
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn umpolung strategy for intermolecular [2 + 2 + 1] cycloaddition of aryl aldehydes and nitriles: a facile access to 2,4,5-trisubstituted oxazoles†
Deevi
Basavaiah
 *,
Gangadhararao
Golime
,
Shivalal
Banoth
and
Saidulu
Todeti
*,
Gangadhararao
Golime
,
Shivalal
Banoth
and
Saidulu
Todeti
School of Chemistry, University of Hyderabad, Hyderabad-500046, India
First published on 11th June 2022
Abstract
We have described the first example of an umpolung strategy for intermolecular [2 + 2 + 1] cycloaddition between two aryl aldehydes and a nitrile under the influence of TMSOTf that proceeds through the formation of N–C, O–C and C–C bonds providing a simple synthetic protocol for obtaining 2,4,5-trisubstituted oxazoles.
Introduction
Umpoled reactivity of aldehydes originating from the benzoin condensation1 has grown extensively through the concepts of Corey–Seebach dithiane chemistry2 and catalytic N-heterocyclic carbene (NHC) chemistry,3 and has become a well-defined powerful synthetic tool in organic chemistry with enormous applications.4 In recent years there has been an increasing interest in intermolecular [2 + 2 + 1] cycloaddition strategies for the synthesis of five-membered carbo and heterocyclic compounds because this strategy proceeds through the construction of multiple bonds in a single operation.5–7 The umpolung concept has uncovered equivalents that address the need for acyl anion reactivity but, to the best of our knowledge, there is no report in the literature on the utilization of umpoled reactivity of aldehydes for the formation of C–C bonds in intermolecular [2 + 2 + 1] cycloaddition reactions.8 Therefore development of strategies employing the umpoled reactivity of aldehydes in [2 + 2 + 1] cycloadditions represents a challenging endeavor as such protocols will further expand the synthetic wealth of the umpoled chemistry of aldehydes.Highly substituted oxazole frameworks occupy a special place in the heterocyclic compounds because of their occurrence in various natural products9 as well as remarkable biological activities10 and also wide applications in fluorescence dyes11 and synthetic chemistry.12 Therefore development of facile and simple methodologies for obtaining oxazole frameworks continues to be a challenging area in synthetic chemistry.9b,10c,13 In recent years, the [2 + 2 + 1] annulation strategy for obtaining oxazole frameworks has attracted the attention of organic chemists because this process constructs multiple bonds in a single operation.7 Zhang and co-workers reported the first example of a [2 + 2 + 1] cycloaddition protocol for obtaining oxazole frameworks via a gold-catalyzed intermolecular reaction between an alkyne, a nitrile and an oxygen source (Scheme 1a).7a Subsequently, Jiang7b and Saito7c,d independently reported similar strategies via copper catalyzed and iodine mediated/catalyzed reactions, respectively, for obtaining 2,4,5-trisubstituted oxazoles. Later on few similar protocols using gold catalysts were reported in the literature.7e,f
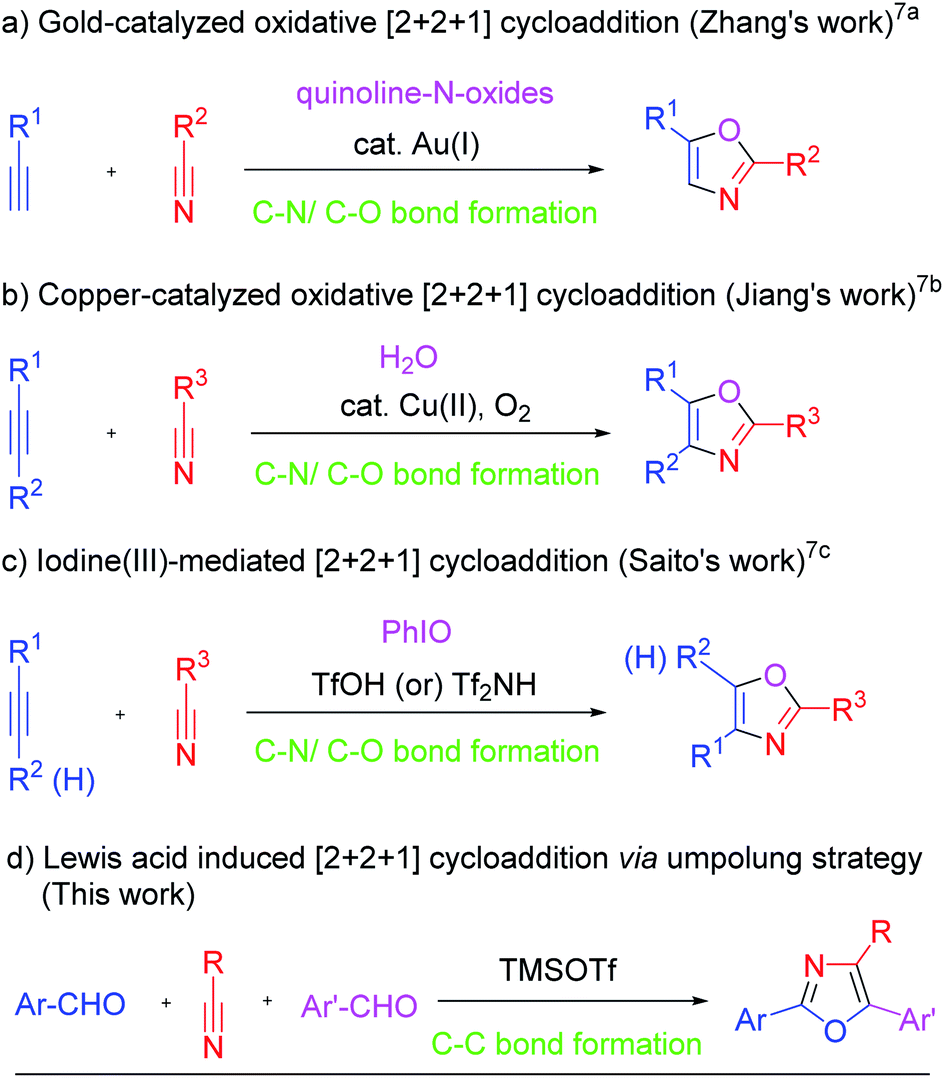 | ||
| Scheme 1 Strategies for oxazoles via [2 + 2 + 1] cycloadditions. | ||
All the known reports7 using [2 + 2 + 1] strategies for synthesis of oxazoles utilize similar starting materials (an alkyne, a nitrile and an oxygen source) and proceed through the formation of new C–N and C–O bonds. It is interesting to note that these protocols neither use aldehydes as substrates nor proceed through the formation of a C–C bond. Therefore we felt that development of such [2 + 2 + 1] cycloaddition protocols, utilizing aldehydes as reaction partners and involving the construction of C–C bonds via an umpolung strategy, for obtaining oxazoles, will not only expand the scope of cycloaddition reactions but also will be highly useful and of great interest in synthetic chemistry. Accordingly, we have been working in this direction and now report a Lewis acid (TMSOTf) induced umpolung strategy for [2 + 2 + 1] cycloaddition between two aryl aldehydes and a nitrile providing a facile synthetic protocol for obtaining 2,4,5-trisubstituted oxazoles (Scheme 1d).
