
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile synthesis of amides via acceptorless dehydrogenative coupling of aryl epoxides and amines†
Yaoyu
Liang
,
Jie
Luo
and
David
Milstein
 *
*
Department of Molecular Chemistry and Materials Science, Weizmann Institute of Science, Rehovot, 76100, Israel. E-mail: david.milstein@weizmann.ac.il
First published on 26th April 2022
Abstract
The synthesis of amides is significant in a wide variety of academic and industrial fields. We report here a new reaction, namely acceptorless dehydrogenative coupling of epoxides and amines to form amides catalyzed by ruthenium pincer complexes. Various aryl epoxides and amines smoothly convert into the desired amides in high yields with the generation of H2 gas as the only byproduct. Control experiments indicate that amides are generated kinetically faster than side products, possibly because of the facile activation of epoxides by metal–ligand cooperation, as supported by the observation of a ruthenium-enolate species. No alcohol or free aldehyde are involved. A mechanism is proposed involving a dual role of the catalyst, which is responsible for the high yield and selectivity of the new reaction.
Introduction
Amides are a very important class of compounds due to the wide presence of their structural units in peptides, natural products, pharmaceuticals, and polymers.1 Hence, the efficient synthesis of amides is of great interest in organic synthesis. Traditionally, amides are synthesized via the condensation of carboxylic acids and their derivatives with amines using activation reagents.2 To meet the requirement of environmentally friendly synthesis, various catalytic methods have been established in the past decades by employing various starting materials.3,4 In this respect, epoxides are potentially attractive candidates toward the preparation of amides. As versatile and useful intermediates in organic synthesis, epoxides have been applied to the preparation of various functional molecules through straightforward and atom economical methods.5–7 In that sense, discovery of direct, waste-free amidation of epoxides with amines can offer an attractive environmentally benign procedure for the synthesis of amides. In addition, since epoxides are generally accessed from alkenes via industrialized procedures and numerous other methods,8,9 the amidation of epoxides also provides a two-step strategy for transforming alkenes into amides.10 Nonetheless, to our knowledge, only one case of amidation of epoxides has been reported, based on the Willgerodt reaction which consumes large amounts of sulfur powder and ammonium and generates copious toxic waste (Scheme 1a).11 The main challenges to successfully achieve the waste-free amidation of epoxides might include the following issues: (i) amines are strong nucleophiles that could lead to nucleophilic ring-opening of epoxides to generate amino alcohols as byproducts, counteracting the occurrence of the major reaction;7 (ii) the highly reactive epoxide easily decomposes and thus results in side reactions;5,6 (iii) and most importantly, regioselective ring-opening of epoxides is required for the amidation to take place. Therefore, developing a new strategy to overcome these challenges and efficiently realize the coupling of epoxides and amines is necessary. | ||
| Scheme 1 Synthesis of amides from epoxides. | ||
Metal–ligand cooperation via dearomatization/aromatization is a useful tool for the activation of chemical bonds.12 To our knowledge, the activation of epoxides in such a manner has not been realized so far. As a continuation of our research interest in pincer complex catalyzed dehydrogenative coupling reactions,4b,f,13 we herein report the ruthenium pincer complex catalyzed acceptorless dehydrogenative coupling of epoxides and amines to form amides (Scheme 1b). The epoxides were found to efficiently convert into amide products with generally excellent yields in a single step with the generation of H2 gas as the only byproduct. Side reactions were efficiently suppressed by utilizing the bipyridine- and pyridine-based PNN ruthenium complexes as catalysts. The high regioselectivity of the ring-opening of epoxides was guaranteed by the unique activation pattern by the metal complex, which results in a Ru-enolate intermediate. This amidation reaction of epoxides offers a facile and atom economical two-step strategy for transforming alkenes into amides.
Results and discussion
Amidation of epoxides
