
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCrystallization induced room-temperature phosphorescence and chiral photoluminescence properties of phosphoramides†
Satyam
Jena
a,
Jusaina
Eyyathiyil
 a,
Santosh Kumar
Behera
a,
Maho
Kitahara
b,
Yoshitane
Imai
*b and
Pakkirisamy
Thilagar
*a
a,
Santosh Kumar
Behera
a,
Maho
Kitahara
b,
Yoshitane
Imai
*b and
Pakkirisamy
Thilagar
*a
aDepartment of Inorganic and Physical Chemistry, Indian Institute of Science Bangalore, India – 560012. E-mail: thilagar@iisc.ac.in
bDepartment of Applied Chemistry, Faculty of Science and Engineering, Kindai University, 3-4-1 Kowakae, Higashi-Osaka, Osaka 577-8502, Japan. E-mail: y-imai@apch.kindai.ac.jp
First published on 20th April 2022
Abstract
We report the design and synthesis of a series of room temperature phosphorescent phosphoramides TPTZPO, TPTZPS, and TPTZPSe with a donor (phenothiazine)–acceptor (P = X, X = O, S, and Se) architecture. All the compounds show structureless fluorescence with a nanosecond lifetime in dilute solutions. However, these compounds show dual fluorescence and room temperature phosphorescence (RTP) in the solid state. Both the intensity and energy of luminescence depend on the heteroatom attached to the phosphorus center. For example, compound TPTZPO with the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O unit exhibits fluorescence at a higher energy region than TPTZPS and TPTZPSe with the PS and PSe groups, respectively. Crystalline samples of TPTZPO, TPTZPS, and TPTZPSe show stronger RTP than the amorphous powder of respective compounds. Detailed steady-state, time-resolved photoluminescence and computational studies established that the 3n–π* state dominated by the phenothiazine moiety is the emissive state of these compounds. Although TPTZPS and TPTZPSe crystallized in the chiral space group, only TPTZPSe showed chiroptical properties in the solid state. The luminescence dissymmetry factor (glum) value of TPTZPS is small and below the detection limit, and a CPL spectrum could not be observed for this compound.
O unit exhibits fluorescence at a higher energy region than TPTZPS and TPTZPSe with the PS and PSe groups, respectively. Crystalline samples of TPTZPO, TPTZPS, and TPTZPSe show stronger RTP than the amorphous powder of respective compounds. Detailed steady-state, time-resolved photoluminescence and computational studies established that the 3n–π* state dominated by the phenothiazine moiety is the emissive state of these compounds. Although TPTZPS and TPTZPSe crystallized in the chiral space group, only TPTZPSe showed chiroptical properties in the solid state. The luminescence dissymmetry factor (glum) value of TPTZPS is small and below the detection limit, and a CPL spectrum could not be observed for this compound.
1. Introduction
Room temperature phosphorescent (RTP) materials have attracted considerable research attention owing to their potential applications in organic light-emitting diodes (OLEDs),1–9 sensing,10–13 and time-gated bio-imaging applications.14–21 Phosphorescence is the spin forbidden radiative decay of triplet excitons to the ground singlet state (T1→S0). The efficiency of RTP is dependent on the Sn→Tn intersystem crossing (ISC); the efficiency of ISC depends on the spin–orbit coupling (SOC). Thus, in the past, the development of RTP materials was mainly limited to only transition metal complexes (e.g. iridium(III), platinum(II), gold(III), ruthenium(II), and copper(I)).22–33Recently, enormous research efforts have been devoted to developing metal-free organic phosphors because of their low toxicity and environmentally benign nature compared to their inorganic counterparts. However, organic phosphors exhibit weak luminescence with low phosphorescence quantum yield under ambient conditions due to weak SOC, inefficient intersystem crossing, and poor radiative T1→S0 transition.34,35 Thus, developing organic phosphors with improved phosphorescence quantum yield is an important and challenging area of research. Innovative techniques such as crystallization-induced phosphorescence, doping in a rigid host (polymer matrices), aggregates, and multi-component molecular hybrids have been successfully explored for developing efficient RTP materials by reducing the non-radiative deactivation of triplet excitons.36–47
On the other hand, incorporating main-group elements such as B, Si, N, P, O, S, and halogens in organic backbones provides a new strategy to achieve functional molecules.35,48–76 Recently, finetuning the interactions between orbitals of different symmetry in heteroatom doped organic luminophores has attracted considerable attention for developing RTP materials. Not long ago, Huang and co-workers developed strongly emissive RTP materials by finetuning the interactions between sulfur and oxygen in oxidized phenothiazine derivatives.77 Despite these developments, bright phosphorescence at room temperature is rarely observed in molecules with lighter elements.
We have been involved in developing multi-functional molecules by constraining their molecular conformations.78–81 During these studies, we observed that the conformation of the molecule plays a significant role in controlling the energy and mixing of the electronically excited states, thereby curbing the undesirable non-radiative decay channels that are detrimental to the PL quantum yield.82,83 As part of the ongoing program, we designed and synthesized phosphoramides (TPTZPO, TPTZPS and TPTZPSe), keeping in view that the nonplanar heterocycle phenothiazine would enhance spin–orbit coupling through the mixing of molecular orbitals with different symmetry.84–94 Further, covalently linking a P = X (X = O/S/Se) group to phenothiazine can enhance the spin–orbit coupling (SOC) and intersystem crossing via n–π* interactions. We also envisioned that the nonplanar structure of phenothiazine and the tetrahedral geometry imposed by the phosphorus center may curb the unwarranted intermolecular interactions and lead to molecules with improved RTP characteristics. As anticipated, RTP was realized with these compounds. Compounds TPTZPS and TPTZPSe crystallized in the enantiomerically pure chiral space group P21, and their chiroptical properties were measured. All these results are reported in this article.
2. Results and discussion
Compounds TPTZPO, TPTZPS, and TPTZPSe were synthesized following the literature procedures.63,95,96 The addition of n-BuLi to anhydrous THF solution of phenothiazine under stirring conditions generated a N-centered anion, and the subsequent trapping of the anion with phosphorus trichloride gave tris-phenothiazinephosphine as a colorless solid. Oxidation of tris-phenothiazinephosphine with H2O2/S-powder/Se-powder yielded TPTZPO, TPTZPS, and TPTZPSe, respectively (Scheme 1). Analytically pure TPTZPO, TPTZPS and TPTZPSe were obtained by repeated recrystallization in a hexane and ethyl acetate solvent mixture (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5). All the compounds were stable under ambient conditions. Compound TPTZPO melts at 192–195 °C; in contrast, TPTZPS and TPTZPSe decompose at 190 °C.
:5). All the compounds were stable under ambient conditions. Compound TPTZPO melts at 192–195 °C; in contrast, TPTZPS and TPTZPSe decompose at 190 °C.
 | ||
| Scheme 1 Synthesis of TPTZPO, TPTZPS and TPTZPSe. (i) n-BuLi, −78 °C, anhydrous THF, 1 h; (ii) PCl3, temperature: 298 K; (iii) 30% H2O2 in dichloromethane for TPTZPO, sulfur powder in dichloromethane for TPTZPS and selenium powder in toluene for TPTZPSe. | ||
All compounds were characterized by NMR (1H, 31P), HRMS, elemental analysis, and high-performance liquid chromatography (HPLC) (Fig. 1 and S1–S8†). The molecular structures of all the compounds were confirmed by single-crystal X-ray diffraction studies.
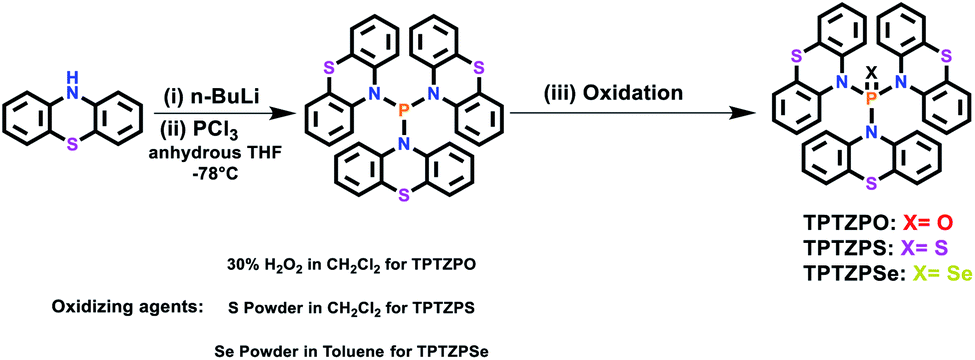
Compounds TPTZPO, TPTZPS, and TPTZPSe show broad 1H NMR resonance at 25 °C in CDCl3, CD2Cl2, DMSO-d6, and THF-d8. Upon decreasing the temperature, the 1H resonances are resolved for compounds TPTZPS and TPTZPSe, while those of TPTZPO are not resolved even at −90 °C in CD2Cl2. The two peaks observed for TPTZPO at 25 °C merged to a single broad peak at −10 °C, and as temperature decreased further, it started to split (Fig. S1†). In total twelve 1H resonances appeared at −90 °C for TPTZPO, indicating the asymmetry in the system.
