
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhosphorylation of covalent organic framework nanospheres for inhibition of amyloid-β peptide fibrillation†
Linli
Yao
a,
Zhe
Zhou
b,
Suxiao
Wang
 a,
Qichao
Zou
a,
Hang-Xing
Wang
*a,
Li-Xin
Ma
c,
Shengfu
Wang
a and
Xiuhua
Zhang
*a
a,
Qichao
Zou
a,
Hang-Xing
Wang
*a,
Li-Xin
Ma
c,
Shengfu
Wang
a and
Xiuhua
Zhang
*a
aCollaborative Innovation Center for Advanced Organic Chemical Materials Co-constructed by the Province and Ministry, College of Chemistry and Chemical Engineering, Hubei University, Wuhan, 430062, China. E-mail: wanghx0917@163.com; zhangxh@hubu.edu.cn
bDepartment of Neurology, The First Hospital of Lanzhou University, Lanzhou, 730000, China
cState Key Laboratory of Biocatalysis and Enzyme Engineering, School of Life Science, Hubei University, Wuhan, 430062, China
First published on 22nd April 2022
Abstract
The development and exploration of new nanostructural inhibitors against Alzheimer's disease (AD)-associated amyloid-β (Aβ) fibrillation have attracted extensive attention and become a new frontier in nanomedicine. However, focusing on finding an effective nanostructure is one of the most challenging parts of the therapeutics task. Herein, nanoscale spherical covalent organic frameworks (COFs) via post-synthetic functionalization with sodium phosphate (SP) groups on the channel networks were found to efficiently inhibit Aβ fibrillation. The as-prepared uniform SP-COF nanospheres with high surface area, good crystallinity, and chemical stability were characterized by multifarious microscopic and spectroscopic techniques. Moreover, molecular dynamics simulation together with fibrillation kinetics and cytotoxicity assay experiments shows that there were restricted-access adsorption channels in the SP-COFs which were formed by the cavities with size and functional groups accommodated to the Aβ peptide sequence and significantly affected the fibrillation and cytotoxicity of Aβ. Transmission electron microscopy (TEM), dynamic light scattering (DLS) monitoring, isothermal titration calorimetry (ITC), Fourier transform infrared (FT-IR) and circular dichroism (CD) spectra measurements, and confocal imaging observation were performed to understand the inhibition mechanism and influencing factors of the SP-COFs. To our knowledge, our strategy is the first exploration of COF-based anti-amyloidogenic nanomaterials with high affinity and specific targeting, which are crucial for the inhibition of Aβ fibrillation for AD prevention and treatment.
Introduction
AD is a progressive and fatal neurodegenerative disease, which poses a substantial socio-economic burden on society and families because of the aging of the world population and the lack of effective treatments.1 In the process of exploring the nature of toxic substances in vivo, there is converging evidence that the cytotoxic aggregation of Aβ is suspected to be the main culprit behind the development of AD.2–5 The mainstream viewpoint holds that soluble and structured Aβ oligomers and mature amyloid fibrils are crucial to the onset and progression of AD,6–8 which lead to disturbing the neural function and apoptosis of neurons.9,10 Therefore, the rational design of potent inhibitors of amyloid formation is considered the primary therapeutic strategy for AD.11Nanostructures represent a rapidly emerging instrument in the biomedical tool kit and are promising candidates to entrap/capture Aβ species, owing to their regulatable physicochemical properties (size, shape, surface charge, hydrophobicity, etc.), superiority of chemical modification, capability of crossing the BBB, and good biosafety.12,13 As a new step in this direction, some pioneering studies reported the efficient inhibition of Aβ fibrillation by inorganic or organic nanoparticles,14,15 nanosheets,16 nanovesicles,17 polyoxometalates,18 zeolites,19 metal–organic frameworks,20 and so on,21 which are capable of inhibiting the aggregation and toxicity of multiple amyloidogenic proteins by adsorption and/or locking Aβ species via disrupting hydrophobic and electrostatic interactions.12 Despite these developments, focusing on finding effective nanostructural inhibitors remains one of the most challenging parts of the therapeutics against the Aβ fibrillation task. Generally, these inhibitors have been limited by major issues including (1) the lack of a sufficient adsorption capacity because the large size of the involved surface results in low binding affinity;22–24 and (2) binding to the “wrong” target sites owing to the dynamic nature of the on-/off-pathway assembly processes of Aβ.8,25–27 In this regard, the applicability of an Aβ fibrosis inhibitor requires at least two key structural aspects: sufficient capacity and specific targeting. To our knowledge, nanoporous anti-amyloidogenic materials with specific recognition of Aβ are seldom researched.
Based on the discussion above, for the first time, we report the synthesis, characteristics, and evaluation of sodium phosphate (SP) group functionalized nanoscale spherical covalent organic frameworks (COFs) as an inhibitor against Aβ fibrillation. The design rationale for our SP-COF-based inhibitors was based on three findings. First, COFs as an emerging class of organic crystalline polymers with well-defined molecular geometry and regulable porosity,28 coupled with the predesigned structures that allow fruitful post-synthetic modifications to introduce new properties and functions,29,30 making them excellent candidates for a plethora of applications ranging from energy to biomedical sciences.31 Among COFs, nanoscale spherical COFs have garnered special attention due to their ultra-high surface area, uniform spherical morphology, controllable particle size, and mesoporous microenvironment.32,33 Moreover, COFs have plentiful and adjustable cavities with specific shapes and sizes that are distinct from the above-mentioned nanostructures, which in principle could host–guest molecules with appropriate size and topology and exploit aromatic interactions.34 Second, pioneering studies reported lysine-specific molecular tweezers with the attachment of two phosphate groups to their periphery renders on both sides of molecular skeleton,35,36 which are capable of inhibiting the fibrillation and toxicity of multiple amyloidogenic proteins by binding to lysine sites and disrupting hydrophobic and electrostatic interactions.37,38 Finally, several studies have suggested that the stronger the ligand-binding affinity of nanostructures, the slower are the rates of the Aβ fibrillation process.39–41 The cavities with size accommodated to the Aβ peptide sequence and functional groups that interact with specific sites on the Aβ chain could permit synergetic inhibitory effects on Aβ fibrillation.12,42,43 Overall, our prepared SP-COFs may be superior in terms of adsorption capacity and target specificity, which are capable of enhancing the restricted-access adsorption force on Aβ through host–guest recognition and specifically capturing the positively charged amino acid sites on the Aβ sequence.