We initiated the investigation using the pyridine-based ruthenium complex Ru-1 as the catalyst with a catalytic amount of tBuOK. The combination of Ru-1 and base results in a dearomatized complex (Ru-7, vide infra), which was reported as the catalytically active species in the acceptorless dehydrogenative coupling of alcohols with amines to yield amides.4b 2-phenyloxirane (1a) and secondary amine 2a were chosen as the model substrates, the latter was chosen in order to avoid potential side reactions due to possible dehydrogenation under the reaction conditions (Table 1).4h Upon heating at 120 °C in toluene for 12 h, low conversion of the epoxide was observed, with no desired amide formed (entry 1). Only 3% of the amino alcohol byproduct 3a′ was detected. 3a′ is likely generated by direct nucleophilic ring-opening of the epoxide with amine 2a. Next, we screened other pincer complexes. The acridine-based Ru-2 did not give the desired product either but produced more byproduct 3a′ than Ru-1 (entry 2). Interestingly, a higher conversion of 1a was observed, and 42% of amide product 3a was formed using Ru-3 as the catalyst (entry 3). The result prompted us to adjust the steric hindrance of the complex to improve the reaction yield further; however using the smaller iPr2P substituted ligand instead of tBu2P was less effective (entry 4). Other metal complexes based on the bipyridine PNN structure were also tested. For example, using Mn and Co complexes as catalysts, only byproduct 3a′ was formed (entries 5 and 6). An improved yield of 3a using Ru-3 was obtained upon increasing the reaction temperature to 135 °C (entry 7). Gratifyingly, further temperature increase to 150 °C resulted in 91% yield of product and the amount of byproduct 3a′ kept low (entry 8). As reported,5c Lewis acids can assist the isomerization of epoxide to aldehydes, which might lead to further amidation of the aldehyde in the current case. Therefore, the Lewis acids Zn(OTf)2 and BF3·Et2O were added as cocatalysts to the reaction to accelerate the isomerization step. However, only byproduct 3a′ was formed (entries 9 and 10). Next, the effect of solvent was explored as well. Solvents with different boiling points and polarities including xylene, chlorobenzene, benzene, dioxane, and DMF did not give higher yields of 3a (entries 11–15). It should be mentioned that the amide was not produced without a catalytic amount of base, (entry 16). Prolonging the reaction time to 24 h or 36 h did not improve the yield (entries 17 and 18).|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat | Solvent | Temp (°C) | Time (h) | Yield (%)b |
| 3a/3a′ | |||||
| a Conditions: 1a (0.5 mmol), 2a (0.5 mmol), cat. (1 mol%), tBuOK (1.2 mol%), solvent (1 mL). b NMR yield using mesitylene as the internal standard. c 1 mol% of NaBEt3H was added. d With 1 mol% Zn(OTf)2. e With 1 mol% BF3·Et2O. f Without tBuOK. | |||||
| 1 | Ru-1 | Toluene | 120 | 12 | nd/3 |
| 2 | Ru-2 | Toluene | 120 | 12 | nd/6 |
| 3 | Ru-3 | Toluene | 120 | 12 | 42/3 |
| 4 | Ru-4 | Toluene | 120 | 12 | 30/5 |
| 5 | Mn-1 | Toluene | 120 | 12 | nd/86 |
| 6c | Co-1 | Toluene | 120 | 12 | nd/72 |
| 7 | Ru-3 | Toluene | 135 | 12 | 61/4 |
| 8 | Ru-3 | Toluene | 150 | 12 | 91/4 |
| 9d | Ru-3 | Toluene | 150 | 12 | nd/96 |
| 10e | Ru-3 | Toluene | 150 | 12 | nd/77 |
| 11 | Ru-3 | Xylene | 150 | 12 | 81/4 |
| 12 | Ru-3 | PhCl | 150 | 12 | 12/15 |
| 13 | Ru-3 | Benzene | 150 | 12 | 52/11 |
| 14 | Ru-3 | Dioxane | 150 | 12 | 83/8 |
| 15 | Ru-3 | DMF | 150 | 12 | nd/12 |
| 16f | Ru-3 | Toluene | 150 | 12 | nd/3 |
| 17 | Ru-3 | Toluene | 150 | 24 | 91/4 |
| 18 | Ru-3 | Toluene | 150 | 36 | 91/4 |
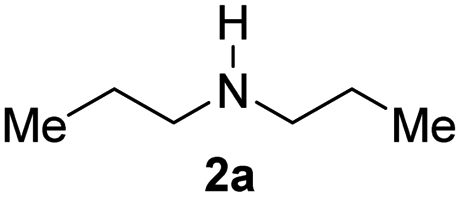
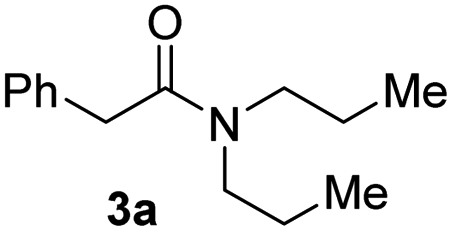
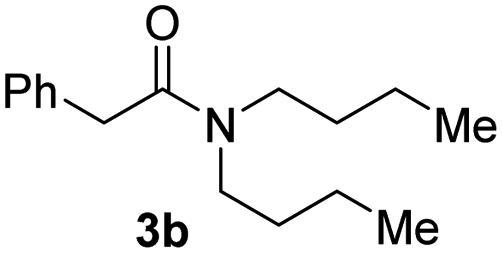
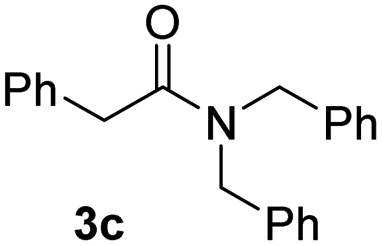
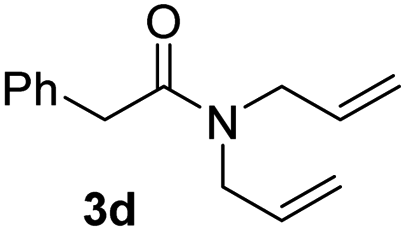
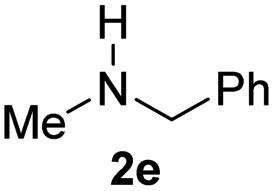
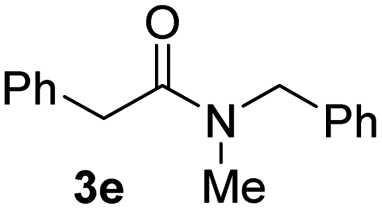
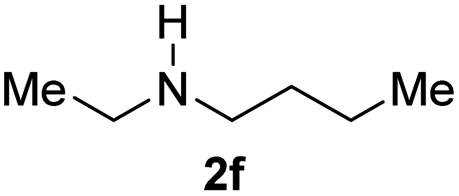
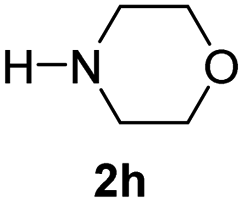
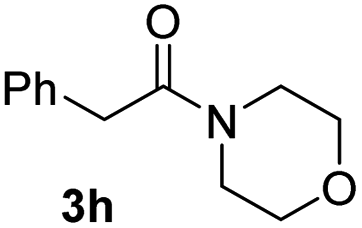
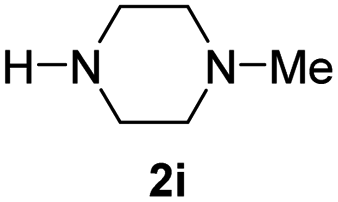
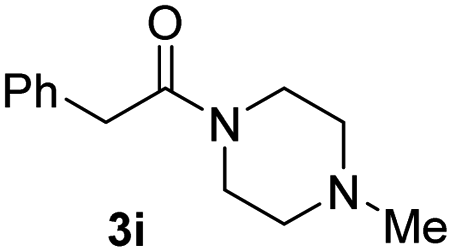
With the optimal conditions in hand, the scope of amine substrates was firstly evaluated (Table 2). Secondary amines with different chain lengths gave the desired amides with excellent isolated yields (3a–3b, 91–95%). Steric hindrance of the employed amines has an impact on the reaction yield. For example, in the case of the dibenzyl substituted amine 2c (entry 3), amide 3c was generated in less than 50% yield under the optimal conditions. Nevertheless, using an open system with the release of the generated H2, 84% of isolated yield was obtained after extending the reaction time to 48 h (entry 3). In addition, the amidation reaction can proceed with an amine bearing C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds, generating the targeted amide product (3d) in 62% yield. Notably, the CC double bond on the amide product provides good opportunities for its further functionalization. Asides from symmetric secondary amines, other secondary amines such as N-methylbenzylamine (2e) and ethylbutylamine (2f) also worked well to give the corresponding products under the optimal conditions (3e–3f). In addition, cyclic secondary amines 2g–2i were also evaluated, the reaction yields being just slightly affected (71–83%).