Based on our long term experience14 in the area of Baylis–Hillman reaction15,16 utilizing aldehydes as electrophiles and on the concept of the umpoled reactivity of aldehydes, it occurred to us that two aryl aldehydes and a nitrile might, under the influence of a Lewis acid, constitute a workable three-component system for designing a facile umpolung protocol for [2 + 2 + 1] cycloaddition involving C–C bond formation for obtaining an oxazole framework as described in Scheme 2. If such a reaction is designed, what would be its profile? Careful examination of such a visionary reaction reveals that there might be three competing possible routes (paths 1–3, Scheme 2). In all these paths the first step is same and provides an intermediate Avia the addition of nitrile onto aldehyde in the presence of a Lewis acid. Afterwards path-1 proceeds through carbanion C to provide oxazoles-I while path-2 gives oxazoles-II through carbanion D and oxyanion E. Path-3 also provides the oxazole-I through carbanions D and F. It is interesting to note that if the reaction proceeds through path-3 it will be a unique reaction which involves reversal of polarity at aldehyde and nitrile groups.
 | ||
| Scheme 2 Our hypothesis: competing cycloaddition reactions. | ||
Results and discussion
With a view to realize our hypothesis towards the development of [2 + 2 + 1] cycloaddition, we first selected the reactive pyridine-2-carboxaldehyde (1a) and benzonitrile (2a) as reaction partners with TMSOTf as a promoter. Thus we performed a reaction between 1a (1 mmol) and 2a (1 mmol) in the presence of TMSOTf (1 mmol) in dichloromethane (3 mL) under reflux for 24 h (Table 1, entry 1). We noticed that there was no reaction and the starting materials were found to be intact. However, when the same reaction was performed in dichloroethane (DCE) under reflux for 24 h, to our pleasant surprise, 4-phenyl-2,5-di(pyridin-2-yl)oxazole (3aa) was obtained in 16% isolated yield (Table 1, entry 2). We have established the structure by spectral data (1H, 13C NMR, and HRMS) and also confirmed it by a single crystal X-ray data analysis (see the ESI, Fig. S1†). We observed that the yield of oxazole (3aa) was increased to 57%, when we used 1a (1 mmol), 2a (3 mmol) and TMSOTf (2 mmol) (Table 1, entry 4). Fascinated by this result, we optimized the reaction conditions by changing the solvents and Lewis acids as shown in Table 1. Solvents like THF, dioxane, toluene, DMF and DMSO did not give satisfactory results (Table 1, entries 5–9). Surprisingly, when we used ethyl acetate as a solvent, the yield of oxazole (3aa) was increased to 75% (Table 1, entry 10). The yield of 3aa was further improved to 81%, when n-propyl acetate was used as a solvent, in a short reaction time (6 h) (Table 1, entry 11). After careful observation of this reaction by varying the quantities of reacting partners (Table 1, entries 13–18), the best result was obtained when 1a (1 mmol) was treated with 2a (2 mmol) in the presence of TMSOTf (1.5 mmol) in n-propyl acetate (2 mL) under reflux for 4 h, to provide oxazole (3aa) in 89% isolated yield (Table 1, entry 18). Other Lewis acids such as Zn(OTf)2, Cu(OTf)2, ZnCl2, FeCl3, Sc(OTf)3, AgOTf, and Ln(OTf)3 did not provide satisfactory results (Table 1, entries 19–25).|
|
|||||
|---|---|---|---|---|---|
| Entry | 2a | Lewis acid (mmol) | Solvent (mL) | Temp.c (°C)/time (h) | Yieldd (%) |
| a All reactions were performed with 1 mmol of pyridine-2-carboxaldehyde (except entry 15). b Number of mmol used. c Oil bath temperature. d Isolated yields based on 1a. e 2 mmol of 1a was used. f Reaction is slow and not complete. NR = no reaction. ND = no product was detected. | |||||
| 1 | 1 | TMSOTf (1) | DCM (3) | 60 °C/24 h | NR |
| 2 | 1 | TMSOTf (1) | DCE (3) | 100 °C/24 h | 16 |
| 3 | 2 | TMSOTf (2) | DCE (3) | 100 °C/24 h | 40 |
| 4 | 3 | TMSOTf (2) | DCE (3) | 100 °C/24 h | 57 |
| 5 | 3 | TMSOTf (2) | THF (3) | 90 °C/24 h | ND |
| 6 | 3 | TMSOTf (2) | Dioxane (3) | 120 °C/24 h | ND |
| 7 | 3 | TMSOTf (2) | Toluene (3) | 130 °C/24 h | 14 |
| 8 | 3 | TMSOTf (2) | DMF (3) | 110 °C/24 h | ND |
| 9 | 3 | TMSOTf (2) | DMSO (3) | 110 °C/24 h | ND |
| 10 | 3 | TMSOTf (2) | EtOAc (3) | 100 °C/24 h | 75 |
| 11 | 3 | TMSOTf (2) | n-PrOAc (3) | 120 °C/6 h | 81 |
| 12 | 3 | TMSOTf (2) | n-BuOAc(3) | 140 °C/2 h | 63 |
| 13 | 1 | TMSOTf (1) | n-PrOAc (3) | 120 °C/6 h | 68 |
| 14 | 1 | TMSOTf (1) | n-PrOAc (2) | 120 °C/6 h | 68 |
| 15e | 1 | TMSOTf (2) | n-PrOAc (2) | 120 °C/8 h | 45 |
| 16 | 2 | TMSOTf (1) | n-PrOAc (2) | 120 °C/5 h | 84 |
| 17 | 2 | TMSOTf (1) | Neat | 120 °C/4 h | 33 |
| 18 | 2 | TMSOTf (1.5) | n-PrOAc (2) | 120 °C/4 h | 89 |
| 19f | 2 | Zn(OTf)2 (1.5) | n-PrOAc (2) | 120 °C/6 h | 16 |
| 20 | 2 | Cu(OTf)2 (1.5) | n-PrOAc (2) | 120 °C/6 h | NR |