Results and discussion
Synthesis of SP-COFs
As illustrated in Scheme 1, 1,3,5-tris(4-aminophenyl) benzene (TPB) and 2,5-dihydroxyterephthalaldehyde (DHTP) with a predesigned hydroxyl unit ligand convenient for phosphorylation modification were employed as building blocks to construct SP-COFs. In brief, TPB and DHTP in the molar ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 were added to acetonitrile (ACN) containing acetic acid (HAc, 0.7 mL, 12 M).32,44,45 After the solution stood at room temperature for 12 h, a yellow precipitate was obtained. The collected precipitate was rinsed with MeOH and then activated in a vacuum, which gave the preliminary product (TD-COFs).46 The predesigned hydroxyl unit ligand in TD-COFs was further modified by phosphorylation to form SP-COFs via POCl3 followed by soaking treatment in basic solution.47 The raw SP-COFs were further rinsed by MeOH which was used to remove the adsorbed solvents and impurities. Eventually, the SP-COFs were obtained via vacuum activation treatment. More details are introduced in the experimental procedures in the ESI.†
:3 were added to acetonitrile (ACN) containing acetic acid (HAc, 0.7 mL, 12 M).32,44,45 After the solution stood at room temperature for 12 h, a yellow precipitate was obtained. The collected precipitate was rinsed with MeOH and then activated in a vacuum, which gave the preliminary product (TD-COFs).46 The predesigned hydroxyl unit ligand in TD-COFs was further modified by phosphorylation to form SP-COFs via POCl3 followed by soaking treatment in basic solution.47 The raw SP-COFs were further rinsed by MeOH which was used to remove the adsorbed solvents and impurities. Eventually, the SP-COFs were obtained via vacuum activation treatment. More details are introduced in the experimental procedures in the ESI.†
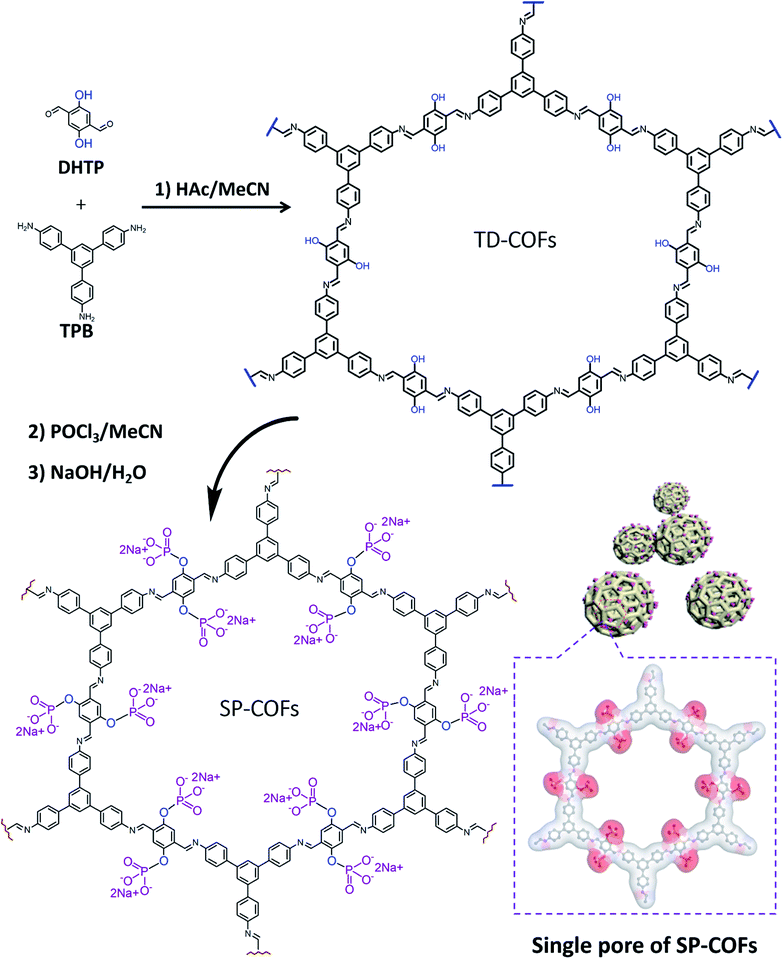 | ||
| Scheme 1 Schematic illustration of the synthesis of SP-COFs via phosphorylation of TD-COFs. Inside the dotted box is the chemical molecular charge surface diagram of a single pore of SP-COFs. | ||
Characterization of SP-COFs
The morphology and size of SP-COFs were firstly investigated by transmission electron microscopy (TEM) and dynamic light scattering (DLS) analysis. As shown in Fig. 1A, TEM images of the prepared COFs via phosphorylation modification showed a small spherical morphology with a smooth surface and a uniform size of ca. 81.4 ± 0.9 nm, which were highly consistent with the result of DLS analysis (hydrated particle size is 94.6 ± 0.8 nm), as shown in Fig. 1B. The narrow distribution of the size statistics based on TEM and DLS demonstrated the high uniformity of the prepared SP-COFs. Besides, the sphere size of SP-COFs was slightly increased as compared with the TD-COFs, which have a particle size distribution of ca. 80.4 ± 0.5 nm as shown in Fig. S1.†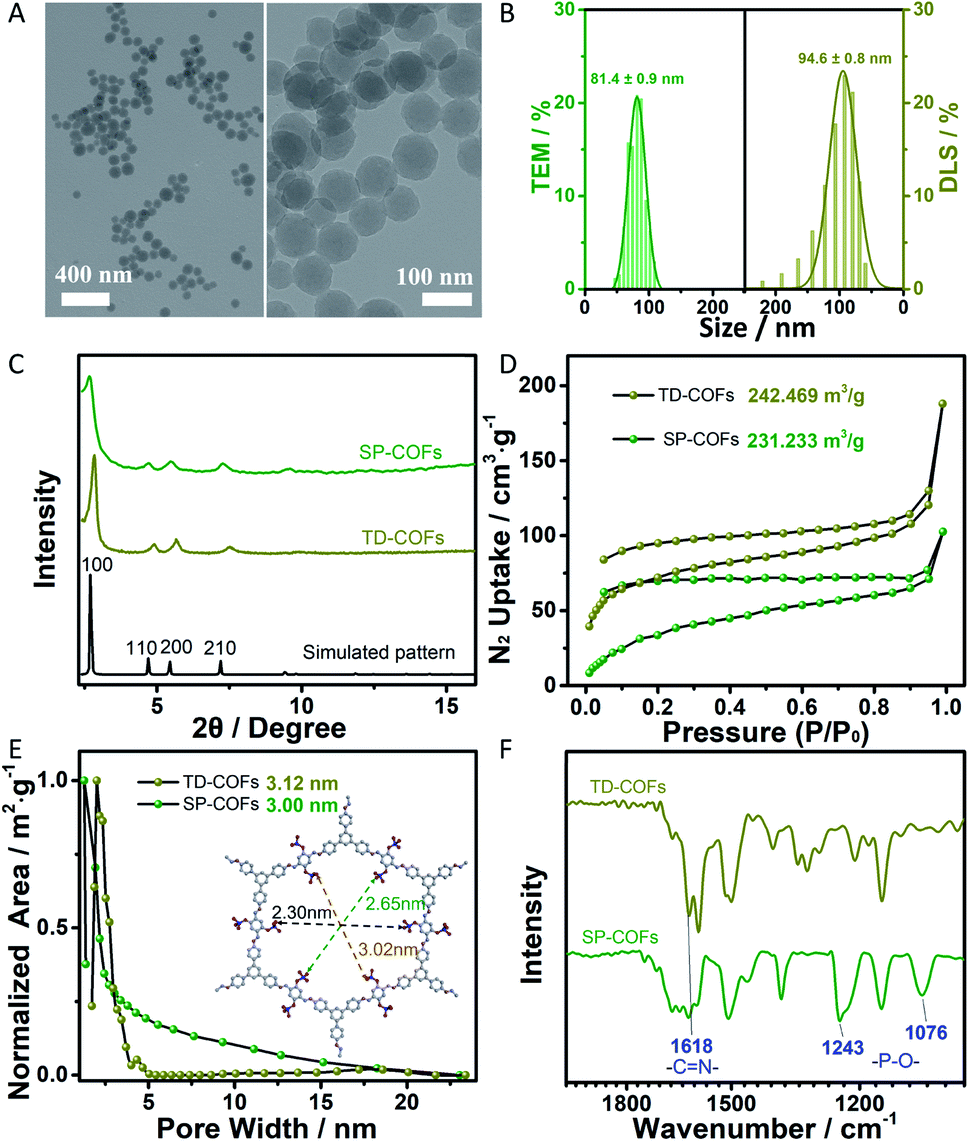 | ||
| Fig. 1 TEM images (A), particle size distribution (B) of SP-COFs. (C) Comparison of the experimental PXRD patterns of TD-COFs and SP-COFs with the simulated eclipsed stacking model. N2 sorption isotherms were measured at 77 K (D) and pore size distribution (E) of TD-COFs and SP-COFs. The inset is the simulated pore size of SP-COFs. (F) FTIR of TD-COFs and SP-COFs. | ||
Powder X-ray diffraction (PXRD) was conducted to confirm the crystallinity of the spherical COFs (Fig. 1C). Both TD-COFs and SP-COFs exhibited similar diffraction patterns, their peak locations exhibited a strong peak at 2.83° accompanied by several relatively weak peaks at 4.91°, 5.66°, and 7.52°, respectively, which were assigned to (100), (110), (200) and (210) diffractions, confirming the good crystallinity of the as-prepared COFs.32 Moreover, the experimental PXRD patterns of TD-COFs and SP-COFs match well with the simulated patterns obtained using the AA stacking mode.48,49 The surface area and porosity of TD-COFs and SP-COFs were measured by N2 adsorption–desorption analysis at 77 K. The Brunauer–Emmett–Teller (BET) surface area of TD-COFs and SP-COFs were measured to be 242.469 m3 g−1 and 231.233 m3 g−1, respectively (Fig. 1D). According to the non-local density functional theory (NLDFT) model,50 their pore size were evaluated to be 3.12 nm and 3.00 nm, respectively (Fig. 1E). These results firmly confirmed the effectiveness of the phosphorylation strategy to obtain the nanoscale spherical SP-COFs with uniform size, good crystallinity, and an obvious nanoscale mesoporous structure.
The physical properties and chemical structure of SP-COFs were further characterized in detail. The FT-IR spectra of the COFs before (TD-COFs) and after modification by phosphorylation (SP-COFs) in Fig. 1F show that the peak of –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N at 1618 cm−1 appeared in the TD-COFs and SP-COFs, indicating successful polymerization between amino groups of TPB and aldehyde groups of DHTP.51,52 Moreover, SP-COFs exhibited obvious characteristic peaks at 1076 cm−1 and 1243 cm−1, which were attributed to the stretching vibration peak of the phosphate group.53 According to the energy-dispersive X-ray (EDX) spectroscopy spectra of SP-COFs, as shown in Fig. S2,† the elemental mapping images revealed that there are C, O, P, and N elements that are uniformly distributed on the SP-COFs.
N at 1618 cm−1 appeared in the TD-COFs and SP-COFs, indicating successful polymerization between amino groups of TPB and aldehyde groups of DHTP.51,52 Moreover, SP-COFs exhibited obvious characteristic peaks at 1076 cm−1 and 1243 cm−1, which were attributed to the stretching vibration peak of the phosphate group.53 According to the energy-dispersive X-ray (EDX) spectroscopy spectra of SP-COFs, as shown in Fig. S2,† the elemental mapping images revealed that there are C, O, P, and N elements that are uniformly distributed on the SP-COFs.