However, broad NMR signals started to split at 0 °C, and a total of thirteen well-resolved peaks appeared at −90 °C for TPTZPS and TPTZPSe, respectively (Fig. 1 and S2†). These results indicated that the phenothiazine rings in TPTZPO, TPTZPS, and TPTZPSe undergo dynamic flip-flap motion at the NMR timescale. The 1H resonances are broad peaks even at −90 °C for TPTZPO, indicating the greater extent of phenothiazine ring dynamics. The shorter PO bond distance may be the root cause for the observed larger ring flipping in TPTZPO. The free energy of activation for the hindered rotation was calculated using the Gutowsky–Holm equation. ΔG# = 0.00457Tc(9.97 + log(Tc/Δδ)).97 Where Δδ is the maximum difference between the two peaks at low temperature and Tc is the coalescence temperature (273 K) (calculation details are given in the ESI†). The free energy calculated for the rotational barrier in TPTZPS and TPTZPSe was 16.78 kcal mol−1 and 16.50 kcal mol−1, respectively.
The 31P NMR chemical shifts for TPTZPO (0.6 ppm), TPTZPS (54.5 ppm), and TPTZPSe (49.4 ppm) are in the region expected for compounds with P = X (O, S, and Se) moieties reported in the literature.98,99 Compounds TPTZPO, TPTZPS, and TPTZPSe show the (M + Na)+ peak at 664.0718, 680.0489, and 727.9936, respectively, under HRMS conditions, and these values are consistent with their molecular compositions.
2.1 Crystal structure
The molecular structures of TPTZPO, TPTZPS, and TPTZPSe were confirmed by single-crystal X-ray diffraction (SCXRD) studies. Compound TPTZPO crystallized in the monoclinic crystal system with the centrosymmetric C2/c space group, while TPTZPS and TPTZPSe crystallized in the monoclinic chiral P21 space group (Table S1†). In all the compounds, the phosphorus centers adopted distorted tetrahedral geometries, and the P = X (X = O, S or Se) fragment is surrounded by three phenothiazine units (Fig. 1). The N–P bond lengths are in the range of 1.668–1.689 Å which are longer than the typical NP bond (1.58 Å) and shorter than the P–N single bond (1.76 Å) (Table S2†), a feature typical of P–N resonance double bond distances observed in phosphoramides.62,63,99,100
 | ||
| Fig. 1 Molecular structures of TPTZPO, TPTZPS and TPTZPSe with atom colour code (hydrogen atoms are removed for clarity) (top). Variable temperature 1H NMR spectra of compound TPTZPS in the range of 25 °C to −90 °C (10 °C interval) recorded in CD2Cl2 (bottom). | ||
Interestingly, compounds TPTZPO, TPTZPS, and TPTZPSe with nonplanar phenothiazine (PTZ) donors show shorter P–N bonds than phosphoramides with planar carbazole (CBZ) donors reported elsewhere.62,63 This can be attributed to the strong electron-donating capacity of PTZ compared to CBZ. In all the compounds, the phenothiazine units were nonplanar, and each phenothiazine moiety was puckered uniquely against the P = X axis and showed a distinct puckering angle. The P–N bond distances were influenced by the degree of puckering of the phenothiazine moiety in these compounds; the more significant the puckering angle the shorter the P–N bond (Table S2†). Compounds TPTZPO, TPTZPS, and TPTZPSe generate supramolecular structures facilitated via intermolecular P = X⋯H–C and S⋯H–C interactions between neighboring molecules in the solid-state. In the case of TPTZPS and TPTZPSe, the supramolecular structures are further augmented by intermolecular C–H⋯π interactions between the phenothiazine moieties of the neighboring molecules (Fig. S9–S11†). Such interactions are absent in TPTZPO. As anticipated, intermolecular face-to-face π⋯π interactions were not found in TPTZPO, TPTZPS, and TPTZPSe. Thus, these compounds are expected to show strong luminescence in the solid state.
2.2 Optical properties
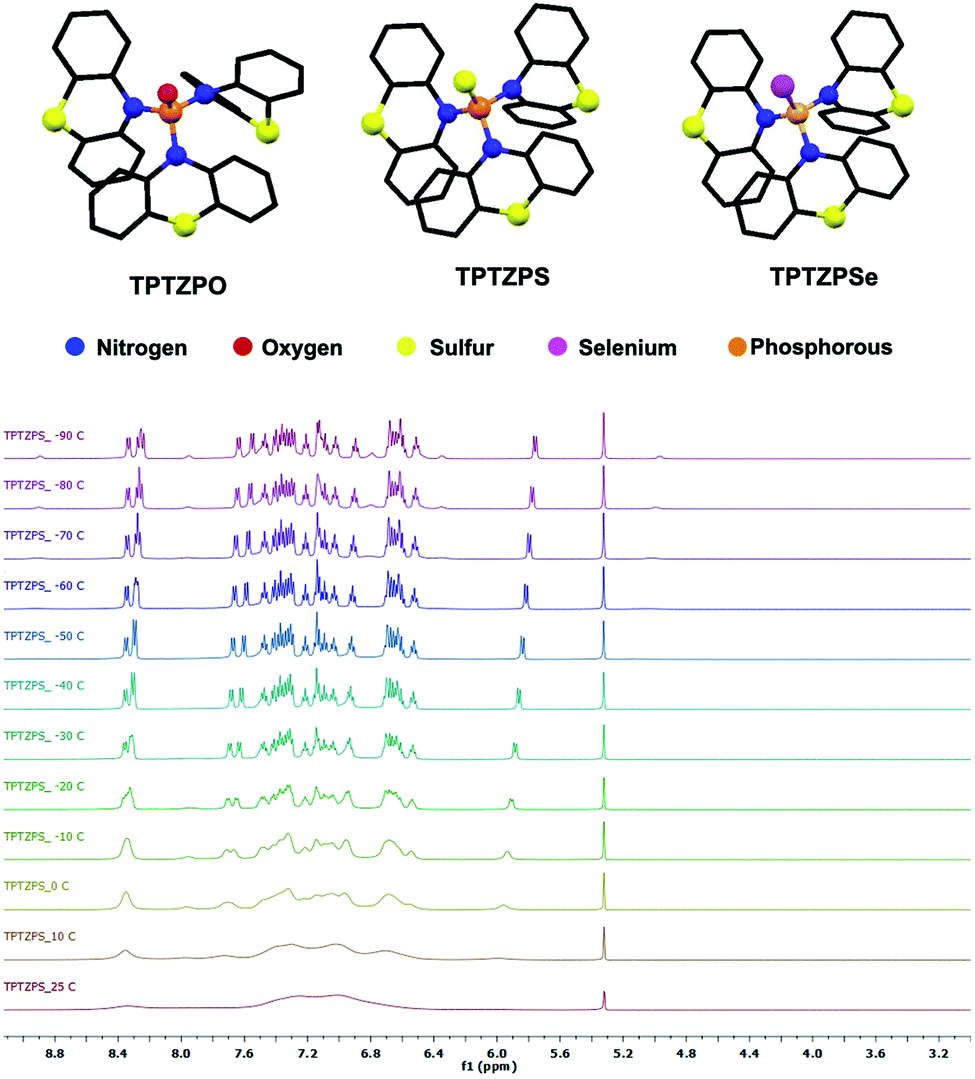
Dichloromethane (DCM) solutions (conc. 10−5 M) of TPTZPO, TPTZPS, and TPTZPSe exhibit a broad absorption band in the region 250–330 nm (Fig. 2a). No shift in the absorption maxima concerning PO, PS, and PSe units indicated that the electronic communication between the phenothiazine and P = X unit is negligible in the ground state. This observation parallels the UV-Vis spectra of tris-carbazole phosphoramides reported by Huang et al.95,98 The absorption process in TPTZPO, TPTZPS, and TPTZPSe is insensitive to the solvent polarity indicating the nonpolar electronic ground state in these compounds.
