
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMe2(CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) CH)SiCN: a bifunctional ethylene equivalent for Diels–Alder reaction based controllable tandem synthesis†
CH)SiCN: a bifunctional ethylene equivalent for Diels–Alder reaction based controllable tandem synthesis†
Wen-Biao
Wu
a,
Bo-Shuai
Mu
a,
Jin-Sheng
Yu
 *ab and
Jian
Zhou
abc
*ab and
Jian
Zhou
abc
aShanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, Shanghai Key Laboratory of Green Chemistry and Chemical Processes, East China Normal University, Shanghai 200062, China. E-mail: jsyu@chem.ecnu.edu.cn
bKey Laboratory of Tropical Medicinal Resource Chemistry of Ministry of Education, Hainan Normal University, Haikou 571158, China
cInstitute of Eco-Chongming, Shanghai 202162, China
First published on 24th February 2022
Abstract
A bifunctional silyl reagent Me2(CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH)SiCN has been developed as a novel ethylene equivalent for the Diels–Alder (DA) reaction. The use of this reagent enables the controllable synthesis of value-added cyclohexenyl ketones or 2-acyl cyclohexancarbonitrile derivatives through a five- or six-step tandem sequence based on a Wittig/cyanosilylation/DA reaction/retro-cyanosilylation/isomerization sequence that involves a temporary silicon-tethered intramolecular DA reaction.
CH)SiCN has been developed as a novel ethylene equivalent for the Diels–Alder (DA) reaction. The use of this reagent enables the controllable synthesis of value-added cyclohexenyl ketones or 2-acyl cyclohexancarbonitrile derivatives through a five- or six-step tandem sequence based on a Wittig/cyanosilylation/DA reaction/retro-cyanosilylation/isomerization sequence that involves a temporary silicon-tethered intramolecular DA reaction.
Introduction
The Diels–Alder (DA) reaction is one of the most important C–C bond-forming reactions for building up molecular complexity.1 Since its discovery, many types of functionalized dienes and dienophiles have been designed and applied for the synthesis of structurally diverse cyclohexenes with control of facial, regio- and stereoselectivity.2 Usually, DA reactions proceed smoothly when the dienophiles are activated by electron-withdrawing groups. When it comes to cycloaddition of ethylene, which is a commonly required synthetic step, the conditions entail very high temperature and pressure,3 which are often too harsh to be used easily (Scheme 1a). Therefore, the development of ethylene equivalents that can be used for DA reactions under milder conditions has received great attention. To date, vinyl sulfide4 or sulfone,5 nitroethylene,6 vinyldihaloboranes,7 acrolein,8 and selenoacrylate9 have been utilized as ethylene surrogates for DA reactions (Scheme 1b). The electron-withdrawing groups of the corresponding adducts can be removed subsequently through a single or multi-step transformation.8 Despite these advances, it is still important to develop new ethylene surrogates to facilitate the cycloaddition and subsequent removal of activating groups, and, in particular, to allow structural reorganization following the DA reaction, which can enable access to cyclohexene derivatives that are inaccessible by the direct [4+2] process.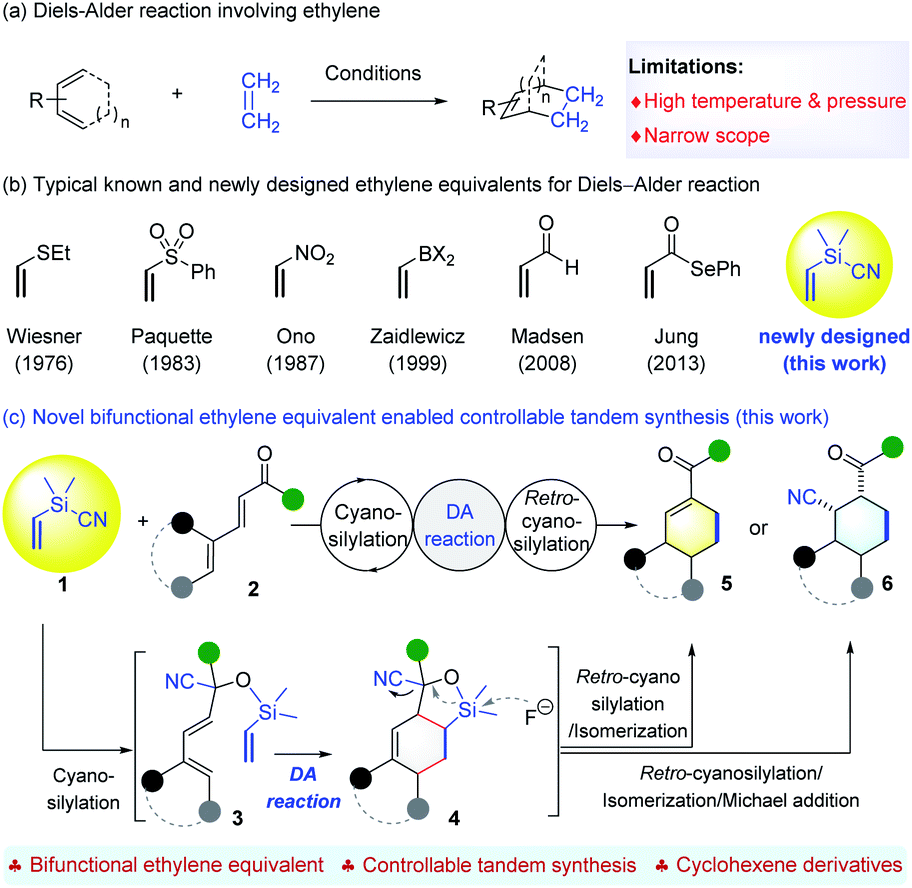 | ||
| Scheme 1 State-of-the-art and present work. | ||
On the other hand, the use of a temporary silicon tether to connect two reaction partners and induce an intramolecular reaction has been established as a powerful strategy to enhance both the reactivity and selectivity, owing to the innate benefits of intramolecularity.10 Since the independent pioneering studies of Nishiyama11 and Stork,12 this strategy has found numerous applications in organic synthesis,10 including diverse synthesis of cyclohexenes through DA cycloaddition of silicon-tethered dienes and dienophiles.13 These elegant advances encouraged us to develop silicon-tethered ethylene equivalents for DA reactions. It is worth mentioning that traditional methods to introduce silicon tethers and labile Si–O bonds rely on the protection of the O–H bond using the corresponding chlorosilyl reagents. Instead, we aim to develop bifunctional silyl reagents (BSR) that can add to CO double bonds to generate the Si–O bond tethering two functionalities for further transformations. This strategy proved to be workable because our reagent, Me2(CH2Cl)SiCN,14 enabled facile access to α-chloroacetyl tertiary alcohols through a tandem asymmetric cyanosilylation-chloromethyl transferring sequence.
