
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbon dioxide reduction by an Al–O–P frustrated Lewis pair†
Lucas
Wickemeyer
 a,
Niklas
Aders
a,
Andreas
Mix
a,
Beate
Neumann
a,
Hans-Georg
Stammler
a,
Jorge J.
Cabrera-Trujillo
b,
Israel
Fernández
b and
Norbert W.
Mitzel
*a
a,
Niklas
Aders
a,
Andreas
Mix
a,
Beate
Neumann
a,
Hans-Georg
Stammler
a,
Jorge J.
Cabrera-Trujillo
b,
Israel
Fernández
b and
Norbert W.
Mitzel
*a
aLehrstuhl für Anorganische Chemie und Strukturchemie, Centrum für Molekulare Materialen CM2, Fakultät für Chemie, Universität Bielefeld, Universitätsstraße 25, Bielefeld 33615, Germany. E-mail: mitzel@uni-bielefeld.de
bDepartamento de Química Orgánica I, Centro de Innovación en Química Avanzada (ORFEO-CINQA), Facultad de Ciencias Químicas, Universidad Complutense de Madrid, Madrid 28040, Spain
First published on 2nd June 2022
Abstract
The reaction of tBu2P(O)H with Bis2AlH (Bis = CH(SiMe3)2) afforded the adduct tBu2P(H)–O–Al(H)Bis2 (3). It slowly releases H2 to form the first oxygen-bridged geminal Al/P frustrated Lewis pair tBu2P–O–AlBis2. It is capable of reversibly binding molecular hydrogen to afford 3, shown by NMR and H/D scrambling experiments, and forms a 1,2-adduct with CO2. Importantly, the H2 adduct 3 reduces CO2 in a stoichiometric reaction leading to the formic acid adduct tBu2P(H)–O–Al(CO2H)Bis2. The formation of the different species was explored by density functional theory calculations which provide support for the experimental results. All products were characterized by NMR spectroscopy as well as X-ray diffraction experiments and elemental analyses.
Introduction
The persisting high level of interest in small molecule activation with frustrated Lewis pairs (FLPs) has led to an enormous variety of different combinations of acid and base functions, the chemistry and applications of which have been investigated in detail.1 Most typically, boranes with electronegative C6F5 substituents are used as Lewis acids.Among the most intensely studied reactions of FLP are the activation of molecular hydrogen and carbon dioxide or a combination of both, that is a reduction of carbon dioxide. This was achieved with different systems containing boron compounds as Lewis acids.2–5 One early example for this is the system TMP/B(C6F5)3 (TMP = 2,2,6,6-tetramethylpiperidine), which cleaves molecular hydrogen to form [TMP–H]+[H–B(C6F5)3]−.6 This ammonium borate complex reduces CO2 to the formate stage when used in equimolar amounts and to methanolate when used in excess.1c
Despite its intrinsically higher Lewis acidity7 the higher homologue aluminum was significantly less frequently used. FLP systems containing aluminum demonstrated their great potential through the large variety of reaction types,8 including C–H activation,9,10 hydrogen activation,11 and carbon dioxide reduction12 or fixation,10,13 as well as their use as catalysts for polymerization reactions.14 A convenient method to prepare geminal Al/P FLPs is the hydroalumination of alkynylphosphines. The obtained products tend to quench themselves by dimerization or the formation of adducts with the alane reagent, even though it was shown that dimeric species are also able to form adducts with CO2.15
Geminal linker motifs other than carbon-based ones like alkylidene10,16 or methylene17 are rarely found for neutral group 13/P FLP. Köster et al. described the formation of different P–O–B species from reactions of diphenylphosphine oxide with trialkylboranes or dialkylhydridoboranes as early as 1987. However, they did not explicitly investigate adduct formations in the sense of FLP chemistry but only reported adducts with an additional hydridodialkylborane being coordinated to the phosphorus atom.18 Only very recently were a few examples of neutral oxygen-bridged P–O–B systems presented, which show typical reactivity towards CO2 and molecular hydrogen. However, these reactions require untypically harsh conditions or only reversibly form adducts despite the strongly electronegative substituents at boron.19,20 Other reactivities of oxygen-bridged FLP include the reversible binding of SO2 by a P/B system derived from phosphinoboranes and the activation of B–H and O–H bonds by an N/B FLP which is formed in the reaction of TEMPO with HB(C6F5)2.21