C bonds, generating the targeted amide product (3d) in 62% yield. Notably, the CC double bond on the amide product provides good opportunities for its further functionalization. Asides from symmetric secondary amines, other secondary amines such as N-methylbenzylamine (2e) and ethylbutylamine (2f) also worked well to give the corresponding products under the optimal conditions (3e–3f). In addition, cyclic secondary amines 2g–2i were also evaluated, the reaction yields being just slightly affected (71–83%).
|
|
|||
|---|---|---|---|
| Entry | Amine | Product | Isolated yield (%) |
| a Conditions: 1a (0.5 mmol), 2 (0.5 mmol), Ru-3 (1 mol%), tBuOK (1.2 mol%), toluene (1 mL), 150 °C, 12 h. b Reaction time is 48 h with mesitylene as solvent in an open system. c Ru-1 (1 mol%) was used as catalyst instead of Ru-3. | |||
| 1 |
|
|
91 |
| 2 |
|
|
95 |
| 3b |

|
|
84 |
| 4 |
|
|
62 |
| 5 |
|
|
78 |
| 6 |
|

|
95 |
| 7 |

|

|
71 |
| 8 |
|
|
80 |
| 9 |
|
|
83 |
| 10c |
|

|
66 |
| 11c |
|
|
44 |
| 12c |

|

|
51 |
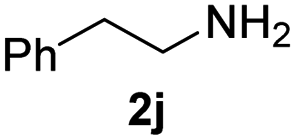
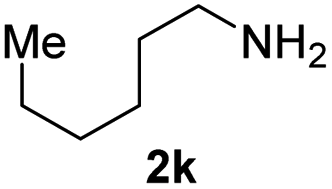
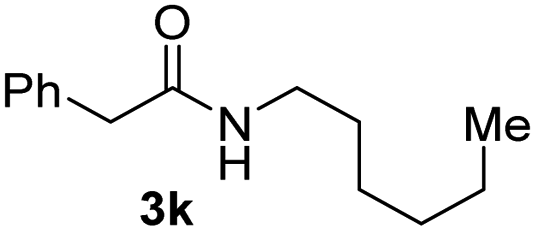
To further extend the scope of amines, we turned our attention to examining primary amines. However, when 2-phenylethylamine 2j was used, diphenethylamine was formed as the major product. Mechanistically speaking, this result could be attributed to the easy dehydration of the hemiaminal intermediate that led to the formation of imine, which subsequently underwent hydrogenation to produce the secondary amine under the current system. To access the desired amide product from primary amines, further optimization was carried out (see Table S2†). Interestingly, using Ru-1 as a catalyst, the reaction of epoxide 1a and amine 2j resulted in formation of amide 3j in 66% isolated yield under the optimal conditions, but the formation of the secondary amine and amino alcohol was inevitable in this case. The conditions were also suitable for other primary amines. Hexylamine 2k gave the amide product, although in only 44% isolated yield, presumably because of its relatively low boiling point. Benzylamine 2l transformed into the corresponding amide with medium isolated yield (51%). These results indicate the compatibility of this method for both secondary and primary amines.
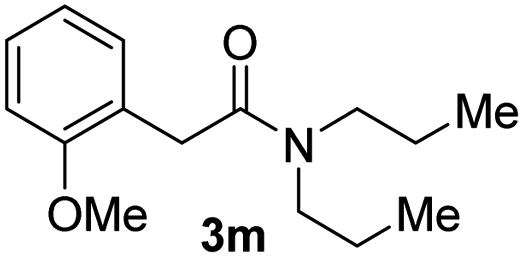
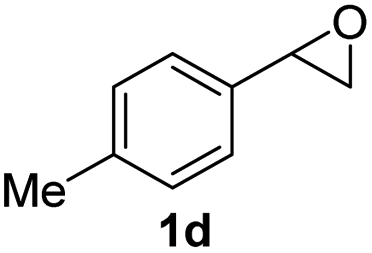
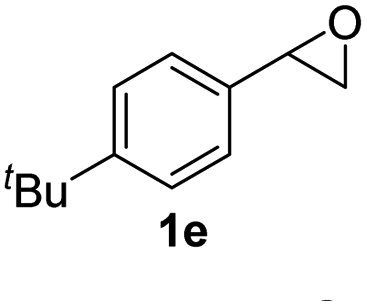
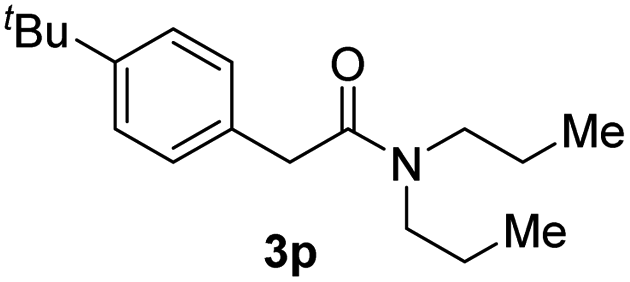
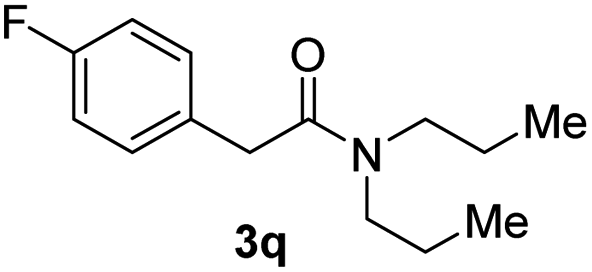
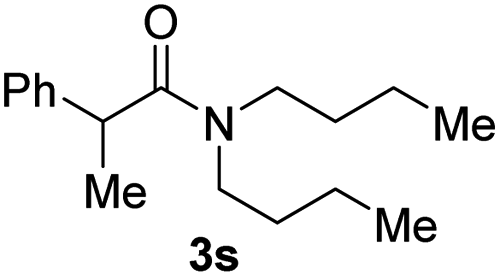
Next, the scope of epoxides was explored under the standard conditions (Table 3). The steric hindrance of the aryl-substituted epoxide had no impact on the reaction yield. For example, ortho-substituted epoxide 1b smoothly produced the desired amide 3m in 89% yield. As expected, meta-methyl substituted epoxide 1c gave an excellent result (3n, 92%). When a variety of epoxides bearing functional groups, i.e., Me-, tBu-, F-, and CF3 at the para-position of the phenyl ring were utilized, the reactions gave the corresponding products with high yields (3o–3r, 80–90%). The electron density of the epoxides slightly affected the yield of the reaction. Electron-rich epoxides (1b–1d) gave higher yields compared to the electron-deficient epoxides (1f–1g). To our surprise, 1,1-disubstituted epoxide 1h also generated the desired amide with good yield (3s, 65%) when mesitylene was used as solvent in an open system. It is worth mentioning that amides bearing a tertiary carbon stereogenic center on the α-position are important in many areas.1 Unfortunately, use of aliphatic epoxides resulted in low conversion and only provided a trace amount of product under the reaction conditions.