X-ray photoelectron spectroscopy (XPS) was further conducted to confirm the phosphorylation of the SP-COFs. The typical XPS survey spectrum showed that SP-COFs are mainly composed of C, N, O, and P elements (Fig. 2A), which is consistent with the results of EDX characterization. The high-resolution XPS spectrum of P2p (Fig. 2B) can be split into two closing peaks at 134.5 and 135.7 eV, the major peak of 134.5 eV is from the phosphate group.54 The high-resolution XPS spectrum of N1s (Fig. 2C) can be split into two closing peaks at 399.6 and 400.3 eV, the major peak of 399.6 eV is from the –CN band.53 The result indicates the covalent formation of the Schiff base reaction between amino groups and aldehyde groups. Moreover, the high-resolution XPS spectrum of O1s (Fig. 2D) can be split into three closing peaks, the major peak of 532.0 eV is that of the phosphate group.55 By the combination of the FT-IR, EDS, and XPS results, it was concluded that the SP group was successfully conjugated to COFs.
 | ||
| Fig. 2 (A) Energy-dispersive X-ray spectroscopy spectra of C, O, N, and P in SP-COFs. The high-resolution XPS spectra of P2p (B), N1s (C), and O1s (D). | ||
Molecular dynamics simulation
The Aβ42 (molecular structure shown in Fig. S3†) peptide was chosen as a target as it has been identified as a potent cytotoxin linked to AD.56 Molecular dynamics simulation was carried out to explore the mechanism of action between a single pore of SP-COFs (denoted as SP-COF) and Aβ42. SP-COF and Aβ42 peptides were docked in an aqueous solution to predict the binding site, binding mode, binding energy value, and molecular conformation for subsequent analysis, as shown in Fig. 3 and S4.† According to the RCSB PDB crystal database, the Aβ42 peptide contains a helix spiral crystal structure with a quasi-circular in shape. Moreover, the total length of the static morphology of the Aβ42 peptide is 50.9 Å, and the three transverse maximum radii calculated for the helical circular transverse plane are 16.1 Å, 16.0 Å, and 16.5 Å respectively (Fig. 3A), which is in good agreement with the simulated pore size of SP-COFs (ca. 2.3 nm). By comparison, the cavity diameter of SP-COF is calculated to be 23.0 Å, 26.5 Å, and 30.2 Å, respectively (Fig. 3B), which are slightly larger than the transverse radius of Aβ42. Moreover, the radius comparison front view (Fig. 3C) and radius comparison top view (Fig. 3D) between the single pore of SP-COFs and Aβ42 directly show that the Aβ42 peptide sequence has the possibility of being adsorbed by SP-COF. | ||
| Fig. 3 The role of the SP-COF (single pore of SP-COFs) in binding Aβ42 peptides by host–guest recognition and targeting specific amino acid sites of the Aβ42 sequence. Simulated Aβ42 peptide transverse radius (A) and SP-COF (B); (C) Aβ42 and SP-COF radius comparison front view; (D) Aβ42 and SP-COF radius comparison top view. (E) The phase of the structure of Aβ42 and SP-COF at 0–5 ns time points in the molecular dynamics process. (F) Interaction of Aβ42 and SP-COF at 0–5 ns time points in the molecular dynamics process. | ||
The overall dynamic process of Aβ42 peptide in aqueous solution from one side to the other side of the SP-COF was further investigated by tensile molecular dynamics simulation (Table S1 and Fig. S4†). Considering that the Aβ42 peptide perforation of the SP-COF mesopore channel belongs to a holistic process, the binding state between the two in real-time was monitored and nine structures were selected to display, which can be divided into three stages: Aβ42 entered the SP-COF (0–4 ns), the most stable state of Aβ42 binding by SP-COF (4–5 ns), and Aβ42 passed through the SP-COF (5–9 ns). As shown in Fig. 3E and F, when co-incubated with SP-COF, the head of the helical Aβ42 peptide structure began to get inserted into SP-COF (0–1 ns), resulting in the formation of a strong salt bridge between lysine (K28) and the SP group (1–2 ns), which makes the Aβ42 peptide pull closer to the SP-COF side. With the passage of Aβ42 into the SP-COF, two hydrogen bonds were formed between asparagine (N27) and serine (S26) on the Aβ42 chain and two phosphate groups on the inner pore of SP-COF (2–3 ns), which maintained the contact between Aβ42 and SP-COF. When half of the body position of Aβ42 has entered the inner pore of SP-COF, histidine (H14) and glutamine (Q15) sites on the Aβ42 chain formed three hydrogen bonds with phosphate groups on SP-COF (3–4 ns). Notably, nearly 3/4 of the body position of Aβ42 passed through the inner pore of SP-COF, and a salt bridge was formed among histidine (H13) and lysine (K16) sites and a phosphate group. At this moment, the whole system reaches the most stable state (4–5 ns) (Fig. S5†). Thereafter, the Aβ42 peptide gradually passed through the SP-COF (5–8 ns) until it completely traversed the SP-COF (8–9 ns). The above simulation results support our design rationale; thus SP-COFs possess sufficient capacity and specific targeting for binding Aβ42 peptides. The former was derived from the pore structure of COFs which can enhance the restricted-access adsorption force on Aβ through host–guest recognition sequences, the latter was benefited by the negatively charged phosphate sites which could specifically capture the positively charged amino acid sites on the Aβ sequence. The theoretical study provided insights into the synergy between the nano-scale mesopores and functional groups on the interaction with Aβ42 through host–guest recognition and specifically capturing the positively charged amino acid sites on the Aβ42 peptide sequence, these functions on SP-COF adsorption and capturing of Aβ42 may play a key role in the specific target-binding affinity with Aβ42.
Evaluation of SP-COFs as an inhibitor against Aβ42 peptide fibrillation
TEM was first used to study the morphology changes and size distribution of the Aβ42 affected by SP-COFs.56 As for the Aβ42 sample alone, TEM images in Fig. 4A–C revealed that morphologies of Aβ42 species at different aggregated states were observed after 0 h, 24 h, and 96 h of cultivation, respectively, which corresponded to certain characteristic features of the Aβ42 fibrillation process (from Aβ42 monomers to oligomers to fibrils).54 In particular, non-branched amyloid fibrils with a diameter of ca. 2–3 nm and lengths of up to several micrometers were observed for the Aβ42 sample undergoing 96 h cultivation, whereas in the case of the SP-COFs and Aβ42 mixture undergoing 0–96 h of fibrillation reaction, TEM images in Fig. 4D–F revealed the coexistence of SP-COFs and small size and amorphous Aβ42 species but the COF-free sample presents thick segments formed by the association of long fibrils that are not observed. Notably, there is a slight aggregation between the COF spheres, and the size of SP-COFs was slightly increased, indicating that Aβ42 species were captured by SP-COFs. | ||
| Fig. 4 TEM images of fibrillation time courses in the absence and presence of SP-COFs at 60 μg mL−1 at Aβ42 concentration of 5 μM. The top row shows images in the absence of SP-COFs, and the bottom row includes images of samples with SP-COFs obtained after 0 h, 24 h, and 96 h of the fibrillation reaction. At 37 °C, buffer solution: 50 μM PBS, pH = 7.40. | ||
Furthermore, the overall Aβ42 fibrillation process and the impact of SP-COFs on the size distribution of Aβ42 were also determined by DLS analysis (the inset of Fig. S6†). In detail, the Aβ42 monomeric state with an average hydrodynamic diameter (Dh) of ca. 37.6 nm was presented for the fresh Aβ42 aqueous solution. With 24 h cultivation, the average Dh increased to 166.4 nm, which was consistent with the size of the Aβ42 oligomeric state.57 Moreover, the average Dh reached up to 968.0 nm for the mature fiber undergoing 96 h cultivation. As a comparison, for the Aβ42 sample incubated for 96 h at 37 °C in the presence of SP-COFs, the scattering intensity of the particle species sharply decreased and the size distribution of Aβ42 species was smaller (ca. 395.8 nm) than that of Aβ42 species without SP-COFs (ca. 968.0 nm). The species size of Aβ42 statistically analyzed by DLS was consistent with the fibrillation process of Aβ42 determined by TEM. All these observations revealed that SP-COFs could effectively inhibit Aβ42 fibrillation.
Isothermal titration calorimetry (ITC) was used to quantify the stoichiometry and binding constant between SP-COFs and Aβ42. TD-COFs as a control group were also tested. As shown in Fig. 5A and B, the studies of entropy and enthalpy (ΔH) changes obtained from ITC data revealed that both SP-COFs and TD-COFs exhibit pure exothermic interactions with the Aβ42 monomers. In detail, they interacted with Aβ42 at a stoichiometric ratio of 1:1 stoichiometry with binding constants Kd(TD-COFs) = 1.38 × 104 M−1 and Kd(SP-COFs) = 3.79 × 107 M−1, respectively. Moreover, the enthalpy changes are ΔH(TD-COFs) = −4.55 kJ mol−1 and ΔH(SP-COFs) = −51.80 kJ mol−1, respectively. The ITC data suggest that SP-COFs have a stronger binding force with Aβ42 (3 orders of magnitude) than that of TD-COFs because of their specific recognition of the SP group. Moreover, the zeta potential was tested to verify the electrostatic interaction between SP-COFs and Aβ42 monomers (Fig. S7†). After binding to Aβ42 monomers, the electric potential of SP-COFs was changed from −32.23 mV to −21.50 mV, indicating that there are obvious coulombic interactions between SP-COFs and Aβ42 monomers. The above results proved that the phosphorylation allowed specific targeting of Aβ42 and synergistically enhanced binding force with Aβ42.