 | ||
| Fig. 2 (a) Normalized UV-visible absorption spectra (conc. 10−5 M). (b) Normalized photoluminescence spectra of TPTZPO, TPTZPS, and TPTZPSe in dichloromethane (conc. 10−4 M), λex = 280 nm. (c) Normalized photoluminescence spectra of pristine solids of these compounds (λex = 360 nm). (d) Solid-state photoluminescence decay plot (λex = 375 nm and λem = 550 nm) of these compounds. The incomplete decay of TPTZPS and TPTZPSe indicates the existence of a long-lived species. | ||
DCM solutions of these compounds showed a structureless broad peak in the region 330–500 nm (Fig. 2b). Compounds TPTZPS (λem = ∼470 nm) and TPTZPSe (λem = ∼470 nm) showed red-shifted emission compared to TPTZPO (λem = ∼380 nm). The PL peak position in these compounds is independent of the excitation wavelength; in contrast, the PL intensity depends on the excitation wavelength, and the maximum intensity was observed for λex = 280 nm (Fig. S12†). The excitation spectra of these compounds reproduced the corresponding absorption spectra (Fig. S13†). Furthermore, the PL peak maxima of these compounds are unchanged as the concentration of the solutions increased from 10−6 molar to 10−4 molar (Fig. S14†), and the maximum intensity was observed for 10−4 molar solutions. Like absorption spectra, the emission spectrum of TPTZPO was insensitive to the solvent polarity (Fig. S15†). However, the λem values of TPTZPS and TPTZPSe are sensitive to the dielectrics of the solvent medium; as the solvent polarity increased from hexane to methanol, the emission peaks were red-shifted (Fig. S15†). All the compounds showed single exponential decay with nanosecond lifetime in the solution state, confirming the emission originating from the singlet excited state (Table S3†). In the case of TPTZPO, the lifetime of the species is insensitive to the polarity of the solvent matrix as observed in the solvent dielectric dependent PL spectrum (Fig. S16†). However, the PL lifetimes of TPTZPS and TPTZPSe increased with increasing solvent polarity, and a longer lifetime was observed for DCM solutions (Table S3†). Based on these results, we concluded that these compounds are weakly emissive in solutions and the PL features of these compounds have their origin from single molecular species. Further, the excited state of TPTZPO is nonpolar, while TPTZPS and TPTZPSe emit from a polar excited state. TD-DFT results are also supportive of these claims (vide infra).
Like in the solution state, the pristine solids of TPTZPO, TPTZPS, and TPTZPSe showed a broad PL with λmax at 424, 476, and 463 nm, respectively. In contrast, the solid-state PL spectra of TPTZPO and TPTZPS red-shifted compared to their respective solution spectra except for TPTZPSe, in which the PL maximum is blue-shifted compared to its solution spectra. Compounds TPTZPS and TPTZPSe show larger Stokes shift than TPTZPO, which can be attributed to the greater P = X (X = S and Se) resonance length in the former than that in the latter (PO) (Fig. 2c). The excitation spectra of these compounds show a band at ∼360 nm, which is ∼60 nm red-shifted compared to the solution absorption maxima (Fig. S17†), indicating that the emissive species in the solid-state is different from that in solution.
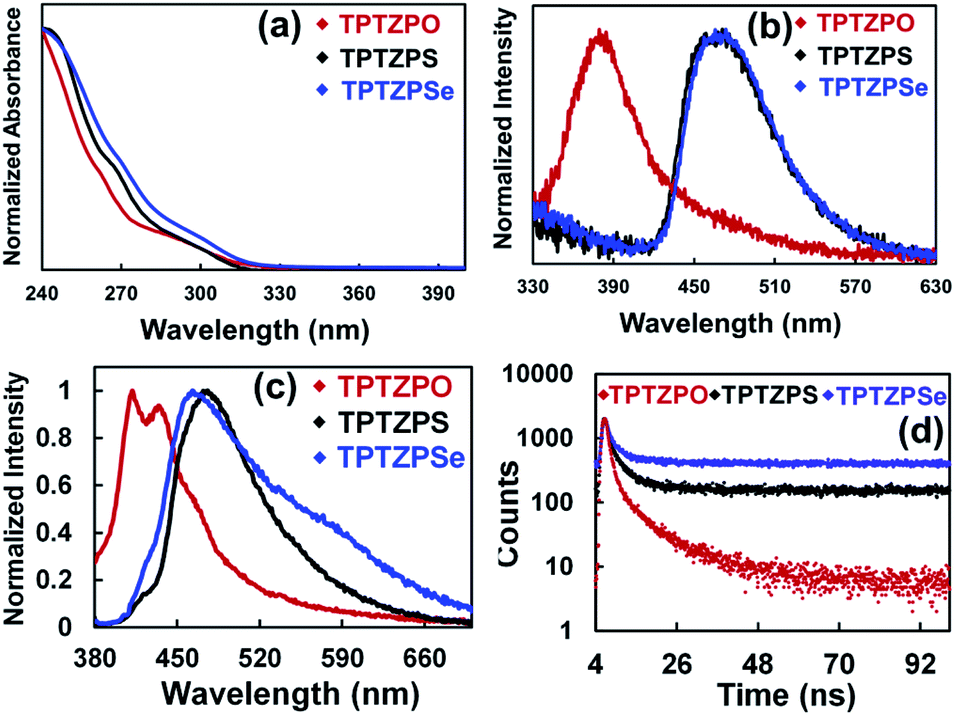
Crystalline solids of these compounds showed two PL bands, one at the blue end of the spectra as observed in the pristine solid and a new strong band at the green end of the spectrum (Fig. 3a–c and S18†). The lower energy bands of TPTZPS and TPTZPSe are red-shifted compared to those of TPTZPO. The ground solids of these compounds did not show the lower energy band at the green end of the spectrum and showed the PL band corresponding to their pristine solids. Higher PL quantum yield (PLQY) was measured for the crystals of TPTZPS (19.14%) and TPTZPSe (7.51%) compared to their pristine solids (TPTZPS, 6.53% and TPTZPSe, 0.60%). Surprisingly, TPTZPO crystals (PLQY = 0.01%) and pristine solids (PLQY = 0.57%) are less emissive than TPTZPS and TPTZPSe. Further, TPTZPS and TPTZPSe exhibited considerably higher PLQY than previously reported RTP co-crystals.101,102
 | ||
| Fig. 3 Photoluminescence spectra (λex = 360 nm) of crystals, ground samples, and pristine samples of (a) TPTZPO, (b) TPTZPS and (c) TPTZPSe (Ground 1 indicates grinding with a mortar and pestle for 5 minutes, and Ground 2 indicates heavy grinding for 15 minutes). Prompt and phosphorescence spectra of pristine solids of (d) TPTZPO, (e) TPTZPS, and (f) TPTZPSe, λex = 360 nm with 30 μs delay time. | ||
Powder X-ray diffraction (PXRD) traces of these compounds before and after grinding were recorded (Fig. S19†). The crystals of these compounds showed strong peaks, while the ground samples showed few diffused diffraction peaks. Based on the PL spectra, PLQY, and PXRD studies, it is appropriate to say that the phase change from crystalline to amorphous is responsible for the emission color changes in these compounds.