Results and discussion
Synthesis and solid-state structure
We now synthesized the first geminal oxygen-bridged Al/P FLP using the sterically extremely demanding hydridoalane Bis2AlH (Bis = CH(SiMe3)2) (1) and tBu2P(O)H (2). Initially they form the phosphane–alane adduct tBu2P(H)–O–Al(H)Bis2 (3). This compound then slowly releases H2 to give the geminal FLP tBu2P–O–AlBis24 in quantitative yield (Scheme 1).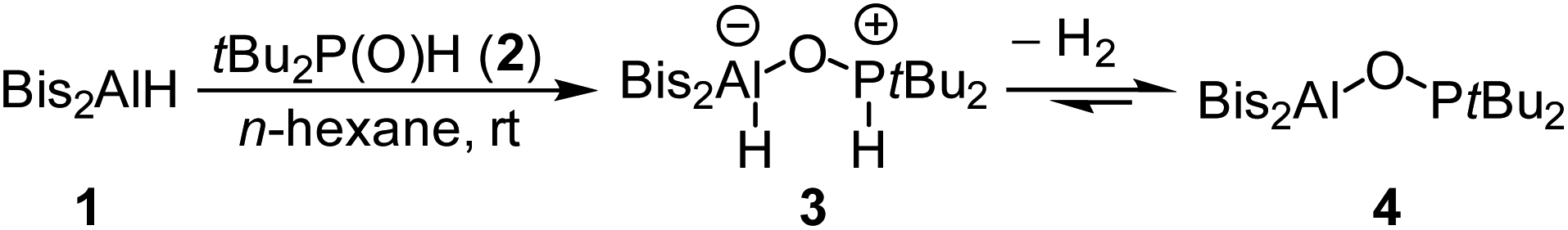 | ||
| Scheme 1 Synthesis of FLP 4 and reversible formation of the H2 adduct 3. | ||
The NMR spectra show the formation of compound 3 to be completed immediately after mixing the substances. A doublet at 5.76 ppm in the 1H NMR spectrum shows a characteristic P–H coupling constant of 457 Hz. The 31P{1H} NMR spectrum contains a signal at 72.6 ppm. The conversion of 3 into 4 under release of hydrogen is rather slow. It was monitored by NMR in a closed tube and is not completed even after two days (conversion of about 92%). Complete conversion to 4 is achieved by removal of all volatiles in vacuo. The 31P{1H} NMR spectrum of 4 shows a singlet at 142.8 ppm; this corresponds well to the comparable P–O–B system tBu2P–O–Bcat (159.3 ppm).20 Resonances of the methine protons of the CH(SiMe3)2 groups are very sensitive to the coordination number at aluminum; the signal for 4 at −0.38 ppm is in a typical range for tri-coordinated aluminum.22
The determination of the solid-state structure by single-crystal X-ray diffraction confirms the monomeric structure of 4 (Fig. 1). The molecular structure exhibits a large Al(1)–O(1)–P(1) angle of 138.1(1)° and an Al(1)⋯P(1) distance of 3.122(1) Å. The aluminum atom features a slightly distorted trigonal-planar coordination with O(1)–Al(1)–C(1/2) angles of 117.6(1)°/127.7(1)° and a C(1)–Al(1)–C(2) angle of 114.6(1)°. The P(1)–O(1) bond (1.635(1) Å) is clearly longer than the P–O bond in tBu2P(O)H (1.482(2) Å).23
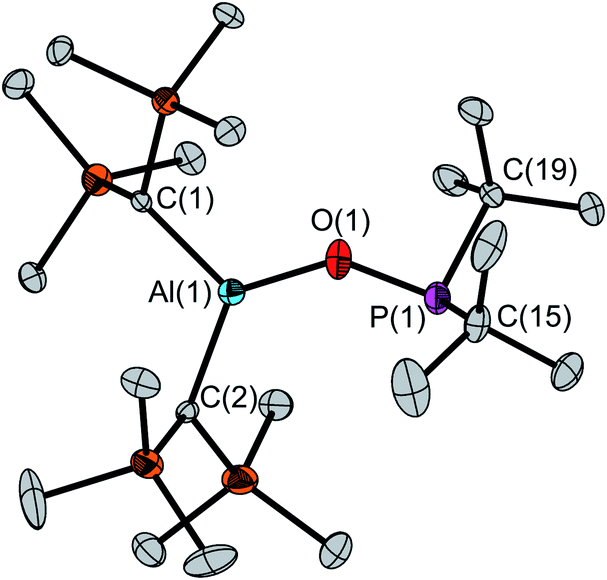 | ||
| Fig. 1 Molecular structure of 4 in the solid state. Hydrogen atoms and a disordered molecule (occupancy 7%) are omitted for clarity. Ellipsoids are set at 50% probability. Selected distances [Å] and angles [°]: P(1)–O(1) 1.635(1), P(1)–C(15) 1.890(4), P(1)–C(19) 1.874(3), Al(1)–O(1) 1.708(1), Al(1)–C(1) 1.939(3), Al(1)–C(2) 1.954(2), P(1)⋯Al(1) 3.122(1); O(1)–P(1)–C(15) 101.4(1), O(1)–P(1)–C(19) 99.2(1), C(1)–P(1)–C(15) 109.4(2), O(1)–Al(1)–C(1) 117.6(1), O(1)–Al(1)–C(2) 127.7(1), C(1)–Al(1)–C(2) 114.6(1), P(1)–O(1)–Al(1) 138.1(1). | ||
Reactivity towards hydrogen and carbon dioxide
Strikingly, H/D-scrambling experiments with H2/D2 mixtures at one atmosphere of pressure showed a reversible binding of molecular hydrogen to 4. However, this reaction is rather slow under the given conditions. At first, the formation of small amounts of H2 and D2 adducts of 4 was observed after five days. Another week later, the characteristic triplet for HD was observed in the 1H NMR spectrum at 4.57 ppm with a 1JD,H coupling constant of 42.6 Hz. Density Functional Theory (DFT) calculations (PCM(hexane)-M06-2X/def2-TZVPP//PCM(hexane)-M06-2X/def2-SVP) confirmed for this binding of hydrogen a free energy difference between 3 and 4 + H2 of only 1.6 kcal mol−1 (at 298 K), with 3 being the more energetically favorable species (Fig. 2). Considering a possible error in this number of a few kcal mol−1, it is consistent with the experimental observation of a slow and incomplete formation of 4 in the presence of hydrogen. This equilibrium indicates a reversible nature of this process and finds its confirmation by the computed interconversion barrier of ΔG‡ = 24.3 kcal mol−1 (from 3).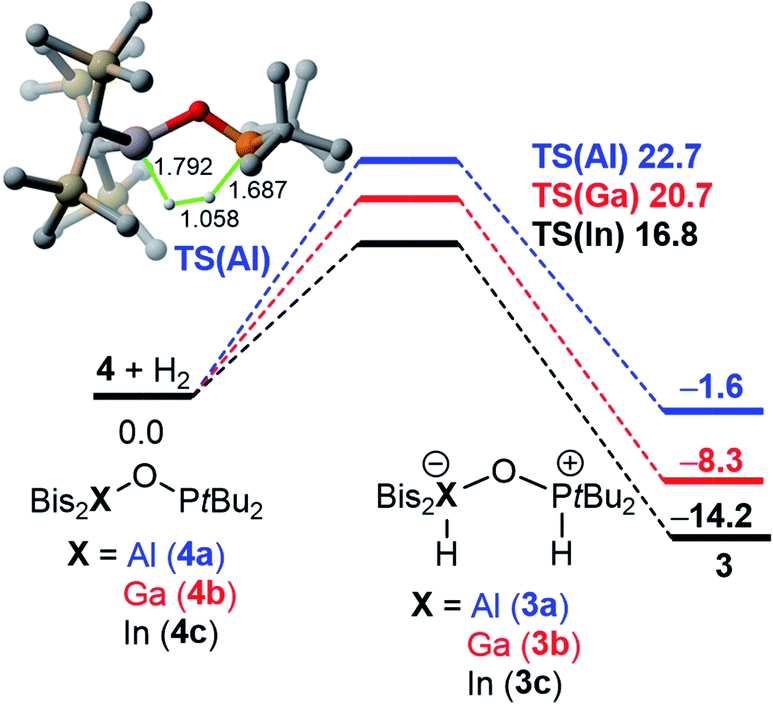 | ||
| Fig. 2 Computed reaction profile for the interconversion of 3 into 4 + H2. Relative free energies (ΔG, at 298 K) and bond lengths (hydrogen atoms are omitted) are given in kcal mol−1 and Å, respectively. All data have been computed at the PCM(hexane)-M06-2X/def2-TZVPP//PCM(hexane)-M06-2X/def2-SVP level. | ||
Interestingly, compared to the analogous systems containing the heavier elements of group 13, gallium and indium, the hydrogen adducts 3_b/c are thermodynamically favored and the barriers for the reactions 4_b/c + H2 → 3_b/c are lower for the heavier FLPs.