Accordingly, a new BSR reagent, Me2(CH2CH)SiCN (1), was designed as an ethylene equivalent for the following reasons. First, it can enable the use of acyclic α,β,γ, andδ-unsaturated aldehydes or ketones 2 that have not been used as dienes to react with ethylene or known surrogates (Scheme 1c). The initial cyanosilylation15 affords cyanohydrins 3 featuring a Si–O bond tethered diene and a vinyl moiety for the DA reaction. A further retro-cyanosilylation,16 with simultaneous cleavage of the C–Si bond under mild conditions, readily releases the ketone moiety from adduct 4 to fulfill formal cycloaddition using dienones 2. Second, it allows tandem synthesis of cyclohexene derivatives that are unattainable by using the normal [4+2] route. Once retro-cyanosilylation occurs, it is possible to control the reaction conditions to achieve the selective synthesis of enones 5 resulting from the conjugation of the ketone moiety with the alkene, or cyclohexanes 6 substituted by the cyano and acetyl group. Both cyclohexene derivatives 5 and 6 are useful synthons17 with two functionalities. Herein, we wish to report that the development of ethylene equivalent 1 enables a controllable synthesis of valuable cyclohexene derivatives with structural diversity through a tandem cyanosilylation/DA reaction/retro-cyanosilylation sequence, by taking advantage of a temporary silicon-tethered DA reaction (Scheme 1c).
Results and discussion
Optimization of the reaction conditions
We began this study by exploring the synthesis of BSR 1. To our delight, we found that it could be readily obtained in 80% yield from NaCN and Me2(CH2CH)SiCl on a 740 mmol scale using the procedure for preparing Me2(CH2Cl)SiCN14a (Scheme 2a). Furthermore, the cyanosilylation of α,β,γ, andδ-unsaturated enone 2a using 1 was found to work well in the presence of 10 mol% acetone-derived P-ylide 7a,18a giving the desired silyl cyanohydrin 3a in 85% yield (Scheme 2b). With the silicon-tethered triene 3a, bearing both a diene and a vinyl group at hand, we next investigated its intramolecular DA reaction, and found that it proceeded well under thermal conditions. Full conversion of 3a was observed in toluene at 120 °C by GC-MS and 1H NMR analysis of the crude reaction mixture, giving the desired DA adduct 4a as a mixture of diastereomer (for details, see Section 3.2 of the ESI†). This finding paved the way for the desired merging of a retro-cyanosilylation step for tandem synthesis of cyclohexene derivatives. The evaluation of typical desilylation conditions to convert adduct 4a into cyclohexenyl methyl ketone 5a revealed that the use of a stoichiometric amount of TBAF was the best choice, but the fluoride that was formed in situ might lead to side Michael addition to give 2-acetyl-5-phenylcyclohexancarbonitrile 6a.19 Further studies focused on the controllable synthesis of 5a and 6a given that both are useful synthons.17 For the selective synthesis of 5a, we tried adding metal salts to mask the cyanide,20 and Mn(OAc)2·4H2O proved to be optimal (for details, see the ESI†). Accordingly, the synthesis of 5a was achieved in 73% yield in toluene by using TBAF and Mn(OAc)2·4H2O (Scheme 2c). On the other hand, the use of EtOAc as the solvent, along with TBAF (70 wt% in H2O) led to the generation of 6a with 64% yield and >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr as the major product.