| 21 | 2 | ZnCl2 (1.5) | n-PrOAc (2) | 120 °C/6 h | 42 |
| 22 | 2 | FeCl3 (1.5) | n-PrOAc (2) | 120 °C/6 h | Trace |
| 23f | 2 | Sc(OTf)3 (1.5) | n-PrOAc (2) | 120 °C/6 h | 18 |
| 24 | 2 | AgOTf (1.5) | n-PrOAc (2) | 120 °C/6 h | 24 |
| 25f | 2 | Ln(OTf)3 (1.5) | n-PrOAc (2) | 120 °C/6 h | 15 |

Having the optimal reaction conditions in hand, with a view to understand the generality of this new reaction, we employed various substituted pyridine-2-carboxaldehydes (1a–d) and aryl nitriles (2a–u) as reaction partners. We were pleased to see that in most cases expected oxazoles (3) were obtained in high yields (Scheme 3). Thiophenecarbonitriles (2s and 2t) and naphthalene-2-carbonitrile (2u) also furnished the corresponding oxazoles (3as-3au) in moderate to good yields. This strategy was successfully extended to quinoline-2-carboxaldehyde (1e) to furnish the corresponding oxazoles (3ea and 3eb) up to 61% yield (Scheme 3). All these results providing oxazoles 3 clearly indicate that this reaction proceeds through carbanion C as shown in path-1 but not through oxyanion E as indicated in path-2 (Scheme 2). Alternatively, this reaction might also proceed through path-3 involving umpoled anions D and F which is very rare in organic chemistry.
 | ||
| Scheme 3 Synthesis of oxazoles (3): substrate scopea,b. aReaction conditions: all reactions were carried out with carboxaldehyde (1) (1 mmol), nitrile (2) (2 mmol), and TMSOTf (1.5 mmol) in n-PrOAc (2 mL) under reflux (oil bath temperature 120 °C) for 2–8 h. bIsolated yields (based on carboxaldehyde). cRemaining starting materials recovered. dThis reaction was performed with quinoline-2-carboxaldehyde. | ||
Unfortunately, we noticed that this strategy failed in the case of less reactive aryl aldehydes such as benzaldehyde and 4-nitrobenzaldehyde when treated with p-tolunitrile (Scheme 3). We also observed that similar reactions of thiophene-2-carboxaldehyde, pyridine-4-carboxaldehyde and pyridine-3-carboxaldehyde with p-tolunitrile did not provide the expected products. In all these cases, the aldehydes (except pyridine-4-carboxaldehyde) were mostly intact while p-tolunitrile underwent some reaction, probably conversion into the corresponding amide.17 In the case of pyridine-4-carboxaldehyde the reaction was not clean. The failure of non-pyridine/quinoline-2-carboxaldehydes (benzaldehyde, 4-nitrobenzaldehyde, pyridine-3-carboxaldehyde etc.) to participate in this strategy to provide similar oxazoles (3) certainly throws some light on the mechanism of this reaction.
We reasoned that this failure might be due to the fact that RCN is not nucleophilic enough to add onto the carbonyl of these aldehydes to form the corresponding oxyanion A (Scheme 2). These results clearly show that since RCN is a very poor nucleophile, the reaction requires a very highly electrophilic aldehyde. RCN is able to add onto the carbonyl of pyridine-2-carboxaldehyde (1a) because of its high electrophilicity (it is a very reactive aldehyde by itself and the presence of TMSOTf makes it even more electrophilic). Initially 1a forms a salt with TMSOTf (HRMS evidence intermediate I1 in Scheme 8) which makes the carboxaldehyde carbon more electrophilic because the electron withdrawing group (C = NTMS) is in α-position to the aldehyde. The high electrophilicity of pyridine-2-carboxaldehyde in comparison to that of pyridine-4-carboxaldehyde might also be due to the formation of a five membered ring via rigid complexation with TMSOTf which makes it more electrophilic because of its easy accessibility to nucleophilic attack by RCN as shown in Scheme 4. Due to our curiosity to look at the chemical shift differences (if any) between the carbonyl carbons of the aldehydes and their corresponding TMSOTf salts,18 we recorded 13C NMR spectra (see the ESI†) of pyridine-2-carboxaldehyde (1a), quinoline-2-carboxaldehyde (1e), pyridine-4-carboxaldehyde and benzaldehyde and their salts/complex with TMOTf (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in a mixture of CDCl3 and CH3CN (2:1) (CH3CN is added for the purpose of solubility of salts and uniformity). These results are presented in Scheme 4. From these results it is clear that there is a remarkable difference of 9.6 ppm (towards upfield) between carbonyl carbons of 1a and its TMSOTf salt. Almost a similar difference (9.5 ppm towards upfield) was observed in the case of carbonyl carbons of 1e and its salt. However the differences are small in the case of pyridine-4-carboxaldehyde (2.9 ppm towards upfield) and benzaldehyde (0.9 ppm towards downfield). Although we cannot presently arrive at any conclusion, we felt that it is appropriate to mention these values in view of significant differences in the case of 1a, 1e and their salts.