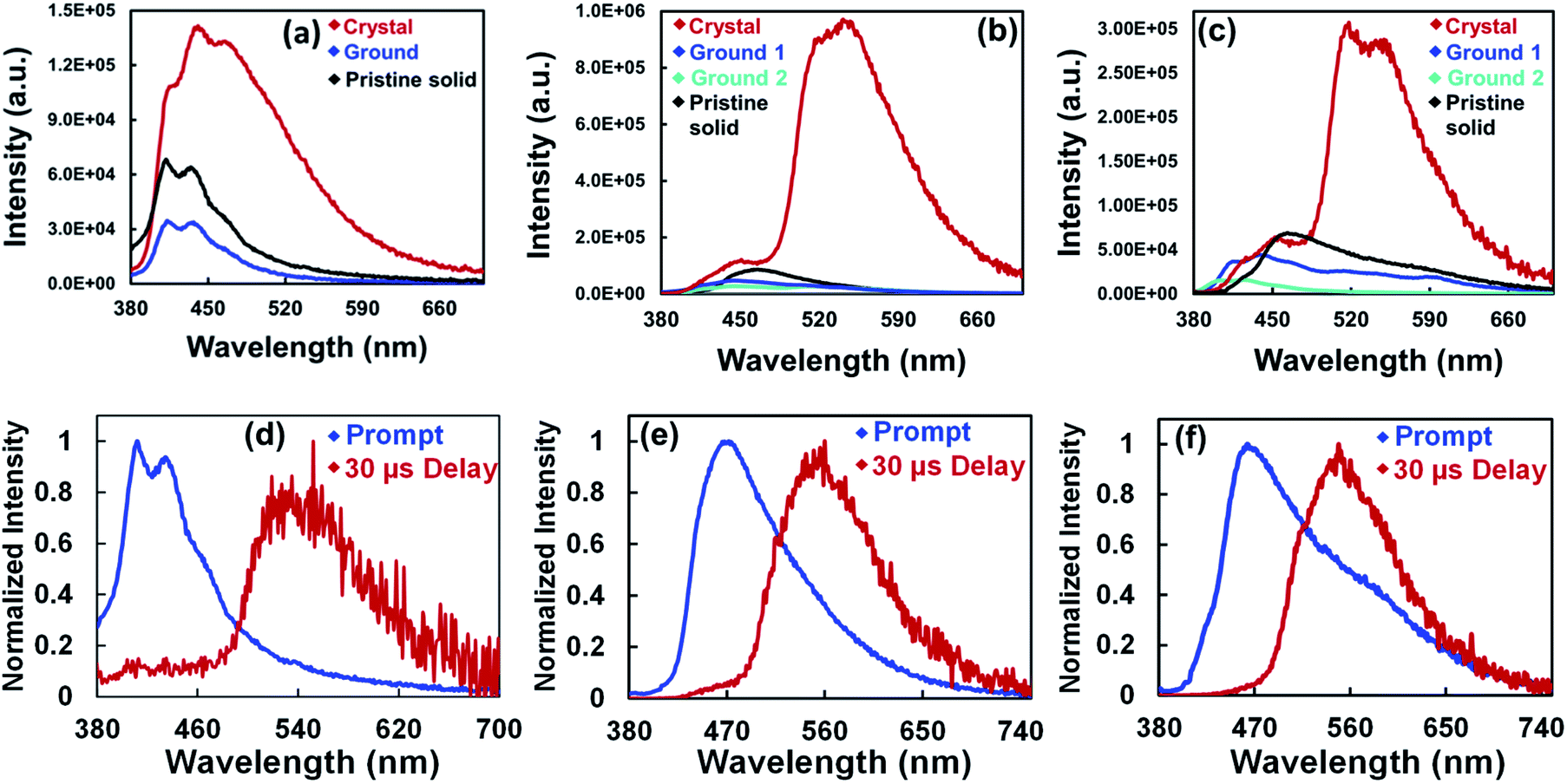
The fluorescence and phosphorescence spectra of all the compounds were recorded at both ambient and low temperature. DCM solutions of these compounds did not show delayed spectra under ambient conditions. However, the pristine solids, crystals, ground samples, and thin films of these compounds showed distinct prompt and delayed spectra under ambient conditions. The delayed spectra of these compounds are 100 nm red-shifted compared to their fluorescence spectra (Fig. 3d–f, 5 and S26†). The delayed peak position of these compounds smoothly overlapped with the intense lower energy band of the respective crystals.
Nanosecond lifetime was obtained for the higher energy fluorescence bands (Fig. S20, Table S4†). In contrast, incomplete decay was observed for the lower energy band, suggesting long-lived emissive species (Fig. 2d). Furthermore, under an O2 atmosphere, the delayed emission band nearly vanished, and the prompt spectra remains unchanged. The delayed PL band was restored when the oxygenated samples were purged with N2 for 5 minutes (Fig. 4b, c and S21†). Furthermore, the delayed emission band decays much faster under an oxygen atmosphere than under a N2 atmosphere (Fig. S22, Table S5†). However, the fluorescence lifetime showed little change under N2 and O2 conditions (Fig. S24, Table S6†). These results indicated that the triplet excited state is involved in the radiative process of these compounds.
 | ||
| Fig. 4 (a) Prompt spectra, λex = 360 nm and (b) phosphorescence spectra of the thin film of TPTZPS, with 30 μs delay obtained using a microsecond flash lamp, λex = 360 nm. (c) Digital photographs of the thin film of TPTZPS under N2 and O2 atmospheres under a 365 nm UV lamp. (d) Digital photographs of crystals of TPTZPS under 365 nm UV light under on and off conditions (photographs are extracted from a video recorded in a REDMI NOTE 8 PRO mobile). | ||
To get further insight into the nature of the excited electronic states, the PL spectra of these compounds were collected in the temperature range from 77 K to 298 K. The PL intensity of these compounds progressively increased with lowering the sample temperature from 298 to 77 K. The PL lifetime of these compounds increased significantly at 77 K compared to that at 298 K, which directly corroborates the strong luminescence observed for these compounds at low temperature (Fig. 5b–d and Table S7†). The crystalline solids showed a longer lifetime than the corresponding pristine solids (Fig. 5 and S25†). However, the triplet excited state lifetimes for these phosphoramides are found to be less than those of the previously reported RTP co-crystals.101,102
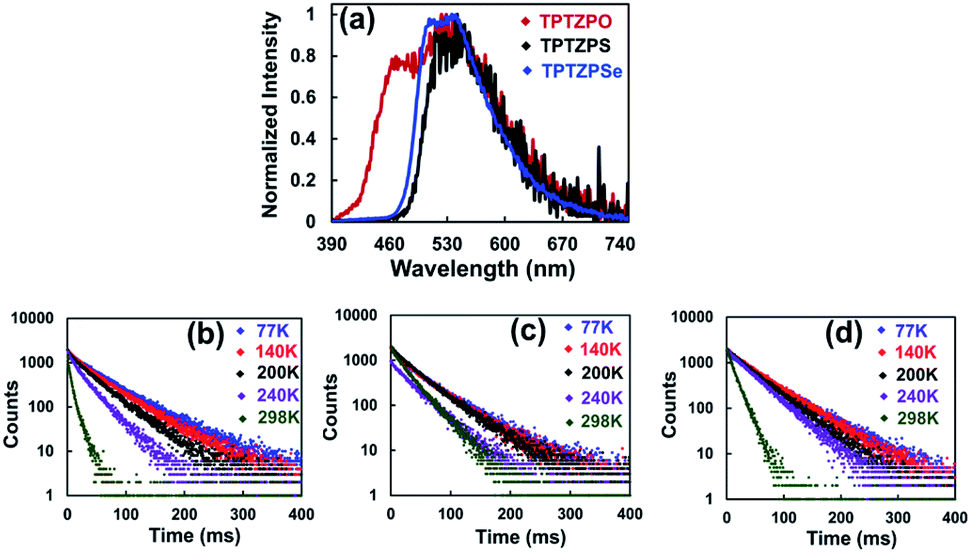 | ||
| Fig. 5 (a) Phosphorescence spectra of crystals of TPTZPO, TPTZPS and TPTZPSe, λex = 360 nm with 30 μs delay obtained using a microsecond flash lamp. Phosphorescence decay profiles of (b) TPTZPO, (c) TPTZPS and (d) TPTZPSe in the crystalline state at different temperatures with 30 μs delay (λex = 360 nm and λem = 550 nm). The lifetimes obtained for crystals of TPTZPO, TPTZPS, and TPTZPSe are 61.03, 53.33, and 58.38 ms, respectively, at 77 K. The lifetime at different temperatures are listed in the ESI, Table S6.† | ||
The crystalline solid of TPTZPS showed yellowish-green fluorescence under 365 nm UV light illumination followed by green phosphorescence emission after removing the light source, as shown in Fig. 4d. The following conclusions were drawn based on the steady-state and time-resolved emission studies. In the solution state, these compounds exhibit only fluorescence. In the solid-state (both pristine and crystalline solids), they exhibit dual fluorescence and phosphorescence at room temperature.