:1 dr as the major product.
 | ||
| Scheme 2 Synthesis of 1 and its application in the tandem sequence of cyanosilylation and the DA reaction. | ||
Having established the optimal conditions for the controllable synthesis of cyclohexene derivatives 5a and 6avia DA reaction/retro-cyanosilylation based transformations, we tried combining the Wittig synthesis of α,β,γ, andδ-unsaturated enone 2a and cyanosilylation to form a five- or six-step tandem sequence starting from P-ylide 7a, enal 8a, and 1. Notably, although very few tandem reactions consisting of five steps and more are known,21 our sequences could work very efficiently (Scheme 3).
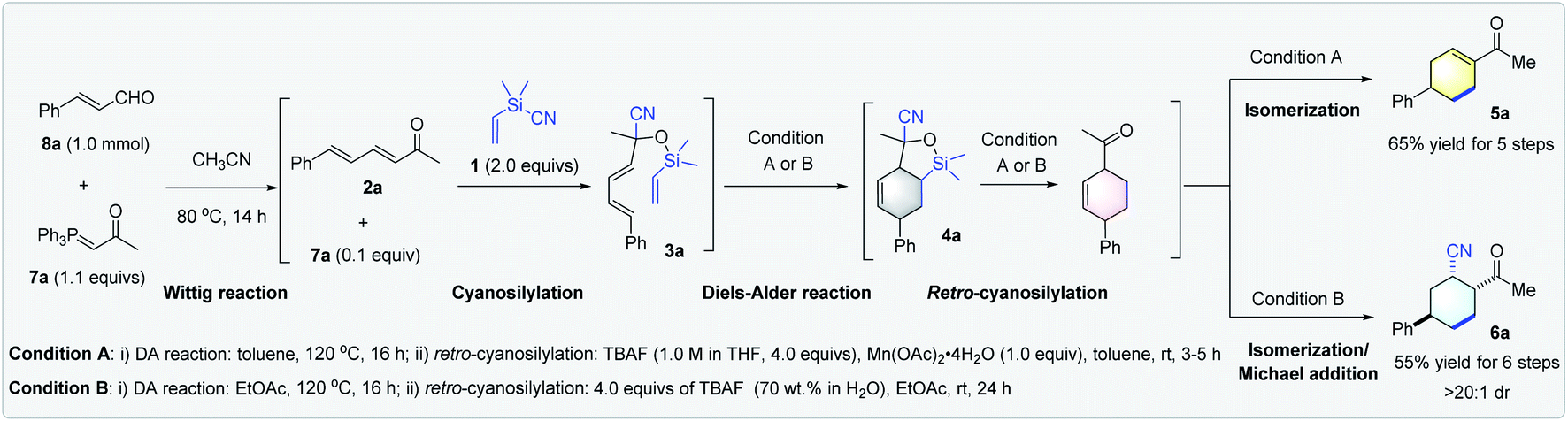 | ||
| Scheme 3 Switchable divergent tandem synthesis of cyclohexenyl methyl ketone 5a and 2-acetyl-5-phenylcyclohexancarbonitrile 6a. The overall isolated yield was reported. The dr values were determined by 1H NMR analysis of the crude reaction mixture. | ||
The initial Wittig reaction of P-ylide 7a and enal 8a proceeded smoothly at 80 °C in MeCN; a solvent required for the next cyanosilylation mediated by the excess P-ylide.18a Once the cyanosilylation completed, the crude silyl cyanohydrins were collected and subjected to the DA reaction/retro-cyanosilylation sequence in either toluene or EtOAc. After heating at 120 °C for 16 h to allow the intramolecular DA reaction to complete, TBAF (1.0 M in THF) and Mn(OAc)2·4H2O or TBAF (70 wt% in H2O) was added at room temperature to facilitate the following retro-cyanosilylation/isomerization or retro-cyanosilylation/isomerization/Michael addition, giving 5a in 65% overall yield in five steps or 6a in 55% overall yield in six steps with >20:1 dr, respectively (Scheme 3).
Evaluation of substrate scope
Next, we examined the generality of the synthesis of cyclohexenyl ketones 5 (Scheme 4). A variety of 3-aryl and 3-alkyl α,β-unsaturated aldehydes were compatible with the tandem Wittig/cyanosilylation/DA/retro-cyanosilylation/isomerization sequence, and worked well with ylide 7a and Me2(CH2CH)SiCN 1 to afford the corresponding cyclohexenyl methyl ketones 5a–o in moderate to high yields. The use of β-(2-furyl) substituted enal delivered the product 5k in 77% yield, and β-alkyl enals gave the desired products 5l–o with slightly lower yields. Ethyl-, isopropyl-, cyclopropyl-, and phenyl-substituted P-ylides 7b–e all worked well to afford the corresponding cyclohexenyl ketones 5p–s in 50–75% yields. However, the low reactivity of isopropyl, cyclopropyl or phenyl substituted ylides required the addition of 1.0 mol% Ph3PCHCONEt2 (7f)18a as the catalyst for the cyanosilylation step. Additionally, 5-methyl-2-phenyl-2-hexenal was also workable, giving multi-substituted cyclohexenyl ketone 5t in 32% yield with 1.5:1 dr. Notably, a range of five- and six-membered cyclic enals were also viable substrates, delivering valuable polycyclic compounds 5u–5aa, featuring a cyclohexenyl ketone moiety in moderate to good yields with good to high dr values.
