
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStronger together for in-cell translation: natural and unnatural base modified mRNA†
Lisa
Bornewasser
 ,
Christof
Domnick
and
Stephanie
Kath-Schorr
*
,
Christof
Domnick
and
Stephanie
Kath-Schorr
*
Institute of Organic Chemistry, Department of Chemistry, University of Cologne, Greinstrasse 4, 50939 Cologne, Germany. E-mail: skathsch@uni-koeln.de
First published on 3rd March 2022
Abstract
The preparation of highly modified mRNAs and visualization of their cellular distribution are challenging. We report in-cell application of in vitro transcribed mRNA containing natural base modifications and site-specifically introduced artificial nucleotides. Click chemistry on mRNA allows visualization in cells with excellent signal intensities. While non-specific introduction of reporter groups often leads to loss in mRNA functionality, we combined the benefits from site-specificity in the 3′-UTR incorporated unnatural nucleotides with the improved translation efficiency of the natural base modifications Ψ and 5mC. A series of experiments is described to observe, quantify and verify mRNA functionality. This approach represents a new way to visualize mRNA delivery into cells and monitor its spread on a cellular level and translation efficiency. We observed increased protein expression from this twofold chemically modified, artificial mRNA counterbalancing a reduced transfection rate. This synergetic effect can be exploited as a powerful tool for future research on mRNA therapeutics.
Introduction
In vitro transcribed (IVT) mRNAs are emerging, powerful therapeutics in fighting the COVID-19 pandemic.1 For future precise tuning of IVT mRNA delivery, stability, and expression, a deeper understanding of cellular processes of artificial mRNAs is essential. This requires new methods for IVT mRNA functionalization, chemical modification and visualization in cells.Natural RNA modifications such as pseudouridine (Ψ) and 5-methylcytidine (5mC) increase protein translation and expression from IVT mRNA.2–4 These modifications also decrease immunostimulation compared to unmodified RNA.5 Referring to fundamental work of Karikó and coworkers,2–5 Ψ and 5mC are substantial factors for mRNA vaccine efficacy.6,7 Upon substituting all cytidines (C) by 5mC or all uridines (U) by Ψ in mRNA, not only protein expression but also cell viability is improved, and concurrently, interferon signaling is reduced.8 Complete replacement of both C and U by 5mC and Ψ even outperforms these findings.8 A current explanation is altered RNA secondary structures induced by these modifications9–13 enhancing translation initiation and increasing polysome complexity as well as translational robustness by means of ribosome recycling.13 Active stabilization of IVT mRNA to improve and preserve translational activity is highly desirable, due to the fact that frequently translated mRNAs undergo degradation faster than non-translating mRNA.14
Thus, controlling the structure, stability, and translational activity is a mandatory prerequisite for future development of mRNA therapeutics. Other crucial characteristics comprise cell delivery, spatiotemporal distribution, binding interactions, and conformational dynamics of IVT mRNAs in cells.15 To study these characteristics, visualization of nucleic acids inside the cell is required which is usually achieved by fluorescent labeling of the nucleic acids. Long RNAs such as mRNAs with several hundred to several thousand nucleotides in length are generally synthesized via in vitro transcription, and site-specific introduction of fluorescent reporter groups into such RNA sequences is challenging.16 Random introduction of functionalized nucleoside triphosphates during IVT in the coding region of the mRNA influences its expression pattern.17 Post-transcriptional fluorescent mRNA functionalization applying copper-catalyzed azide–alkyne click reactions (CuAAC) allows mRNA visualization in cells17 but is not suitable for live cell applications due to cell toxicity18 and is further limited by promoting RNA hydrolysis.19 Expanding the approach to strain-promoted azide–alkyne click reactions (SPAAC)17,20 enables click functionalization inside cells17,20 but was so far only applied in protocols modifying an mRNA's poly(A) tail either at a single terminal position17 or multiple times at random positions within the poly(A) sequence.20 For the latter, Rentmeister and coworkers reported not only efficient fluorescent visualization of mRNA but also an improved translation of modified mRNAs.20 Expansion of the approach to different types of RNA other than mRNA is however complicated as the functionalized nucleotides are installed post-transcriptionally into the poly(A) tail, which represents an inherent feature of mRNA. Thus, site-specific and co-transcriptional introduction of functionalized nucleotides in combination with subsequent attachment of several reporter groups in untranslated regions of an IVT mRNA is highly desirable.