The phosphorescence lifetime and the PL intensity observed for crystalline solids are significantly larger than those of the corresponding pristine samples (Fig. 5 and S25†). Furthermore, for the crystalline solids, both the lifetime and intensity of the phosphorescence decreased significantly upon ball-milling (Fig. S26 and S27†). These results suggest that the intermolecular interactions in the solid-state effectively inhibit the vibrational deactivation of triplet states; consequently, intense phosphorescence was observed for crystalline solids (Fig. S9–S11†). These characteristics of TPTZPO, TPTZPS and TPTZPSe are in line with the crystallization-induced phosphorescence observed for organic/organometallic compounds reported in the literature.34,101–103
It is imperative to note that the positions of the phosphorescence band for the crystals of TPTZPO, TPTZPS, and TPTZPSe are the same, except for a higher energy shoulder band of TPTZPO (Fig. 5a). These results indicate that the luminescence of these compounds is centered on the phenothiazine moiety. The shoulder band decays much faster than the phosphorescence band in the lower energy region. The most rapid decay of the blue shoulder phosphorescence of TPTZPO may be attributed to the TTA (triplet–triplet annihilation) emission.104–106 These observations are in line with the PL characteristics of other phenothiazine derivatives reported by Dias et al.86,87
2.3 Circularly polarized luminescence (CPL) studies
Circularly polarized luminescent (CPL) materials have attracted much attention in recent years because of their potential applications in niche technologies such as 3D displays, information storage, spintronics devices, biological probes, etc.107–112 Compounds TPTZPS and TPTZPSe crystallized in enantiomerically pure form, and their CPL characteristics were studied. The CPL spectra of these compounds were collected in the fluorescence mode. Upon excitation with 360 nm light, solids of TPTZPO show no CPL signals as it is crystallized in the centrosymmetric crystal lattice. However, ground samples of TPTZPSe exhibit strong CPL bands at ∼460 nm under identical conditions (Fig. S28†). The luminescence dissymmetry factor (glum) values obtained for TPTZPSe (2.7 × 10−3) are comparable to those obtained for CPL active organic luminophores reported elsewhere.The crystalline solids of TPTZPSe (glum value = 3.4 × 10−3) showed the CPL band; surprisingly, no CPL signal was observed for the crystals and ground samples of TPTZPS (Fig. 6). Theoretically, both TPTZPS and TPTZPSe should exhibit CPL properties. However, as the glum value of TPTZPS is small and below the detection limit, a CPL spectrum could not be observed for this compound. In crystalline form, both TPTZPS and TPTZPSe show intense phosphorescence, while the ground samples of these compounds show strong fluorescence. As the CPL spectra were collected in the fluorescence mode, no CPL signal was observed for strongly phosphorescent crystalline solids of TPTZPS. Intense CPL activity was observed for the ground sample of TPTZPSe, which was strongly fluorescent. However, phosphorescence emission being inherently weak could not be detected in the CPL. No CPL signal was observed for the solutions of these compounds due to fast racemization.
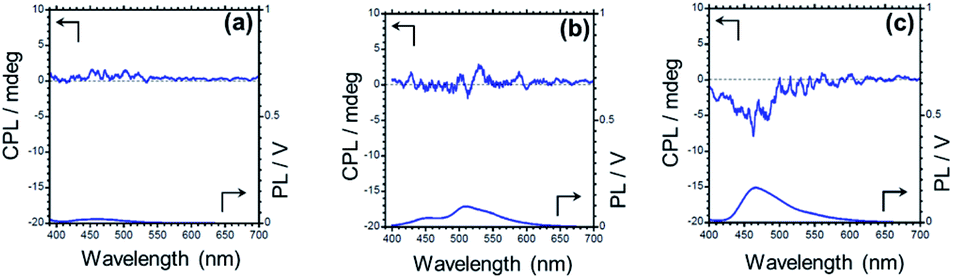 | ||
| Fig. 6 Circularly polarized luminescence (CPL) spectra and the corresponding photoluminescence spectra of crystals of TPTZPO (a), TPTZPS (b), and TPTZPSe (c), λex = 360 nm (CPL spectra – top, PL spectra – bottom) (gCPL = 3.4 × 10−3 for TPTZPSe). | ||
2.4 Theoretical calculations
Computational studies were carried out to gain further insight into the electronic structure and the observed optical properties of these compounds. Both ground (S0) and excited state (S1 and T1) geometries of TPTZPO, TPTZPS, and TPTZPSe were optimized using DFT (B3LYP, 6-31G (d,p)) and TDDFT (CAM-B3LYP and 6-31G(d,p)), respectively. Frontier molecular orbital (HOMO and LUMO) pictures suggested that n–π* and π–π* are the major transitions in these compounds (n orbital of phenothiazine sulfur and heteroatom (O/S/Se) attached to the P center to the π* orbital of phenothiazine) (Fig. 7a). Geometries and bonding parameters of these molecules in the S0 and S1 states are very similar to those of the structure obtained by single-crystal X-ray diffraction studies. The T1 state geometries of TPTZPO and TPTZPSe molecules are very different from the S0 and S1 state geometries (Table S8–S10, Fig. S29†).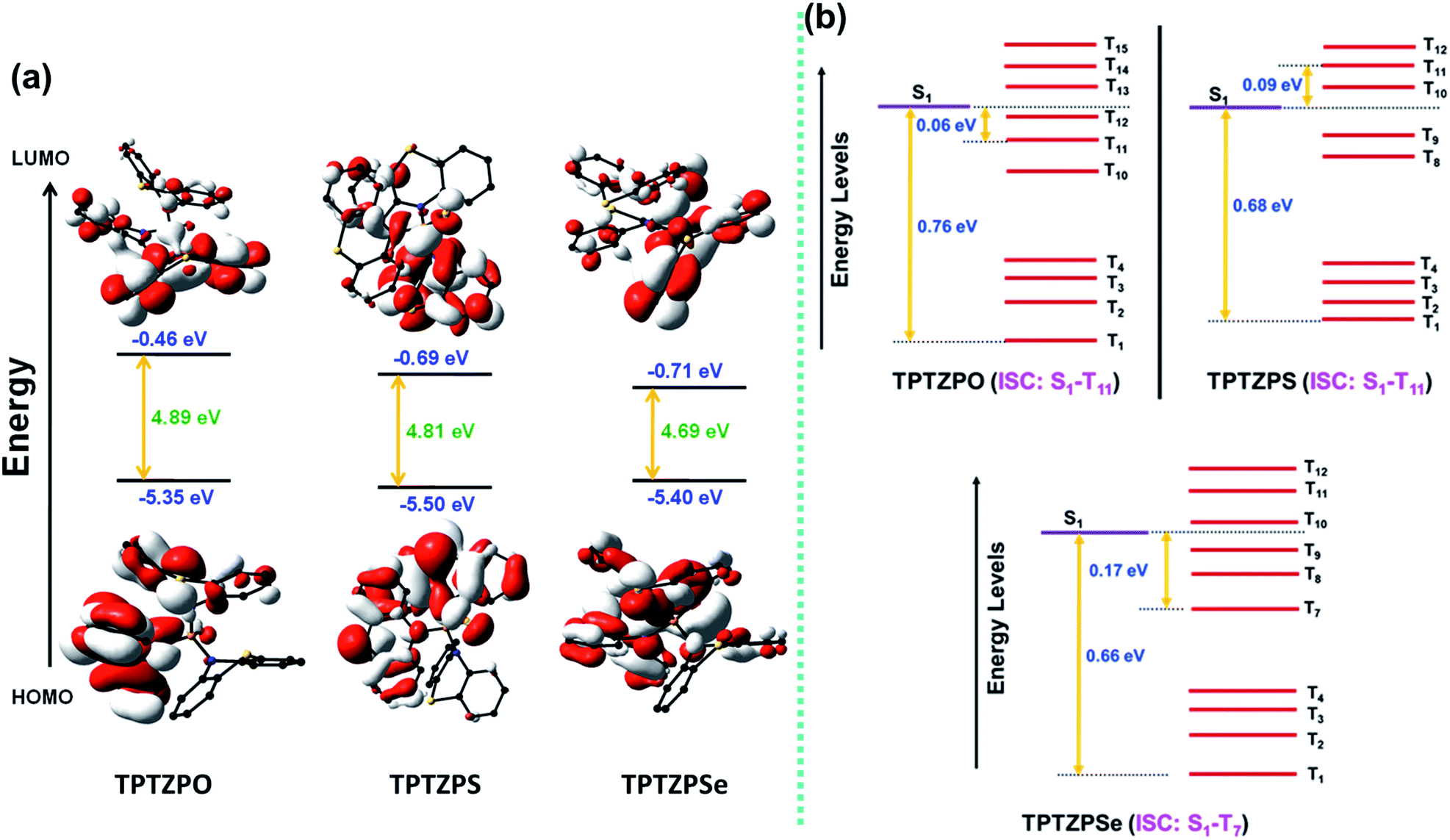 | ||
| Fig. 7 (a) Frontier molecular orbital energy level diagrams of TPTZPO, TPTZPS, and TPTZPSe (DFT, B3LYP functional with 6-31G (d,p) basis set). (b) Energy level diagram showing the singlet–triplet gap and possible ISC channel of TPTZPO, TPTZPS and TPTZPSe (energy levels in the diagram are not up to scale). | ||
In S0 and S1 all the phenothiazine units are symmetrically puckered, however, in the T1 state, one of the three phenothiazines is unsymmetrically puckered; in contrast, the geometry of the other two rings remains unchanged. To accommodate these conformational changes, the P–N bond length is elongated in the T1 state (Tables S8–S10†) compared to that in S0 and S1. However, in the case of TPTZPS, the geometries of all three phenothiazine moieties remain unchanged when it goes from the S1 to T1 state.