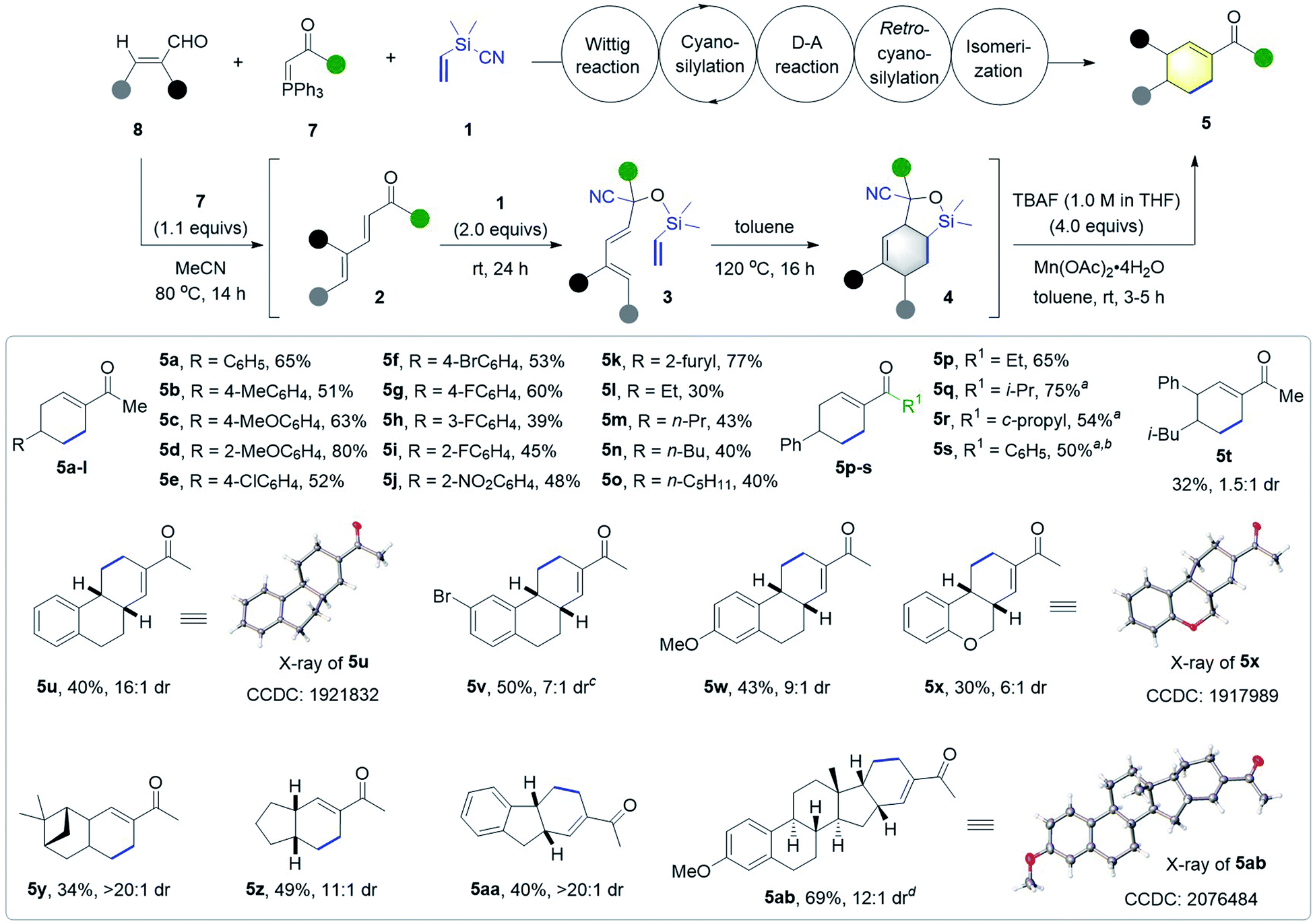 | ||
| Scheme 4 Scope of the tandem synthesis of substituted cyclohexenyl ketones 5. Reaction conditions: 1 (2.0 mmol), 7 (1.1 mmol), 8 (1.0 mmol), TBAF (4.0 mL, 1.0 M in THF) and Mn(OAc)2·H2O (1.0 mmol). The dr values were determined by 1H NMR analysis of the crude reaction mixture. aPh3PCHCONEt27f (1.0 mol%) was added in the cyanosilylation step. b5s was obtained from 1,5-diphenylpenta-2,4-dien-1-one via a cyanosilylation/Diels–Alder/retro-cyanosilylation/isomerization tandem reaction. cRun on a 0.5 mmol scale. dRun on a 0.3 mmol scale. | ||
Moreover, this method was successfully applied to late-stage modification of estrone through the introduction of a cyclohexenyl ketone moiety, affording the pentacyclic product 5ab in 69% yield and 12:1 dr. The structure and relative configuration were confirmed by X-ray analysis of products 5u, 5x, and 5ab.