The reaction of 4 with carbon dioxide afforded adduct 5 in nearly quantitative yield (Scheme 2). Unlike in previously reported P–O–B systems, the binding is stable at room temperature and an equilibrium is not formed.19,20 Compared to the free FLP 4, the 31P NMR signal at 64.8 ppm is clearly high field shifted, while the characteristic 13C{1H} NMR doublet for the bound CO2 appears at 166.0 ppm with a 1JP,C coupling constant of 98.6 Hz. According to our DFT calculations, the formation of 5 occurs in a concerted manner through the asynchronous transition state depicted in Scheme 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 24 and with an activation barrier of only 5.5 kcal mol−1 in a highly exergonic (ΔGR = −22.9 kcal mol−1) transformation.
24 and with an activation barrier of only 5.5 kcal mol−1 in a highly exergonic (ΔGR = −22.9 kcal mol−1) transformation.
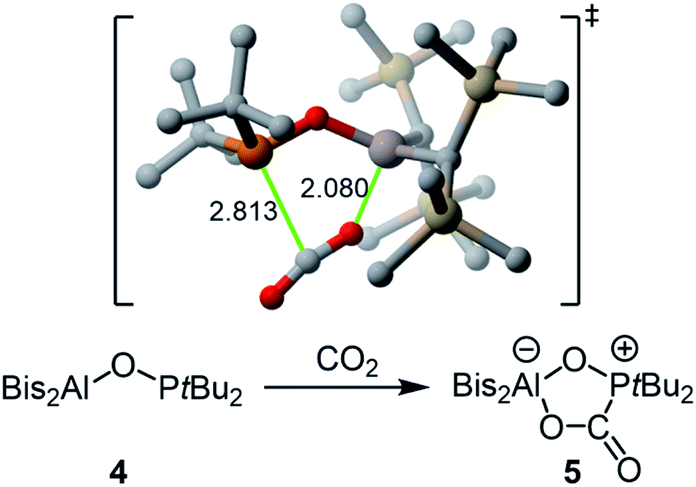 | ||
| Scheme 2 Reaction of FLP 4 with carbon dioxide to form adduct 5 and above the computed transition state (bond lengths are given in Å; hydrogen atoms are omitted for clarity). | ||
The molecular structure of 5 in the crystal shows the formation of a typical planar five-membered ring with an exocyclic C–O moiety (Fig. 3). Its P–O bond at 1.540(2) Å is significantly shorter than in the free FLP 4 (1.635(1) Å) while the Al–O bond is longer at 1.855(2) Å (4: 1.708(1) Å). With 117.1(1)° the P(1)–O(1)–Al(1) angle is noticeably narrower than in 4 (138.1(1)°). The sum of angles at C(1) of 360.0(2)° indicates trigonal planar coordination.
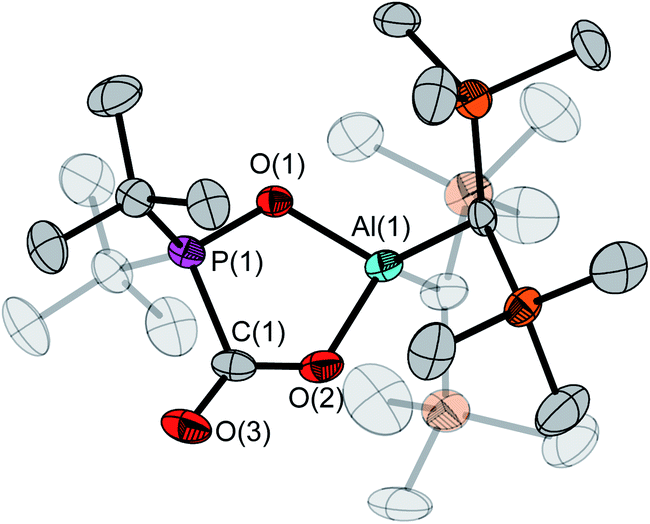 | ||
| Fig. 3 Molecular structure of the CO2 adduct 5 in the solid state. Ellipsoids are set at 30% probability. Hydrogen atoms as well as the minor occupied disordered part of the –CH(Si(CH3)3)2 groups are omitted for clarity. Selected bond lengths, distances [Å] and angles [°]: P(1)–O(1) 1.540(2), P(1)–C(1) 1.868(2), Al(1)–O(1) 1.855(2), Al(1)–O(2) 1.848(2), O(2)–C(1) 1.278(3), O(3)–C(1) 1.215(3), P(1)⋯Al(1) 2.901(1); P(1)–O(1)–Al(1) 117.1(1), O(1)–P(1)–C(1) 101.3(1), C(1)–O(2)–Al(1) 122.0(1), O(2)–C(1)–P(1) 109.6(2), O(3)–C(1)–P(1) 122.8(2), O(3)–C(1)–O(2) 127.6(2). | ||
CO2 reduction