Mechanistic investigation
To gain insight into the mechanism of the amidation reaction, control experiments were conducted. As demonstrated in Table 1, epoxide 1a can undergo nucleophilic ring-opening with amine 2a to produce the amino alcohol byproduct 3a′. According to previous works, 3a′ can be formed spontaneously without catalyst.7a,c Therefore, when epoxide 1a was treated with amine 2a at 150 °C with toluene as solvent, 51% yield of 3a′ was obtained after heating for 20 h (Fig. 1a, top). 3a′ was proved unable to transform into the corresponding amide under the standard reaction conditions, indicating that 3a′ is not an intermediate toward formation of amide 3a. Meanwhile, ester 4 was observed under the catalysis of Ru-3 and tBuOK using the epoxide in the absence of amine (Fig. 1a, bottom, 14% yield after 20 h). As reported, esters can convert into amides with Ru-3 and tBuOK as catalysts under refluxing conditions, according to our previous results.4f However, only a trace amount of 3a was observed when ester 4 was subjected to the current conditions in a closed system (Fig. 1a, bottom). The results suggest that ester 4 is not an intermediate toward amide formation in this reaction either. These observations led us to investigate why 3 could be generated as the major products rather than amino alcohols or esters under the developed reaction. The rate of formation of 3a, 3a′, and 4 was compared, and the results are exhibited in Fig. 1b. It was found that the formation of amide 3a was relatively rapid under the standard conditions, and the reaction was finished in about 8 to 10 h (blue curve). In contrast, the formation of amino alcohol 3a′ in the absence of catalyst, and the formation of ester 4 in the absence of amine were both slower than the formation of amide 3a at the same temperature (red and black curves). Thus, almost all of the epoxides would quickly convert into amides in the actual catalysis.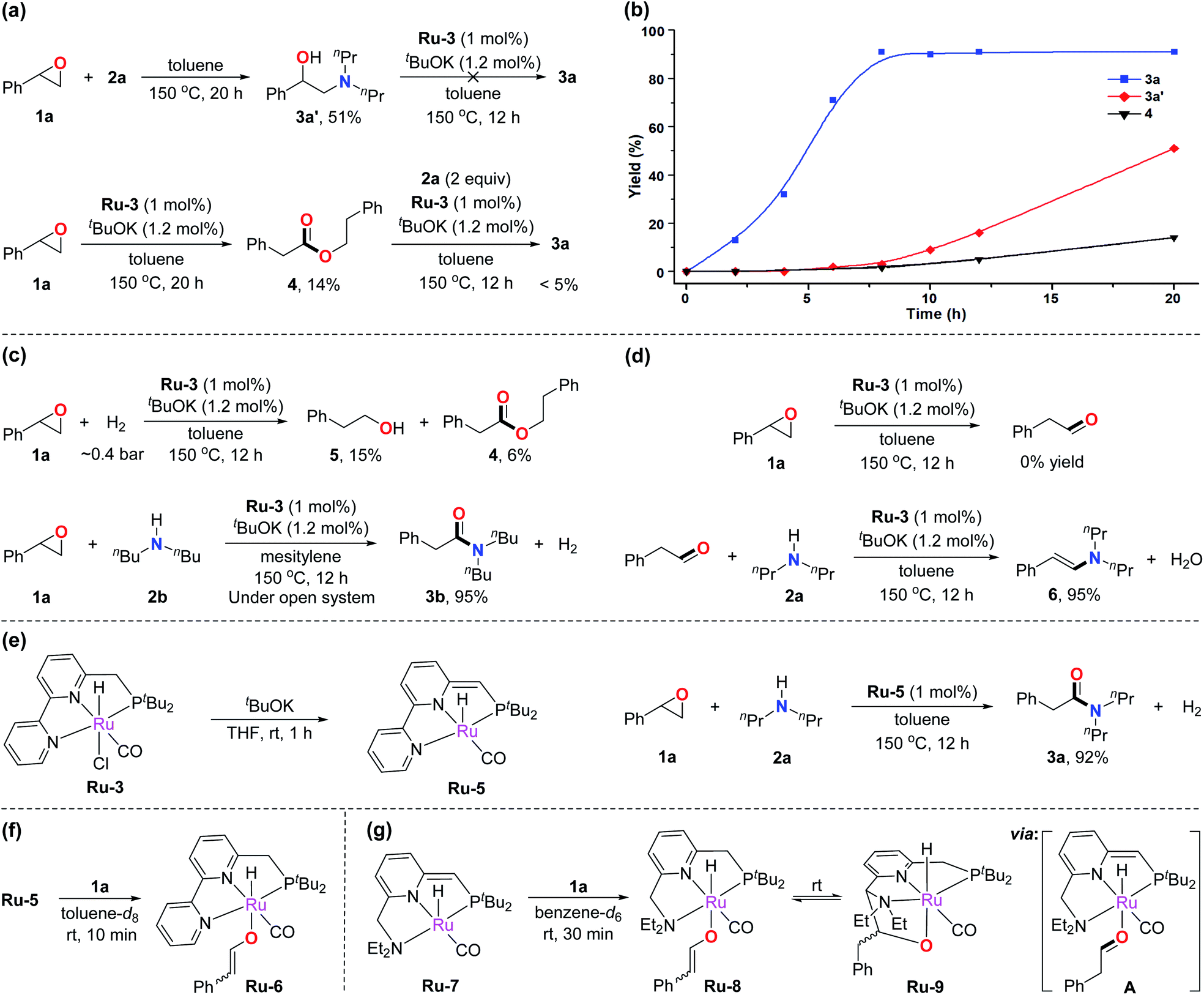 | ||
| Fig. 1 Mechanistic aspects of the reaction. (a) Investigation of the generation of byproducts 3a′ and 4, and the possibility of converting into 3a. (b) The correlation between reaction time and yields of 3a (standard conditions), 3a′ (without catalyst), and 4 (without amine). (c) Control experiments for excluding the participation of H2. (d) Control experiments for excluding the generation of free aldehyde. (e) The active catalytic species of amidation reaction. (f) Formation of Ru-enolate intermediates Ru-6 in the reaction. (g) Formation of Ru-8 and its reversible conversion into Ru-9. | ||
In a closed system, epoxides can potentially undergo hydrogenation by the generated H2 in the current reaction to produce primary alcohols,6g,h which could further couple with amines to generate amides catalyzed by the employed ruthenium pincer complexes.4b,14 To explore this possibility, epoxide 1a was treated with ∼0.4 bar of H2 (in a 90 mL Fischer–Porter tube, relevant to the amount of generated H2 in the amidation reaction) at 150 °C, forming only 15% of primary alcohol 5 along with 6% of ester 4 under similar conditions (Fig. 1c, top). This result suggests that the hydrogen gas generated by the amidation reaction was inefficient in converting the epoxide into primary alcohol. Moreover, the amidation reaction was carried out in an open system to further exclude the effect of the generated hydrogen gas. Considering the boiling points of the employed components, mesitylene and dibutylamine were selected as the solvent and amine, respectively. It was found that the amide product was formed in the same yield as that in the closed system (Fig. 1c, bottom). This result proves that H2 is not involved in the catalytic cycle. Thus, an alcohol does not serve as an intermediate in this transformation.