As shown in Scheme 5, the scope of the six-step tandem synthesis of 2-acyl cyclohexancarbonitriles 6 was also evaluated under the optimized conditions. A series of 3-aryl substituted α,β-unsaturated aldehydes 8 were viable substrates, furnishing the desired products 6a–g in 51–77% yield with 2.5:1 to >20:1 dr. 2-Furyl substituted enal provided the corresponding product 6h in 83% yield with modest dr. Phenyl substituted phosphorus ylide 7e also reacted smoothly with aryl- or alkyl-substituted enals to give 2-benzoylcyclohexane-1-carbonitrile derivatives 6i–l in good yields and dr values. Interestingly, this protocol could be extended to access trifluoromethylated cyclohexane derivatives when CF3-substituted enal was used, as shown by the formation of 6m in 90% yield with 5.0:1 dr. The scope of the reaction with substituted ylides was also examined, and it was found that isopropyl and cyclopropyl phosphoranes worked well to give the desired products 6n and 6o in 58% and 73% yield with 6.4:1 and 4.8:1 dr, respectively. To our delight, tricyclic compounds 6p–r, with a 2-acetyl cyclohexancarbonitrile moiety, could be readily generated through this sequence from the corresponding cyclic enals. X-ray analysis of both 6i and 6r confirmed their structure and the cis relative configuration of the cyano and carbonyl group; those of other products 6 were tentatively assigned.
 | ||
| Scheme 5 Scope of the tandem synthesis of 2-acyl cyclohexancarbonitriles 6. Reaction conditions: 1 (2.0 mmol), 7 (1.1 mmol), 8 (1.0 mmol), and TBAF (4.0 mmol, 70 wt% in H2O). The dr values were determined by 1H NMR analysis of the crude reaction mixture. aPh3PCHCONEt27f (1.0 mol%) was added as catalyst in the cyanosilylation reaction. b6m was obtained from (2E,4E)-6,6,6-trifluoro-1-phenylhexa-2,4-dien-1-one via a cyanosilylation/Diels–Alder/retro-cyanosilylation/isomerization/Michael addition sequence. cRun on a 0.5 mmol scale. | ||
Synthetic utility
It should be noted that the cyclohexenyl ketone moiety is found widely in natural products and bioactive molecules. Although four major synthetic strategies have been developed to access this group,22 most are based on the conversion of cyclohexanone derivatives, which limits the structural diversity of the products. By contrast, our approach provides a facile and flexible route for the synthesis of structurally diverse cyclohexenyl ketones and their derivatives from simple reagents and α,β-unsaturated enals with either an acyclic or cyclic backbone. The synthetic utility of the cyclohexenyl ketones thus obtained was further highlighted by the application of various diversifying reactions (Scheme 6). For instance, trans-1-(4-phenylcyclohexyl)-1-ethanone 9 could be obtained in 68% yield with 7:1 dr via the selective hydrogenation of cyclohexenyl methyl ketone 5a and sequential base-mediated epimerization (Scheme 6). Under the action of NaOH, 5a worked with thiosemicarbazide or PhCHO to furnish thiosemicarbazone 10 and divinyl ketone 11via imine formation or aldol condensation, respectively. Upon treatment with TIPSOTf and Et3N, 5a was converted into the corresponding silyl enol ether, which further underwent a DA reaction with ethyl propiolate to furnish bicyclic tetrahydronaphthalene derivative 12 in 40% yield after oxidation.
 | ||
| Scheme 6 Synthetic transformations. | ||
Finally, we attempted the synthesis of chiral cyclohexenyl ketone 5a by integrating an asymmetric cyanosilylation into the sequence. However, while enantioenriched 3a could be obtained in 88% yield with 90% ee by using the three-component catalyst system of (R,R)-salen(AlCl) 15/ylide/Ph3PO that we previously developed,18c the relay of the chiral information of 3a to the desired cyclohexenyl ketone was not efficient under the standard conditions, as evidenced by the access of 5a with 42% ee and 76% yield (Scheme 7). Nevertheless, the result establishes the feasibility of using such a tandem sequence to access chiral cyclohexenyl ketone derivatives, and further work in this direction is now in progress in our lab.
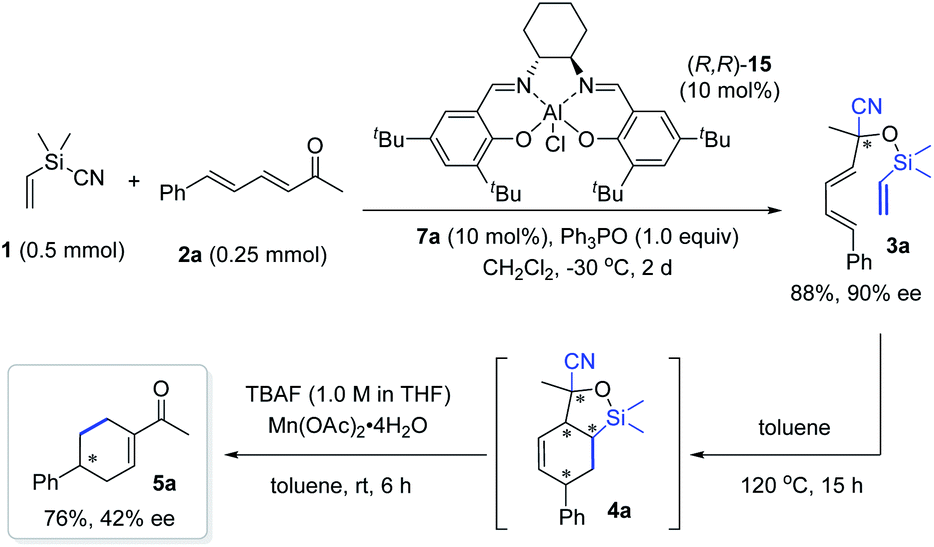 | ||
| Scheme 7 Attempt to access chiral cyclohexenyl ketone 5a. | ||
Conclusions
We have developed a novel bifunctional silyl reagent, Me2(CH2CH)SiCN, as an ethylene equivalent capable of undergoing a temporary silicon-tethered intramolecular DA reaction through the addition to the CO double bond of enones. The value of this reagent is demonstrated by an unprecedented five- or six-step tandem synthesis involving Wittig/cyanosilylation/DA reaction/retro-cyanosilylation, paving the way for controllable and diverse synthesis of cyclohexenyl ketones and 2-acyl cyclohexancarbonitrile derivatives. The development of an asymmetric variant of this tandem synthesis and further application of this reagent are ongoing in our lab.