Transcription methods with an expanded genetic alphabet21–27 enable the site-specific introduction of modified unnatural nucleotides in RNA. Groundbreaking studies have been published by Ichiro Hirao and Floyd Romesberg. Both groups published unnatural base pairs that can be amplified and transcribed with high fidelity and efficiency.28,29 Based on the work of Romesberg and coworkers, we evolved protocols and adapted established methods to site-specifically and enzymatically introduce functionalized unnatural nucleosides in long RNAs as demonstrated recently.30–32
Here, we present a new and efficient way to insert reactive handles into the 3′-untranslated region (UTR) for post-transcriptional labeling of IVT mRNA. Genetic alphabet expansion transcription21–27 (GENAEXT) with its site-specificity along with smallness and reactivity of cyclopropene moieties (CP)32,33 are the key features of this novel technique. GENAEXT is compatible with all desirable features of an enzymatic mRNA synthesis3,34 such as a combined application of naturally occurring RNA base modifications Ψ and 5mC and the artificial nucleobases TPT3:NaM.35 Our technique fulfills all requirements of a powerful tool to observe intracellular mRNA distribution over time.
The concept of mRNA live cell click labeling and GENAEXT mRNA function analysis is shown in Fig. 1. GENAEXT mRNA32,33 is obtained by in vitro transcription: site-specific unnatural nucleobases (UBs) are incorporated in its 3′-UTR, and Ψ and 5mC2–4 modifications are randomly distributed throughout the entire mRNA to improve translational activity, and a 5′ cap analog and a 3′ poly(A) tail are introduced.34 In detail, we describe the preparation of mRNA that bears site-specific cyclopropene moieties in its 3′-UTR and codes for the red fluorescent mCherry reporter protein.36 We analyze mRNA levels after liposome-based transfection into mammalian cells and compare translation efficiencies of the CP modified GENAEXT mRNA to unmodified mRNA. Levels of the mRNA are analyzed by reverse transcription coupled with quantitative PCR (RT-qPCR). mRNA function is examined by quantification of the mCherry reporter protein expression. We further show that mRNA with combined modifications of partially substituted and randomly positioned natural RNA modifications (Ψ and 5mC) together with site-specifically introduced unnatural bases with cyclopropene moieties leads to a synergetic effect on mRNA translation in cells.
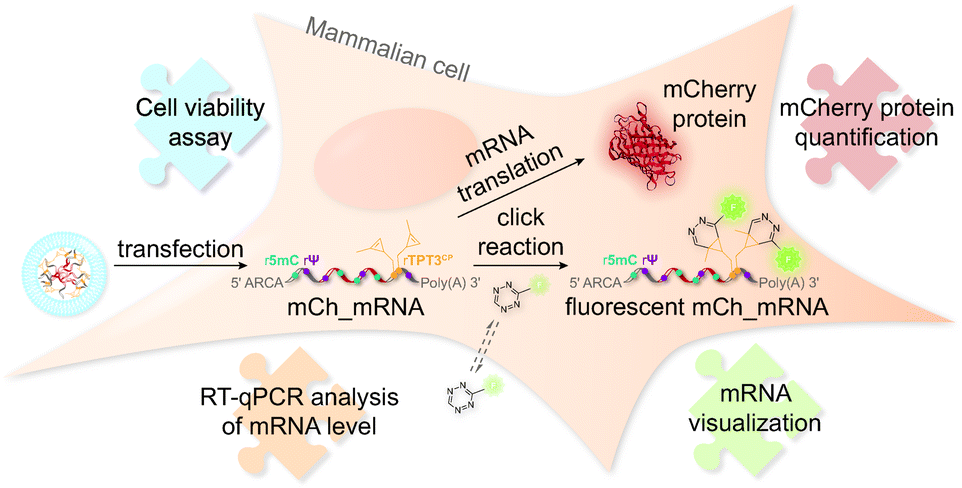 | ||