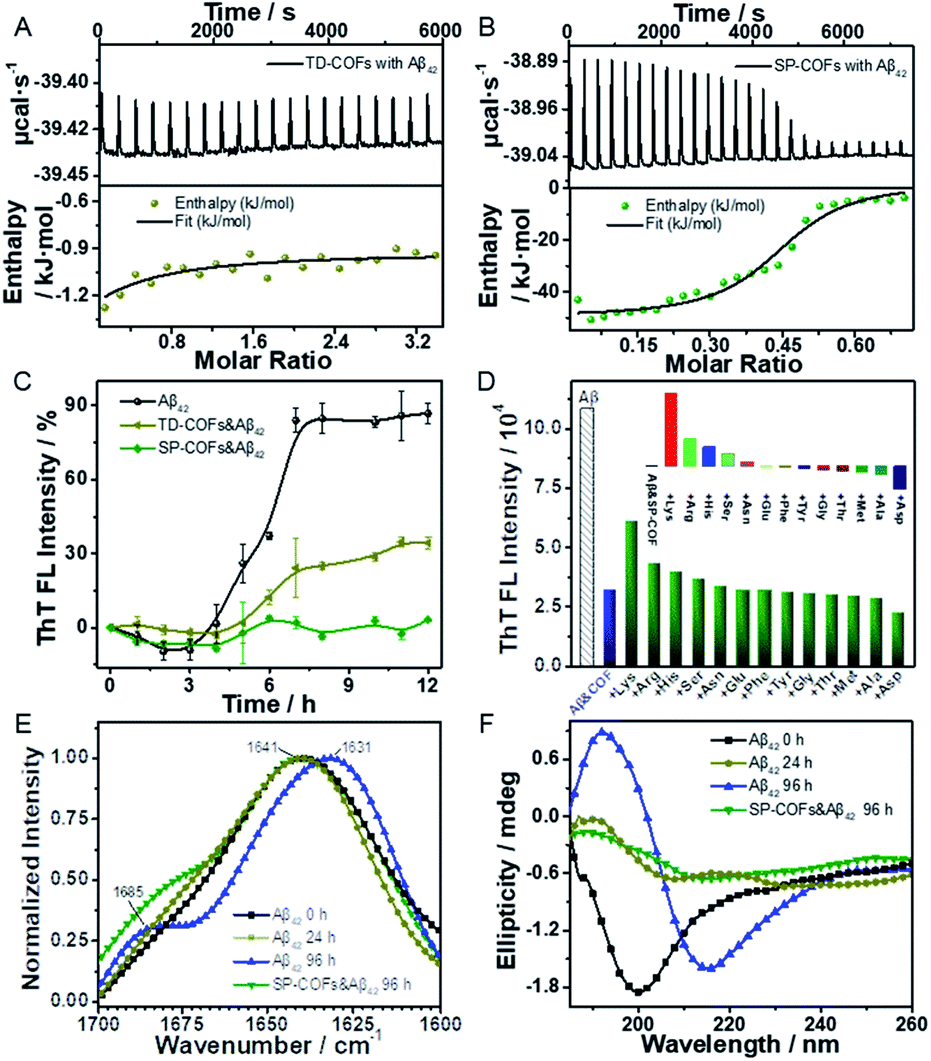 | ||
| Fig. 5 Influence of SP-COFs on the Aβ42 fibrosis process. (A and B) ITC curves of Aβ42 incubated with TD-COFs (A) and SP-COFs (B). (C) Fibrillation kinetics of Aβ42 (5 μM) with addition of TD-COFs and SP-COFs (60 μg mL−1) (buffer: 50 μM PBS containing 0.02% NaN3, pH = 7.40). (D) The binding specificity of SP-COFs. ThT fluorescence assay (co-incubated for 12 h) of the SP-COFs&Aβ42 co-incubation system added with a single amino acid (50 μM, 10 times of Aβ42), respectively. (E) FT-IR spectra of fibrillation time courses of Aβ42 in the presence of SP-COFs. (F) CD spectra of Aβ42 banding to SP-COFs. | ||
Fibrillation kinetics analysis was exploited to verify the achieved inhibitory effect of SP-COFs on Aβ42 fibrillation. TD-COFs as a control group was also tested. As shown in Fig. 5C, fibrillation kinetics of Aβ42 with SP-COFs is monitored by the thioflavin-T (ThT) fluorescence assay (λex = 437 nm, λem = 485 nm) which revealed the formation of amyloid aggregates.58 The ThT fluorescence of Aβ42 was followed over 12 h in the absence and presence of SP-COFs incubated at 37 °C. The fluorescence spectra of Aβ42 alone incubated at 37 °C showed that the ThT fluorescence of Aβ42 (black curve) gradually increased, after an initial lag time of 3 h; the ThT fluorescence of Aβ42 reached the plateau phase at 7 h, and the relative fluorescence intensity was significantly increased by 86.8%. In the case of Aβ42 incubated with TD-COFs at 37 °C, the fluorescence intensity (pale yellow curve) showed a significant increase in the lag phase of Aβ42 (lag time of 4 h) but does not completely inhibit fibres formation, and the relative fluorescence intensity was increased by 34.4%. When SP-COFs were co-incubated with Aβ42, the initial lag time (green curve) prolonged to 5 h, the fluorescence enhancement effect of ThT diminished, and the relative fluorescence intensity slightly increased by 3.4%. This remarkable difference in the ThT fluorescence assay was direct evidence that phosphorylation can improve the inhibition efficiency of Aβ42 fibrillation. Besides, SP-COFs were found to not affect the fluorescence intensity change of ThT when SP-COFs were co-incubated with ThT (Fig. S8†). The above results not only demonstrated that SP-COFs can effectively delay and inhibit the fibrillation of Aβ42, but also proved that phosphorylation played an important role in enhancing the inhibition efficiency of Aβ42 fibrillation. In addition, we used UV-Vis absorption spectroscopy to explore the concentration changes of Aβ42 before and after mixing with SP-COFs. According to the analysis of absorbance change, ca. 16.0% of the protein was adsorbed by SP-COFs when they were mixed, as shown in Fig. S9.†
Furthermore, the binding specificity of SP-COFs towards amino acids was evaluated by monitoring the kinetic curves of the SP-COFs&Aβ42 co-incubation system by adding different amino acids including acidic amino acids and basic amino acids, hydrophilic amino acids and hydrophobic amino acids. In this experiment, the SP-COFs&Aβ42 co-incubation system was added with a single amino acid, and the fibrillation kinetic curves by the ThT fluorescence assay were monitored, respectively. It can be seen from Fig. 5D that the fluorescence intensity of ThT was partially recovered by adding Lys, Arg, and His, whereas it was decreased by adding Asp and Ala, thus the inhibitory effect of SP-COFs was weakened by the addition of basic amino acids, especially Lys, while it was enhanced by the addition of acidic Asp and hydrophobic Ala. In addition, the aggregation kinetic curves of the independent Aβ42 with amino acids were also adopted as control experiments to investigate the effect of the surface properties of SP-COFs on Aβ42 fibrillation. Notably, the fluorescence intensity of ThT in the SP-COF-free systems showed no discernible difference by adding acidic amino acids, basic amino acids, and hydrophobic amino acids (Fig. S10†). These results together with molecular dynamics simulation results revealed that the pore structure of COF is capable of enhancing the restricted-access adsorption force on Aβ through host–guest recognition sequences, and the negatively charged SP sites could specifically capture the positively charged amino acid sites on the Aβ42 sequence.59
FT-IR spectroscopy was applied to reveal the secondary structural distribution of Aβ42 aggregated species affected by SP-COFs. As shown in Fig. 5E, the infrared amide I absorbance band between 1700 and 1600 cm−1 which originates from the CO stretching vibration of the backbone of the protein was analyzed.60 For the independent Aβ42via incubation at 37 °C from 0 h to 24 h to 96 h, the corresponding characteristic peaks were shifted towards lower wavenumbers from 1639 cm−1, to 1637 cm−1, to 1631 cm−1, along with the sub-band around 1685 cm−1 was increased, revealing that the number of β-sheets was increased along with the Aβ42 fibrosis process.61 By comparison, in the case of Aβ42 incubated with SP-COFs at 37 °C for 96 h, the right flank of the band was high-degree and overlapped with that of the fresh Aβ42 sample. Moreover, for the SP-COFs&Aβ42 sample, the band at 1685 cm−1 decreased and the amide I band appeared at 1641 cm−1, which is far from the characteristic peak of the independent Aβ42via incubation for 96 h, suggesting that SP-COFs could decrease the number of β-sheet-rich frameworks, thus inhibiting Aβ42 fibrillation.62
Circular dichroism (CD) spectroscopy was employed for analyzing the structural features of the inhibited Aβ42 products,63 and the BeStSel webserver was used for quantitative analysis (Table S2†).64Fig. 5F shows that the fresh Aβ42 sample alone adopted a predominant α-helical structure with an obvious negative band at 199 nm.65 After 24 h incubation, the CD spectrum displayed a weak positive peak at 190 nm and a negative peak at 206 nm, respectively. According to quantitative analysis, the content of helical species decreased from 19.9% to 1.3%, while the antiparallel species increased from 1.8% to 15.8%. After incubation for 96 h at 37 °C, the CD spectrum for independent Aβ42 adopted a predominant β-folded structure with a typical positive peak at 192 nm and a negative minimum at 216 nm, respectively, which are characteristics of the β-sheet-rich framework corresponding to Aβ42 fibrils.40 The parallel structure content was calculated to be 12.0% from 4.1%. In contrast, for the Aβ42 sample incubated with SP-COFs at 37 °C for 96 h, the CD spectrum exhibited significantly reduced representative peaks at 216 nm and 192 nm with the lowest parallel species content of 4.6%, indicating that there is very little β-sheet structure formed.66 The FT-IR and CD results strongly suggested that the SP-COFs efficiently prevented Aβ42 fiber production.