Data availability
All of the experimental data have been included in the ESI.† Crystallographic data can be obtained from the CCDC (1921832, 1917989, 2076484, 2083519, 2101568 and 2089395).Author contributions
W.-B. Wu and B.-S. Mu performed the experiments; W.-B. Wu collected and analyzed the data. J.-S. Yu and J. Zhou directed the project and co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21725203 and 22171087), the Shanghai Science and Technology Innovation Action Plan (21N41900500 and 20JC1416900), the Ministry of Education (PCSIRT), the Fundamental Research Funds for the Central Universities, and the open foundation of Key Laboratory of Tropical Medicinal Resource Chemistry of Ministry of Education (rdzh2020003).Notes and references
- (a) O. Diels and K. Alder, Justus Liebigs Ann. Chem., 1928, 460, 98–122 CrossRef CAS; (b) E. J. Corey, Angew. Chem., Int. Ed., 2002, 41, 1650–1667 CrossRef CAS; (c) K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS; (d) K. Takao, R. Munakata and K. Tadano, Chem. Rev., 2005, 105, 4779–4807 CrossRef CAS PubMed; (e) M. Juhl and D. Tanner, Chem. Soc. Rev., 2009, 38, 2983–2992 RSC.
- (a) D. Craig, Chem. Soc. Rev., 1987, 16, 187–238 RSC; (b) F. Fringuelli and A. Taticchi, The Diels–Alder Reaction—Selected Practical Methods, Wiley, Chichester, 2002 Search PubMed.
- (a) L. M. Joshel and L. W. Butz, J. Am. Chem. Soc., 1941, 63, 3350–3351 CrossRef CAS; (b) P. G. Gassman and P. G. Pape, J. Org. Chem., 1964, 29, 160–163 CrossRef CAS; (c) D. Bartlett and K. E. Schueller, J. Am. Chem. Soc., 1968, 90, 6071–6077 CrossRef; (d) R. Golden and L. M. Stock, J. Am. Chem. Soc., 1972, 94, 3080–3088 CrossRef CAS; (e) H. Takeshita, T. Hatsui, I. Shimooda and H. Mametsuka, Bull. Chem. Soc. Jpn., 1982, 55, 2291–2292 CrossRef CAS; (f) H. Takeshita, H. Kawakami, Y. Ikeda and A. Mori, J. Org. Chem., 1994, 59, 6490–6492 CrossRef CAS; (g) J. J. McNally and J. B. Press, J. Org. Chem., 1991, 56, 245–251 CrossRef CAS.
- K. Wiesner, T. Y. R. Tsai, G. I. Dmitruenko and K. P. Nambiar, Can. J. Chem., 1976, 54, 3307–3309 CrossRef CAS.
- (a) R. V. C. Carr and L. A. Paquette, J. Am. Chem. Soc., 1980, 102, 853–855 CrossRef CAS; (b) C. V. R. Carr, R. V. Williams and L. A. Paquette, J. Org. Chem., 1983, 48, 4976–4986 CrossRef; (c) W. A. Kinney, G. D. Crouse and L. A. Paquette, J. Org. Chem., 1983, 48, 4986–5000 CrossRef CAS . For the application in total synthesis, see: ; (d) J. R. Bull and K. Bischofberger, J. Chem. Soc., Perkin Trans. 1, 1991, 2859–2865 RSC; (e) T. Omodani and K. Shishido, J. Chem. Soc., Chem. Commun., 1994, 2781–2782 RSC; (f) S. Dalai, V. N. Belov, S. Nizamov, K. Rauch, D. Finsinger and A. de Meijere, Eur. J. Org. Chem., 2006, 2753–2765 CrossRef CAS.
- N. Ono, H. Miyake, A. Kamimura and A. Kaji, J. Chem. Soc., Perkin Trans. 1, 1987, 1929–1935 RSC.
- M. Zaidlewicz, J. R. Binkul and W. Sokol, J. Organomet. Chem., 1999, 580, 354–362 CrossRef CAS.
- E. Taarning and R. Madsen, Chem.–Eur. J., 2008, 14, 5638–5644 CrossRef CAS PubMed.
- M. E. Jung, F. Perez, C. F. Regan, S. W. Yi and Q. Perron, Angew. Chem., Int. Ed., 2013, 52, 2060–2062 CrossRef CAS PubMed.