The computed ΔES1T1 values match well with the experimental ΔES1T1 values calculated from the maxima of fluorescence and phosphorescence spectra (Table S11†). Furthermore, high ΔES1T1 values (0.66–0.76 eV) rule out the S1←T1 RISC and TADF (Fig. 7b, Table S12†). To get insight into the possible ISC pathways, S0→Tn vertical transitions were calculated. The results showed that higher triplet states are placed close to the S1 state. Moreover, the commonality of the orbitals involved in the S0→S1 and S0→TnTPTZPO (S0→T1, S0→T11), TPTZPS (S0→T1, S0→T11), and TPTZPSe (S0→T7) transitions suggests that the ISC transition occurs via higher triplet states (Tables S13–S15†). Frontier molecular orbital pictures indicated that both S1 and T1 states are of n–π* nature (Fig. S30–31†).
To investigate more about excited state dynamics, we calculated the spin density of these molecules in the ground, singlet, and triplet excited states. The spin density pictures of S0 and S1 look similar for all three compounds, but the scenario is different in the triplet state (T1) (Fig. S32–S34†). In the triplet state, for TPTZPO, the spin density is localized on one of the three phenothiazine moieties (the ring distorted in the triplet state). However, the spin density is delocalized over both phenothiazine and P = X (X = S and Se) in TPTZPS and TPTZPSe. Thus, TPTZPS and TPTZPSe show similar phosphorescence features involving n–π* triplet states. However, in TPTZPO, the localization of spin density on a single phenothiazine unit and the types of orbitals involved in S0–T1 transitions indicate that the phosphorescence is from the triplet having hybridized LE and n–π* states. Thus, TPTZPO shows a structured phosphorescence band at a lower wavelength than TPTZPS and TPTZPSe.
3. Conclusions
In summary, we have designed and synthesized three phosphoramides, TPTZPO, TPTZPS, and TPTZPSe. These compounds show fluorescence in solution and dual fluorescence and RTP in the solid-state (crystalline and amorphous solids). Detailed experimental and computational studies demonstrate that the phosphorescence of these compounds originates from the triplet n–π* state dominated by the phenothiazine moiety. All the compounds exhibit crystallization-induced room-temperature phosphorescence, a phenomenon quite rarely observed for this class of compounds. Moreover, both TPTZPS and TPTZPSe crystallized in the chiral space group; however, only TPTZPSe showed CPL signals in the solid-state.Data availability
Publicly available datasets were analyzed in this study. This data can be found here: (https://www.ccdc.cam.ac.uk/structures/) CCDC 1977279, 1916761 and 1916789.†Author contributions
SJ did compound synthesis, DFT calculations and PL characterization. SB did preliminary PL studies. Both JE and SJ contributed to data analysis. YI and MK did CPL studies. All the authors commented on the manuscript. PT organized the entire project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to the IISc, DST and SERB for financial support. SJ thanks the Indian Institute of Science Bangalore, JE acknowledges the support from Prime Minister's Research Fellowship (PMRF) and SKB thanks Kothari postdoctoral fellowship of India, The authors thank Prof. Debasis Das for providing the HPLC facility and Kalpana Rajendran for doing the measurements.References
- C. W. Tanga and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913 CrossRef.
- N. J. Turro, Modern Molecular Photochemistry, University Science Books, Sausalito, CA, 1991,Benjamin/Cummings, Menlo Park, NJ, 1978 Search PubMed.
- C. Adachi, M. A. Baldo, M. E. Thompson and S. R. Forrest, J. Appl. Phys., 2001, 90, 5048–5051 CrossRef CAS.
- B. Song, W. Shao, J. Jung, S.-J. Yoon and J. Kim, ACS Appl. Mater. Interfaces, 2020, 12, 6137–6143 CrossRef CAS PubMed.
- Y. Kawamura, K. Goushi, J. Brooks, J. J. Brown, H. Sasabe and C. Adachi, Appl. Phys. Lett., 2005, 86, 071104 CrossRef.
- E. Holder, B. M. W. Langeveld and U. S. Schubert, Adv. Mater., 2005, 17, 1109–1121 CrossRef CAS.
- S. J. Su, T. Chiba, T. Takeda and J. Kido, Adv. Mater., 2008, 20, 2125–2130 CrossRef CAS.
- H. F. Higginbotham, M. Okazaki, P. de Silva, S. Minakata, Y. Takeda and P. Data, ACS Appl. Mater. Interfaces, 2021, 13, 2899–2907 CrossRef CAS PubMed.
- S. Hu, J. Zeng, X. Zhu, J. Guo, S. Chen, Z. Zhao and B. Z. Tang, ACS Appl. Mater. Interfaces, 2019, 11, 27134–27144 CrossRef CAS.
- J. M. Vanderkooi, G. Maniara, T. J. Green and D. F. Wilson, J. Biol. Chem., 1987, 262, 5476–5482 CrossRef CAS.
- R. Gao, D. G. Ho, B. Hernandez, M. Selke, D. Murphy, P. I. Djurovich and M. E. Thompson, J. Am. Chem. Soc., 2002, 124, 14828–14829 CrossRef CAS PubMed.
- M. C. DeRosa, D. J. Hodgson, G. D. Enright, B. Dawson, C. E. B. Evans and R. J. Crutchley, J. Am. Chem. Soc., 2004, 126, 7619–7626 CrossRef CAS PubMed.
- S. M. Borisov, G. Zenkl and I. Klimant, ACS Appl. Mater. Interfaces, 2010, 2, 366–374 CrossRef CAS PubMed.
- A. Pfister, G. Zhang, J. Zareno, A. F. Horwitz and C. L. Fraser, ACS Nano, 2008, 2, 1252–1258 CrossRef CAS.
- N. Gan, H. Shi, Z. An and W. Huang, Adv. Funct. Mater., 2018, 28, 1802657 CrossRef.
- G. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. Fraser, Nat. Mater., 2009, 8, 747–751 CrossRef CAS PubMed.
- Q. Zhao, C. Huang and F. Li, Chem. Soc. Rev., 2011, 40, 2508–2524 RSC.
- X. Chen, C. Xu, T. Wang, C. Zhou, J. Du, Z. Wang, H. Xu, T. Xie, G. Bi, J. Jiang, X. Zhang, J. N. Demas, C. O. Trindle, Y. Luo and G. Zhang, Angew. Chem., Int. Ed., 2016, 55, 9872–9876 CrossRef CAS PubMed.
- N. Gan, H. Shi, Z. An and W. Huang, Adv. Funct. Mater., 2018, 28, 1802657 CrossRef.
- R. Kabe and C. Adachi, Nature, 2017, 550, 384–387 CrossRef CAS PubMed.
- R. Kumar, T. Y. Ohulchanskyy, I. Roy, S. K. Gupta, C. Borek, M. E. Thompson and P. N. Prasad, ACS Appl. Mater. Interfaces, 2009, 1, 1474–1481 CrossRef CAS.
- A. Tsuboyama, H. Iwawak, M. Furugori, T. Mukaide, J. Kamatani, S. Igawa, T. Moriyama, S. Miura, T. Takiguchi, S. Okada, M. Hoshino and K. Ueno, J. Am. Chem. Soc., 2003, 125, 12971–12979 CrossRef CAS PubMed.
- Y. J. Su, H. L. Huang, C. L. Li, C. H. Chien, Y. T. Tao, P. T. Chou, S. Datta and R. S. Liu, Adv. Mater., 2003, 15, 884–888 CrossRef CAS.