The formation of the CO2 adduct 5 in combination with the reversible binding of molecular hydrogen raised the question of how H2 adduct 3 would react with CO2. Since 3 releases hydrogen slowly and is only formed in small amounts when 4 is treated with hydrogen, handling is only possible for a very limited time. We therefore mixed alane 1 and phosphine 2 to generate 3 which could be reacted directly before the release of hydrogen. Upon complete dissolution of the solids, this mixture was cooled to −196 °C, and the reaction flask was degassed and backfilled with CO2. By this, we achieved stoichiometric reduction of carbon dioxide to afford the formic acid adduct 6 (Scheme 3). The characteristic 1H NMR signal for the formate moiety appears at 8.45 ppm, while the 31P{1H} NMR shift of 72.2 ppm is similar to that of 3. This unexpected reactivity makes FLP 4 a versatile tool with highly useful abilities. Being able to (reversibly) bind both CO2 and H2 individually on one hand and at the same time unite both reactivities to reduce carbon dioxide with previously activated molecular hydrogen almost instantly at ambient temperature and atmospheric pressure is a unique combination in the field of FLP chemistry. As mentioned in the introduction, this reactivity has been observed for boron containing systems before. However, the reduction was only achieved at higher temperatures or elevated CO2 pressure.1c,2–5 However, the nature of the individual reactions of 3 with H2 (equilibrium on the side of free H2) and CO2 (formation of a stable adduct) make the system unlikely to be used as a catalyst for the reduction of CO2.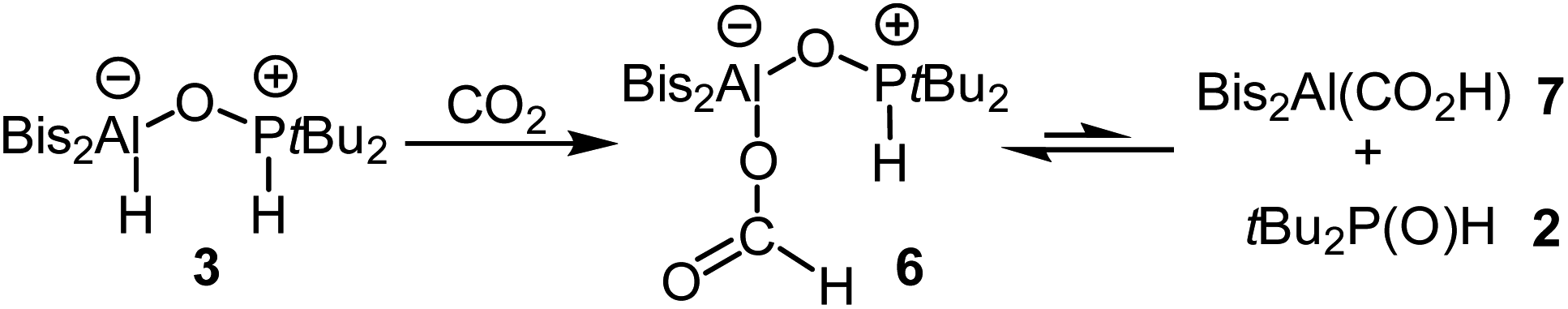 | ||
| Scheme 3 Reduction of CO2 with 3 to form the formic acid adduct 6 and its equilibrium with alane 7 and tBu2P(O)H (2). | ||
Compound 6 forms an equilibrium in solution: small amounts of phosphine 2 and the alane Bis2Al(CO2H) (7) are observed by NMR spectroscopy (Scheme 3). Characteristic 1H resonances are the doublet at 5.84 ppm for 2 and the signal of the formate group of 7 at 7.35 ppm.
The asymmetric unit of the unit cell of crystalline 6 contains two independent molecules, whose structural parameters differ mainly in the O–Al–O–P torsion angles (107.6(3)°/41.6(12)°); the Al–O–P angles (168.8(1)°/154.3(1)°) are noticeably wider than in 4. Moreover, the bond of aluminum to the bridging oxygen atom is significantly longer (1.834(1)/1.814(1) vs. 1.708(1) Å) while the corresponding P–O bond is shorter (1.529(1)/1.519(1) Å vs. 1.635(1) Å). The aluminum atom exhibits a slightly distorted tetrahedral geometry, with a 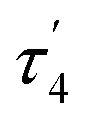 parameter25 of 0.93 (Fig. 4).
parameter25 of 0.93 (Fig. 4).