:1) in a mixture of CDCl3 and CH3CN (2:1) (CH3CN is added for the purpose of solubility of salts and uniformity). These results are presented in Scheme 4. From these results it is clear that there is a remarkable difference of 9.6 ppm (towards upfield) between carbonyl carbons of 1a and its TMSOTf salt. Almost a similar difference (9.5 ppm towards upfield) was observed in the case of carbonyl carbons of 1e and its salt. However the differences are small in the case of pyridine-4-carboxaldehyde (2.9 ppm towards upfield) and benzaldehyde (0.9 ppm towards downfield). Although we cannot presently arrive at any conclusion, we felt that it is appropriate to mention these values in view of significant differences in the case of 1a, 1e and their salts.
 | ||
| Scheme 4 13C NMR carbonyl chemical shifts of 1a, 1e, pyridine-4-carboxaldehyde and benzaldehyde and their TMSOTf salts/complex. | ||
At this juncture, it occurred to us that if one of the less reactive aldehydes (say benzaldehyde) is used along with pyridine-2-carboxaldehyde (1a), this reaction might provide first oxyanion A, which might react with benzaldehyde through either path-1 or path-3 to provide a mixed oxazole 5aab containing both pyridine-2-carboxaldehyde and benzaldehyde units. Accordingly, we carried out the reaction of 1a (1 mmol) with p-tolunitrile (2b) (2 mmol) in the presence of benzaldehyde (4a) (1 mmol) under the influence of TMSOTf (1.5 mmol) in n-propyl acetate (2 mL) (see ESI Table S1†). As expected, we obtained the mixed oxazole 5aab in 37% isolated yield along with oxazole 3ab in 12% yield (Table S1,† entry 1). Fascinated by this result, we optimized the reaction conditions (Table S1†). We obtained the best result when 1a (1 mmol) was treated with 2b (3 mmol) in the presence of 4a (2 mmol) under the influence of TMSOTf (1.5 mmol) in n-propyl acetate (2 mL) under reflux for 3 h, thus providing oxazole 5aab in 56% isolated yield along with another oxazole 3ab in 8% yield (Table S1,† entry 4). To understand the generality of our strategy, we employed representative substituted benzaldehydes (4), pyridine-2-carboxaldehydes/quinoline-2-carboxaldehyde (1), and nitriles (2) in this reaction sequence. The resulting oxazoles (5) were obtained in 30–88% yields along with other oxazoles (3) in minor amounts (Scheme 5). These results are indeed encouraging in the sense that the failure of less reactive aldehydes such as benzaldehyde and 4-nitrobenzaldehyde, etc., in participating directly in this strategy has turned out to be a positive failure because this led to the development of a protocol for obtaining oxazoles having different substituents at 2,5 positions which otherwise is not possible.
 | ||
| Scheme 5 Synthesis of mixed oxazoles (5): substrate scopea,b. aReaction conditions: all reactions were carried out with carboxaldehyde (1) (1 mmol), aryl aldehyde (4) (2 mmol), nitrile (2) (3 mmol), and TMSOTf (1.5 mmol) in n-PrOAc (2 mL) under reflux (oil bath temperature 120 °C) for 2–6 h. Other oxazoles (3) were not isolated as they were obtained in minor amounts (less than 10%). bIsolated yields [based on carboxaldehyde (1)]. cOther possible oxazoles (3) were not observed in these reactions. | ||
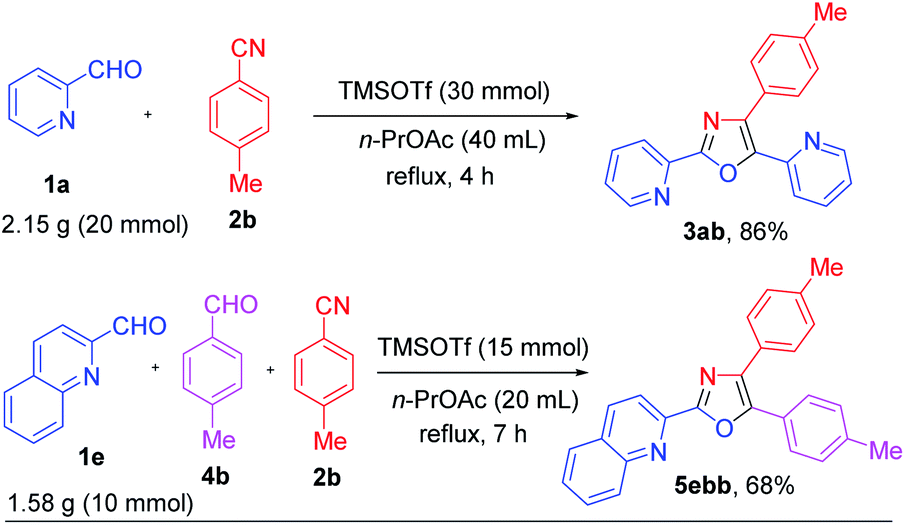
We next examined the applicability of various aliphatic nitriles (2v-z, 2z′) in this strategy and were pleased to realize that the resulting oxazoles (3av-3az, 3az′, 3ev, and 3ez′) and mixed oxazoles (5acz, 5afv, and 5egw) were obtained in 21–93% isolated yields (Scheme 6). From these results it is clear that oxazoles 3 were obtained in better yields (up to 93%) in comparison to mixed oxazoles 5. For gram scale synthesis, we performed the reactions in 20 mmol scale of pyridine-2-carboxaldehyde (1a) and in 10 mmol scale of quinoline-2-carboxaldehyde (1e) to provide the corresponding oxazoles 3ab and 5ebb, respectively. We were pleased to see that 3ab and 5ebb were obtained in 86% and 68% isolated yields, respectively, almost the same as in 1.0 mmol scale reactions (Scheme 7).