The Meinwald rearrangement of epoxides is a well-known procedure for converting epoxides into aldehydes,6e,15 hence the possibility of an aldehyde intermediate in amide formation under the current catalytic system should be considered. The ruthenium pincer complex Ru-3 may also act as a Lewis acid catalyst to facilitate the rearrangement of epoxide 1a to phenylacetaldehyde. However, no aldehyde product was observed when 1a was subjected to the standard conditions in the absence of amine (Fig. 1d, top). Besides, when phenylacetaldehyde was employed as an alternative substrate under the standard conditions, enamine 6 was generated as the major product (Fig. 1d, bottom). These results suggest that no free aldehyde is produced in this reaction.
Next, we turned our attention to investigating the actual ruthenium catalytic species in the developed reaction. According to our previous studies,16Ru-3 undergoes dearomatization upon treatment with base to produce the dearomatized complex Ru-5 as the catalytically active species (Fig. 1e, left). Indeed, utilizing the independently prepared Ru-5 as the catalyst in the amidation reaction, the desired amide 3a was obtained in 92% yield under similar conditions (Fig. 1e, right). Interestingly, mixing of Ru-5 and epoxide 1a in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio at room temperature led to the formation of new complexes, which were identified by 31P NMR and 1H NMR as two isomers of Ru-6 in a 1:2 ratio with an enolate (Z and E isomers) coordinated to the aromatic ruthenium complex (Fig. 1f). The 31P NMR spectrum exhibits two new peaks at 105.2 and 104.7 ppm in a 2:1 ratio; the chemical shifts of the signals indicate the formation of aromatized complexes. In the 1H NMR spectrum, two characteristic doublets corresponding to the alkene were observed at 7.74 and 4.79 ppm. H–H COSY spectrum confirms their correlations (J = 5.1 Hz) (see ESI page S17† for more characterization data). The formation of Ru-6 presumably stems from ring-opening of the epoxide followed by proton transfer to the side arm of Ru-5. We believe that generation of the relatively stable vinylbenzene enolate intermediate is an important driving force for the activation of the epoxide. Such evidence can also explain why the aliphatic epoxides were less effective in the amidation reaction.
:3 ratio at room temperature led to the formation of new complexes, which were identified by 31P NMR and 1H NMR as two isomers of Ru-6 in a 1:2 ratio with an enolate (Z and E isomers) coordinated to the aromatic ruthenium complex (Fig. 1f). The 31P NMR spectrum exhibits two new peaks at 105.2 and 104.7 ppm in a 2:1 ratio; the chemical shifts of the signals indicate the formation of aromatized complexes. In the 1H NMR spectrum, two characteristic doublets corresponding to the alkene were observed at 7.74 and 4.79 ppm. H–H COSY spectrum confirms their correlations (J = 5.1 Hz) (see ESI page S17† for more characterization data). The formation of Ru-6 presumably stems from ring-opening of the epoxide followed by proton transfer to the side arm of Ru-5. We believe that generation of the relatively stable vinylbenzene enolate intermediate is an important driving force for the activation of the epoxide. Such evidence can also explain why the aliphatic epoxides were less effective in the amidation reaction.