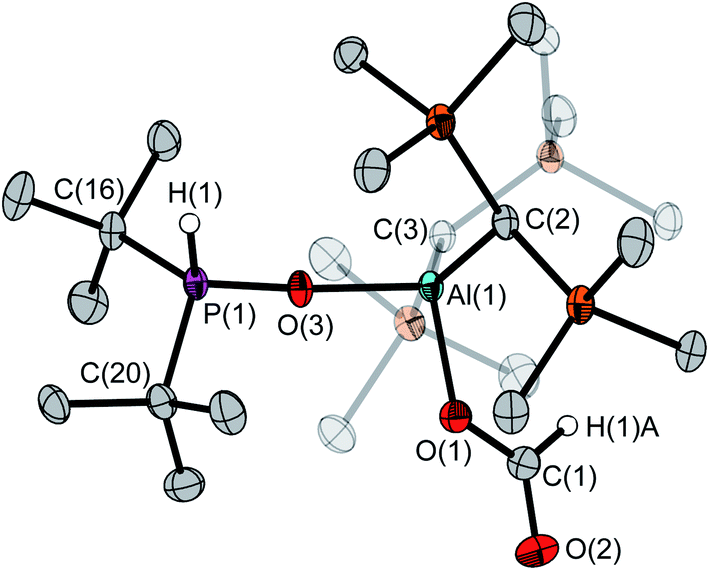 | ||
| Fig. 4 Molecular structure of 6 in the crystalline state. Only one of two independent molecules in the asymmetric unit is shown. H(1) was refined isotropically while H(1)A was taken into account using a riding model; all other hydrogen atoms were omitted for clarity. Ellipsoids are set at 50% probability. Selected bond lengths [Å] and angles [°]: P(1)–O(3) 1.519(1); P(1)–H(1) 1.278(1), P(1)–C(16) 1.838(1), P(1)–C(20) 1.828(1), Al(1)–O(1) 1.810(1), Al(1)–O(3) 1.814(1), O(1)–C(1) 1.295(1), O(2)–C(1) 1.210(2); O(3)–P(1)–C(16) 111.6(1), O(3)–P(1)–C(20) 109.0(1), C(20)–P(1)–C(16) 117.0(1), P(1)–O(3)–Al(1) 168.8(1), O(1)–Al(1)–O(3) 99.3(1), O(1)–Al(1)–C(2) 108.6(1), O(1)–Al(1)–C(3) 110.2(1), C(1)–O(1)–Al(1) 127.4(1), O(2)–C(1)–O(1) 125.8(1). | ||
We observed that the formic acid adduct 6 is in equilibrium with small amounts of phosphine oxide 2 and alane 7 in solution. This raised the question of whether CO2 could also be directly reduced with Bis2AlH and then treated with tBu2P(O)H to give 6. When alane 1 was reacted directly with CO2, the 1H NMR spectrum of the dissolved solid obtained (7d, identified by XRD, see below) contained two sets of signals, those of the monomer 7 and dimer 7d. Signals for the formate protons are found at 8.84 (7d) and 7.33 ppm (7). After several days, practically only one species (7.33 ppm, 7) remained observable. Inconsistent with this experimental observations, the DFT calculations predict the dimerization of 7 to 7d to be rather exergonic (ΔGR = −19.8 kcal mol−1).
The identification of these species is based on diffusion ordered spectroscopy (DOSY). Using the Stokes–Einstein equation, the diffusion coefficients of 4.65 × 10−10 m2 s−1 (8.84 ppm, 7d) and 5.70 × 10−10 m2 s−1 (7.33 ppm, 7) were converted into hydrodynamic radii and volumes: 7.21 Å/1570 Å3 for 7d and 5.87 Å/850 Å3 for 7. Compared to the empirically calculated values26 of 6.29 Å/1043 Å3 and 5.01 Å/527 Å3, the values in solution are increased due to a solvation shell. Still, the volume of the species at 8.84 ppm is about twice the size of the one at 7.33 ppm, which confirms that monomeric alane 7 forms from an initially dimeric species 7d upon dissolution (Scheme 4).
 | ||
| Scheme 4 Reduction of CO2 by Bis2AlH, equilibria between monomeric and dimeric alanes and formation of formic acid adduct 6. | ||
Solely the dimeric species was found in the solid state, as determined by X-ray diffraction studies of a single crystal (Fig. 5). It has an almost planar eight-membered ring with an inversion center. Each aluminum atom is coordinated by two oxygen atoms from different formate groups in a distorted tetrahedral manner 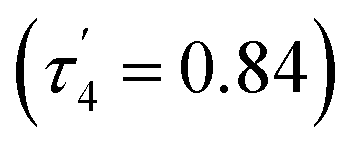 . The Al–O distances differ only slightly at 1.848(1) and 1.837(1) Å, and so do the C–O distances (1.255(2), 1.239(2) Å). In contrast, two very different Al–O–C angles are found at 138.4(1)° and 167.9(1)°.
. The Al–O distances differ only slightly at 1.848(1) and 1.837(1) Å, and so do the C–O distances (1.255(2), 1.239(2) Å). In contrast, two very different Al–O–C angles are found at 138.4(1)° and 167.9(1)°.
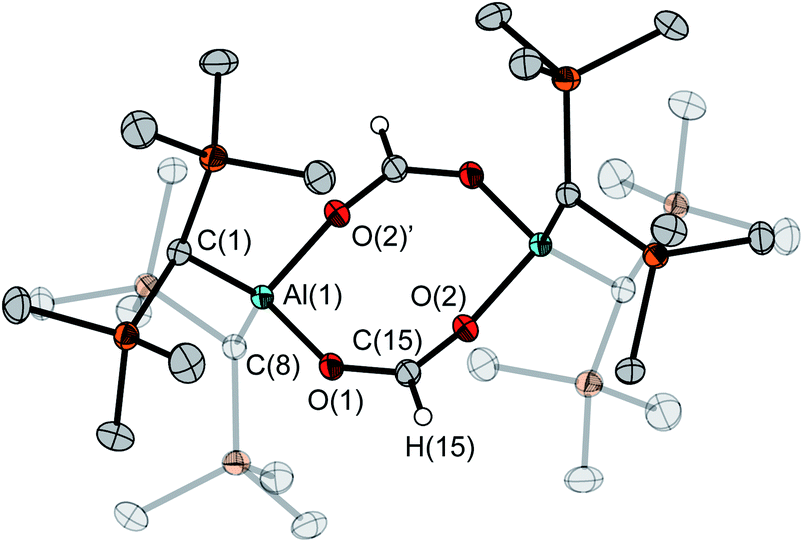 | ||
| Fig. 5 Molecular structure of 7d in the crystal. Ellipsoids are set at 50% probability; hydrogen atoms except for the ones of the formate units are omitted for clarity. Selected bond lengths [Å] and angles [°]: Al(1)–O(1) 1.848(1), Al(1)–O(2)′ 1.837(1), O(1)–C(15) 1.255(2), O(2)–C(15) 1.239(2); O(2)′–Al(1)–O(1) 99.9(1), C(15)–O(1)–Al(1) 138.3(1), C(15)–O(2)–Al(1)′ 167.9(1). | ||
When adding phosphine oxide 2 to a solution of Bis2Al(CO2H) (7), the formic acid adduct 6 is formed. However, as described above, the reaction remains incomplete, and small amounts of phosphine oxide 2 and alane 7 are detectable in solution, which further confirms the proposed equilibrium described above. DFT calculations were also carried out to gain more insight into the formation of 6 in solution (Fig. 6). We found that 1 and 2 form adduct 3 in a barrier-free and highly exergonic reaction (ΔGR = −25.0 kcal mol−1). Subsequent reaction of 3 with CO2 leads to the highly exergonic (ΔGR = −27.0 kcal mol−1) formation of the formic acid adduct 6via the transition state TS3–6, a saddle point mainly associated with the nucleophilic addition of the aluminum-hydride to the electrophilic carbon atom of CO2. Both the computed free energy of reaction and activation barrier (ΔG‡ = 19.3 kcal mol−1) are compatible with a process occurring at room temperature.