- Y. You and S. Y. Park, Dalton Trans., 2009, 1267–1282 RSC.
- J. Brooks, Y. Babayan, S. Lamansky, P. I. Djurovich, I. Tsyba, R. Bau and M. E. Thompson, Inorg. Chem., 2002, 41, 3055–3066 CrossRef CAS PubMed.
- Y. Y. Lin, S. C. Chan, M. C. W. Chan, Y. J. Hou, N. Zhu, C. M. Che, Y. Liu and Y. Wang, Chem.–Eur. J., 2003, 9, 1263–1272 CrossRef CAS PubMed.
- X. L. Yang, C. L. Yao and G. J. Zhou, Platinum Met. Rev., 2013, 57, 2–16 CrossRef CAS.
- W. Lu, N. Zhu and C. M. Che, J. Am. Chem. Soc., 2003, 125, 16081–16088 CrossRef CAS.
- M. Osawa, M. Hoshino, M. Akita and T. Wada, Inorg. Chem., 2005, 44, 1157–1159 CrossRef CAS PubMed.
- H. Rudmann, S. Shimada and M. F. Rubner, J. Am. Chem. Soc., 2002, 124, 4918–4921 CrossRef CAS PubMed.
- H. V. R. Dias, H. V. K. Diyabalanage, M. A. R. Omary, M. A. Franzman and M. A. Omary, J. Am. Chem. Soc., 2003, 125, 12072–12073 CrossRef CAS PubMed.
- C.-L. Ho, W.-Y. Wong, Z.-Q. Gao, C.-H. Chen, K.-W. Cheah, B. Yao, Z. Xie, Q. Wang, D. Ma, L. Wang, X.-M. Yu, H.-S. Kwok and Z. Lin, Adv. Funct. Mater., 2008, 18, 319–331 CrossRef CAS.
- A. B. Alemayehu, N. U. Day, T. Mani, A. B. Rudine, K. E. Thomas, O. A. Gederaas, S. A. Vinogradov, C. C. Wamser and A. Ghosh, ACS Appl. Mater. Interfaces, 2016, 8, 18935–18942 CrossRef CAS PubMed.
- J. Yang, X. Gao, Z. Xie, Y. Gong, M. Fang, Q. Peng, Z. Chi and Z. Li, Angew. Chem., Int. Ed., 2017, 56, 15299–15303 CrossRef CAS PubMed.
- Y. Xiong, Z. Zhao, W. Zhao, H. Ma, Q. Peng, Z. He, X. Zhang, Y. Chen, X. He, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2018, 57, 7997–8001 CrossRef CAS PubMed.
- O. Bolton, K. Lee, H. J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205–210 CrossRef CAS PubMed.
- M. Baroncini, G. Bergamini and P. Ceroni, Chem. Commun., 2017, 53, 2081–2093 RSC.
- D. Lee, O. Bolton, B. C. Kim, J. H. Youk, S. Takayama and J. Kim, J. Am. Chem. Soc., 2013, 135, 6325–6632 CrossRef CAS PubMed.
- Y. Xie, Y. Ge, Q. Peng, C. Li, Q. Lia and Z. Li, Adv. Mater., 2017, 29, 1606829 CrossRef PubMed.
- S. Hirata, K. Totani, J. Zhang, T. Yamashita, H. Kaji, S. R. Marder, T. Watanabe and C. Adachi, Adv. Funct. Mater., 2013, 23, 3386–3397 CrossRef CAS.
- W. Zhao, Z. He and B. Z. Tang, Nat. Rev. Mater., 2020, 5, 869–885 CrossRef CAS.
- Kenry, C. Chen and B. Liu, Nat. Commun., 2019, 10, 2111 CrossRef CAS PubMed.
- H. Wu, L. Gu, G. V. Baryshnikov, H. Wang, B. F. Minaev, H. Ågren and Y. Zhao, ACS Appl. Mater. Interfaces, 2020, 12, 20765–20774 CrossRef CAS PubMed.
- T. Virgili, A. Forni, E. Cariati, D. Pasini and C. Botta, J. Phys. Chem. C, 2013, 117, 27161–27166 CrossRef CAS.
- R. Gao, M. S. Kodaimati and D. Yan, Chem. Soc. Rev., 2021, 50, 5564–5589 RSC.
- X. Yang and D. Yan, Chem. Sci., 2016, 7, 4519–4526 RSC.
- D. Yan and D. G. Evans, Mater. Horiz., 2014, 1, 46–57 RSC.
- S. Mukherjee and P. Thilagar, Chem. Commun., 2015, 51, 10988–11003 RSC.
- G. Zhang, J. Chen, S. J. Payne, S. E. Kooi, J. N. Demas and C. L. Fraser, J. Am. Chem. Soc., 2007, 129, 8942–8943 CrossRef CAS PubMed.
- S. Kawai, S. Saito, S. Osumi, S. Yamaguchi, A. S. Foster, P. Spijker and E. Meyer, Nat. Commun., 2015, 6, 1–6 Search PubMed.
- Z. Chai, C. Wang, J. Wang, F. Liu, Y. Xie, Y. Z. Zhang, J. R. Li, Q. Li and Z. Li, Chem. Sci., 2017, 8, 8336–8344 RSC.
- Y. Shoji, Y. Ikabata, Q. Wang, D. Nemoto, A. Sakamoto, N. Tanaka, J. Seino, H. Nakai and T. Fukushima, J. Am. Chem. Soc., 2017, 139, 2728–2733 CrossRef CAS PubMed.
- Y. Ren and F. Jakle, Dalton Trans., 2016, 45, 13996–14007 RSC.
- M. Shimizu, R. Shigitani, M. Nakatani, K. Kuwabara, Y. Miyake, K. Tajima, H. Sakai and T. Hasobe, J. Phys. Chem. C, 2016, 120, 11631–11639 CrossRef CAS.
- V. Chandrasekhar, M. D. Pandey, B. Das, B. Mahanti, K. Gopal and R. Azhakar, Tetrahedron, 2011, 67, 6917–6926 CrossRef CAS.
- V. Chandrasekhar, R. Azhakar, J. F. Bickley and A. Steiner, Cryst. Growth Des., 2006, 6, 910–914 CrossRef CAS.
- T. Baumgartner and R. Réau, Chem. Rev., 2006, 106, 4681–4727 CrossRef CAS PubMed.
- M. Stolar and T. Baumgartner, Chem.–Asian J., 2014, 9, 1212–1225 CrossRef CAS PubMed.
- F. M. Xie, Q. Ou, Q. Zhang, J. K. Zhang, G. L. Dai, X. Zhao and H. X. Wei, Beilstein J. Org. Chem., 2018, 14, 869–874 CrossRef CAS PubMed.
- J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng, K. Pu and Z. Li, Nat. Commun., 2018, 9, 1–10 CrossRef PubMed.
- Y. Ren, M. Sezen, F. Guo, F. Jakle and Y. L. Loo, Chem. Sci., 2016, 7, 4211–4219 RSC.
- Y. Tao, J. Xiao, C. Zheng, Z. Zhang, M. Yan, R. Chen, X. Zhou, H. Li, Z. An, Z. Wang, H. Xu and W. Huang, Angew. Chem., Int. Ed., 2013, 52, 10491–10495 CrossRef CAS PubMed.
- T. Ye, L. Xu, Z. Zhang, R. Chen, H. Li, H. Xu, C. Zheng and W. Huang, J. Am. Chem. Soc., 2016, 138, 9655–9662 CrossRef PubMed.
- F. Gao, R. Du, C. Han, J. Zhang, Y. Wei, G. Lu and H. Xu, Chem. Sci., 2019, 10, 5556–5567 RSC.
- Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685–690 CrossRef CAS PubMed.
- W. Z. Yuan, X. Y. Shen, H. Zhao, J. W. Y. Lam, L. Tang, P. Lu, C. Wang, Y. Liu, Z. Wang, Q. Zheng, J. Z. Sun, Y. Ma and B. Z. Tang, J. Phys. Chem. C, 2010, 114, 136090–136099 Search PubMed.
- P. Pander, A. Swist, J. Soloducho and F. B. Dias, Dyes Pigm., 2017, 142, 315–322 CrossRef CAS.
- P. Pander, A. Swist, R. Turczyn, S. Pouget, D. Djurado, A. Lazauskas, R. Pashazadeh, J. V. Grazulevicius, R. Motyka, A. Klimash, P. J. Skabara, P. Data, J. Soloducho and F. B. Dias, J. Phys. Chem. C, 2018, 122, 24958–24966 CrossRef CAS.