Cell imaging and cytotoxicity assay of SP-COFs
A remarkable inhibition effect was observed when the Aβ42 aggregated on SP-COFs, which encouraged us to further study of the performances of rescuing Aβ42-induced cytotoxicity. Herein, PC12 cells were used as the neuronal model to evaluate the effect of SP-COFs on cell viability and Aβ42-induced cytotoxicity.67 Before the PC12 cell experiment, the stability in the cell sap of SP-COFs was investigated by monitoring the morphology change of SP-COFs before and after co-incubation with the cell sap for 96 h. The TEM images revealed that the morphology of SP-COFs is consistent with that in the aqueous solution system and has not been degraded (Fig. S11†). In addition, the stability evaluation in Fig. S12 and S13† revealed that the SP-COFs in PBS buffer and cell fluid had stable morphology and crystal structure after incubation for 96 h.Furthermore, the adsorption effect of SP-COFs in the co-incubation of SP-COFs with Aβ42in vitro was investigated by confocal laser scanning microscopy (CLSM).68,69 Since SP-COFs and Aβ42 peptides show no fluorescence emission, a stilbazolium-based NIR fluorescent dye (4-N,N-dimethylamino-4′-N-methyl-1,3-butadienyl-pyridinium dimer, Fig. S14†) was embedded in the SP-COFs (denoted as DMBPD&SP-COFs),70 and a commercial fluorescein 5-isothiocyanate dye (FITC) was introduced to label the Aβ42 peptide (denoted as FITC-Aβ42) for fluorescence imaging, respectively. Based on the above results, the DMBPD&SP-COFs were co-incubated with PC12 cells in a confocal Petri dish for 12 h in the absence or presence of Aβ42, and confocal imaging was then performed under excitation of a 488 nm laser. For the DMBPD&SP-COFs and PC12 cell co-culture system in the absence of Aβ42 as shown in Fig. 6A and S15,† obvious red fluorescence was observed in the cytoplasm, indicating that SP-COFs can enter PC12 cells. In contrast, the DMBPD&SP-COFs and PC12 cell co-culture system in the presence of Aβ42 gave no fluorescence in the cytoplasm but obvious red fluorescence outside the PC12 cells (Fig. S16†). Moreover, for the FITC-Aβ42 and PC12 cell co-culture system without DMBPD&SP-COFs, the confocal imaging in Fig. 6B and S17† showed that there was no fluorescence inside PC12 cells but obvious green fluorescence outside the cells, revealing that none of the Aβ42 was taken up by the cells. Furthermore, it is important to point out that, in agreement with the images of the Aβ42 and DMBPD&SP-COFs co-incubated system, the co-incubated system of FITC-Aβ42 and DMBPD&SP-COFs, which were further co-cultured with PC12 cells as shown in Fig. 6C and S18†, presented red and green fluorescence outside the PC12 cells, but gave no fluorescence in the cytoplasm of cells, indicating that SP-COFs could not enter the cells and none of the peptides is taken up by the cell in this case. The above results indicated that SP-COFs have an excellent extracellular adsorption effect and would not release Aβ42 after specific binding.
 | ||
| Fig. 6 The CLSM images of 60 μg mL−1 DMBPD&SP-COFs (A), 5 μM FITC-Aβ42 (B), and 60 μg mL−1 DMBPD&SP-COFs and 5 μM FITC-Aβ42 co-culture system (C) with an excitation light of 488 nm. Darkfield (left), bright field (center), and superposition field (right). | ||
Finally, the cytotoxicity of SP-COFs and their effect on amyloid fibrillization in vitro were investigated. Herein, a standard 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) method was used to assess cell proliferation and cytotoxicity.71,72 As shown in Fig. 7A, negligible cytotoxicity was observed for SP-COFs with varying concentrations from 5 to 60 μg mL−1, suggesting the great biocompatibility of SP-COFs. The cell viability of PC12 cells was as high as about 93.33 ± 8.88% in the presence of SP-COFs with a concentration range of 5 to 60 μg mL−1. When PC12 cells were incubated with Aβ42 alone, the cell viability was reduced significantly with the increase of Aβ42 concentration (0–20 μM), indicating the remarkable cytotoxicity of Aβ42 aggregates (Fig. 7B). As shown in Fig. 7C, the addition of SP-COFs (0–40 μg mL−1) exhibited obvious and significant mitigation of toxicity in amyloids (Aβ42, 5 μM); the viability of PC12 cells sharply increased from 45.02 ± 0.58% to 88.52 ± 0.80%, and there was no significant difference in the inhibitory effect when the concentration of SP-COFs was in the range of 40–60 μg mL−1. Furthermore, it is important to point out that, although SP-COFs reduced the neurotoxicity of Aβ42 in a concentration-dependent manner, there was still an obvious increase in the cell viability (above 81.43 ± 4.98%) for the SP-COFs (60 μg mL−1) and PC12 cell co-culture system co-preincubated with high Aβ42 concentrations (5–20 μM), as shown in Fig. 7D. All these observations demonstrated that our SP-COFs not only showed an excellent ability to inhibit Aβ42 fibrillation but also could substantially decrease Aβ42-induced cytotoxicity. Moreover, cytotoxicity experiments of three other kinds of cells including HeLa, HUVECs, and A549 cells were performed to evaluate the effect of SP-COFs on cell viability and Aβ-induced cytotoxicity, as shown in Fig. S18.†
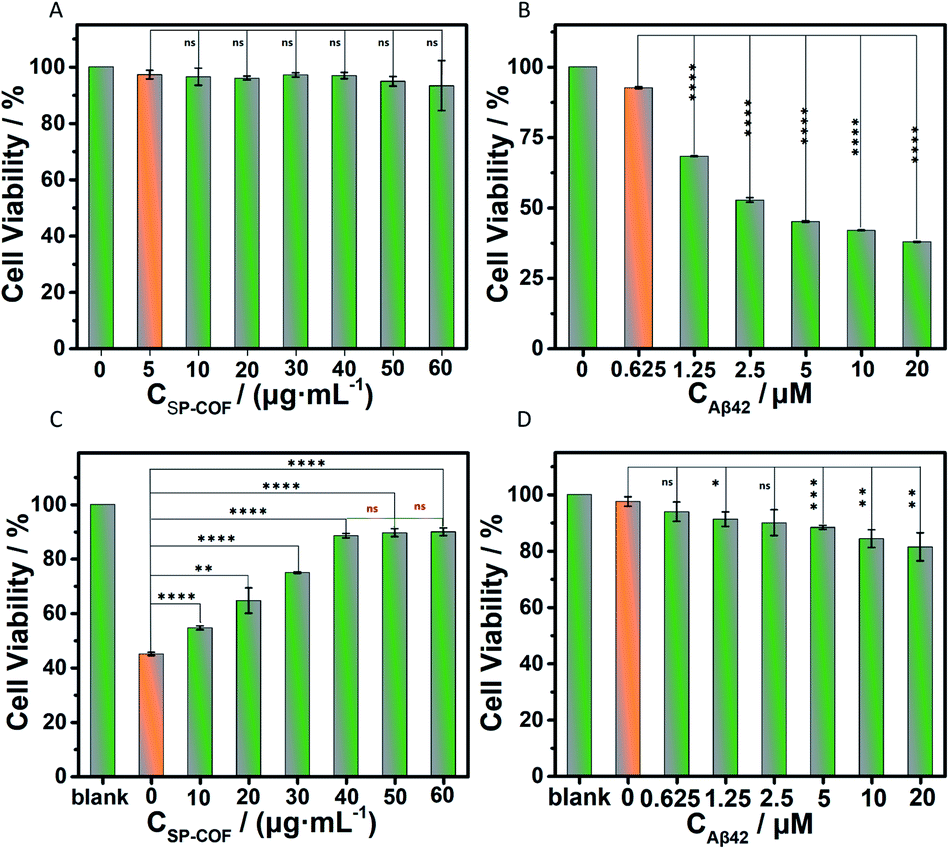 | ||
| Fig. 7 The cell viability was measured by a standard MTT assay. The viability of PC12 cells without any treatment for 48 h was specified as 100%. (A) The cells were preincubated with SP-COFs (0–60 μg mL−1); (B) the cells were preincubated with Aβ42 (0–20 μM); (C) the cells were preincubated with SP-COF (0–60 μg mL−1) and Aβ42 (5 μM); (D) the cells were preincubated with SP-COFs (60 μg mL−1) and Aβ42 (0–5 μM). Experiments were performed in triplicate and error bars in the graph represent the mean ± SD (n = 3). Statistical analysis was carried out by means of the t-test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns: not significant). | ||
Conclusions