| Fig. 1 Tracking and function analysis of GENAEXT mRNA in cells. Site-specifically cyclopropene labeled mRNA bearing additional Ψ and 5mC modifications and coding for the mCherry protein is transfected into mammalian cells. Relative amounts of transfected mCh_mRNA are quantified and analyzed by RT-qPCR. Translation of the IVT mRNA is investigated by quantification of its encoded reporter protein, the red fluorescent protein mCherry. mCh_mRNA is visualized after fluorescence labeling in living cells by bioorthogonal iEDDA cycloaddition with the CP modification in the 3′-UTR of the GENAEXT mRNA. Cell health in the course after mRNA transfection is studied by a cell viability assay. | ||
Furthermore, CP modified mRNA is fluorescently labeled by inverse electron-demand Diels–Alder (iEDDA) cycloaddition in live cells.37,38 This state-of-the-art click reaction18 allows temporal and spatial tracking of GENAEXT mRNA after transfection. As 1,2,4,5-tetrazines hold a characteristic absorption between 500 and 525 nm,39 green-light emitting fluorophores are quenched most efficiently, when conjugated to tetrazines.40 Upon reaction with a strained double bond, dihydropyridazines are formed18, and a turn-on in fluorescence is gained. Used fluorophore-tetrazine conjugates are cell-permeable, stable under physiologic conditions and gain a very high turn-on in fluorescence upon reaction completion.39,40 By this, an excellent intracellular signal-to-noise ratio for spatiotemporal mRNA visualization is achieved. Further advantages of iEDDA chemistry are fast reaction rates and lack of cytotoxic catalysts, such as e.g. copper(I).18,39,40 To show compatibility of our technique for in-cell applications, a cell viability assay was performed in order to assess cell health in the course of mRNA transfection and live-cell click reaction.
Results
In vitro synthesis of GENAEXT mRNA with chemical modifications in the 3′-UTR
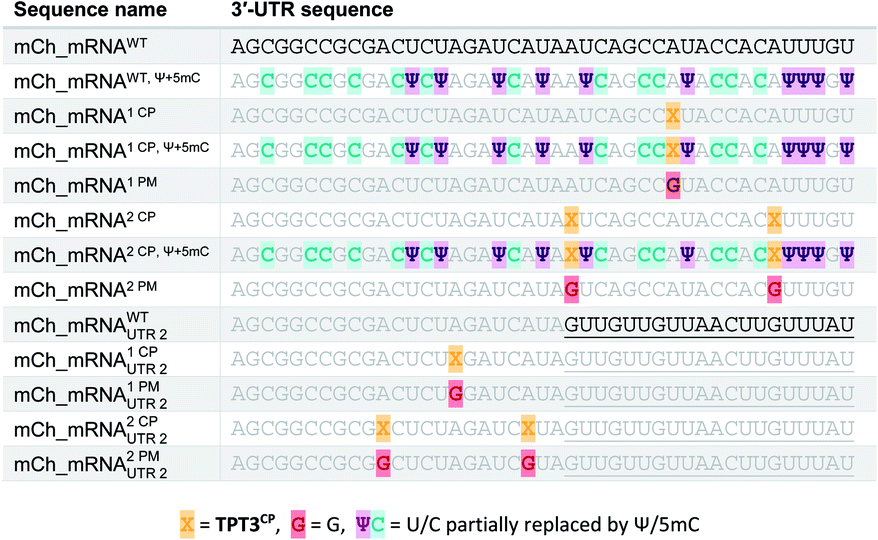
The general procedure for preparation of site-specifically cyclopropene labeled mCherry coding mRNA (mCh_mRNA) (Fig. 2a) is outlined as follows: a double stranded DNA (dsDNA) template is amplified from the pmCherry-N1 plasmid in a six-letter polymerase chain reaction (PCR). Thereby, either a single or double unnatural dTPT3:dNaM35 base pair (Fig. 2b) modification is introduced site-specifically into the dsDNA template. The unnatural base pair (UBP) is placed in the 3′-UTR downstream of the mCherry protein coding region of the mRNA. For this purpose, we apply the six-letter PCR method as described earlier by our group.33,41 During PCR, a dNaM-modified reverse primer hybridizes to the 3′-UTR of the mCherry gene tolerating dNaM:dA mismatches between the primer and plasmid. Simultaneously, a forward primer including the T7 RNA polymerase promotor as an overhang sequence hybridizes to the 5′-UTR of the mCherry gene. Using GENAEXT, the CP functionalized unnatural triphosphate rTPT3CP (Fig. 2d) is incorporated opposite to dNaM in a template-directed manner. Co-transcriptionally, the anti-reverse cap analog (ARCA, see ESI Fig. 34† for structure) is introduced into the 5′-end. Thus, using this approach, either a single or double cyclopropene modified mCherry protein coding mCh_mRNA1 CP and mCh_mRNA2 CP is prepared (Fig. 2e). Unmodified mCh_mRNAWT is obtained in an analogous manner by standard T7 in vitro transcription from an unmodified dsDNA template generated under standard PCR conditions. Electively, partial substitution of Ψ for U and 5mC for C at random positions is performed during transcription (Fig. 2f). A poly(A) tail of approximately 150 nt length is added to the RNA post-transcriptionally. Full length dsDNA and full length RNA products were verified by agarose gel analysis (Fig. 2c, e, f and ESI Fig. 4–9†). Sequence identities of UBP modified and unmodified PCR products (Fig. 2c) were confirmed via Sanger sequencing (sequencing results listed in the ESI†). A summary of all different mCh_mRNA sequences used in this study is listed in Table 1. Two sets of different mCh_mRNA sequences, mCh_mRNA and mCh_mRNAUTR 2, were prepared. Each set consists of an unmodified (WT), a single and a double CP modified (1 CP and 2 CP) as well as a single and a double canonical base point mutated (1 PM and 2 PM) mRNA sequence. For CP modified and unmodified mCh_mRNA sequences, we also prepared mRNA with additional Ψ and 5mC modifications.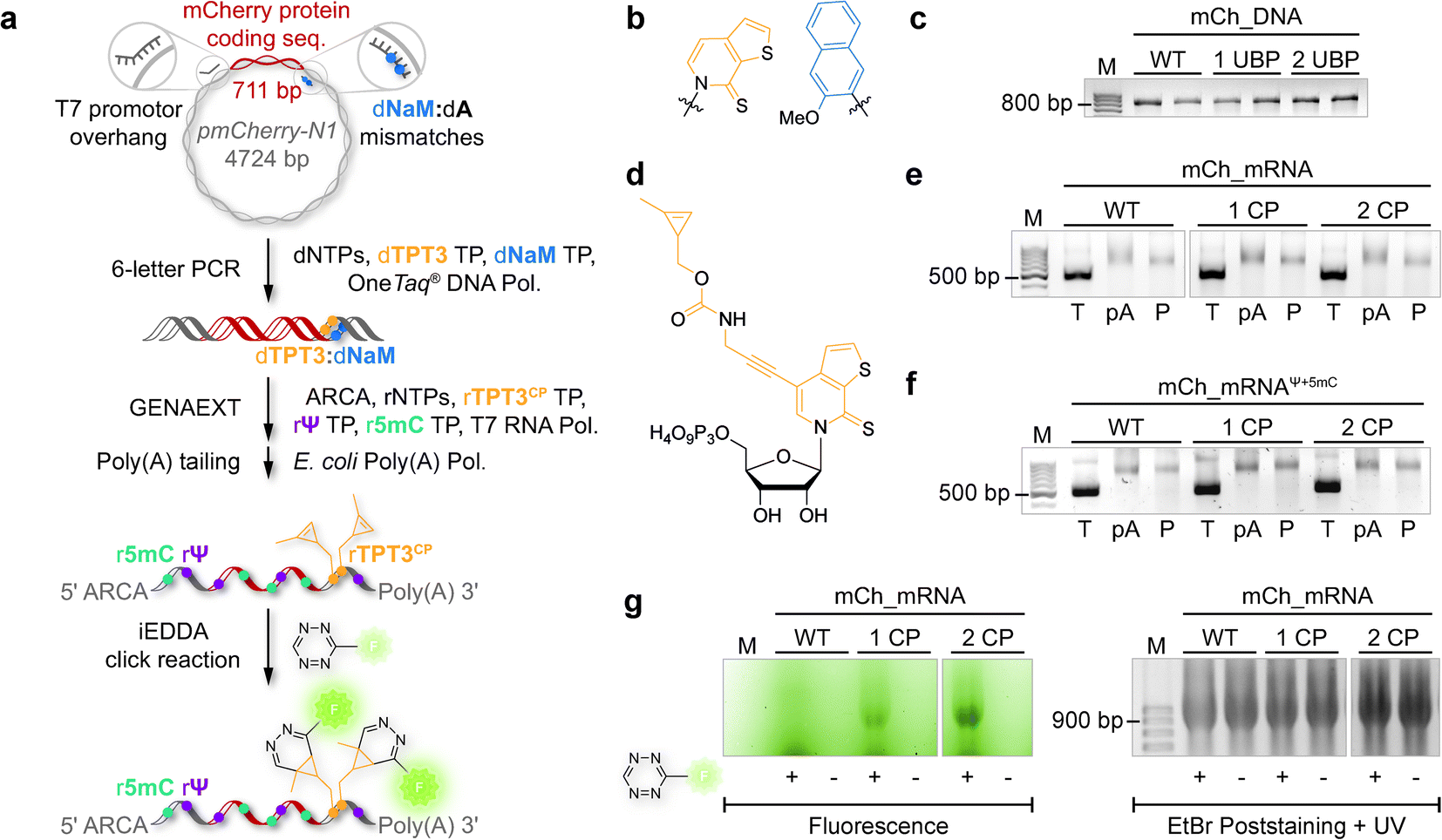 | ||
| Fig. 2 Preparation of site-specifically CP modified mRNA. (a) Principle for generation of site-specifically labeled mRNA. In a six-letter PCR, the mCherry protein coding region is amplified from a plasmid using a dNaM-modified reverse primer. During GENAEXT, the unnatural nucleotide rTPT3CP is site-specifically introduced into the RNA transcript (817 nt). Co-transcriptionally, the ARCA cap is installed at the 5′-end of the RNA; additionally, natural base modifications such as Ψ and 5mC are included in the nascent mRNA. The transcribed RNA is poly(A) tailed post-transcriptionally. iEDDA cycloaddition with a tetrazine-fluorophore conjugate results in fluorescently labeled mCh_mRNA. (b) Unnatural base pair (UBP) TPT3:NaM. Sugar residues omitted for clarity. (c) Agarose gel of mCh_DNA PCR products. WT: unmodified mCh_DNAWT; 1 UBP: single UBP modified mCh_DNA1 UBP; 2 UBP: double UBP modified mCh_DNA2 UBP; M: marker. (d) Methyl-cyclopropene modified unnatural base (UB) rTPT3CP. (e) Agarose gel of mCh_RNA transcripts. WT: unmodified mCh_mRNAWT; 1 CP: mCh_mRNA1 CP; 2 CP: mCh_mRNA2 CP. (T): transcribed RNA; (pA): after poly(A) tailing; (P) after purification; (M): marker. (f) Agarose gel of mCh_RNA transcripts with additional Ψ and 5mC modifications. WT: mCh_mRNAWT, Ψ+5mC, 1 CP: mCh_mRNA1 CP, Ψ+5mC, and 2 CP: mCh_mRNA2 CP, Ψ+5mC. (T): transcribed RNA; (pA): after poly(A) tailing; (P) after purification; (M): marker. (g) Agarose gel of mCh_mRNAin vitro click reaction. WT: mCh_mRNAWT; 1 CP: mCh_mRNA1 CP; 2 CP: mCh_mRNA2 CP. With or without addition of TET-FL (indicated as + or −). Left panel: fluorescence scan. Right panel: UV scan after EtBr post-staining. | ||
|
Visualization of IVT mRNA in cells by live-cell click labeling
mRNA visualization in vitro and in cells is enabled by fluorescent labeling of CP modified mRNA. For this, a tetrazine-conjugated fluorophore is covalently reacted with the methyl cyclopropene moiety32 of modified GENAEXT mRNA via iEDDA click chemistry. Efficient mRNA labeling using a tetrazine-fluorophore conjugate was first tested in an in vitro iEDDA click reaction. All sequences, either a modified or wild type of mCh_mRNA (Fig. 2g and ESI Fig. 1†) and mCh_mRNAUTR 2 (ESI Fig. 2†), were incubated with an excess of the tetrazine-conjugated fluorophore AF 488 (TET-FL) followed by agarose gel separation and fluorescent readout. mCh_mRNA1 CP and mCh_mRNA2 CP show a strong fluorescence signal (Fig. 2g on the left), whereas the control RNA mCh_mRNAWT, prepared in the presence of rTPT3CP TP from an unmodified DNA template, is only visualized by post-staining (ESI Fig. 3†). This confirms the successful introduction of rTPT3CP unnatural nucleotides into the transcribed RNAs and further excludes non-specific incorporation of rTPT3CP TP.33For in-cell mRNA visualization, the modified mCh_mRNA was transfected into human HeLa cells and subsequently subjected to fluorescent labeling via iEDDA click reactions in live cells. This gives a measure of the location of the transfected mRNA. The functionality of the mCh_mRNA can be monitored via fluorescence measurements of in-cell translation and expression of the encoded mCherry protein. UB modification sites within the mCherry coding mRNAs are exclusively placed in the mRNA's 3′-UTR. We anticipate that site-specific rTPT3CP modifications within the 3′-UTR do not impair cellular translational processes. Our site-specific approach avoids possible mutating readthroughs,42 truncation or inhibition during protein translation which might be caused by unnatural nucleotides positioned in the open reading frame. This makes it a facile straightforward procedure to analyze the functionality of modified mRNA.