 | ||
| Scheme 6 Scope of aliphatic nitrilesa,b. aReaction conditions: all reactions were carried out with carboxaldehyde 1 (1 mmol). bIsolated yields (based on 1). cAcetonitrile was also used as a solvent (2 mL) under reflux (oil bath temperature 120 °C) for 24 h. dn-Propyl acetate was used as a solvent for the reaction of 1a with acetonitrile (4 mmol). | ||
With a view to understanding the mechanistic pathway we thought that it would be appropriate to monitor the reaction by HRMS and to look for the molecular ion peaks at different intervals (after 5, 15, 30, 60, 120, 180, and 240 minutes) (Scheme 8). Accordingly we performed the reaction between pyridine-2-carboxaldehyde (1a) with benzonitrile (2a) in the presence of TMSOTf using n-propyl acetate as a solvent. After 5 minutes, HRMS (ESI-MS) (see ESI† Mass Spectrum-1) of the reaction mixture showed a major molecular ion peak at m/z 210.1518 (calcd m/z 210.0793) and a minor ion peak at 317.1872 (calcd m/z 317.1164) indicating the presence of intermediates I2 and I4 or I4*, respectively, in addition to the molecular ion peaks of aldehyde 1a and the ion of salt I1. After 15 minutes HRMS (see ESI† Mass Spectrum-2) showed major ion peaks at m/z 253.1368 (calcd m/z 253.1209) (M + H) and at m/z 359.1629 (calcd m/z 359.1503) which are indicative of the presence of intermediates I3 and I5 (or I5*), respectively. After 30 minutes (see ESI† Mass Spectrum-3) we observed major molecular ion peaks at m/z 300.1174 (calcd m/z 300.1137) (M + H) for the product (3aa) and m/z 432.2124 (calcd m/z 432.1976) (M + H) for I6 (or I6*) in addition to the other major ion peaks of I3 and I5 (or I5*). After 1 h, 2 h, 3 h and 4 h (see ESI† Mass Spectra 4–7) we noticed major molecular ion peaks at m/z 164.1080 (calcd m/z 164.1075) (M + H) for N-n-propylbenzamide (I7)17 and m/z 599.2194 (calcd m/z 599.2196) for the dimeric molecular ion (I8) of the product 3aa along with the major peak of the product at m/z 300.1158 (3aa). The formation of N-n-propylbenzamide as a side product also explains the necessity of employing excess benzonitrile in this reaction.
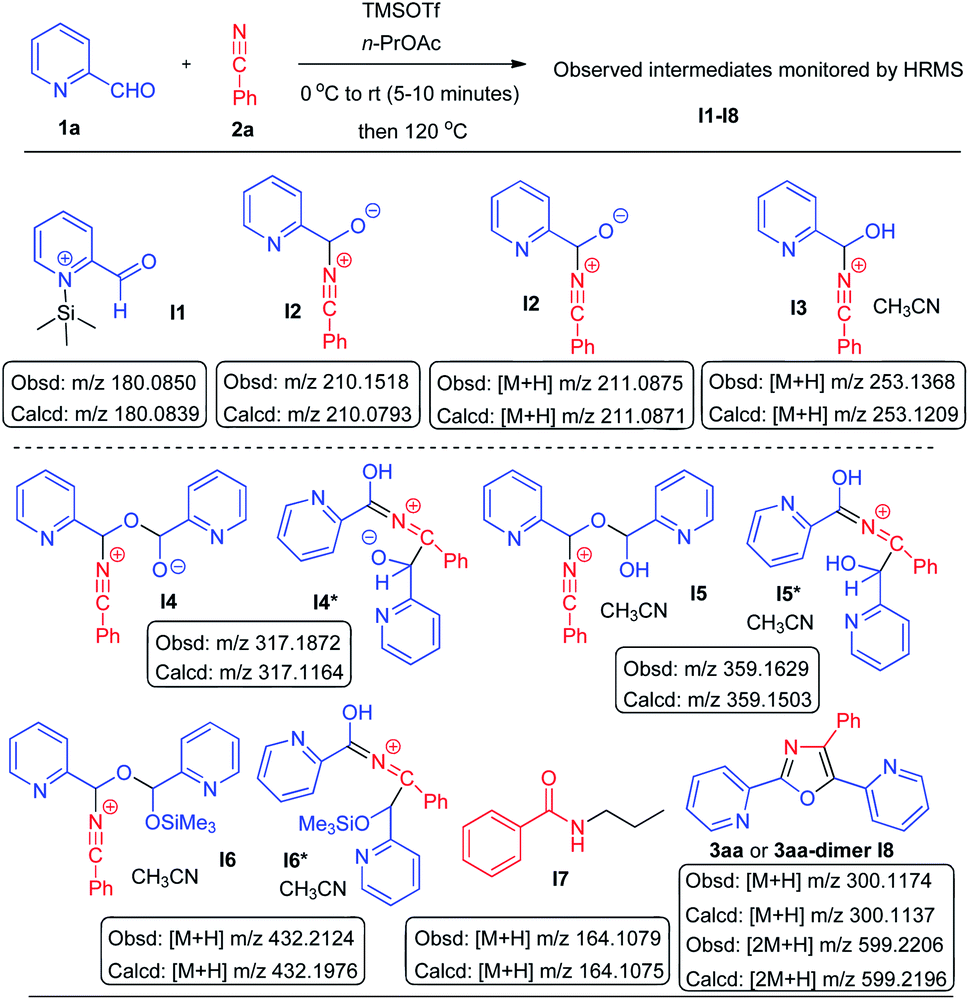 | ||
| Scheme 8 HRMS mechanistic studies: observed intermediates. [obsd = observed, calcd = calculated]. | ||
The above-mentioned mechanistic experiments by HRMS indicate the presence of intermediates proposed in path-1 as well as in path-3 (Scheme 2) thus supporting the mechanism to a reasonable extent. Accordingly, two plausible mechanistic pathways for obtaining 3 and 5 are presented in Scheme 9 which clearly demonstrates the formation of the C–C bond through the umpoled reaction. It is very interesting to note that the oxygen of oxazole-I (for obtaining oxazole 5) ring through path-1 arises from the carbonyl oxygen of pyridine-2-carboxaldehydes (1), while path-3 shows that the same oxygen comes from the carbonyl oxygen of aryl aldehyde (4). Since two moles of the same aldehyde are utilized for the synthesis of oxazoles 3 it does not matter whether path-1 is operating or path-3. For compounds 5 there is a remarkable difference in path-1 and path-3 (Scheme 2) regarding the origin of the oxygen of the oxazole ring. It needs to be mentioned here that path-3 presents the umpoled reactions at both aldehyde and nitrile groups (Schemes 2 and 9). Our efforts are currently going on to understand the exact mechanism of this reaction. Although the exact mechanism is not clearly known, this protocol represents the first example of the umpolung strategy for [2 + 2 + 1] cycloaddition which will have tremendous impact in synthetic and mechanistic organic chemistry. Formation of oxazole-I clearly states that the reaction does not proceed through path-2 in Scheme 2. It needs to be mentioned here that there is a report in the literature on the reaction of 2-cyano hetero arenes with aryl aldehydes in the presence of acetic acid providing oxazole-II derivatives as presented in Scheme 2 with a different kind of reaction mechanism.19 It is appropriate to mention here the work of Khalafi-Nezhad and coworkers who reported a simple synthesis of tetrasubstituted imidazoles via the trifluoroacetic acid catalyzed reaction of benzoin with amine and nitrile under microwave conditions.20 To have an insight into the mechanistic understanding, we performed a reaction of benzoin with benzonitrile under our experimental conditions with a view to see the possibility of obtaining either oxazole-I or oxazole-II.21 