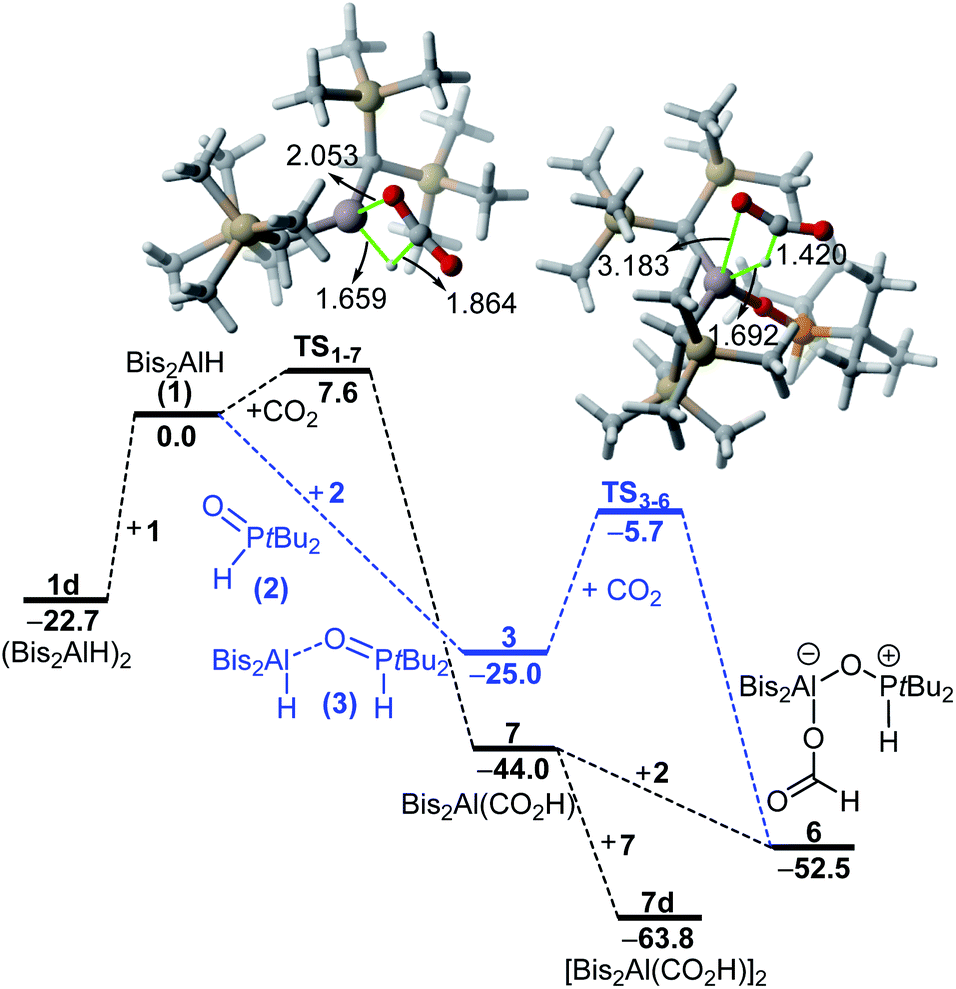 | ||
| Fig. 6 Computed reaction profiles for the formation of the formic acid adduct 6. Relative free energies (at 298 K) are given in kcal mol−1, bond lengths in Å. All data have been computed at the PCM(hexane)-M06-2X/def2-TZVPP//PCM(hexane)-M06-2X/def2-SVP level. | ||
The alternative process involving the initial reaction of alane 1 with CO2 was also explored. This transformation leads to the exergonic (ΔGR = −44.0 kcal mol−1) formation of 7 with a low activation barrier of only 7.6 kcal mol−1 through the transition state TS1–7, a saddle point associated again with the concomitant formation of the new C–H and Al–O bonds. Subsequent barrier-free addition of phosphine oxide 2 leads to the formation of 6 again in an exergonic transformation (ΔGR = −8.5 kcal mol−1).
The computed free energies of reaction and relatively low barriers therefore confirm the feasibility of both alternative processes, the formic acid adduct 6 being the most stable species in both computed profiles. Our calculations also predict that the possible release of formic acid from 6 is highly endergonic (ΔGR = +29.2 kcal mol−1) and thus highly unlikely (see Fig. S19 in the ESI†).