- S. O. Jeon, K. S. Yook, C. W. Joo and J. Y. Lee, Adv. Funct. Mater., 2009, 19, 3644–3649 CrossRef CAS.
- C. Fan, C. Duan, Y. Wei, D. Ding, H. Xu and W. Huang, Chem. Mater., 2015, 27, 5131–5140 CrossRef CAS.
- C. Fan, C. Duan, C. Han, B. Han and H. Xu, ACS Appl. Mater. Interfaces, 2016, 8, 27383–27393 CrossRef CAS PubMed.
- C. Zhou, S. Zhang, Y. Gao, H. Liu, T. Shan, X. Liang, B. Yang and Y. Ma, Adv. Funct. Mater., 2018, 28, 1802407 CrossRef.
- X. Bi, Y. Shi, T. Peng, S. Yue, F. Wang, L. Zheng and Q.-E. Cao, Adv. Funct. Mater., 2021, 31, 2101312 CrossRef CAS.
- W. Zhao, T. S. Cheung, N. Jiang, W. Huang, J. W. Y. Lam, X. Zhang, Z. He and B. Z. Tang, Nat. Commun., 2019, 10, 1595 CrossRef PubMed.
- L. Ma, S. Sun, B. Ding, X. Ma and H. Tian, Adv. Funct. Mater., 2021, 31, 2010659 CrossRef CAS.
- R. Chen, Y. Guan, H. Wang, Y. Zhu, X. Tan, P. Wang, X. Wang, X. Fan and H.-L. Xie, ACS Appl. Mater. Interfaces, 2021, 13, 41131–41139 CrossRef PubMed.
- S. Tian, H. Ma, X. Wang, A. Lv, H. Shi, Y. Geng, J. Li, F. Liang, Z. M. Su, Z. An and W. Huang, Angew. Chem., Int. Ed., 2019, 58, 6645–6649 CrossRef CAS PubMed.
- P. C. A. Swamy, S. Mukherjee and P. Thilagar, Chem. Commun., 2013, 49, 993–995 RSC.
- S. K. Sarkar and P. Thilagar, Chem. Commun., 2013, 49, 8558–8560 RSC.
- P. C. A. Swamy, S. Mukherjee and P. Thilagar, Inorg. Chem., 2014, 53, 4813–4823 CrossRef PubMed.
- K. N. Kalluvettukuzhy, S. Pagidi, D. Kumbhar and P. Thilagar, Chem. Commun., 2017, 53, 3641–3644 RSC.
- S. Mukherjee and P. Thilagar, Phys. Chem. Chem. Phys., 2014, 16, 20866–20877 RSC.
- S. Mukherjee and P. Thilagar, Chem.–Eur. J., 2014, 20, 8012–8023 CrossRef CAS PubMed.
- Z. Xei, C. Chen, S. Xu, J. Li, Y. Zhang, S. Liu, J. Xu and Z. Chi, Angew. Chem., Int. Ed., 2015, 54, 7181–7184 CrossRef PubMed.
- J. Guo, X. L. Li, H. Nie, W. Luo, R. Hu, A. Qin, Z. Zhao, S. J. Su and B. Z. Tang, Chem. Mater., 2017, 29, 3623–3631 CrossRef CAS.
- F. B. Dias, J. Santos, D. R. Graves, P. Data, R. S. Nobuyasu, M. A. Fox, A. S. Batsanov, T. Palmeira, M. N. Berberan-Santos, M. R. Bryce and A. P. Monkman, Adv. Sci., 2016, 3, 1600080 CrossRef PubMed.
- J. S. Ward, R. S. Nobuyasu, A. S. Batsanov, P. Data, A. P. Monkman, F. B. Dias and M. R. Bryce, Chem. Commun., 2016, 52, 2612–2615 RSC.
- M. Okazaki, Y. Takeda, P. Data, P. Pander, H. Higginbotham, A. P. Monkman and S. Minakata, Chem. Sci., 2017, 8, 2677–2686 RSC.
- S. Gan, W. Luo, B. He, L. Chen, H. Nie, R. Hu, A. Qin, Z. Zhao and B. Z. Tang, J. Mater. Chem. C, 2016, 4, 3705–3708 RSC.
- C. Chen, R. Huang, A. S. Batsanov, P. Pander, Y. T. Hsu, Z. Chi, F. B. Dias and M. R. Bryce, Angew. Chem., Int. Ed., 2018, 130, 16645–16649 CrossRef.
- H. Tanaka, K. Shizu, H. Nakanotani and C. Adachi, J. Phys. Chem. C, 2014, 118, 15985–15994 CrossRef CAS.
- P. Pander, A. Swist, R. Motyka, J. Soloducho, F. B. Dias and P. Data, J. Mater. Chem. C, 2018, 6, 5434–5443 RSC.
- J. Yang, H. Gao, Y. Wang, Y. Yu, Y. Gong, M. Fang, D. Ding, W. Hu, B. Z. Tang and Z. Li, Mater. Chem. Front., 2019, 3, 1391–1397 RSC.
- Y. Wang, J. Yang, M. Fang, Y. Gong, J. Ren, L. Tu, B. Z. Tang and Z. Li, Adv. Funct. Mater., 2021, 31, 2101719 CrossRef CAS.
- Z. Zhao, G. Yu, Q. Chang, X. Liu, Y. Liu, L. Wang, Z. Liu, Z. Bian, W. Liu and C. Huang, J. Mater. Chem. C, 2017, 5, 7344–7351 RSC.
- A. D. Burrows, G. K. Köhn, M. F. Mahon and M. Varrone, C. R. Chim., 2006, 9, 111–119 CrossRef CAS.
- H. S. Gutowsky and C. H. Holm, J. Chem. Phys., 1956, 25, 1228–1234 CrossRef CAS.
- H. Jiang, L. Jia, Y. Li, S. Liu, R. Chen, L. Jin, J. Jin, C. Zheng, F. Fan and W. Huang, Chem. Commun., 2017, 53, 3641–3644 RSC.
- J. P. Majoral and A. M. Caminade, Chem. Rev., 1999, 99, 845–880 CrossRef CAS PubMed.
- T. Nishimoto, T. Yasuda, S. Y. Lee, R. Kondo and C. Adachi, Mater.Horiz., 2014, 1, 264–269 RSC.
- B. Zhou, Q. Zhao, L. Tanga and D. Yan, Chem. Commun., 2020, 56, 7698–7701 RSC.
- B. Zhou and D. Yan, Adv. Funct. Mater., 2019, 29, 1807599 CrossRef.
- A. Nitti, C. Botta, A. Forni, E. Cariati, E. Lucenti and D. Pasini, CrystEngComm, 2020, 22, 7782–7785 RSC.
- H. Sternlicht, G. C. Nieman and G. W. Robinson, J. Chem. Phys., 1963, 38, 1326 CrossRef CAS.
- M. Shao, L. Yan, M. Li, I. Ilia and B. Hu, J. Mater. Chem. C, 2013, 1, 1330–1336 RSC.
- Y. Zhang and S. R. Forrest, Chem. Phys. Lett., 2013, 590, 106–110 CrossRef CAS.
- J. J. Maki, T. Verbiest, M. Kauranen, S. V. Elshocht and A. Persoons, J. Chem. Phys., 1996, 105, 767 CrossRef CAS.
- R. Tempelaar, A. Stradomska, J. Knoester and F. C. Spano, J. Phys. Chem. B, 2011, 115, 10592 CrossRef CAS PubMed.
- W. B. Sparks, J. Hough, T. A. Germer, F. Chen, S. DasSarma, P. DasSarma, F. T. Robb, N. Manset, L. Kolokolova, N. Reid, F. D. Macchetto and W. Martin, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 7816 CrossRef CAS PubMed.
- S. W. Huo, P. F. Duan, T. F. Jiao, Q. M. Peng and M. H. Liu, Angew. Chem., Int. Ed., 2017, 56, 12174–12178 CrossRef CAS PubMed.
- X. Yang, X. Lin, Y. Zhao, Y. S. Zhao and D. Yan, Angew. Chem., Int. Ed., 2017, 56, 7853–7857 CrossRef CAS PubMed.
- A. Nitti and D. Pasini, Adv. Mater., 2020, 32, 1908021 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1977279, 1916761 and 1916789. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc00990k |
| This journal is © The Royal Society of Chemistry 2022 |