. Gu and X. Zhang, Org. Lett., 2015, 17, 5384 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: General comments, general procedures, analytic data, and NMR spectra. CCDC 2144265 and 2144260. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2sc00546h |
| ‡ These two authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |