
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium-catalyzed chemoselective direct α-arylation of carbonyl compounds with chloroaryl triflates at the C–Cl site†
Zicong
Chen
a,
Changxue
Gu
a,
On Ying
Yuen
a and
Chau Ming
So
 *ab
*ab
aState Key Laboratory of Chemical Biology and Drug Discovery, Department of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Kowloon, Hong Kong SAR, China. E-mail: chau.ming.so@polyu.edu.hk
bThe Hong Kong Polytechnic University Shenzhen Research Institute, Shenzhen 518057, Guangdong, China
First published on 16th March 2022
Abstract
This study described palladium-catalyzed chemoselective direct α-arylation of carbonyl compounds with chloroaryl triflates in the Ar–Cl bond. The Pd/SelectPhos system showed excellent chemoselectivity toward the Ar–Cl bond in the presence of the Ar–OTf bond with a broad substrate scope and excellent product yields. The electronic and steric hindrance offered by the –PR2 group of the ligand with the C2-alkyl group was found to be the key factor affecting the reactivity and chemoselectivity of the α-arylation reaction. The chemodivergent approach was also successfully employed in the synthesis of flurbiprofen and its derivatives (e.g., –OMe and –F).
Introduction
Palladium-catalyzed cross-coupling reactions are versatile tools in organic synthesis for constructing new bonds between two motifs.1 The functionalization of aryl (pseudo)halides provides practical synthetic routes for academia and industry for new chemical entities.2 However, in previous studies of cross-coupling reactions, substrates frequently bear only a single electrophile.1 Despite the fact that substrates bearing two or more reactive electrophilic sites could introduce a huge challenge to both reactivity and chemoselectivity,3 many natural products and materials with multiple electrophilic sites exist. The success in controlling chemoselectivity sequences allows iterative or sequential cross-couplings4 for the construction of more complicated molecules with optimal pathways and steps.Aryl halides and triflates are the most widely used electrophiles, and they have a generally accepted reactivity order of I > Br ≈ OTf > Cl.5 However, the reactivity order is sometimes substrate dependent and affected by the reaction conditions, which has resulted in a considerable challenge in the practical usage of these reactions. Therefore, much effort has been devoted to understanding the effects of catalysts and developing a substrate-independent chemoselective reaction following the conventional reactivity order (Scheme 1A).6
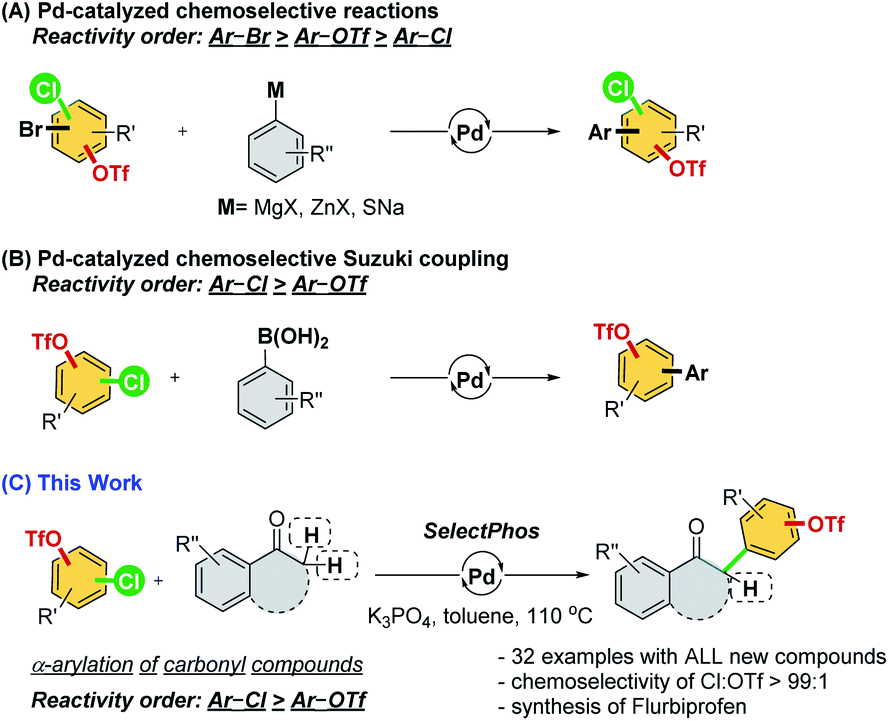 | ||
| Scheme 1 Palladium-catalyzed chemoselective reaction of aryl (pseudo)halides. | ||
As ligands are an indispensable factor in cross-coupling reactions, developing appropriate ligands that can directly manipulate the reactivity sequence (i.e., –Cl > –OTf) is an incredibly attractive strategy and would significantly broaden the choice of synthetic routes in actual applications.7 However, only a few reports demonstrating Suzuki coupling and a single example of the Stille reaction have demonstrated the possibility of inverting the conventional reactivity order in cross-coupling reactions, which is still highly difficult to achieve (Scheme 1B).7
α-Arylated carbonyl motifs are commonly found in natural products and pharmaceutically active molecules.8 The palladium-catalyzed α-arylation of ketones was initiated by Miura,9 Buchwald,10 and Hartwig,11 which constituted a milestone in this field of research.12 We envision that the palladium-catalyzed chemoselective α-arylation of carbonyl compounds with multi-(pseudo)halogenated electrophiles could offer a robust and attractive methodology for synthesizing a new series of these important compounds. Herein, we attempt to develop an α-arylation reaction of carbonyl compounds through the chemoselective cross-coupling of chloroaryl triflates (Scheme 1C).
To achieve this reaction, several issues must be tackled (Scheme 2). First, the oxidative addition step is considered critical in determining the chemoselectivity of cross-coupling reactions, which has been proposed to be affected by the ligation state of the metal center in previous studies.7d,13 However, the reductive elimination step was also reported to be the rate-determining step in direct α-arylation reactions, which are affected by the electron nature of the ligand's –PR2 group.14 In fact, the fundamental phosphine ligands, such as PCy3 and Pt-Bu3, have been commonly employed in the study of the oxidative addition step to account for –Cl/–OTf chemoselectivity.7 As such, the factors of ligand design with mechanistic insights for handling a chemoselective reaction involving the potential reductive elimination-demanding step remain unexplored. Second, the catalytic system must offer excellent chemoselectivity toward the Ar–Cl bond and leave the Ar–OTf bond intact to prevent the occurrence of multiple arylation reactions from generating non-separable reaction mixtures. Third, the catalytic system should be promoted using a mild base to prevent triflate hydrolysis. To the best of our knowledge, there have been no studies on the transition metal-catalyzed chemoselective C–Cl (over C–OTf) direct α-arylation of carbonyl compounds using chloroaryl triflates.
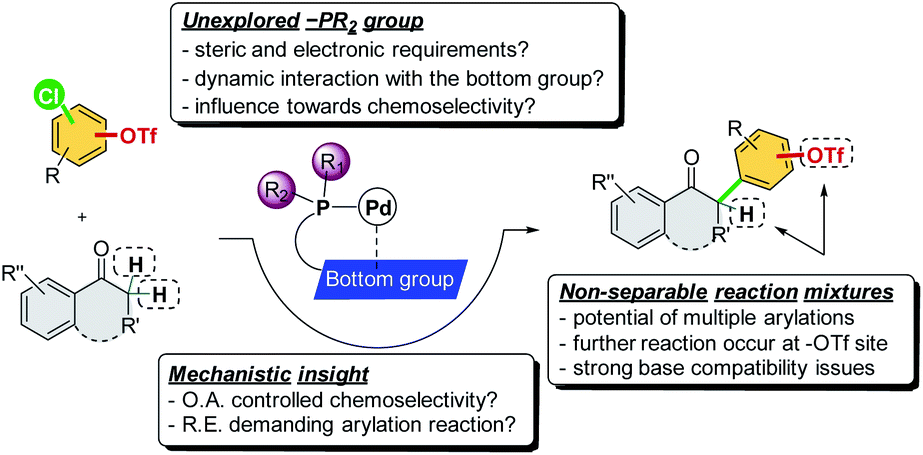 | ||
| Scheme 2 Challenges of the chemoselective direct α-arylation reaction. | ||
Results and discussion
We commenced our study by initially examining the ligand effect using oxindoles, which correspond to a class of privileged structural motifs in natural products and pharmaceutical agents.15 The cross-coupling of 3-chlorophenyl triflate 1a and 1-methylindolin-2-one 2a was selected as the reaction model to probe the feasibility of the commercially available and sophisticated ligands for chemoselective coupling reactions (Scheme 3). PCy3 (L2) and Pt-Bu3 (L3) reportedly provide good chemoselectivity for the Ar–OTf of the former and Ar–Cl bond of the latter in the Suzuki coupling of 4-chlorophenyl triflate.7a However, in this context, PCy3 (L2) yielded a mixture of 3a/3a′, and Pt-Bu3 (L3) was not effective toward this reaction. PPh3 (L1) along with Buchwald-type biphenyl ligands (L4–L7), which have Pd-arene π-coordination with its ortho-aryl group and act as the bis-coordinated ligand, prefers to react with the Ar–OTf bond and follows the conventional reactivity order of Ar–OTf > Ar–Cl to yield 3a′. The CataCxium series (L8–L11) showed activity toward the Ar–Cl bond. However, this series of ligands suffers from low activity (L8–L9) and the formation of the reaction mixture of 3a/3a′ (L10–L11). P,N-type ligand L12, which acts as a bis-coordinated ligand, has also shown chemoselectivity toward the Ar–OTf bond.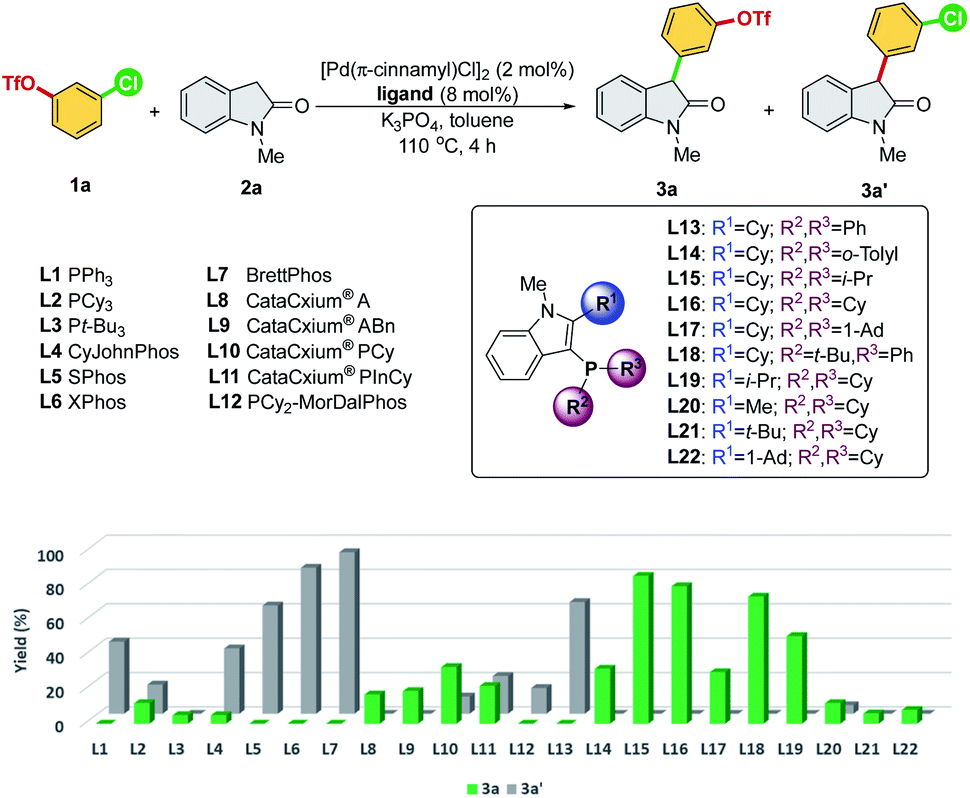 | ||
| Scheme 3 Ligand screening of chemoselectivity in α-arylation of N-methyl oxindole. Reaction conditions: 1a (0.20 mmol), 2a (0.20 mmol), K3PO4 (0.40 mmol), toluene (1.0 mL), and under N2. The yield was calibrated by GC-FID using dodecane as the internal standard. | ||
Recently, we developed a novel phosphine ligand, SelectPhos (L15), with a C2-cyclohexyl-substituted indole skeleton, which was found to be active toward the chemoselective Suzuki–Miyaura coupling reaction.7f The preagostic interaction and the steric hindrance offered by the C2-alkyl group enabled the inversion of the conventional selectivity order of Ar–Cl > Ar–OTf. Thus, we investigated the feasibility of alkyl-indole-based phosphines (L13–L22)16 in the chemoselective α-arylation reaction. We found that L15 (SelectPhos) and L16 (CySelectPhos) inverted the chemoselectivity (–Cl over –OTf), affording the desired product with excellent yields through the Ar–Cl bond cleavage. The structure of the C2-alkyl group has a critical influence on the reaction. Either replacing the cyclohexyl group with the smaller –Me (L20) or more steric hindered the tertiary alkyl group of –t-Bu (L21) or –Ad (L22), but lacking methine hydrogen significantly lowered the chemoselectivity and reactivity of the catalyst.17 To further probe the importance of the C2-secondary alkyl group, a ligand with the C2-i-Pr group (L19) was newly synthesized. The Pd/L19 system afforded a slightly lower yield of 3a in the α-arylation reaction with unchanged chemoselectivity at the Ar–Cl bond.
In addition to the C2-alkyl groups, extra attention was paid to evaluating the effects of dynamic interactions between –PR2 modules and the C2-alkyl group of alkyl-indole-based phosphines (L13–L18), which has been unexplored to date. The –PR2 was found to be critical and able to alternate the rate-determining steps of the α-arylation reaction when only a single electrophile was involved.18 In general, the –PR2 moiety with an alkyl group, such as –i-Pr (L15), –Cy (L16), –Ad (L17), and –t-Bu (L18), demonstrated chemoselectivity toward the Ar–Cl bond. Surprisingly, palladium with the ligand bearing the –PPh2 group (L13) could chemoselectively react with the Ar–OTf bond to yield product 3a′. We suspected that L13 with a relatively small –PPh2 might be able to bis-ligate the palladium center to form Pd(L)2 species. Thus, we further synthesized L14 with a larger –P(o-tolyl)2 group, which demonstrated a preference toward the Ar–Cl bond and yielded the product 3a selectively.19
To further investigate the ligand effects on the chemoselectivity of the –PR2 moiety, we performed a density functional theory (DFT) study of the oxidative addition process for the bis-ligated Pd(L13)2 to evaluate the effects of the –PPh2 on the chemoselectivity (Scheme 4). The calculations were performed at the SMD(toluene) B3PW91-D3(BJ)/6-31G*//B3PW91-D3(BJ)/Def2-TZVP level of theory (see ESI for details†). The calculated results indicated that Pd(L13)2 favored the oxidative addition of C–OTf (7C-TS-OTf) over C–Cl (7B-TS-Cl) by 8.9 kcal mol−1, which also agrees with the previous finding that the bis-coordinated palladium complex preferred the C–OTf bond activation (Scheme 4A).7d To examine whether chemoselectivity may be governed by the electron-deficient property of –PPh2 to react with the C–Cl bond, a simulation of the oxidative addition of 4-chlorophenyl triflate using Pd-L13 was attempted (Scheme 4A), and the result shows that mono-ligated Pd-L13 favored the oxidative addition of C–Cl (7E-TS-Cl) over C–OTf (7G-TS-OTf) by 11.2 kcal mol−1. Furthermore, the rotational energy barrier study of Pd/L13 and Pd/L15via the constrained-relaxation scan was performed (see ESI, Fig. S6† for details). The calculated energy barrier for the –PPh2 group (L13) rotation along the indole skeleton was about 3.8 kcal mol−1 (Scheme 4B), which should be at least 8.1 kcal mol−1 lower than the rotational energy barrier of the –Pi-Pr2 group (L15). The low rotational energy barrier of the –PPh2 group might be a possible reason for the facial bis-ligation to form the Pd(L)2 species and the C–OTf selectivity. These results indicate that in addition to the effects of the C2-substituted group of the ligand core, the steric effect offered by the –PR2 group is also an important factor in chemoselectivity.
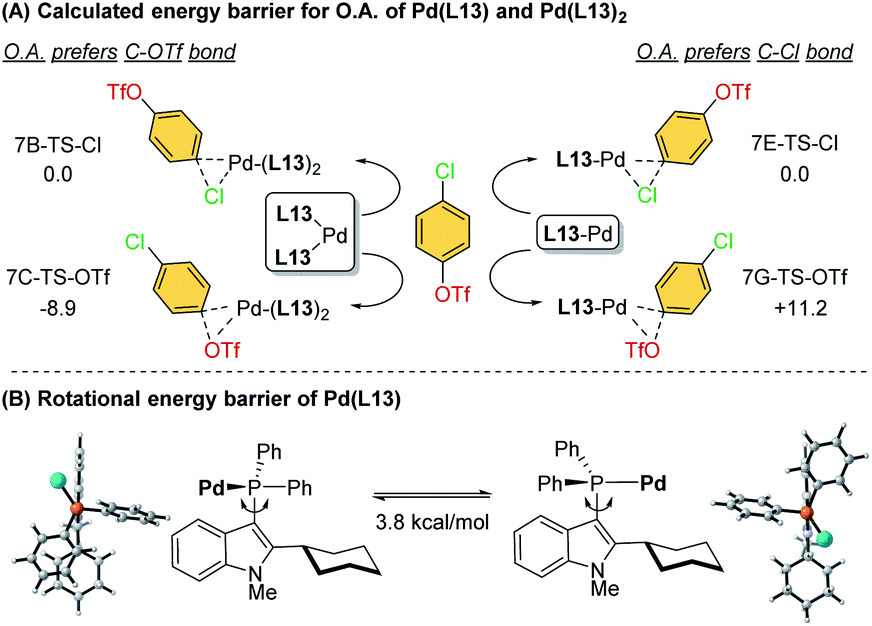 | ||
| Scheme 4 (A) Calculated energy barrier for the oxidative addition step of Pd(L13) and Pd(L13)2 and (B) rotational energy barrier of Pd(L13). | ||
We then optimized the reaction conditions using SelectPhos (L15) for the direct α-arylation reaction (see ESI, Table S2† for details). The replacement of palladium sources had no effect on the chemoselective outcome (Table S2,† entries 1–3), and neither did the ligand ratio (Table S2,† entry 10). Milder bases were found to be inferior to this reaction, while the triflate group was decomposed by the strong base (Table S2,† entries 5–6). Toluene was found to be the best of the solvents tested (Table S2,† entries 3 and 7–8). Based on the chemoselective Ar–Cl bond (over Ar–OTf bond) functionalization enabled by the Pd/L15 system, we explored both electrophiles and nucleophiles (Scheme 5). The Pd/L15 system demonstrated high efficiency in the cross-coupling of substituted chloroaryl triflates, including electron-rich/deficient substitutions (–OMe, –Me, –F, –CF3, and –Cl) in meta/para-positions (3c–3f, 3h, 3i). Substrates with the ketone carbonyl were also compatible under these conditions (3i). When a fluoro group was substituted at the C5 position of the oxindole, a good product yield was obtained (3m). Other N-substituted oxindoles, such as N-benzyl (3n, 3o) and N-phenyl (3p), were also feasible substrates and afforded the corresponding products in a good yield. Although L18 generally showed inferior results compared to L15, interestingly, Pd/L18 was found to be more effective in the reaction of sterically hindered substrates (3g, 3k, and 3l) and complementary to L15 in expanding the substrate scope.
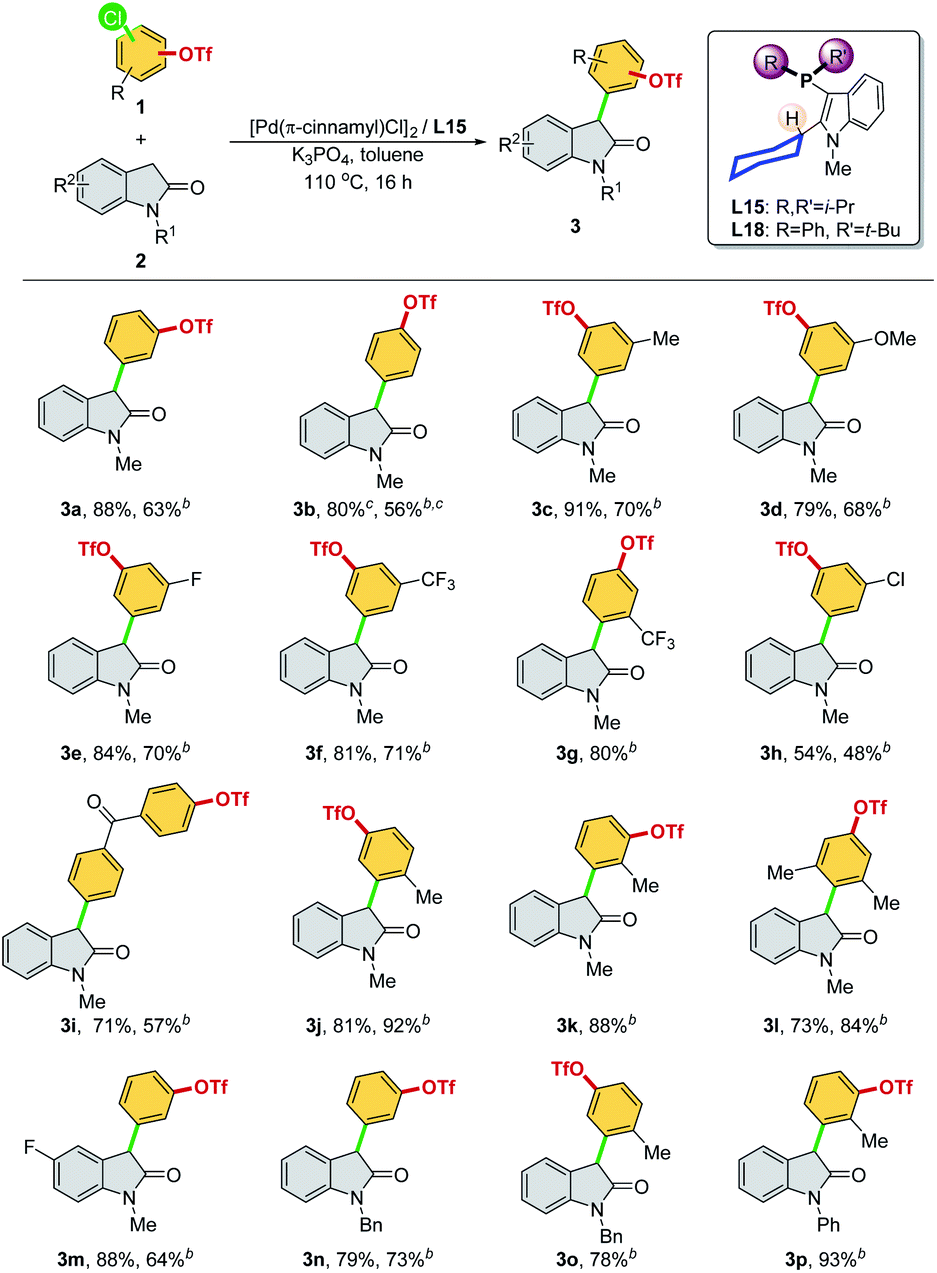 | ||
Scheme 5 Palladium-catalyzed α-arylation of oxindoles through chemoselective C–Cl bond cleavagea (aReaction conditions: 1 (0.20 mmol), 2 (0.20 mmol), [Pd(π-cinnamyl)Cl]2 (2 mol%), L15 (8 mol%), Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :L = 1:2, K3PO4 (0.40 mmol), toluene (1.0 mL), 110 °C, and 16 h. Isolated yields were reported. The chemoselectivity ratio between Ar–Cl and Ar–OTf was >99/1. bL18 was used. cReaction conditions: 80 °C and 14 h). :L = 1:2, K3PO4 (0.40 mmol), toluene (1.0 mL), 110 °C, and 16 h. Isolated yields were reported. The chemoselectivity ratio between Ar–Cl and Ar–OTf was >99/1. bL18 was used. cReaction conditions: 80 °C and 14 h). | ||
We attempted buried volume and steric map analyses to understand the steric properties of the L15 and L18 in the catalysis reaction (Fig. 1).20 In addition to the %Vbur of L15 with –Pi-Pr2 larger than that of L18 with –Pt-BuPh, as well as the large steric contributions of the cyclohexyl group of both ligands at the SE quadrant, the steric map showed that the steric impact of the two isopropyl P-substituents of L15 was evenly spread over the SW, NW, and NE quadrants, while the steric impact of –Pt-BuPh L18 was biased toward the NE quadrants (Ph, NW and t-Bu, NE), which might facilitate the coordination of hindered substrates to yield the products.21
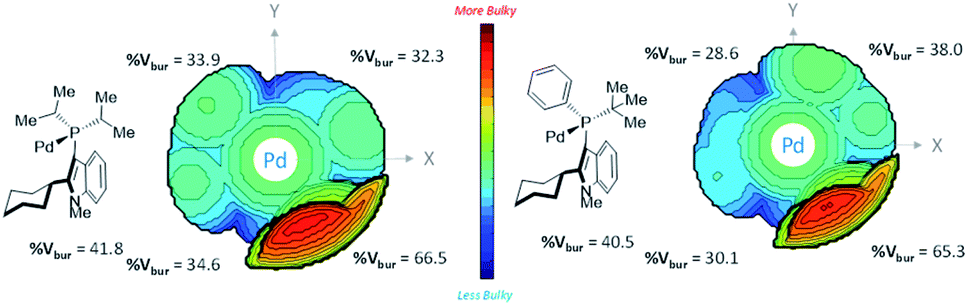 | ||
| Fig. 1 %Vburvs. steric map representation of Pd-L15 and Pd-L18. | ||
A series of experiments were conducted to further investigate the origin of the ligand effects on reactivity and chemoselectivity. 3-Chloro-5-(trifluoromethyl)phenyl triflate and 3-chloro-5-methylphenyl triflate were selected to probe the electronic effect with respect to the initial rate of α-arylation (Scheme 6).
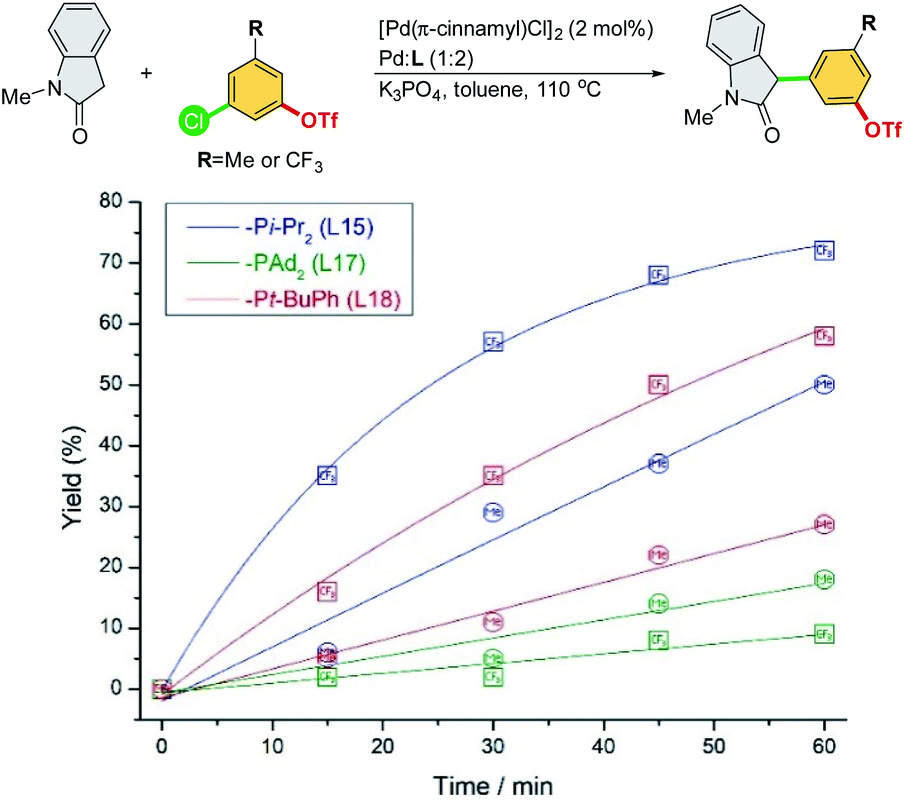 | ||
| Scheme 6 Reaction rate study of electronically different chloroaryl triflates in the cross-coupling of N-methyl-oxindole. | ||
Electron-deficient 3-chloro-5-(trifluoromethyl)phenyl triflate proceeded faster than electron-rich 3-chloro-5-methylphenyl triflate, suggesting that oxidative addition could be the rate-limiting step for both Pd/L15 and Pd/L18 systems. The faster reaction rate of Pd/L15 may be attributed to its dialkyl phosphine group (–Pi-Pr2), which should be more electron-rich than the aryl-alkyl phosphine group (–Pt-BuPh) of the Pd/L18 system. Interestingly, the Pd/L17 system with the most electron-rich –PAd2 group showed the slowest reaction rate compared to the Pd/L15 and Pd/L18 systems. Moreover, electron-deficient 3-chloro-5-(trifluoromethyl)phenyl triflate, which is more prone to oxidative addition, proceeded at a slower rate than electron-rich 3-chloro-5-methylphenyl triflate with the Pd/L17 system. Among the Pd/L15, Pd/L17, and Pd/L18 systems tested, the reactions undergo the C–Cl pathway chemoselectively.
A set of individual and competition experiments were further conducted for Pd/L15, Pd/L17, and Pd/L18 systems (Scheme 7). For Pd/L15 and Pd/L18 systems, both the individual experiments (Scheme 7A) and competition experiments (Scheme 7B) showed that the electron-poor 3-chloro-5-(trifluoromethyl)phenyl triflate 2f offered the corresponding product in a higher yield than the electron-rich 3-chloro-5-methylphenyl triflate 2c. These results suggested that the oxidative addition step could be the rate-limiting step for both Pd/L15 and Pd/L18 systems.
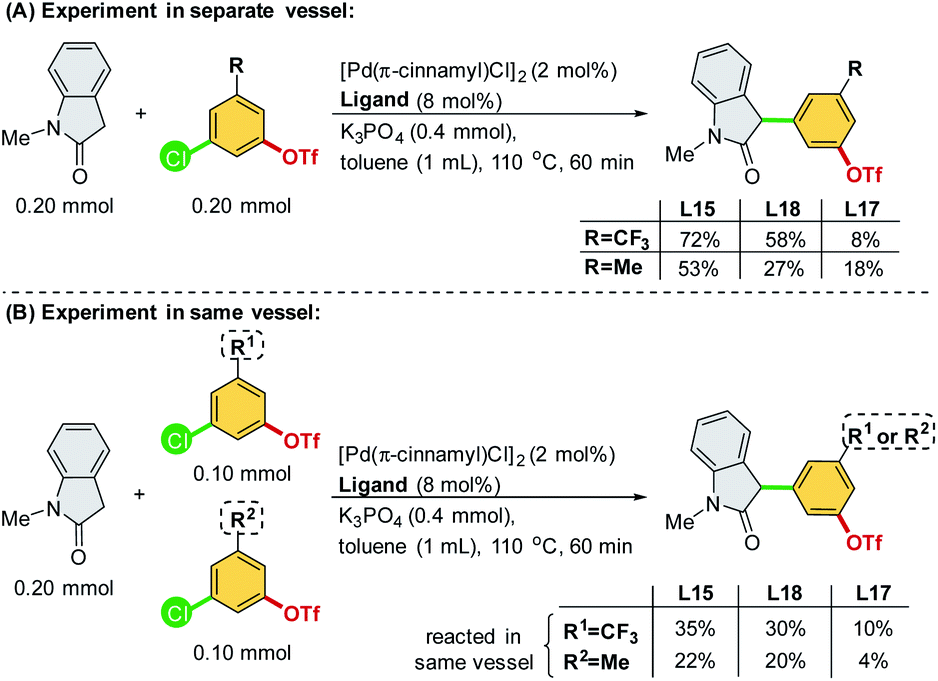 | ||
| Scheme 7 Individual and competition experiments of different chloroaryl triflates. | ||
However, for the Pd/L17 system, the results of the individual experiments (Scheme 7A) showed that the electron-rich 3-chloro-5-methylphenyl triflate 2c gives the corresponding product in a higher yield than the electron-poor 3-chloro-5-(trifluoromethyl)phenyl triflate 2f. In contrast, the competition experiments of Pd/L17 (Scheme 7B) showed that the electron-poor 3-chloro-5-(trifluoromethyl)phenyl triflate 2f offered the corresponding product in a higher yield than the electron-rich 3-chloro-5-methylphenyl triflate 2c. The results of the competition reaction may be due to a more rapid oxidative addition of the electron-poor 3-chloro-5-(trifluoromethyl) phenyl triflate 2f followed by a slow reductive elimination step, thus inhibiting the catalyst from reacting with electron-rich 3-chloro-5-methylphenyl triflate 2c. These experimental results suggest that reductive elimination is likely to be the rate-limiting step in the catalytic cycle involving the Pd/L17 system.14,22
The experimental results (Schemes 6 and 7) of Pd/L15 and Pd/L18 systems suggested that the oxidative addition step could be the rate-limiting step in the α-arylation reaction, accompanied by the previous DFT study results7f of Pd/L15 which prefers oxidative addition of the C–Cl bond over the C–OTf bond, which should account for the chemoselective formation of the C–Cl reacted product.
For the Pd/L17 system, chemoselectivity of the Pd/L17 system remained unchanged (Ar–Cl > Ar–OTf), even if the oxidative addition of Ar–Cl was not the rate-limiting step. To have a better insight into the reasons of the Pd/L17 system why the C–OTf pathway is not preferable, a series of DFT calculations were conducted (Fig. 2).
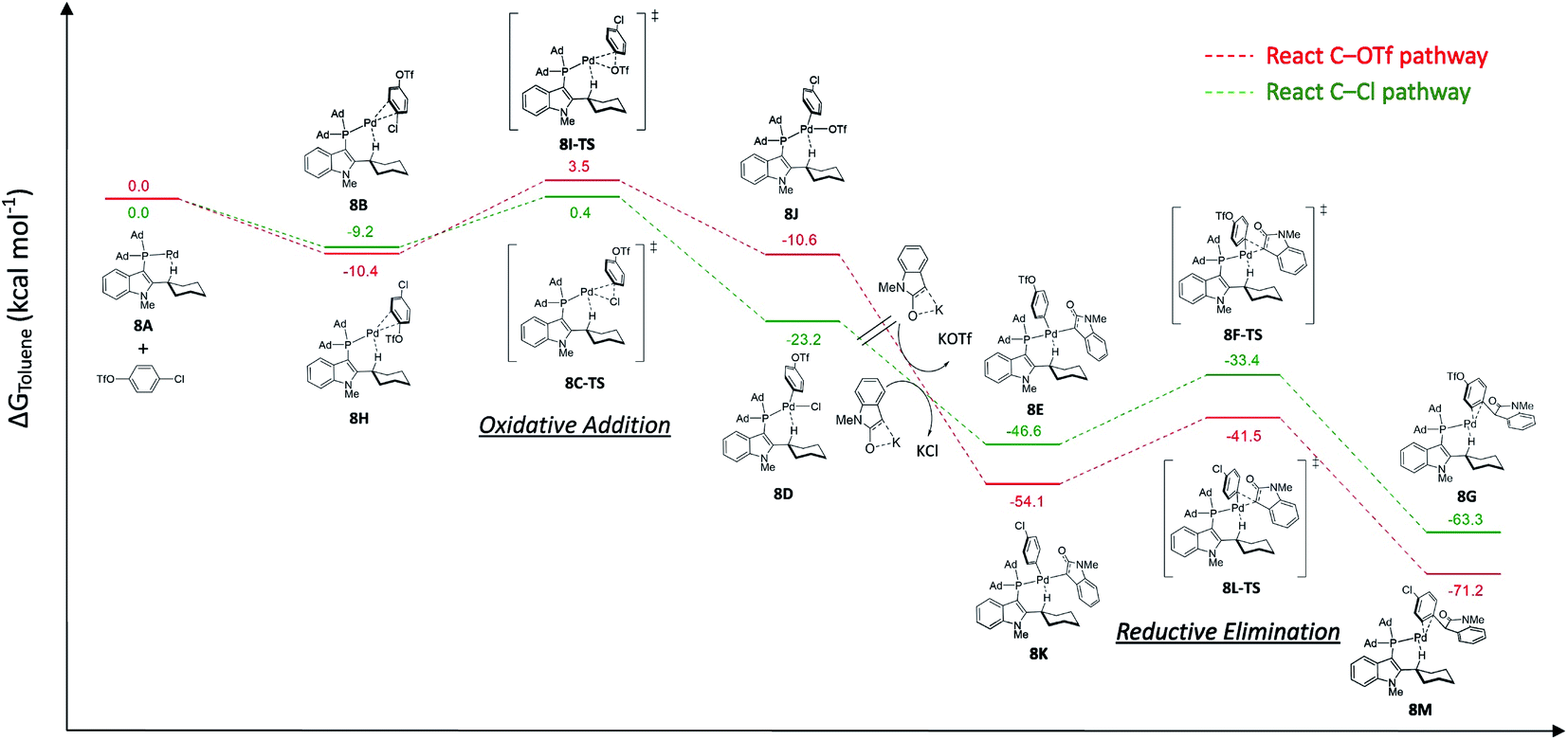 | ||
| Fig. 2 Free energy profiles calculated for the chemoselective Pd-L17 cross-coupling reaction of 4-chlorophenyl triflate with 1-methyloxindole. | ||
The monoligated Pd-L17 reacted with 4-chlorophenyl triflate, and oxindole was used for the study. In the reaction of the C–Cl pathway, aligned with the kinetic study, the calculated results suggested that the energy barrier of the reductive elimination step (8F-TS, 13.2 kcal mol−1) was higher than the oxidative addition of C–Cl (8C-TS, 9.6 kcal mol−1). In the reaction of the C–OTf pathway, the oxidative addition of the C–OTf (8I-TS, 13.9 kcal mol−1) showed a higher energy barrier than the reductive elimination step (8L-TS, 12.6 kcal mol−1). The higher energy barrier of the oxidative addition step of C–OTf (8I-TS, 13.9 kcal mol−1) than the C–Cl (8C-TS, 9.6 kcal mol−1) may disfavor the C–OTf pathway of the reaction, which might account for the preference of chemoselectivity toward the C–Cl bond in the Pd/L17 system.
We then investigated other ketones as cross-coupling partners in the chemoselective α-arylation reaction (Scheme 8). The –Cl site was selectively reacted, regardless of whether the C–Cl and C–OTf bonds were positioned meta (5a–5e, 5i–5l, and 5o) or para (5f, 5g, 5n, and 5p) to each other and whether functional groups were present (5h and 5m). α-Tetralone with different substitutions were applicable coupling partners, affording the desired α-arylated products with exclusive chemoselectivity (5a–5e). A diversity of aryl ethyl ketones was examined as substrates (5f–5p). Electron-neutral (–H, and 3-Me), rich (4-OMe), and poor (4-F, 3-CF3, and 4-CF3) aryl ethyl ketones were α-arylated smoothly in good product yields with excellent chemoselectivity at the –Cl site. It is worth noting that all of the products (Schemes 5 and 8, 3a–3p and 5a–5p) synthesized with this chemoselective reaction were new compounds, which offered a convenient pathway to access the new class of synthetically valuable molecules.23
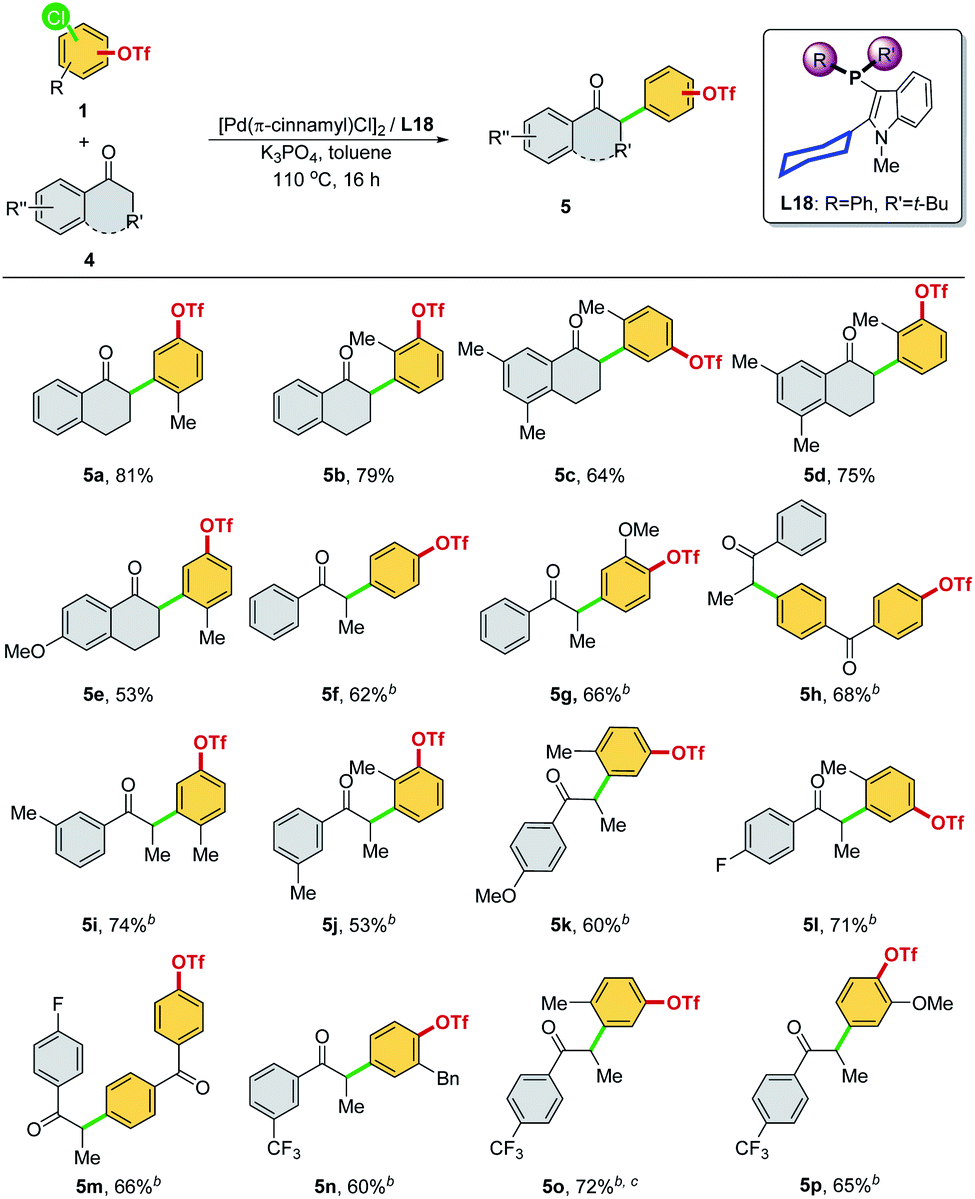 | ||
| Scheme 8 Palladium-catalyzed α-arylation of ketones through chemoselective C–Cl bond cleavagea (aReaction conditions: 1 (0.20 mmol), 4 (0.20 mmol), [Pd(π-cinnamyl)Cl]2 (2 mol%), L18 (8 mol%), Pd:L = 1:2, K3PO4 (0.40 mmol), toluene (1.0 mL), 110 °C, and 16 h. Isolated yields were reported. The chemoselectivity ratio between Ar–Cl and Ar–OTf was >99/1. bK3PO4 (0.50 mmol) was used. cThe chemoselectivity ratio between Ar–Cl and Ar–OTf was >25/1). | ||
Based on the successful attempt to functionalize ketones, we are encouraged to expand the scope of nucleophiles further and apply this chemoselective cross-coupling system to synthesize new pharmaceutical substances.24 Flurbiprofen is a commercial nonsteroidal anti-inflammatory drug, and the optimization of its synthetic methods has been widely studied.10d,25 Flurbiprofen has two functional groups attached to the fluorobenzene linker; thus, it was considered an appropriate final product for examining the practicability of our palladium-catalyzed chemoselective system (Scheme 9). In view of the α-arylation of esters, reactions generally required strong bases such as NaHMDS,11c LiHMDS,26 and LiNCy227 in previous reports, which may not be tolerated by the triflate electrophile. To minimize the potential hydrolysis of aryl triflates with a strong base and further enhance the potential usage of carbonyl derivatives in the chemoselective reaction, we attempted to use the ester enolate28 as a convenient alternative to tert-butyl propionate. The mild reaction conditions required for trimethylsilyl enolates in Pd-catalyzed transformations28b offered a good advantage for our current study.