To further determine the formation of Ru-enolate intermediate, we turned our attention to the dearomatized Ru-7, obtained from Ru-1 by treatment of base.13a As mentioned above, Ru-1 catalyzed the current amidation reaction when primary amines were utilized. Mixing of Ru-7 and epoxide 1a in a 1:1 ratio, a Ru-enolate species Ru-8 was also observed in a 1:3 ratio of its isomers (Fig. 1g). In the 1H NMR spectrum, two doublets corresponding to the enolate were observed at 8.07 and 5.34 ppm. The proton chemical shift of the methyne group (CH) connected to the ORu appears at the low field (8.07 ppm), in the range characteristic of corresponding enol ethers. The correlation of these two signals was observed in the H–H COSY spectrum. The DEPTQ spectrum further confirms their adjacent carbons belong to sp2 CH units (see ESI† for more characterization). All these evidences are similar to the observation in Ru-6, thus confirming the formation of Ru-enolate intermediate. In addition, Ru-8 slowly converted into another new complex reaching a chemical equilibrium. The 1H NMR of the solution showed four different Ru-H signals due to the existence of isomers in both complexes (see Fig. S16†). We propose that the enolate on Ru-8 isomerizes to an aldehyde intermediate A, via a keto–enol equilibrium, which rapidly converted to Ru-9via electrophilic attack of the aldehyde on the side arm of intermediate A. Aldehyde attack on the side arm of Ru-7 analogous to the generation of Ru-9 was previously reported.17 The chemical shifts of phosphine and Ru-H in the NMR spectra match the previously reported works, which further confirm our assumption.
Based on the above results and previous studies,4b,f,13,16 a plausible reaction pathway is depicted in Scheme 2. The epoxide is firstly activated by catalyst Ru-5 to form the active adduct intermediate Int-I, which subsequently undergoes ring-opening and β-H extraction of the epoxide by metal–ligand cooperation to afford the enolate intermediate Ru-6 with high regioselectivity. Ru-6 reversibly converts into the Ru-aldehyde adduct Int-II, which transforms into the off-cycle intermediate Int-III in the absence of amines. Notably, free aldehyde was not generated at this stage, avoiding the formation of the enamine byproduct. In addition, the epoxide activation process via metal–ligand cooperation is distinct from the Lewis acid activated Meinwald rearrangement. Next, the nucleophilic amine attacks the bound aldehyde in Int-II to yield the hemiaminal intermediate Int-IV, which further undergoes β-H elimination to release the amide product and a molecule of H2, regenerating the active catalyst Ru-5 to enter the second catalytic cycle. Clearly, the mechanism demonstrates that Ru-5 plays a dual role in the current reaction. On the one hand, it facilitates epoxide activation to produce the Ru-enolate complex, which rapidly and reversibly transforms into the aldehyde intermediate Int-II; on the other hand, Ru-5 catalyzes the well-known dehydrogenative process via metal–ligand cooperation, namely amidation of intermediate aldehydes with amines.
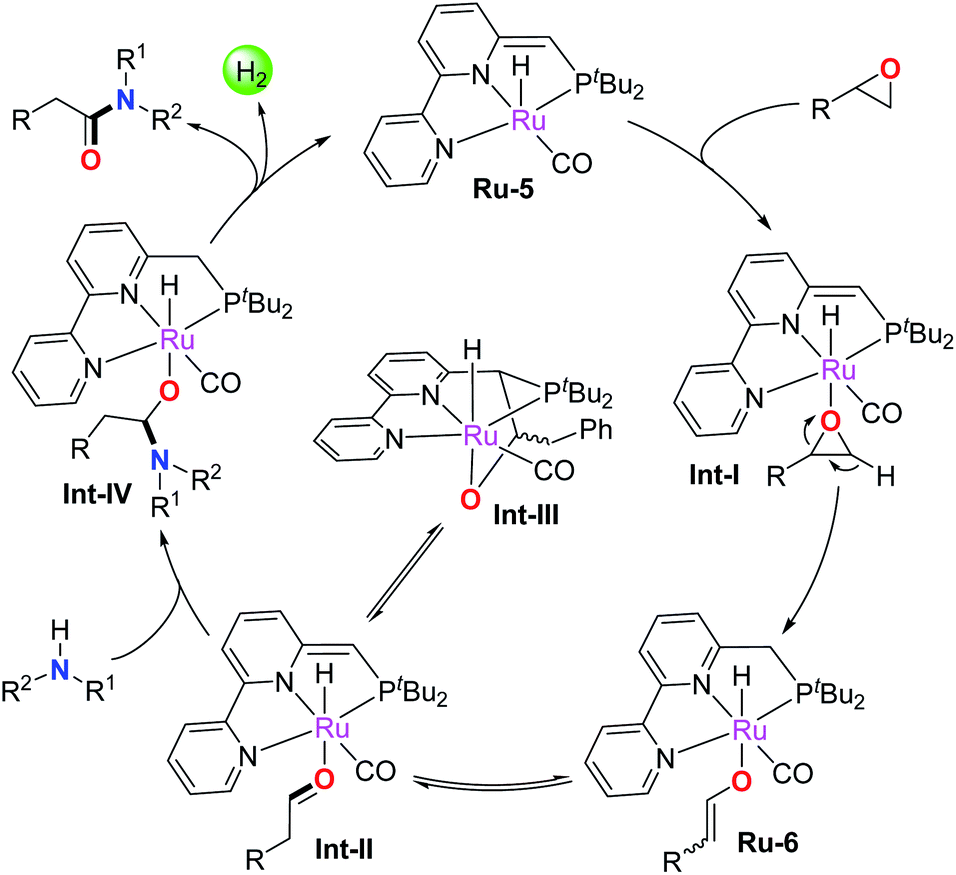 | ||
| Scheme 2 Proposed mechanism. | ||
Conclusion
In conclusion, we have developed a novel strategy for the synthesis of amides based on ruthenium pincer complex catalyzed acceptorless dehydrogenative coupling of terminal aryl epoxides and amines. The reaction yields are generally high. Various functional groups are tolerated under the reaction conditions. Control experiments demonstrate that the epoxide is activated by the ruthenium pincer complex through metal–ligand cooperation, leading to an observed Ru-enolate intermediate formed by reaction of the dearomatized ruthenium complex with the epoxide. Noteworthy, no alcohol or free aldehyde are involved in this reaction. The ruthenium pincer complex plays a dual role in the whole transformation. Notably, the reaction provides an environmentally friendly and convenient two-step procedure for transforming alkenes into amides. Further studies about the transformation of epoxides are underway in our laboratory.Author contributions
D. M. and Y. L. conceived the project and designed the experiments. Y. L. performed the experiments and analyzed the data. J. L. synthesized part of catalysts and provided helpful discussions on the project. Y. L., J. L., and D. M. prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
Experimental details, compound characterization data, NMR spectra.Acknowledgements
This research was supported by the European Research Council (ERC AdG 692775). D. M. holds the Israel Matz Professorial Chair of Organic Chemistry. J. L. is thankful to the Feinberg Graduate School of Weizmann Institute of Science for a Senior Postdoctoral Fellowship.Notes and references
- (a) The Amide Linkage: Structural Significance in Chemistry, Biochemistry and Material Science, ed. A. Greenberg, C. M. Breneman and J. F. Liebman, Wiley, New York, 2000 Search PubMed; (b) J. M. Humphrey and A. R. Chamberlin, Chem. Rev., 1997, 97, 2243–2266 CrossRef CAS PubMed; (c) B. L. Bray, Nat. Rev. Drug Discovery, 2003, 2, 587–593 CrossRef CAS PubMed; (d) J. W. Clader, J. Med. Chem., 2004, 47, 1–9 CrossRef CAS PubMed; (e) J. R. Dunetz, J. Magano and G. A. Weisenburger, Org. Process Res. Dev., 2016, 20, 140–177 CrossRef CAS.