In summary, this study described the construction of SP groups in functionalized uniform COF nanospheres and employed them to explore the effect of the SP-COFs on amyloid fibrillation. Our results reveal that the SP group follows a double strategy that combines dispersibility improvement of COF and process-specificity function in targeting the Aβ42 sequence. Moreover, the SP-COFs, because of the advantageous nature of their large surface area, nanoscale spherical shape, appropriate cavity size with AA stacking mode, and negatively charged phosphate groups, serve as an efficient inhibitor for enhancing the restricted-access adsorption force on Aβ42 through host–guest recognition and specifically capturing the positively charged amino acid sites on the Aβ42 peptide sequence. The simulation guidance, and multiple biophysical, and biochemical characterization results revealed that SP-COFs specifically targeted the side chains of residues of Aβ42. In detail, the SP groups underwent electrostatic interactions with the positively charged sites (Lys16, Lys28), together with the mesoporous microenvironment of SP-COFs that underwent hydrophobic interactions with hydrophobic residues (Leu17, Val18, Phe19, Phe20), resulting in the binding affinity between SP-COFs and Aβ42 with a binding specificity, which significantly affected the fibrillation and cytotoxicity of Aβ42. Our study compensates well for the disadvantages of pore-free anti-amyloidogenic materials, developing a novel type of Aβ fibrillation inhibitor with superiority in two key structural aspects of sufficient capacity and specific targeting, which indicates a new direction for research on nano-inhibitors. However, this study is only a preliminary exploration, a deeper fundamental understanding of the effects of SP-COFs on amyloidosis processes as well as the biodistribution and toxicity evaluation in vivo will be the subject of our future work.Author contributions
L. Yao synthesized the samples and participated in characterization. H.-X. Wang and X. Zhang supervised the project. H.-X. Wang conceived the idea for this study. L. Yao, Z. Zhou, and H.-X. Wang wrote the manuscript. S. Wang, Q. Zou, and S. Wang helped revise the manuscript. L.-X. Ma provides CD and ICT test equipment. All authors commented on the data and the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Natural Science Foundation of China (NSFC. 21875060, 22076042, and 11674085) and the Applied Basic Research Programs of Wuhan Science and Technology Bureau of Hubei Province (No. 2020020601012269). We thank Dr S. Fang for conducting the molecular dynamics simulation.References
- Z. Du, M. Li, J. Ren and X. Qu, Current Strategies for Modulating Aβ Aggregation with Multifunctional Agents, Acc. Chem. Res., 2021, 54, 2172–2184 CrossRef CAS PubMed.
- S. J. Lee, E. Nam, H. J. Lee, M. G. Savelieff and M. H. Lim, Towards an Understanding of Amyloid-beta Oligomers: Characterization, Toxicity Mechanisms, and Inhibitors, Chem. Soc. Rev., 2017, 46, 310–323 RSC.
- M. G. Savelieff, G. Nam, J. Kang, H. J. Lee, M. Lee and M. H. Lim, Development of Multifunctional Molecules as Potential Therapeutic Candidates for Alzheimer's Disease, Parkinson's Disease, and Amyotrophic Lateral Sclerosis in the Last Decade, Chem. Rev., 2019, 119, 1221–1322 CrossRef CAS PubMed.
- D. Willbold, B. Strodel, G. F. Schröder, W. Hoyer and H. Heise, Amyloid-type Protein Aggregation and Prion-like Properties of Amyloids, Chem. Rev., 2021, 121, 8285–8307 CrossRef CAS PubMed.
- J. Wang, C. Zhao, A. Zhao, M. Li, J. Ren and X. Qu, New Insights in Amyloid Beta Interactions with Human Telomerase, J. Am. Chem. Soc., 2015, 137, 1213–1219 CrossRef CAS PubMed.
- M. Tolar, J. Hey, A. Power and S. Abushakra, Neurotoxic Soluble Amyloid Oligomers Drive Alzheimer's Pathogenesis and Represent a Clinically Validated Target for Slowing Disease Progression, Int. J. Mol. Sci., 2021, 22, 6355 CrossRef CAS PubMed.
- C. Haass and D. J. Selkoe, Soluble Protein Oligomers in Neurodegeneration: Lessons from the Alzheimer's Amyloid β-Peptide, Nat. Rev. Mol. Cell Biol., 2007, 8, 101–112 CrossRef CAS PubMed.
- V. Armiento, A. Spanopoulou and A. Kapurniotu, Peptide-Based Molecular Strategies To Interfere with Protein Misfolding, Aggregation, and Cell Degeneration, Angew. Chem., Int. Ed., 2020, 59, 3372–3384 CrossRef CAS PubMed.
- P. C. Ke, M. A. Sani, F. Ding, A. Kakinen, I. Javed, F. Separovic, T. P. Davis and R. Mezzenga, Implications of Peptide Assemblies in Amyloid Diseases, Chem. Soc. Rev., 2017, 46, 6492–6531 RSC.
- T. C. T. Michaels, A. Šarić, S. Curk, K. Bernfur, P. Arosio, G. Meisl, A. J. Dear, S. I. A. Cohen, C. M. Dobson, M. Vendruscolo, S. Linse and T. P. J. Knowles, Dynamics of Oligomer Populations Formed During the Aggregation of Alzheimer's Aβ42 Peptide, Nat. Chem., 2020, 12, 445–451 CrossRef CAS PubMed.
- X. Han, J. Park, W. Wu, A. Malagon, L. Wang, E. Vargas, A. Wikramanayake, K. N. Houk and R. M. Leblanc, A Resorcinarene for Inhibition of Aβ Fibrillation, Chem. Sci., 2017, 8, 2003–2009 RSC.
- Z. Zhou, Y.-Q. Gu and H.-X. Wang, Artificial Chiral Interfaces against Amyloid-β Peptide Aggregation: Research Progress and Challenges, ACS Chem. Neurosci., 2021, 12, 4236–4248 CrossRef CAS PubMed.
- M. J. Hajipour, M. R. Santoso, F. Rezaee, H. Aghaverdi, M. Mahmoudi and G. Perry, Advances in Alzheimer's Diagnosis and Therapy: The Implications of Nanotechnology, Trends Biotechnol., 2017, 35, 937–953 CrossRef CAS PubMed.
- J. Zhao, K. Li, K. Wan, T. Sun, N. Zheng, F. Zhu, J. Ma, J. Jiao, T. Li, J. Ni, X. Shi, H. Wang, Q. Peng, J. Ai, W. Xu and S. Liu, Organoplatinum-Substituted Polyoxometalate Inhibits β-amyloid Aggregation for Alzheimer's Therapy, Angew. Chem., Int. Ed., 2019, 131, 18200–18207 CrossRef.
- C. Cabaleiro-Lago, F. Quinlan-Pluck, I. Lynch, S. Lindman, A. M. Minogue, E. Thulin, D. M. Walsh, K. A. Dawson and S. Linse, Inhibition of Amyloid β Protein Fibrillation by Polymeric Nanoparticles, J. Am. Chem. Soc., 2008, 130, 15437–15443 CrossRef CAS PubMed.
- G. Qing, S. Zhao, Y. Xiong, Z. Lv, F. Jiang, Y. Liu, H. Chen, M. Zhang and T. Sun, Chiral Effect at Protein/Graphene Interface: A Bioinspired Perspective To Understand Amyloid Formation, J. Am. Chem. Soc., 2014, 136, 10736–10742 CrossRef CAS PubMed.
- D. Maity, M. Howarth, M. C. Vogel, M. Magzoub and A. D. Hamilton, Peptidomimetic-Based Vesicles Inhibit Amyloid-β Fibrillation and Attenuate Cytotoxicity, J. Am. Chem. Soc., 2021, 143, 3086–3093 CrossRef CAS PubMed.
- J. Geng, M. Li, J. Ren, E. Wang and X. Qu, Polyoxometalates as Inhibitors of the Aggregation of Amyloid β Peptides Associated with Alzheimer's Disease, Angew. Chem., Int. Ed., 2011, 50, 4184–4188 CrossRef CAS PubMed.
- M. J. Lucas and B. K. Keitz, Influence of Zeolites on Amyloid-β Aggregation, Langmuir, 2018, 34, 9789–9797 CrossRef CAS PubMed.
- Z. L. Mensinger, B. L. Cook and E. L. Wilson, Adsorption of Amyloid Beta Peptide by Metal–Organic Frameworks, ACS Omega, 2020, 5, 32969–32974 CrossRef CAS PubMed.
- N. Andrikopoulos, Y. Li, L. Cecchetto, A. Nandakumar, T. Da Ros, T. P. Davis, K. Velonia and P. C. Ke, Nanomaterial Synthesis, an Enabler of Amyloidosis Inhibition against Human Diseases, Nanoscale, 2020, 12, 14422–14440 RSC.