We noticed that this reaction is not clean. After careful column chromatography we isolated O-acetylbenzoin (2-acetoxy-1,2-diphenylethanone) in 10% yield (see the ESI†). We also prepared α-pyridoin (which exits in the enol form) following the known procedure22 and carried out the reaction between α-pyridoin and benzonitrile under our reaction conditions.21 In this case, the reaction mixture showed that some of the α-pyridoin was intact and the remaining was oxidized to 2,2′-pyridil (see the ESI†). The failure of these experiments to provide any kind of oxazole (either oxazole-I or oxazole-II) clearly indicates that benzoins are not intermediates in our reactions and also RCN is not nucleophilic enough to add onto either the carbonyl carbon of benzoin or enol carbon of α-pyridoin.
 | ||
| Scheme 9 Plausible mechanistic pathways for the formation of oxazoles 3 and 5. | ||
It is surprising and interesting to see that the reaction works much better in n-propyl acetate as a solvent in comparison with other solvents such as THF, DCE, toluene, dioxane, DMF and DMSO as shown in Table 1. We do not have at present any clear reason to explain this fact. Polar solvents like DMF, DMSO and dioxane (also THF) coordinate very strongly with TMSOTf thereby preventing the interaction between TMSOTf and aldehyde/nitriles leading to the inhibition of the reaction. DCE works to some extent, may be because of its low boiling point it is less efficient. Because of its low dipole moment and dielectric constant toluene probably is not effective for this reaction. Although there is no exact reason for the choice of n-propyl acetate as a solvent, presumably this might be attributed to the matching dielectric constant, dipole moment and boiling point of n-propyl acetate that suits this reaction.
Conclusions
In conclusion, we have demonstrated a facile intermolecular [2 + 2 + 1] cycloaddition between two aryl aldehydes and a nitrile providing a simple methodology for obtaining 2,4,5-trisubstituted oxazoles. This process meticulously utilizes one of the two aryl aldehydes as an acyl anion (or equivalent) thus becoming the first example of an umpolung strategy for [2 + 2 + 1] cycloaddition reactions. The exciting part of this methodology is that it utilizes very common, easily and commercially available chemicals and reagents, thus representing the novelty, simplicity and uniqueness of this reaction. Presently our efforts are underway to understand the reactivity profile and also towards exploring the synthetic applications of the reactive intermediate A (Scheme 2) generated in situ by the treatment of pyridine-2-carboxaldehyde with a nitrile, to provide important heterocyclic frameworks via the reaction with various substrates like activated alkenes, alkynes, imines etc. These results will be reported in due course.Data availability
The X-ray crystallographic data for compounds 3aa, 3bb, 3ea, 5aab, 5aib, and 5afv have been deposited in the Cambridge Crystallographic Data Centre with CCDC numbers 2108509, 2108510, 2108511, 2110001, 2110003, and 2110002, respectively.Author contributions
This reaction was conceived and designed by D. B. All the experiments were performed by G. G., S. B. and S. T. All authors analysed the data and wrote the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We are grateful to the Science and Engineering Research Board (SERB) (New Delhi) for funding this project. D. B. thanks the University of Hyderabad-Institution of Eminence (IoE) program for offering him an IoE Research Chair Professorship in Chemistry. G. G., S. B. and S. T. thank the University Grants Commission (UGC) (New Delhi) for Dr D. S. Kothari Post-Doctoral Fellowships. G. G., S. B. and S. T. also thank the Science and Engineering Research Board (SERB) for their research (RA) fellowships. Financial and infrastructure support from the University of Hyderabad under the Institution of Eminence (IoE) program and UGC (through CAS program) are gratefully acknowledged. We thank Professor Vishwakarma Singh (former Professor, Department of Chemistry, IIT Bombay) for helpful discussions. We also thank Professor Samudranil Pal (School of Chemistry, University of Hyderabad) for helpful discussions regarding the X-ray crystal structures.Notes and references
- (a) F. Wöhler and J. Liebig, Ann. Pharm., 1832, 3, 249–282 CrossRef; (b) N. Zinin, Ann. Pharm., 1839, 31, 329–332 CrossRef; (c) N. Zinin, Ann. Pharm., 1840, 34, 186–192 CrossRef.