- For reviews, see: (a) S.-Y. Han and Y.-A. Kim, Tetrahedron, 2004, 60, 2447–2467 CrossRef CAS; (b) C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827–10852 CrossRef CAS; (c) E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC; (d) R. M. Lanigan and T. D. Sheppard, Eur. J. Org. Chem., 2013, 33, 7453–7465 CrossRef.
- For reviews, see: (a) C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405–3415 RSC; (b) V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed; (c) L. Huang, M. Arndt, K. Goobβn, H. Heydt and L. J. Goobβn, Chem. Rev., 2015, 115, 2596–2697 CrossRef CAS PubMed; (d) R. M. de Figueiredo, J.-S. Suppo and J.-M. Campagne, Chem. Rev., 2016, 116, 12029–12122 CrossRef CAS PubMed.
- For selective examples of catalytic synthesis of amides, see: (a) W.-J. Yoo and C.-J. Li, J. Am. Chem. Soc., 2006, 128, 13064–13065 CrossRef CAS PubMed; (b) C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 317, 790–792 CrossRef CAS PubMed; (c) Y. Nakao, H. Idei, K. S. Kanyiva and T. Hiyama, J. Am. Chem. Soc., 2009, 131, 5070–5071 CrossRef CAS PubMed; (d) N. A. Stephenson, J. Zhu, S. H. Gellman and S. S. Stahl, J. Am. Chem. Soc., 2009, 131, 10003–10008 CrossRef CAS PubMed; (e) B. Shen, D. M. Makley and J. N. Johnston, Nature, 2010, 465, 1027–1032 CrossRef CAS PubMed; (f) B. Gnanaprakasam and D. Milstein, J. Am. Chem. Soc., 2011, 133, 1682–1685 CrossRef CAS PubMed; (g) X. Fang, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 14089–14093 CrossRef CAS PubMed; (h) J. R. Khusnutdinova, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2014, 136, 2998–3001 CrossRef CAS PubMed.
- (a) L. P. C. Nielsen, E. N. Jacobsen, P. Crotti, M. Pineschi, B. Olofsson and P. Somfai, Aziridines and Epoxides in Organic Synthesis, ed. A. K. Yudin, Wiley-VCH, Weinheim, Germany, 2006, pp. 229–343 Search PubMed; (b) C.-Y. Huang and A. G. Doyle, Chem. Rev., 2014, 114, 8153–8198 CrossRef CAS PubMed; (c) J. L. Jat and G. Kumar, Adv. Synth. Catal., 2019, 361, 4426–4441 CrossRef CAS; (d) A. K. Hubbell and G. W. Coates, J. Org. Chem., 2020, 85, 13391–13414 CrossRef CAS PubMed.
- For selective examples of epoxide transformation, see: (a) A. Gansäuer, C.-A. Fan and F. Piestert, J. Am. Chem. Soc., 2008, 130, 6916–6917 CrossRef PubMed; (b) D. K. Nielsen and A. G. Doyle, Angew. Chem., Int. Ed., 2011, 50, 6056–6059 CrossRef CAS PubMed; (c) D. J. Vyas, E. Larionov, C. Besnard, L. Guénée and C. Mazet, J. Am. Chem. Soc., 2013, 135, 6177–6183 CrossRef CAS PubMed; (d) Y. Zhao and D. J. Weix, J. Am. Chem. Soc., 2014, 136, 48–51 CrossRef CAS PubMed; (e) J. R. Lamb, M. Mulzer, A. M. LaPointe and G. W. Coates, J. Am. Chem. Soc., 2015, 137, 15049–15054 CrossRef CAS PubMed; (f) V. Pace, L. Castoldi, E. Mazzeo, M. Rui, T. Langer and W. Holzer, Angew. Chem., Int. Ed., 2017, 56, 12677–12682 CrossRef CAS PubMed; (g) C. Yao, T. Dahmen, A. Gansäuer and J. Norton, Science, 2019, 364, 764–767 CrossRef CAS PubMed; (h) W. Liu, W. Li, A. Spannenberg, K. Junge and M. Beller, Nat. Catal., 2019, 2, 523–528 CrossRef CAS; (i) M. Magre, E. Paffenholz, B. Maity, L. Cavallo and M. Rueping, J. Am. Chem. Soc., 2020, 142, 14286–14294 CrossRef CAS PubMed; (j) J. Xu, Y. Song, J. He, S. Dong, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2021, 60, 14521–14527 CrossRef CAS PubMed; (k) S. Zhang, M. Vayer, F. Noël, V. D. Vuković, A. Golushko, N. Rezajooei, C. N. Rowley, D. Lebœuf and J. Moran, Chem, 2021, 7, 3425–3441 CrossRef CAS.