- For reviews, see: (a) M. Bols, Chem. Rev., 1995, 95, 1253–1277 CrossRef CAS; (b) S. Bracegirdle and E. A. Anderson, Chem. Soc. Rev., 2010, 39, 4114–4129 RSC.
- H. Nishiyama, T. Kitajima, M. Matsumoto and K. Itoh, J. Org. Chem., 1984, 49, 2298–2300 CrossRef CAS.
- G. Stork and M. Kahn, J. Am. Chem. Soc., 1985, 107, 500–501 CrossRef CAS.
- (a) K. Tamao, K. Kobayashi and Y. Ito, J. Am. Chem. Soc., 1989, 111, 6478–6480 CrossRef CAS; (b) K. J. Shea, K. S. Zandi, A. J. Staab and R. Carr, Tetrahedron Lett., 1990, 31, 5885–5888 CrossRef CAS; (c) D. Craig and J. C. Reader, Tetrahedron Lett., 1990, 31, 6585–6588 CrossRef CAS; (d) J. W. Gillard, R. Fortin, E. L. Grimm, M. Maillard, M. Tjepkema, M. A. Bernstein and R. Glaser, Tetrahedron Lett., 1991, 32, 1145–1148 CrossRef CAS; (e) K. J. Shea, A. J. Staab and K. S. Zandi, Tetrahedron Lett., 1991, 32, 2715–2718 CrossRef CAS; (f) D. Craig and J. C. Reader, Tetrahedron Lett., 1992, 33, 4073–4076 CrossRef CAS; (g) S. N. Sieburth and L. Fensterbank, J. Org. Chem., 1992, 57, 5279–5281 CrossRef CAS; (h) G. Stork, T. Y. Chan and G. A. Breault, J. Am. Chem. Soc., 1992, 114, 7578–7579 CrossRef CAS; (i) P. J. Coelho and L. Blanco, Synlett, 2001, 9, 1455–1457 CrossRef.
- Selected examples in developing bifunctional silyl reagents: (a) X.-P. Zeng and J. Zhou, J. Am. Chem. Soc., 2016, 138, 8730–8733 CrossRef CAS PubMed; (b) X. Ye, X.-P. Zeng and J. Zhou, Huaxue Xuebao, 2016, 74, 984–989 CrossRef CAS; (c) C. Chen, W.-B. Wu, Y.-H. Li, Q.-H. Zhao, J.-S. Yu and J. Zhou, Org. Lett., 2020, 22, 2099–2104 CrossRef CAS PubMed; (d) W.-B. Wu, X. Yu, J.-S. Yu, X. Wang, W.-G. Wang and J. Zhou, CCS Chem., 2021, 3, 2168–2180 CrossRef; (e) P.-W. Xu, X.-Y. Cui, C. Chen, F. Zhou, J.-S. Yu, Y.-F. Ao and J. Zhou, Org. Lett., 2021, 23, 8471–8476 CrossRef CAS PubMed.
- For selected recent reviews: (a) F.-X. Chen and X. Feng, Curr. Org. Synth., 2006, 3, 77–97 CrossRef CAS; (b) J.-M. Brunel and I. P. Holmes, Angew. Chem., Int. Ed., 2004, 43, 2752–2778 CrossRef CAS PubMed; (c) W. Wang, X. Liu, L. Lin and X. Feng, Eur. J. Org. Chem., 2010, 2010, 4751–4769 CrossRef; (d) H. Pellissier, Adv. Synth. Catal., 2015, 357, 857–882 CrossRef CAS; (e) N. Kurono and T. Ohkuma, ACS Catal., 2016, 6, 989–1023 CrossRef CAS; (f) X.-P. Zeng, J.-C. Sun, C. Liu, C.-B. Ji and Y.-Y. Peng, Adv. Synth. Catal., 2019, 361, 3281–3305 CrossRef CAS; (g) W.-B. Wu, J.-S. Yu and J. Zhou, ACS Catal., 2020, 10, 7668–7690 CrossRef CAS.
- (a) H. Kunz and H. Waldmann, Protecting Groups, in Comprehensive Organic Synthesis, Pergamon, Oxford, 1991 Search PubMed; (b) D. A. Evans, J. M. Hoffman and L. K. Truesdale, J. Am. Chem. Soc., 1973, 95, 5822–5823 CrossRef CAS; (c) G. Stork and G. Kraus, J. Am. Chem. Soc., 1976, 98, 6747–6748 CrossRef CAS PubMed; (d) T. Ohnuma, N. Hata, H. Fujiwara and Y. Ban, J. Org. Chem., 1982, 47, 4713–4717 CrossRef CAS; (e) D. E. Ward and M. M. Zahedi, Org. Lett., 2012, 14, 6246–6249 CrossRef CAS PubMed.