- (a) E. J. Corey and D. Seebach, Angew. Chem., Int. Ed., 1965, 4, 1075–1077 ( Angew. Chem., Int. Ed. , 1965 , 4 , 1077–1078 ) CrossRef CAS; (b) D. Seebach and E. J. Corey, J. Org. Chem., 1975, 40, 231–237 CrossRef CAS.
- (a) R. Breslow, J. Am. Chem. Soc., 1958, 80, 3719–3726 CrossRef CAS; (b) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS PubMed; (c) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed; (d) D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS PubMed; (e) R. S. Menon, A. T. Biju and V. Nair, Beilstein J. Org. Chem., 2016, 12, 444–461 CrossRef CAS PubMed.
- (a) B.-T. Grobel and D. Seebach, Synthesis, 1977, 357–402 CrossRef; (b) D. Seebach, Angew. Chem., Int. Ed., 1979, 18, 239–258 CrossRef; (c) M. Yus, C. Najera and F. Foubelo, Tetrahedron, 2003, 59, 6147–6212 CrossRef CAS; (d) J. Izquierdo, G. E. Hutson, D. T. Cohen and K. A. Scheidt, Angew. Chem., Int. Ed., 2012, 51, 11686–11698 CrossRef CAS PubMed; (e) X.-J. Dai, C.-C. Li and C.-J. Li, Chem. Soc. Rev., 2021, 50, 10733–10742 RSC.
- For reviews, see: (a) J. Blanco-Urgoiti, L. Añorbe, L. Pérez-Serrano, G. Domínguez and J. Pérez-Castells, Chem. Soc. Rev., 2004, 33, 32–42 RSC; (b) L. V. R. Bonaga and M. E. Krafft, Tetrahedron, 2004, 60, 9795–9833 CrossRef CAS; (c) S. E. Gibson and N. Mainolfi, Angew. Chem., Int. Ed., 2005, 44, 3022–3037 CrossRef CAS PubMed; (d) N. Chatani, Chem. Rec., 2008, 8, 201–212 CrossRef CAS PubMed; (e) S. Kitagaki, F. Inagaki and C. Mukai, Chem. Soc. Rev., 2014, 43, 2956–2978 RSC.
- For selected reports on synthesis of five membered heterocycles, see: (a) N. Chatani, M. Tobisu, T. Asaumi, Y. Fukumoto and S. Murai, J. Am. Chem. Soc., 1999, 121, 7160–7161 CrossRef CAS; (b) Z. W. Gilbert, R. J. Hue and I. A. Tonks, Nat. Chem., 2016, 8, 63–68 CrossRef CAS PubMed; (c) H. Miura, K. Takeuchi and T. Shishido, Angew. Chem., Int. Ed., 2016, 55, 278–282 CrossRef CAS PubMed; (d) K. Kawakita, E. P. Beaumier, Y. Kakiuchi, H. Tsurugi, I. A. Tonks and K. Mashima, J. Am. Chem. Soc., 2019, 141, 4194–4198 CrossRef CAS PubMed; (e) L. Ma, F. Jin, X. Cheng, S. Tao, G. Jiang, X. Li, J. Yang, X. Bao and X. Wan, Chem. Sci., 2021, 12, 9823–9830 RSC; (f) T. Fukuyama, N. Nakashima, T. Okada and I. Ryu, J. Am. Chem. Soc., 2013, 135, 1006–1008 CrossRef CAS PubMed; (g) J. T. M. Correia, G. P. Silva, E. Andre and M. W. Paixao, Adv. Synth. Catal., 2019, 361, 5558–5564 CrossRef CAS.
- For reports on the synthesis of oxazoles via [2 + 2 + 1] cycloaddition strategy, see: (a) W. He, C. Li and L. Zhang, J. Am. Chem. Soc., 2011, 133, 8482–8485 CrossRef CAS PubMed; (b) X. Li, L. Huang, H. Chen, W. Wu, H. Huang and H. Jiang, Chem. Sci., 2012, 3, 3463–3467 RSC; (c) A. Saito, A. Taniguchi, Y. Kambara and Y. Hanzawa, Org. Lett., 2013, 15, 2672–2675 CrossRef CAS PubMed; (d) T. Yagyu, Y. Takemoto, A. Yoshimura, V. V. Zhdankin and A. Saito, Org. Lett., 2017, 19, 2506–2509 CrossRef CAS PubMed; (e) W. Yang, R. Zhang, F. Yi and M. Cai, J. Org. Chem., 2017, 82, 5204–5211 CrossRef CAS PubMed; (f) A. Y. Dubovtsev, D. V. Dar’in and V. Y. Kukushkin, Adv. Synth. Catal., 2019, 361, 2926–2935 CrossRef CAS.