Conclusions
In essence, we have described herein the preparation, full characterization and reactivity of a first geminal oxygen-bridged Al/P FLP tBu2P–O–AlBis2. The reactivity of this system includes the reversible binding of molecular hydrogen and adduct formation with carbon dioxide. Reduction of carbon dioxide was achieved with the FLP–H2 addition product to the formate stage. Its reactivity is unique in the sense of being able to both, to reversibly split molecular hydrogen and to reduce carbon dioxide to the formate stage, with the H2 addition product rapidly at ambient temperature and atmospheric pressure. Our findings encourage us to further explore and investigate this reactivity. Theoretical results for analogous systems involving the heavier elements gallium and indium indicate the possibility of a facilitated uptake of hydrogen by a higher homologue of FLP 4. We also expect an easier release of formic acid from the higher homologues of 6. With this promising outlook we will focus on synthesizing these systems and confirming theoretical predictions with the aim to eventually achieve a catalytic reduction of carbon dioxide.Experimental section
General considerations
All reactions and manipulations with air- and moisture-sensitive compounds were carried out under conventional Schlenk techniques using nitrogen as inert gas or in a glove box using argon as inert gas. Volatile compounds were handled using a vacuum line. n-Hexane and [D6]benzene were dried over a Na/K alloy, distilled and degassed prior to use. CDCl3 was dried over 3 Å molecular sieves, distilled and degassed prior to use. CO2 (99.5%, Linde) was used without further purification. Bis2AlH (Bis = CH(SiMe3)2) was prepared according to a literature procedure.22 NMR spectra were recorded using a Bruker Avance III 300, Avance III 500 or Avance III 500 HD spectrometer at ambient temperature. Chemical shifts were referenced to the residual proton or carbon signal of the solvent (C6D6: 1H: 7.16 ppm, 13C: 128.1 ppm; CDCl3: 1H: 7.26 ppm) or externally (29Si: SiMe4, 31P: 85% H3PO4 in H2O). Elemental analyses were carried out by co-workers of the University of Bielefeld using a EURO EA Elemental Analyzer.Synthetic procedures
Analytical data. 1H NMR (500 MHz, C6D6): δ [ppm] = 5.85 (d, 1JP,H = 419.3 Hz, 1H, P–H), 1.00 (d, 3JP,H = 14.5 Hz, 18H, tBu). 31P{1H} NMR (202 MHz, CDCl3): δ [ppm] = 61.6 Hz. 1H NMR (500 MHz, CDCl3): δ [ppm] = 6.06 (d, 1JP,H = 428.6 Hz, 1H, P–H), 1.26 (d, 3JP,H = 15.1 Hz, 18H, tBu). 31P{1H} NMR (202 MHz, CDCl3): δ [ppm] = 66.6 Hz.
Analytical data. 1H NMR (500 MHz, C6D6): δ [ppm] = 5.76 (d, 1JP,H = 457.0 Hz, 1H, P–H), 4.58 (br s, 1H, Al–H), 0.83 (d, 3JP,H = 16.5 Hz, 18H, tBu), 0.50 (s, 18H, CH(Si(CH3)3)2), 0.43 (s, 18H, CH(Si(CH3)3)2), −1.04 (s, 2H, CH(Si(CH3)3)2). 31P{1H} NMR (202 MHz, C6D6): δ [ppm] = 72.6 Hz.
Analytical data. 1H NMR (300 MHz, C6D6): δ [ppm] = 1.20 (d, 3JP,H = 10.7 Hz, 18H, C(CH3)3), 0.31 (d, 36H, Si(CH3)3), −0.38 (s, 2H, CH(SiMe3)2). 13C{1H} NMR (75 MHz, C6D6): δ [ppm] = 35.0 (d, 1JP,C = 31.7 Hz, C(CH3)3), 28.2 (d, 2JP,C = 16.0 Hz, C(CH3)3), 8.7 (s, CH(Si(CH3)3)2), 4.6 (s, CH(Si(CH3)3)2). 29Si{1H} NMR (60 MHz, C6D6): δ [ppm] = −2.7 (s). 31P{1H} NMR (121 MHz, C6D6): δ [ppm] = 142.6 (s). Elemental analysis calcd (%) for C22H56AlOPSi4 (Mr = 506.98): C 52.12, H 11.13; found: C 52.81, H 11.04.
Analytical data. 1H NMR (500 MHz, C6D6): δ [ppm] = 0.99 (d, 3JP,H = 15.7 Hz, 18H, C(CH3)3), 0.39 (s, 18H, Si(CH3)3), 0.38 (s, 18H, Si(CH3)3), −0.79 (br s, 1H, CH(SiMe3)2), −1.09 (br s, 1H, CH(SiMe3)2). 13C{1H} NMR (126 MHz, C6D6): δ [ppm] = 166.0 (d, 1JP,C = 98.6 Hz, CO2), 34.8 (d, 1JP,C = 42.2 Hz, C(CH3)3), 26.3 (s, C(CH3)3), 5.3 (s, CH(Si(CH3)3)2), 5.1 (s, CH(Si(CH3)3)2), signals of the methine proton were not observed due to high dynamics. 29Si{1H} NMR (99 MHz, C6D6): δ [ppm] = −1.8 (br m). 31P{1H} NMR (202 MHz, C6D6): δ [ppm] = 64.8 (s). Elemental analysis calcd (%) for C23H56AlO3PSi4 (Mr = 550.99): C 50.14, H 10.24; found: C 50.20, H 10.03.
Analytical data. 1H NMR (500 MHz, C6D6): δ [ppm] = 8.45 (s, 1H, CO2H), 6.50 (d, 1JP,H = 485.3 Hz, 1H, P–H), 0.88 (d, 3JP,H = 16.6 Hz, 18H, P(C(CH3)3)2), 0.43 (s, 18H, CH(Si(CH3)3)2), 0.39 (s, 18H, CH(Si(CH3)3)2), −1.00 (s, 2H, CH(Si(CH3)3)2). 13C{1H} NMR (126 MHz, C6D6): δ [ppm] = 162.6 (s, CO2H), 33.7 (d, 1JP,C = 56.1 Hz, C(CH3)3), 26.0 (s, C(CH3)3), 5.6 (s, CH(Si(CH3)3)2), 5.3 (s, CH(Si(CH3)3)2), 2.4 (br s, CH(Si(CH3)3)2). 29Si{1H} NMR (99 MHz, C6D6): δ [ppm] = −1.2 (s), −2.0 (s). 31P{1H} NMR (121 MHz, C6D6): δ [ppm] = 72.3 (s). Elemental analysis calcd (%) for C23H58AlO3PSi4 (Mr = 553.01): C 49.95, H 10.57; found: C 50.19, H 10.85.
Analytical data. 1H NMR (500 MHz, C6D6): δ [ppm] = 8.84 (s, 2H, CO2H of 7d), 7.33 (s, 1H, CO2H), 0.28 (s, 72H, CH(SiCH3)2 of 7d), 0.24 (s, 36H, CH(SiCH3)2), −0.93 (s, 4H, CH(SiMe3)2 of 7d), −1.06 (s, 2H, CH(SiMe3)2). 13C{1H} NMR (126 MHz, C6D6): δ [ppm] = 170.5 (s, CO2H of 7d), 166.8 (s, CO2H), 4.9 (s, CH(SiCH3)2 of 7d), 4.8 (s, CH(SiCH3)2), 1.8 (s, CH(SiCH3)2), 1.5 (s, CH(SiCH3)2 of 7d). 29Si{1H} NMR (99 MHz, C6D6): δ [ppm] = −1.6 (s), −1.7 (s, 7d). Elemental analysis calcd (%) for C15H39AlO2Si4 (Mr = 390.80): C 46.10, H 10.06; found: C 45.98, H 10.31.