- C. P. Shaw, D. A. Middleton, M. Volk and R. Lévy, Amyloid-Derived Peptide Forms Self-Assembled Monolayers on Gold Nanoparticle with a Curvature-Dependent β-Sheet Structure, ACS Nano, 2012, 6, 1416–1426 CrossRef CAS PubMed.
- S. I. Yoo, M. Yang, J. R. Brender, V. Subramanian, K. Sun, N. E. Joo, S.-H. Jeong, A. Ramamoorthy and N. A. Kotov, Inhibition of Amyloid Peptide Fibrillation by Inorganic Nanoparticles: Functional Similarities with Proteins, Angew. Chem., Int. Ed., 2011, 50, 5110–5115 CrossRef CAS PubMed.
- S. Linse, C. Cabaleiro-Lago, W.-F. Xue, I. Lynch, S. Lindman, E. Thulin, S. E. Radford and K. A. Dawson, Nucleation of protein fibrillation by nanoparticles, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8691–8696 CrossRef CAS PubMed.
- Y. S. Eisele, C. Monteiro, C. Fearns, S. E. Encalada, R. L. Wiseman, E. T. Powers and J. W. Kelly, Targeting Protein Aggregation for the Ttreatment of Degenerative Diseases, Nat. Rev. Drug Discovery, 2015, 14, 759–780 CrossRef CAS PubMed.
- R. Maleki, M. Khedri, S. Rezvantalab, F. Afsharchi, K. Musaie, S. Shafiee and M.-A. Shahbazi, β-Amyloid Targeting with Two-Dimensional Covalent Organic Frameworks: Multi-Scale In-Silico Dissection of Nano-Biointerface, ChemBioChem, 2021, 22, 2306–2318 CrossRef CAS PubMed.
- A. J. Dear, G. Meisl, A. Saric, T. C. T. Michaels, M. Kjaergaard, S. Linse and T. P. J. Knowles, Identification of on- and off-pathway oligomers in amyloid fibril formation, Chem. Sci., 2020, 11, 6236–6247 RSC.
- R. Liu, K. T. Tan, Y. Gong, Y. Chen, Z. Li, S. Xie, T. He, Z. Lu, H. Yang and D. Jiang, Covalent Organic Frameworks: an Ideal Platform for Designing Ordered Materials and Advanced Applications, Chem. Soc. Rev., 2021, 50, 120–242 RSC.
- Y. Yusran, X. Guan, H. Li, Q. Fang and S. Qiu, Postsynthetic Functionalization of Covalent Organic Frameworks, Natl. Sci. Rev., 2019, 7, 170–190 CrossRef PubMed.
- Z. Li, T. He, Y. Gong and D. Jiang, Covalent Organic Frameworks: Pore Design and Interface Engineering, Acc. Chem. Res., 2020, 53, 1672–1685 CrossRef CAS PubMed.
- S. Bhunia, K. A. Deo and A. K. Gaharwar, 2D Covalent Organic Frameworks for Biomedical Applications, Adv. Funct. Mater., 2020, 30, 2002046 CrossRef CAS.
- W. Ma, Q. Zheng, Y. He, G. Li, W. Guo, Z. Lin and L. Zhang, Size-Controllable Synthesis of Uniform Spherical Covalent Organic Frameworks at Room Temperature for Highly Efficient and Selective Enrichment of Hydrophobic Peptides, J. Am. Chem. Soc., 2019, 141, 18271–18277 CrossRef CAS PubMed.
- K.-X. Gao, Z. Zhou, L. Yao, S. Wang, Y. Zhang, Q. Zou, L.-X. Ma and H.-X. Wang, Aspartic Acid-Assisted Size-Controllable Synthesis of Nanoscale Spherical Covalent Organic Frameworks with Chiral Interfaces for Inhibiting Amyloid-β Fibrillation, ACS Appl. Bio Mater., 2022, 5, 1210–1221 CrossRef CAS PubMed.
- C. Yuan, X. Wu, R. Gao, X. Han, Y. Liu, Y. Long and Y. Cui, Nanochannels of Covalent Organic Frameworks for Chiral Selective Transmembrane Transport of Amino Acids, J. Am. Chem. Soc., 2019, 141, 20187–20197 CrossRef CAS PubMed.
- S. Sinha, D. H. J. Lopes, Z. Du, E. S. Pang, A. Shanmugam, A. Lomakin, P. Talbiersky, A. Tennstaedt, K. McDaniel, R. Bakshi, P.-Y. Kuo, M. Ehrmann, G. B. Benedek, J. A. Loo, F.-G. Klärner, T. Schrader, C. Wang and G. Bitan, Lysine-Specific Molecular Tweezers Are Broad-Spectrum Inhibitors of Assembly and Toxicity of Amyloid Proteins, J. Am. Chem. Soc., 2011, 133, 16958–16969 CrossRef CAS PubMed.
- P. Talbiersky, F. Bastkowski, F.-G. Klärner and T. Schrader, Molecular Clip and Tweezer Introduce New Mechanisms of Enzyme Inhibition, J. Am. Chem. Soc., 2008, 130, 9824–9828 CrossRef CAS PubMed.
- T. Weil, R. Groß, A. Röcker, K. Bravo-Rodriguez, C. Heid, A. Sowislok, M.-H. Le, N. Erwin, M. Dwivedi, S. M. Bart, P. Bates, L. Wettstein, J. A. Müller, M. Harms, K. Sparrer, Y. B. Ruiz-Blanco, C. M. Stürzel, J. von Einem, S. Lippold, C. Read, P. Walther, M. Hebel, F. Kreppel, F.-G. Klärner, G. Bitan, M. Ehrmann, T. Weil, R. Winter, T. Schrader, J. Shorter, E. Sanchez-Garcia and J. Münch, Supramolecular Mechanism of Viral Envelope Disruption by Molecular Tweezers, J. Am. Chem. Soc., 2020, 142, 17024–17038 CrossRef CAS PubMed.
- T. Schrader, G. Bitan and F.-G. Klärner, Molecular Tweezers for Lysine and Arginine-Powerful Inhibitors of Pathologic Protein Aggregation, Chem. Commun., 2016, 52, 11318–11334 RSC.
- M. H. Viet, S. T. Ngo, N. S. Lam and M. S. Li, Inhibition of Aggregation of Amyloid Peptides by beta-Sheet Breaker Peptides and Their Binding Affinity, J. Phys. Chem. B, 2011, 115, 7433–7446 CrossRef CAS PubMed.
- H. Sun, J. Liu, S. Li, L. Zhou, J. Wang, L. Liu, F. Lv, Q. Gu, B. Hu, Y. Ma and S. Wang, Reactive Amphiphilic Conjugated Polymers for Inhibiting Amyloid β Assembly, Angew. Chem., Int. Ed., 2019, 58, 5988–5993 CrossRef CAS PubMed.
- Q. Luo, Y.-X. Lin, P.-P. Yang, Y. Wang, G.-B. Qi, Z.-Y. Qiao, B.-N. Li, K. Zhang, J.-P. Zhang, L. Wang and H. Wang, A Self-destructive Nanosweeper that Captures and Clears Amyloid β-Peptides, Nat. Commun., 2018, 9, 1802 CrossRef PubMed.
- H. Zhang, D. Yu, S. Liu, C. Liu, Z. Liu, J. Ren and X. Qu, NIR-II Hydrogen-Bonded Organic Frameworks (HOFs) Used for Target-Specific Amyloid-β Photooxygenation in an Alzheimer's Disease Model, Angew. Chem., Int. Ed., 2022, 61, e202109068 CAS.
- D. Yu, Y. Guan, F. Bai, Z. Du, N. Gao, J. Ren and X. Qu, Metal–Organic Frameworks Harness Cu Chelating and Photooxidation Against Amyloid β Aggregation in Vivo, Chem.–Eur. J., 2019, 25, 3489–3495 CrossRef CAS PubMed.
- B. J. Smith, A. C. Overholts, N. Hwang and W. R. Dichtel, Insight into the Crystallization of Amorphous Imine-linked Polymer Networks to 2D Covalent Organic Frameworks, Chem. Commun., 2016, 52, 3690–3693 RSC.
- W. Ma, G. Li, C. Zhong, Y. Yang, Q. Sun, D. Ouyang, W. Tong, W. Tian, L. Zhang and Z. Lin, Room-Temperature Controllable Synthesis of Hierarchically Flower-like Hollow Covalent Organic Frameworks for Brain Natriuretic Peptide Enrichment, Chem. Commun., 2021, 57, 7362–7365 RSC.
- C. H. Feriante, S. Jhulki, A. M. Evans, R. R. Dasari, K. Slicker, W. R. Dichtel and S. R. Marder, Rapid Synthesis of High Surface Area Imine-Linked 2D Covalent Organic Frameworks by Avoiding Pore Collapse During Isolation, Adv. Mater., 2020, 32, 1905776 CrossRef CAS PubMed.
- D. M. C. Schwarz, S. K. Williams, M. Dillenburg, C. R. Wagner and J. E. Gestwicki, A Phosphoramidate Strategy Enables Membrane Permeability of a Non-nucleotide Inhibitor of the Prolyl Isomerase Pin1, ACS Med. Chem. Lett., 2020, 11, 1704–1710 CrossRef CAS PubMed.