- Cyclohexenyl methyl ketones are privileged structures in natural products, see: (a) Y.-Y. Lai, M.-C. Lu, L.-H. Wang, J.-J. Chen, L.-S. Fang, Y.-C. Wu and P.-J. Sung, Mar. Drugs, 2015, 13, 4296–4309 CrossRef CAS PubMed; (b) L.-L. Liu, J.-L. Yang and Y.-P. Shi, J. Asian Nat. Prod. Res., 2011, 13, 920–929 CrossRef CAS PubMed; (c) J. J. Xu, N. H. Tan, J. Xiong, A. H. Adebayo, H. J. Han, G. Z. Zeng, C. J. Ji, Y. M. Zhang and M. J. Zhu, Chin. Chem. Lett., 2009, 20, 945–948 CrossRef CAS; (d) J. Xu, J. Su, Y. Li and N. Tan, Chem. Nat. Compd., 2013, 49, 457–461 CrossRef CAS; (e) P. Chen, P. P. Wang, Z. Z. Jiao and L. Xiang, Helv. Chim. Acta, 2014, 97, 388–397 CrossRef CAS; (f) J. Qu, S. Deng, L. Li, Y. Liu, Y. Li, S. Ma, X. Chen and S. Yu, Phytochemistry, 2016, 132, 115–122 CrossRef CAS PubMed ; cyclohexenyl methyl ketone as an important building block for the total synthesis of natural products, see: ; (g) A. B. Smith III, J. R. Empfield, R. A. Rivero and H. A. Vaccaro, J. Am. Chem. Soc., 1991, 113, 4037–4038 CrossRef . β-Cyano ketones could be used to synthesize γ-amino acids, see: ; (h) M. Winkler, A. C. Knall, M. R. Kulterer and M. Klempier, J. Org. Chem., 2007, 72, 7423–7426 CrossRef CAS PubMed ; β-cyanoketone as an important building block in the synthesis of natural products and biologically active molecules, see: ; (i) A. Siwicka, D. Cuperly, L. Tedeschi, R. Le Vézouët, A. J. P. White and A. G. M. Barrett, Tetrahedron, 2007, 63, 5903–5917 CrossRef CAS; (j) H. Muratake and M. Natsume, Tetrahedron, 2006, 62, 7071–7092 CrossRef CAS; (k) L. N. Mander and R. J. Thomson, J. Org. Chem., 2005, 70, 1654–1670 CrossRef CAS PubMed; (l) S. M. A. Rahman, H. Ohno, H. Yoshino, N. Satoh, M. Tsukaguchi, K. Murakami, C. Iwata, N. Maezaki and T. Tanaka, Tetrahedron, 2001, 57, 127–134 CrossRef CAS; (m) S. M. A. Rahman, H. Ohno, N. Maezaki, C. Iwata and T. Tanaka, Org. Lett., 2000, 2, 2893–2895 CrossRef CAS PubMed.
- (a) W.-B. Wu, X.-P. Zeng and J. Zhou, J. Org. Chem., 2020, 85, 14342–14350 CrossRef CAS PubMed . For several related studies, see: ; (b) J.-J. Cao, F. Zhou and J. Zhou, Angew. Chem., Int. Ed., 2010, 49, 4976–4980 CrossRef CAS PubMed; (c) X.-P. Zeng, Z.-Y. Cao, X. Wang, L. Chen, F. Zhou, F. Zhu, C.-H. Wang and J. Zhou, J. Am. Chem. Soc., 2016, 138, 416–425 CrossRef CAS PubMed.
- See the ESI† for the possible formation pathway of two products.
- F. A. Cotton and G. Wilkinson, Advanced Inorganic Chemistry, Wiley-Interscience, New York, 6th edn, 1992 Search PubMed.
- For selected reviews: (a) D. J. Ramón and M. Yus, Angew. Chem., Int. Ed., 2005, 44, 1602–1634 CrossRef PubMed; (b) J. C. Wasilke, S. J. Obrey, R. T. Baker and G. C. Bazan, Chem. Rev., 2005, 105, 1001–1020 CrossRef CAS PubMed; (c) D. Enders, C. Grondal and R. M. M. Hüttl, Angew. Chem., Int. Ed., 2007, 46, 1570–1581 CrossRef CAS PubMed; (d) K. C. Nicolaou and J. S. Chen, Chem. Soc. Rev., 2009, 38, 2993–3009 RSC; (e) J. Zhou, Chem.–Asian J., 2010, 5, 422–434 CrossRef CAS PubMed.
- (a) M. S. Newman and P. H. Goble, J. Am. Chem. Soc., 1960, 82, 4098–4099 CrossRef CAS; (b) D. F. Taber and B. P. Gunn, J. Org. Chem., 1979, 44, 450–452 CrossRef CAS; (c) P. G. Ciattini, E. Morera and G. Ortar, Tetrahedron Lett., 1991, 32, 6449–6452 CrossRef CAS; (d) M. E. Krafft, K. A. Seibert, T. F. N. Haxell and C. Hirosawa, Chem. Commun., 2005, 5772–5774 RSC; (e) M. E. Krafft, T. F. N. Haxell, K. A. Seibert and K. A. Abboud, J. Am. Chem. Soc., 2006, 128, 4174–4175 CrossRef CAS PubMed; (f) F.-F. Pan, P. Guo, C.-L. Li, P. Su and X.-Z. Shu, Org. Lett., 2019, 21, 3701–3705 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1917989, 1921832, 2076484, 2083519, 2089395 and 2101568. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2sc00147k |
| This journal is © The Royal Society of Chemistry 2022 |