Author contributions
Lucas Wickemeyer: synthesis, spectroscopy, conceptualization, visualization, writing – original draft; Niklas Aders: synthesis of HAlBis2, conceptualization; Beate Neumann and Hans-Georg Stammler: crystallography; Jorge J. Cabrera-Trujillo and Israel Fernández: quantum-chemical calculations, writing – quantum-chemical paragraphs; Norbert W. Mitzel: conceptualization, project administration, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Barbara Teichner for performing CHN analyses. The work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – grant MI 477/44-1, project number 461833739 and the Spanish Ministerio de Ciencia e Innovación/Agencia Estatal de Investigación/10.13039/501100011033 (grants PID2019-106184GB-I00 and RED2018-102387-T).Notes and references
-
(a) T. W. Hudnall, Y.-M. Kim, M. W. P. Bebbington, D. Bourissou and F. P. Gabbaï, J. Am. Chem. Soc., 2008, 130, 10890 CrossRef CAS PubMed
; (b) D. W. Stephan, Dalton Trans., 2009, 3129 RSC
- S. D. Tran, T. A. Tronic, W. Kaminsky, D. M. Heinekey and J. M. Mayer, Inorg. Chim. Acta, 2011, 369, 126 CrossRef CAS
- M.-A. Courtemanche, A. P. Pulis, É. Rochette, M.-A. Légaré, D. W. Stephan and F.-G. Fontaine, Chem. Commun., 2015, 51, 9797 RSC
- T. Zhao, X. Hu, Y. Wu and Z. Zhang, Angew. Chem., Int. Ed., 2019, 58, 722 CrossRef CAS PubMed
- T. Wang, M. Xu, A. R. Jupp, Z.-W. Qu, S. Grimme and D. W. Stephan, Angew. Chem., Int. Ed., 2021, 60, 25771 CrossRef CAS PubMed
- V. Sumerin, F. Schulz, M. Nieger, M. Leskelä, T. Repo and B. Rieger, Angew. Chem., Int. Ed., 2008, 47, 6001 CrossRef CAS PubMed
-
(a) A. Y. Timoshkin and G. Frenking, Organometallics, 2008, 27, 371 CrossRef CAS
-
M. Layh, W. Uhl, G. Bouhadir and D. Bourissou, Organoaluminum Compounds and Lewis Pairs in Patai’s Chemistry of Functional Groups, ed. Z. Rappoport, Wiley, Chichester, 2016, pp. 1–46 Search PubMed
-
(a) M. A. Dureen and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 8396 CrossRef CAS PubMed
- C. Appelt, H. Westenberg, F. Bertini, A. W. Ehlers, J. C. Slootweg, K. Lammertsma and W. Uhl, Angew. Chem., Int. Ed., 2011, 50, 3925 CrossRef CAS PubMed
-
(a) G. Ménard and D. W. Stephan, Angew. Chem., Int. Ed., 2012, 51, 8272 CrossRef PubMed
-
(a) G. Ménard and D. W. Stephan, J. Am. Chem. Soc., 2010, 132, 1796 CrossRef PubMed
-
(a) M.-A. Courtemanche, J. Larouche, M.-A. Légaré, W. Bi, L. Maron and F.-G. Fontaine, Organometallics, 2013, 32, 6804 CrossRef CAS
-
(a) D. Chakraborty and E. Y.-X. Chen, Macromolecules, 2002, 35, 13 CrossRef CAS
-
(a) W. Uhl, C. Appelt, J. Backs, H. Westenberg, A. Wollschläger and J. Tannert, Organometallics, 2014, 33, 1212 CrossRef CAS
-
(a) C. Rosorius, G. Kehr, R. Fröhlich, S. Grimme and G. Erker, Organometallics, 2011, 30, 4211 CrossRef CAS
-
(a) A. Schnurr, H. Vitze, M. Bolte, H.-W. Lerner and M. Wagner, Organometallics, 2010, 29, 6012 CrossRef CAS
- R. Köster, Y.-H. Tsay and L. Synoradzki, Chem. Ber., 1987, 120, 1117 CrossRef
- Y. Wang, Z. H. Li and H. Wang, RSC Adv., 2018, 8, 26271 RSC
- D. Zhu, Z.-W. Qu and D. W. Stephan, Dalton Trans., 2020, 49, 901 RSC
-
(a) N. Szynkiewicz, J. Chojnacki and R. Grubba, Inorg. Chem., 2020, 59, 6332 CrossRef CAS PubMed
- W. Uhl, L. Cuypers, R. Graupner, J. Molter, A. Vester and B. Neumüller, Z. Anorg. Allg. Chem., 2001, 627, 607 CrossRef CAS
- F. Dornhaus, H.-W. Lerner and M. Bolte, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2005, 61, o657–o658 CrossRef CAS
- This resembles related transition states associated with the addition of CO2 to geminal FLPs. See, for instance, ref. 17d and J. J. Cabrera-Trujillo and I. Fernández, Chem. Commun., 2019, 55, 675 RSC
- A. Okuniewski, D. Rosiak, J. Chojnacki and B. Becker, Polyhedron, 2015, 90, 47 CrossRef CAS
- Y. H. Zhao, M. H. Abraham and A. M. Zissimos, J. Org. Chem., 2003, 68, 7368 CrossRef CAS PubMed
- K. A. Smoll, W. Kaminsky and K. I. Goldberg, Organometallics, 2017, 36, 1213 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, crystallographic data and computational details. CCDC 2151906–2151909. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc01870e |
| This journal is © The Royal Society of Chemistry 2022 |