- For selective examples of addition of amines to epoxides, see: (a) U. Das, B. Crousse, V. Kesavan, D. Bonnet-Delpon and J.-P. Bégué, J. Org. Chem., 2000, 65, 6749–6751 CrossRef CAS PubMed; (b) A. K. Chakraborti, S. Rudrawar and A. Kondaskar, Org. Biomol. Chem., 2004, 2, 1277–1280 RSC; (c) N. Azizi and M. R. Saidi, Org. Lett., 2005, 7, 3649–3651 CrossRef CAS PubMed; (d) R. I. Kureshy, S. Singh, N. H. Khan, S. H. R. Abdi, E. Suresh and R. V. Jasra, J. Mol. Catal. A: Chem., 2007, 264, 162–169 CrossRef CAS.
- (a) R. A. Johnson and K. B. Sharpless, Asymmetric Oxidations and Related Reactions: Catalytic Asymmetric Epoxidation of Allylic Alcohols, in Catalytic Asymmetric Synthesis, John Wiley & Sons, Ltd, 2000, pp. 229–280 Search PubMed; (b) B. S. Lane and K. Burgess, Chem. Rev., 2003, 103, 2457–2474 CrossRef CAS PubMed; (c) E. M. McGarrigle and D. G. Gilheany, Chem. Rev., 2005, 105, 1563–1602 CrossRef CAS PubMed; (d) Y. Zhu, Q. Wang, R. G. Cornwall and Y. Shi, Chem. Rev., 2014, 114, 8199–8256 CrossRef CAS PubMed; (e) M. S. Batra, R. Dwivedi and R. Prasad, ChemistrySelect, 2019, 4, 11636–11673 CrossRef CAS; (f) X. Li, Q. Wang, J. Lyu and X. Li, ChemistrySelect, 2021, 6, 9735–9768 CrossRef CAS.
- For selective examples, see: (a) K. Chen, M. Costas, J. H. Kim, A. K. Tipton and L. Que, J. Am. Chem. Soc., 2002, 124, 3026–3035 CrossRef CAS PubMed; (b) A. Murphy, G. Dubois and T. D. P. Stack, J. Am. Chem. Soc., 2003, 125, 5250–5251 CrossRef CAS PubMed; (c) R. Mas-Ballesté and L. Que, J. Am. Chem. Soc., 2007, 129, 15964–15972 CrossRef PubMed; (d) O. Cussó, I. Garcia-Bosch, X. Ribas, J. Lloret-Fillol and M. Costas, J. Am. Chem. Soc., 2013, 135, 14871–14878 CrossRef PubMed.
- To date, direct transformation of C=C double bonds into amides, rather than introducing additional amide groups into the alkenes, still remains elusive. Multistep procedures are usually required. For example, a three-step procedure involving the regioselective hydration of alkenes to produce primary alcohols, which further undergo oxidation to the corresponding carbonyl derivatives, and the final amidation reaction to generate amides Search PubMed.
- R. T. Gerry and E. V. Brown, J. Am. Chem. Soc., 1953, 75, 740–741 CrossRef CAS.
- (a) C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588–602 CrossRef CAS PubMed; (b) C. Gunanathan and D. Milstein, Chem. Rev., 2014, 114, 12024–12087 CrossRef CAS PubMed; (c) J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed; (d) A. Mukherjee and D. Milstein, ACS Catal., 2018, 8, 11435–11469 CrossRef CAS; (e) L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2019, 119, 2681–2751 CrossRef CAS PubMed.
- (a) J. Zhang, G. Leitus, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2005, 127, 10840–10841 CrossRef CAS PubMed; (b) E. Balaraman, E. Khaskin, G. Leitus and D. Milstein, Nat. Chem., 2013, 5, 122–125 CrossRef CAS PubMed; (c) P. Hu, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2016, 138, 6143–6146 CrossRef CAS PubMed; (d) N. A. Espinosa-Jalapa, A. Kumar, G. Leitus, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2017, 139, 11722–11725 CrossRef CAS PubMed; (e) J. Luo, M. Rauch, L. Avram, Y. Diskin-Posner, G. Shmul, Y. Ben-David and D. Milstein, Nat. Catal., 2020, 3, 887–892 CrossRef CAS.
- (a) T. Zweifel, J.-V. Naubron and H. Grützmacher, Angew. Chem., Int. Ed., 2009, 48, 559–563 CrossRef CAS PubMed; (b) N. D. Schley, G. E. Dobereiner and R. H. Crabtree, Organometallics, 2011, 30, 4174–4179 CrossRef CAS; (c) D. Cho, K. C. Ko and J. Y. Lee, Organometallics, 2013, 32, 4571–4576 CrossRef CAS; (d) B. Saha, G. Sengupta, A. Sarbajna, I. Dutta and J. K. Bera, J. Organomet. Chem., 2014, 771, 124–130 CrossRef CAS; (e) J. Kothandaraman, S. Kar, R. Sen, A. Goeppert, G. A. Olah and G. K. S. Prakash, J. Am. Chem. Soc., 2017, 139, 2549–2552 CrossRef CAS PubMed; (f) L. Li, M. Lei, L. Liu, Y. Xie and H. F. Schaefer, Inorg. Chem., 2018, 57, 8778–8787 CrossRef CAS PubMed; (g) G. Choi and S. H. Hong, ACS Sustainable Chem. Eng., 2019, 7, 716–723 CrossRef CAS.
- J. Meinwald, S. S. Labana and M. S. Chadha, J. Am. Chem. Soc., 1963, 85, 582–585 CrossRef CAS.
- E. Balaraman, B. Gnanaprakasam, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2010, 132, 16756–16758 CrossRef CAS PubMed.
- C. A. Huff, J. W. Kampf and M. S. Sanford, Chem. Commun., 2013, 49, 7147–7149 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc01959k |
| This journal is © The Royal Society of Chemistry 2022 |