- There is a recent publication describing the umpoled activity of imine in [2 + 2 + 1] cycloaddition involving N-alkylation. H. Wu, X. Li, Z. Yan, N. Ma, S. Song, G. Zhang and N. Jiao, Org. Lett., 2021, 23, 762–766 CrossRef CAS PubMed.
- (a) P. Wipf, Chem. Rev., 1995, 95, 2115–2134 CrossRef CAS; (b) V. S. C. Yeh, Tetrahedron, 2004, 60, 11995–12042 CrossRef CAS; (c) Z. Jin, Nat. Prod. Rep., 2009, 26, 382–445 RSC; (d) Z. Jin, Nat. Prod. Rep., 2011, 28, 1143–1191 RSC.
- (a) H.-Z. Zhang, Z.-L. Zhao and C.-H. Zhou, Eur. J. Med. Chem., 2018, 144, 444–492 CrossRef CAS PubMed; (b) S. Kakkar and B. Narasimhan, BMC Chem., 2019, 13, 16 CrossRef PubMed; (c) E. Sisco and K. L. Barnes, ACS Med. Chem. Lett., 2021, 12, 1030–1037 CrossRef CAS PubMed.
- (a) T. G. Pavlopoulos and P. R. Hammond, J. Am. Chem. Soc., 1974, 96, 6568–6579 CrossRef CAS; (b) S. Chen, X. Ji, M. Gao, L. M. Dedkova and S. M. Hecht, J. Am. Chem. Soc., 2019, 141, 5597–5601 CrossRef CAS PubMed.
- (a) D. L. Boger, Chem. Rev., 1986, 86, 781–793 CrossRef CAS; (b) G. V. Boyd, Sci. Synth., 2001, 11, 383–480 Search PubMed; (c) G. Blond, M. Gulea and V. Mamane, Curr. Org. Chem., 2016, 20, 2161–2210 CrossRef CAS.
- (a) Oxazoles: Synthesis, Reactions, and Spectroscopy, Part A, ed. D. C. Palmer, J. Wiley & Sons, Hoboken, NJ, 2003, vol. 60 Search PubMed; (b) Oxazoles: Synthesis, Reactions, and Spectroscopy, Part B, ed. D. C. Palmer, J. Wiley & Sons, Hoboken, NJ, 2004, vol. 60 Search PubMed; (c) Chemistry of Heterocyclic Compounds: Oxazoles, ed. I. J. Turchi, Wiley & Sons, 1986, vol. 45 Search PubMed; (d) I. J. Turchi and M. J. S. Dewar, Chem. Rev., 1975, 75, 389–437 CrossRef CAS; (e) T. Fukumoto, Y. Aso, T. Otsubo and F. Ogura, J. Chem. Soc., Chem. Commun., 1992, 1070–1072 RSC; (f) Z. Huizhen, Z. Chenghe, G. Rongxia and J. Qinggang, Chin. J. Org. Chem., 2011, 31, 1963–1976 Search PubMed; (g) A. Ibrar, I. Khan, N. Abbas, U. Farooq and A. Khan, RSC Adv., 2016, 6, 93016–93047 RSC; (h) X. Zheng, W. Liu and D. Zhang, Molecules, 2020, 25, 1594 CrossRef CAS PubMed.
- D. Basavaiah and R. T. Naganaboina, New J. Chem., 2018, 42, 14036–14066 RSC.
- (a) D. Basavaiah and G. Veeraraghavaiah, Chem. Soc. Rev., 2012, 41, 68–78 RSC; (b) D. Basavaiah, B. S. Reddy and S. S. Badsara, Chem. Rev., 2010, 110, 5447–5674 CrossRef CAS PubMed; (c) D. Basavaiah, A. J. Rao and T. Satyanarayana, Chem. Rev., 2003, 103, 811–891 CrossRef CAS PubMed.
- (a) Y. Wei and M. Shi, Chem. Rev., 2013, 113, 6659–6690 CrossRef CAS PubMed; (b) V. Declerck, J. Martinez and F. Lamaty, Chem. Rev., 2009, 109, 1–48 CrossRef CAS PubMed.
- The formation of n-propylbenzamide as a side product might be due to the formation of n-propyl cation which on reaction with benzonitrile provides the corresponding benzamide via hydrolysis. In fact we have carried out the reaction between n-propyl acetate and benzonitrile in the presence of TMSOTf and pleased to see the formation of N-n-propylbenzamide.
- Salts of 1a, 1e and pyridine-4-carboxaldehyde are prepared by treatment with one equivalent of TMSOTf in n-propyl acetate at room temperature (5–10 minutes). 13C NMR spectra of these salts were recorded in a mixture of CDCl3 and CH3CN (2:1). CH3CN is added for solubility. Accordingly to have uniformity we have recorded 13C NMR spectra of 1a, 1e, pyridine-4-carboxaldehyde, benzaldehyde and benzaldehyde-TMSOTf complex in the same solvent system.
- H. Meng, Y. Zi, X.-P. Xu and S.-J. Ji, Tetrahedron, 2015, 71, 3819–3826 CrossRef CAS.
- A. Khalafi-Nezhad, M. Shekouhy, H. Sharghi, J. Aboonajmi and A. Zare, RSC Adv., 2016, 6, 67281–67289 RSC.
- This was suggested by one of the referees and we thank the referee for this suggestion.
- (a) M. Hatanaka, K. Takahashi, S. Nakamura and T. Mashino, Bioorg. Med. Chem., 2005, 13, 6763–6770 CrossRef CAS PubMed; (b) L.-X. Cheng, X. L. Jin, Q.-F. Teng, J. Chang, X.-J. Yao, F. Dai, Y.-P. Qian, J.-J. Tang, X.-Z. Li and B. Zhou, Org. Biomol. Chem., 2010, 8, 1058–1063 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC [2108509–2108511, 2110001–2110003]. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc00046f |
| This journal is © The Royal Society of Chemistry 2022 |