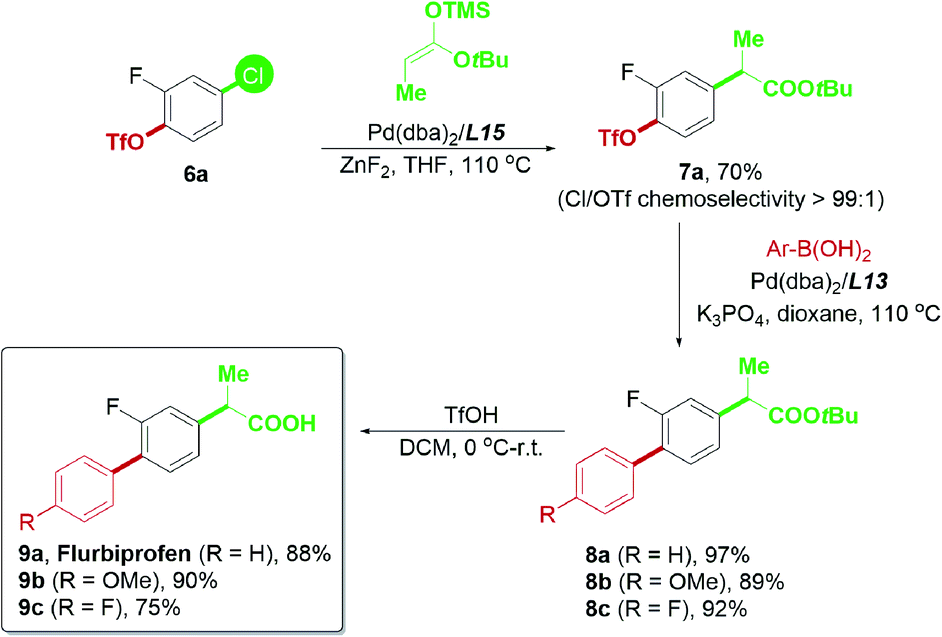 | ||
| Scheme 9 Synthesis of flurbiprofen using the chemoselective strategy. | ||
Using the ester enolate of tert-butyl propionate, 4-chloro-2-fluorophenyl triflate 6a was successfully converted into the key intermediate 7avia Pd/L15-catalyzed chemoselective Ar–Cl bond cleavage, paving the way for subsequent transformations. Subsequent Suzuki–Miyaura coupling enabled by the Pd/L13 system could afford 8a–8c efficiently. Upon acidic hydrolysis, flurbiprofen 9a and its derivatives 9b–9c were prepared with a satisfactory yield (Scheme 9). Thus, the practicability of this chemoselective transformation has proven to be promising and is worthy of further exploration.
Conclusions
In summary, for the first time, we developed a chemoselective palladium-catalyzed α-arylation reaction. The α-arylation reactions of carbonyl compounds, including oxindoles, ketones, and enol ester, were achieved to access a new class of carbonyl compounds that covers a wide range of chloroaryl triflates with an inversion of the common reactivity order of C–Cl > C–OTf. Furthermore, this strategy was successfully applied to synthesize flurbiprofen. We anticipate that this method will provide new insights into the optimal pathways for synthesizing complex molecules.Data availability
NMR and HRMS spectra and characterization data of the compounds and crystallographic data of L17, and experimental and computational details are provided in the ESI.†Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval regarding the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Research Grants Council of the Hong Kong Special Administrative Region, China (PolyU 15302821, 15300220 and 25301819), the National Natural Science Foundation of China (21972122) for the financial support. We also thank Dr Pui-kin So (PolyU), for HRMS analysis, and Dr Siu-cheong Yan, Kenneth (PolyU), for NMR analysis.Notes and references
- (a) J. F. Yang and J. R. Zhou, Org. Chem. Front., 2014, 1, 365–367 RSC; (b) C. C. C. J. Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef PubMed; (c) L. C. Campeau and N. Hazari, Organometallics, 2019, 38, 3–35 CrossRef CAS PubMed.
- (a) A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176–4211 CrossRef CAS; (b) H. Y. Zeng, Z. H. Qiu, A. Dominguez-Huerta, Z. Hearne, Z. W. Chen and C. J. Li, ACS Catal., 2017, 7, 510–519 CrossRef CAS.
- E. K. Reeves, E. D. Entz and S. R. Neufeldt, Chem.–Eur. J., 2021, 27, 6161–6177 CrossRef PubMed.
- P. Dobrounig, M. Trobe and R. Breinbauer, Monatsh. Chem., 2017, 148, 3–35 CrossRef CAS PubMed.
- (a) J. Almond-Thynne, D. C. Blakemore, D. C. Pryde and A. C. Spivey, Chem. Sci., 2017, 8, 40–62 RSC; (b) I. J. S. Fairlamb, Chem. Soc. Rev., 2007, 36, 1036–1045 RSC.
- (a) T. Kamikawa and T. Hayashi, Tetrahedron Lett., 1997, 38, 7087–7090 CrossRef CAS; (b) G. Espino, A. Kurbangalieva and J. M. Brown, Chem. Commun., 2007, 2007, 1742–1744 RSC; (c) J. Z. Wang, M. A. Seefeld and J. Luengo, Tetrahedron Lett., 2011, 52, 6346–6348 CrossRef CAS; (d) X. T. Yang, G. Q. Xu and W. J. Tang, Tetrahedron, 2016, 72, 5178–5183 CrossRef CAS; (e) I. Kalvet, G. Magnin and F. Schoenebeck, Angew. Chem., Int. Ed., 2017, 56, 1581–1585 CrossRef CAS PubMed; (f) S. T. Keaveney, G. Kundu and F. Schoenebeck, Angew. Chem., Int. Ed., 2018, 57, 12573–12577 CrossRef CAS PubMed; (g) T. Scattolin, E. Senol, G. Y. Yin, Q. Q. Guo and F. Schoenebeck, Angew. Chem., Int. Ed., 2018, 57, 12425–12429 CrossRef CAS PubMed; (h) C. Wern, C. Ehrenreich, D. Joosten, T. vom Stein, H. Buchholz and B. Konig, Eur. J. Org. Chem., 2018, 2018, 5644–5656 CrossRef CAS; (i) I. Kalvet, K. Deckers, I. Funes-Ardoiz, G. Magnin, T. Sperger, M. Kremer and F. Schoenebeck, Angew. Chem., Int. Ed., 2020, 59, 7721–7725 CrossRef CAS PubMed.
- (a) A. F. Littke, C. Y. Dai and G. C. Fu, J. Am. Chem. Soc., 2000, 122, 4020–4028 CrossRef CAS; (b) A. F. Littke, L. Schwarz and G. C. Fu, J. Am. Chem. Soc., 2002, 124, 6343–6348 CrossRef CAS PubMed; (c) F. Schoenebeck and K. N. Houk, J. Am. Chem. Soc., 2010, 132, 2496–2497 CrossRef CAS PubMed; (d) Z. L. Niemeyer, A. Milo, D. P. Hickey and M. S. Sigman, Nat. Chem., 2016, 8, 611–618 CrossRef PubMed; (e) E. K. Reeves, J. N. Humke and S. R. Neufeldt, J. Org. Chem., 2019, 84, 11799–11812 CrossRef CAS PubMed; (f) C. M. So, O. Y. Yuen, S. S. Ng and Z. C. Chen, ACS Catal., 2021, 11, 7820–7827 CrossRef CAS.
- (a) P. Vitale, S. Tacconelli, M. G. Perrone, P. Malerba, L. Simone, A. Scilimati, A. Lavecchia, M. Dovizio, E. Marcantoni, A. Bruno and P. Patrignani, J. Med. Chem., 2013, 56, 4277–4299 CrossRef CAS PubMed; (b) C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748–8758 CrossRef CAS PubMed; (c) B. S. Patel, F. M. Chavda and S. G. Mundhava, Int. J. Pharma Sci. Res., 2016, 7, 2174–2180 Search PubMed.
- T. Satoh, Y. Kawamura, M. Miura and M. Nomura, Angew. Chem., Int. Ed., 1997, 36, 1740–1742 CrossRef CAS.