- Y. Peng, W. K. Wong, Z. Hu, Y. Cheng, D. Yuan, S. A. Khan and D. Zhao, Room Temperature Batch and Continuous Flow Synthesis of Water-Stable Covalent Organic Frameworks (COFs), Chem. Mater., 2016, 28, 5095–5101 CrossRef CAS.
- X. Li, C. Zhang, S. Cai, X. Lei, V. Altoe, F. Hong, J. J. Urban, J. Ciston, E. M. Chan and Y. Liu, Facile Transformation of Imine Covalent Organic Frameworks into Ultrastable Crystalline Porous Aromatic Frameworks, Nat. Commun., 2018, 9, 2998 CrossRef PubMed.
- W. Liu, X. Li, C. Wang, H. Pan, W. Liu, K. Wang, Q. Zeng, R. Wang and J. Jiang, A Scalable General Synthetic Approach toward Ultrathin Imine-Linked Two-Dimensional Covalent Organic Framework Nanosheets for Photocatalytic CO2 Reduction, J. Am. Chem. Soc., 2019, 141, 17431–17440 CrossRef CAS PubMed.
- R. Tao, X. Shen, Y. Hu, K. Kang, Y. Zheng, S. Luo, S. Yang, W. Li, S. Lu, Y. Jin, L. Qiu and W. Zhang, Phosphine-Based Covalent Organic Framework for the Controlled Synthesis of Broad-Scope Ultrafine Nanoparticles, Small, 2020, 16, 1906005 CrossRef CAS PubMed.
- Y. Li, X. Guo, X. Li, M. Zhang, Z. Jia, Y. Deng, Y. Tian, S. Li and L. Ma, Redox-Active Two-Dimensional Covalent Organic Frameworks (COFs) for Selective Reductive Separation of Valence-Variable, Redox-Sensitive and Long-Lived Radionuclides, Angew. Chem., Int. Ed., 2020, 59, 4168–4175 CrossRef CAS PubMed.
- C. V. Waiman, J. M. Arroyave, H. Chen, W. Tan, M. J. Avena and G. P. Zanini, The Simultaneous Presence of Glyphosate and Phosphate at the Goethite Surface as Seen by XPS, ATR-FTIR and Competitive Adsorption Isotherms, Colloids Surf., A, 2016, 498, 121–127 CrossRef CAS.
- I. W. Hamley, Peptide Fibrillization, Angew. Chem., Int. Ed., 2007, 46, 8128–8147 CrossRef CAS PubMed.
- S. Zhao, J. Berry-Gair, W. Li, G. Guan, M. Yang, J. Li, F. Lai, F. Corà, K. Holt, D. J. L. Brett, G. He and I. P. Parkin, The Role of Phosphate Group in Doped Cobalt Molybdate: Improved Electrocatalytic Hydrogen Evolution Performance, Adv. Sci., 2020, 7, 1903674 CrossRef CAS PubMed.
- Z. Zhao, L. Zhu, H. Li, P. Cheng, J. Peng, Y. Yin, Y. Yang, C. Wang, Z. Hu and Y. Yang, Antiamyloidogenic Activity of Aβ42-Binding Peptoid in Modulating Amyloid Oligomerization, Small, 2017, 13, 1602857 CrossRef PubMed.
- F. Huang, J. Wang, A. Qu, L. Shen, J. Liu, J. Liu, Z. Zhang, Y. An and L. Shi, Maintenance of Amyloid β Peptide Homeostasis by Artificial Chaperones Based on Mixed-Shell Polymeric Micelles, Angew. Chem., Int. Ed., 2014, 53, 8985–8990 CrossRef CAS PubMed.
- M. Richman, S. Wilk, M. Chemerovski, S. K. T. S. Wärmländer, A. Wahlström, A. Gräslund and S. Rahimipour, In Vitro and Mechanistic Studies of an Antiamyloidogenic Self-Assembled Cyclic d,l-α-Peptide Architecture, J. Am. Chem. Soc., 2013, 135, 3474–3484 CrossRef CAS PubMed.
- M. Fokkens, T. Schrader and F.-G. Klärner, A Molecular Tweezer for Lysine and Arginine, J. Am. Chem. Soc., 2005, 127, 14415–14421 CrossRef CAS PubMed.
- R. Sarroukh, E. Cerf, S. Derclaye, Y. F. Dufrene, E. Goormaghtigh, J. M. Ruysschaert and V. Raussens, Transformation of Amyloid beta(1-40) Oligomers into Fibrils is Characterized by A Major Change in Secondary Structure, Cell. Mol. Life Sci., 2011, 68, 1429–1438 CrossRef CAS PubMed.
- D. Röhr, B. D. C. Boon, M. Schuler, K. Kremer, J. J. M. Hoozemans, F. H. Bouwman, S. F. El-Mashtoly, A. Nabers, F. Grosserueschkamp, A. J. M. Rozemuller and K. Gerwert, Label-free Vibrational Imaging of Different Abeta Plaque Types in Alzheimer’s Disease Reveals Sequential Events in Plaque Development, Acta Neuropathol. Commun., 2020, 8, 222 CrossRef PubMed.
- A. Nabers, J. Ollesch, J. Schartner, C. Kotting, J. Genius, H. Hafermann, H. Klafki, K. Gerwert and J. Wiltfang, Amyloid-beta-Secondary Structure Distribution in Cerebrospinal Fluid and Blood Measured by an Immuno-Infrared-Sensor: A Biomarker Candidate for Alzheimer's Disease, Anal. Chem., 2016, 88, 2755–2762 CrossRef CAS PubMed.
- A. Micsonai, F. Wien, L. Kernya, Y.-H. Lee, Y. Goto, M. Réfrégiers and J. Kardos, Accurate Secondary Structure Prediction and Fold Recognition for Circular Dichroism Spectroscopy, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E3095–E3103 CrossRef CAS PubMed.
- A. Micsonai, F. Wien, É. Bulyáki, J. Kun, É. Moussong, Y.-H. Lee, Y. Goto, M. Réfrégiers and J. Kardos, BeStSel: A Web Server for Accurate Protein Secondary Structure Prediction and Fold Recognition from the Circular Dichroism Spectra, Nucleic Acids Res., 2018, 46, W315–W322 CrossRef CAS PubMed.
- S. Kumar, A. Henning-Knechtel, I. Chehade, M. Magzoub and A. D. Hamilton, Foldamer-Mediated Structural Rearrangement Attenuates Aβ Oligomerization and Cytotoxicity, J. Am. Chem. Soc., 2017, 139, 17098–17108 CrossRef CAS PubMed.
- K. Hou, J. Zhao, H. Wang, B. Li, K. Li, X. Shi, K. Wan, J. Ai, J. Lv, D. Wang, Q. Huang, H. Wang, Q. Cao, S. Liu and Z. Tang, Chiral Gold Nanoparticles Enantioselectively Rescue Memory Deficits in A Mouse Model of Alzheimer's Disease, Nat. Commun., 2020, 11, 4790 CrossRef CAS PubMed.
- Y. J. Chung, K. Kim, B. I. Lee and C. B. Park, Carbon Nanodot-Sensitized Modulation of Alzheimer's beta-Amyloid Self-Assembly, Disassembly, and Toxicity, Small, 2017, 13, 201700983 CrossRef PubMed.
- C. Wang, B. Wu, Y. Wu, X. Song, S. Zhang and Z. Liu, Camouflaging Nanoparticles with Brain Metastatic Tumor Cell Membranes: A New Strategy to Traverse Blood–Brain Barrier for Imaging and Therapy of Brain Tumors, Adv. Funct. Mater., 2020, 30, 1909369 CrossRef CAS.
- S. H. Nasr, H. Kouyoumdjian, C. Mallett, S. Ramadan, D. C. Zhu, E. M. Shapiro and X. Huang, Detection of β-Amyloid by Sialic Acid Coated Bovine Serum Albumin Magnetic Nanoparticles in a Mouse Model of Alzheimer's Disease, Small, 2018, 14, 1701828 CrossRef PubMed.
- Y. Liu, Y. Ma, J. Yang, C. S. Diercks, N. Tamura, F. Jin and O. M. Yaghi, Molecular Weaving of Covalent Organic Frameworks for Adaptive Guest Inclusion, J. Am. Chem. Soc., 2018, 140, 16015–16019 CrossRef CAS PubMed.
- Q. Guan, L.-L. Zhou, Y.-A. Li, W.-Y. Li, S. Wang, C. Song and Y.-B. Dong, Nanoscale Covalent Organic Framework for Combinatorial Antitumor Photodynamic and Photothermal Therapy, ACS Nano, 2019, 13, 13304–13316 CrossRef CAS PubMed.
- M. Li, C. Xu, L. Wu, J. Ren, E. Wang and X. Qu, Self-assembled Peptide-polyoxometalate Hybrid Nanospheres: Two in One Enhances Targeted Inhibition of Amyloid beta-peptide Aggregation Associated with Alzheimer's Disease, Small, 2013, 9, 3455–3461 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, molecular dynamic simulation, morphology, cell imaging, and cytotoxicity assay details are included here. See https://doi.org/10.1039/d2sc00253a |
| This journal is © The Royal Society of Chemistry 2022 |