- (a) H. N. Nguyen, X. H. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2003, 125, 11818–11819 CrossRef CAS PubMed; (b) J. L. Rutherford, M. P. Rainka and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 15168–15169 CrossRef CAS PubMed; (c) J. G. Zhou, Q. Jiang, P. Fu, S. Q. Liu, S. S. Zhang, S. Xu and Q. Zhang, J. Org. Chem., 2017, 82, 9851–9858 CrossRef CAS PubMed; (d) W. A. Moradi and S. L. Buchwald, J. Am. Chem. Soc., 2001, 123, 7996–8002 CrossRef CAS PubMed; (e) J. M. Fox, X. H. Huang, A. Chieffi and S. L. Buchwald, J. Am. Chem. Soc., 2000, 122, 1360–1370 CrossRef CAS; (f) D. W. Old, J. P. Wolfe and S. L. Buchwald, J. Am. Chem. Soc., 1998, 120, 9722–9723 CrossRef CAS; (g) M. Palucki and S. L. Buchwald, J. Am. Chem. Soc., 1997, 119, 11108–11109 CrossRef CAS.
- (a) D. A. Culkin and J. F. Hartwig, Organometallics, 2004, 23, 3398–3416 CrossRef CAS; (b) J. P. Wolkowski and J. F. Hartwig, Angew. Chem., Int. Ed., 2002, 41, 4289–4291 CrossRef CAS; (c) S. Lee, N. A. Beare and J. F. Hartwig, J. Am. Chem. Soc., 2001, 123, 8410–8411 CrossRef CAS PubMed; (d) D. A. Culkin and J. F. Hartwig, J. Am. Chem. Soc., 2001, 123, 5816–5817 CrossRef CAS PubMed; (e) M. Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 1999, 121, 1473–1478 CrossRef CAS; (f) K. H. Shaughnessy, B. C. Hamann and J. F. Hartwig, J. Org. Chem., 1998, 63, 6546–6553 CrossRef CAS; (g) B. C. Hamann and J. F. Hartwig, J. Am. Chem. Soc., 1997, 119, 12382–12383 CrossRef CAS.
- (a) S. T. Sivanandan, A. Shaji, I. Ibnusaud, C. C. C. J. Seechurn and T. J. Colacot, Eur. J. Org. Chem., 2015, 2015, 38–49 CrossRef CAS; (b) J. Schranck and J. Rotzler, Org. Process Res. Dev., 2015, 19, 1936–1943 CrossRef CAS; (c) H. K. Potukuchi, A. P. Spork and T. J. Donohoe, Org. Biomol. Chem., 2015, 13, 4367–4373 RSC; (d) C. C. C. Johansson and T. J. Colacot, Angew. Chem., Int. Ed., 2010, 49, 676–707 CrossRef CAS PubMed; (e) F. Bellina and R. Rossi, Chem. Rev., 2010, 110, 3850 CrossRef CAS; (f) D. A. Culkin and J. F. Hartwig, Acc. Chem. Res., 2003, 36, 234–245 CrossRef CAS PubMed.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- (a) O. Y. Yuen and C. M. So, Angew. Chem., Int. Ed., 2020, 59, 23438–23444 CrossRef CAS PubMed; (b) X. M. Chen, Z. C. Chen and C. M. So, J. Org. Chem., 2019, 84, 6337–6346 CrossRef CAS PubMed; (c) H. Zhang, X. C. Luo, K. Wongkhan, H. Duan, Q. Li, L. Z. Zhu, J. Wang, A. S. Batsanov, J. A. K. Howard, T. B. Marder and A. W. Lei, Chem.–Eur. J., 2009, 15, 3823–3829 CrossRef CAS PubMed; (d) J. F. Hartwig, Inorg. Chem., 2007, 46, 1936–1947 CrossRef CAS PubMed; (e) K. D. Hesp, R. J. Lundgren and M. Stradiotto, J. Am. Chem. Soc., 2011, 133, 5194–5197 CrossRef CAS PubMed.
- (a) P. Siengalewicz, T. Gaich and J. Mulzer, Angew. Chem., Int. Ed., 2008, 47, 8170–8176 CrossRef CAS PubMed; (b) C. Marti and E. M. Carreira, Eur. J. Org. Chem., 2003, 2003, 2209–2219 CrossRef.
- There were no detectable phosphine oxide signals of L15 and L18 from 31P NMR when the solid-form ligand was allowed to stand under air for 2 days. It allows for benchtop reaction mixture preparation under an air atmosphere. However, we recommend storing the ligands under an inert atmosphere for long-term storage..
- There is a preagostic interaction between the methine hydrogen on the cyclohexyl group and Pd center, which may play a role in stabilizing and affecting the activity of the unsaturated Pd complex during oxidative addition. The steric effect induced by the C2-cyclohexyl group may prevent the formation of the L2Pd species. For details, see ref. 7f. For the references describing the role and the importance of C–H…M interactions, see: (a) H.-J. Cao, Q. Zhao, Q.-F. Zhang, J. Li, E. J. M. Hamilton, J. Zhang, L.-S. Wang and X. Chen, Dalton Trans., 2016, 45, 10194–10199 RSC; (b) H. Darmandeh, J. Löffler, N. V. Tzouras, B. Dereli, T. Scherpf, K.-S. Feichtner, S. V. Broeck, K. V. Hecke, M. Saab, C. S. J. Cazin, L. Cavallo, S. P. Nolan and V. H. Gessner, Angew. Chem., Int. Ed., 2021, 60, 21014–21024 CrossRef CAS PubMed.
- W. C. Fu, C. M. So, W. K. Chow, O. Y. Yuen and F. Y. Kwong, Org. Lett., 2015, 17, 4612–4615 CrossRef CAS PubMed.
- Further investigation showed that Pd/L13 with the Pd:L ratio 1:1 yielded the product 3a/3a′ mixture, which may further support the dynamic formation of Pd-L13 and Pd-(L13)2 during the reaction with a low concentration of ligand. Additional NMR and HRMS studies showed that the Pd-(L13)2 can readily be formed (see ESI† for details)..
- The percent buried volumes of L15 and L18 were calculated using SambVca (Salerno molecular buried volume calculation) 2.1 software..
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed.
- A set of isotope experiments was carried out to investigate the dependence of the C–H bond cleavage. The rapid H/D exchange between the D2-oxindole and the H2O present in the system indicated that the deprotonation of the oxindole C3–H should be in a direct manner with K3PO4 as the base..
- We have examined some of the heteroaryl chloride/triflates such as 5-chloropyridin-2-yl triflate, 6-chloropyridin-2-yl triflate, 5-chloropyridin-3-yl triflate. However, the reaction of heteroaryl substrates was not successful, which suffered from the severe decomposition of the starting material, and/or formation of a very low yield of product with the triflate group hydrolyzed to a hydroxyl group. Further study is needed to address this issue..
- F. Richy, V. Rabenda, A. Mawet and J. Y. Reginster, Int. J. Clin. Pract., 2007, 61, 1396–1406 CrossRef CAS PubMed.
- (a) Z. T. He and J. F. Hartwig, J. Am. Chem. Soc., 2019, 141, 11749–11753 CrossRef CAS PubMed; (b) K. W. Quasdorf, M. Riener, K. V. Petrova and N. K. Garg, J. Am. Chem. Soc., 2009, 131, 17748–17749 CrossRef CAS PubMed.
- J. A. Friest, Y. Maezato, S. Broussy, P. Blum and D. B. Berkowitz, J. Am. Chem. Soc., 2010, 132, 5930–5931 CrossRef CAS PubMed.
- T. Hama and J. F. Hartwig, Org. Lett., 2008, 10, 1545–1548 CrossRef CAS PubMed.
- (a) M. Jorgensen, S. Lee, X. X. Liu, J. P. Wolkowski and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 12557–12565 CrossRef CAS PubMed; (b) J. F. Yang and J. R. Zhou, Org. Chem. Front., 2014, 1, 365–367 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2117109. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc06701j |
| This journal is © The Royal Society of Chemistry 2022 |