A strong and increasing red fluorescent signal over time was observed by confocal fluorescence microscopy analysis for all transfected HeLa cells, showing efficient mCherry protein translation and expression in cellulo, independently of variant mCh_mRNA modifications (for different mCh_mRNA 3′-UTR sequences, see Table 1). Single or double rTPT3CP modified GENAEXT mRNA was evidentially shown to be translationally active.
A brighter red fluorescent signal and hence higher amounts of the mCherry protein are seen in cell nuclei for all mCh_mRNA sequence modifications. Analysis of the mCherry coding sequence revealed a nuclear localization sequence (ESI Fig. 35†) accountable for this observation.
mRNA visualization was initiated by adding cell permeable TET-FL to the cell medium for live cell RNA labeling by iEDDA click chemistry18 at different timepoints post transfection. Unspecific interactions of TET-FL with cellular components can be excluded (see control images in ESI Fig. 25†).
Confocal fluorescence microscopy analysis of transfected cells carrying GENAEXT mRNA reveals that a single, iEDDA clicked CP modification is sufficient for excellent visualization of the mRNA (ESI Fig. 19, 20, 22 and 23†). A double CP modification as for mCh_mRNA2 CP improves visualization strength by a brighter signal enhancing the signal-to-noise ratio (Fig. 3, ESI Fig. 19, 20, 22 and 23†). To allow higher resolution and light sensitivity compared to live cell microscopy, confocal fluorescence microscopy was performed on fixed cells. This also enabled the usage of identical laser adjustments for all samples and different time points, additionally allowing direct comparisons of signal intensities. Green spots of accumulated GENAEXT mCh_mRNA in the 6 h post transfection microscopy images arise from incompletely dissolved lipid vesicles from transfection as already reported earlier by us and others.43,44 24 h post transfection, as the mRNA has entirely escaped from the transfection vesicles, the signal for clicked GENAEXT mCh_mRNA is more evenly spread throughout the cell cytoplasm. Increase in cellular autofluorescence45–49 reduced the signal-to-background ratio of clicked mCh_mRNA 48 h post transfection. We further recorded a series of z-stacking images. Live-cell clicked GENAEXT mCh_mRNA is detected within the cells correlating with red fluorescent mCherry protein signals. Both signals show the brightest intensity in the mid-series z-stack images and attenuate towards the cell periphery (ESI Fig. 27†). The close-up images of mCh_mRNA2 CP transfected cells (ESI Fig. 26†) show the power of our approach to visualize subcellular mRNA localization.
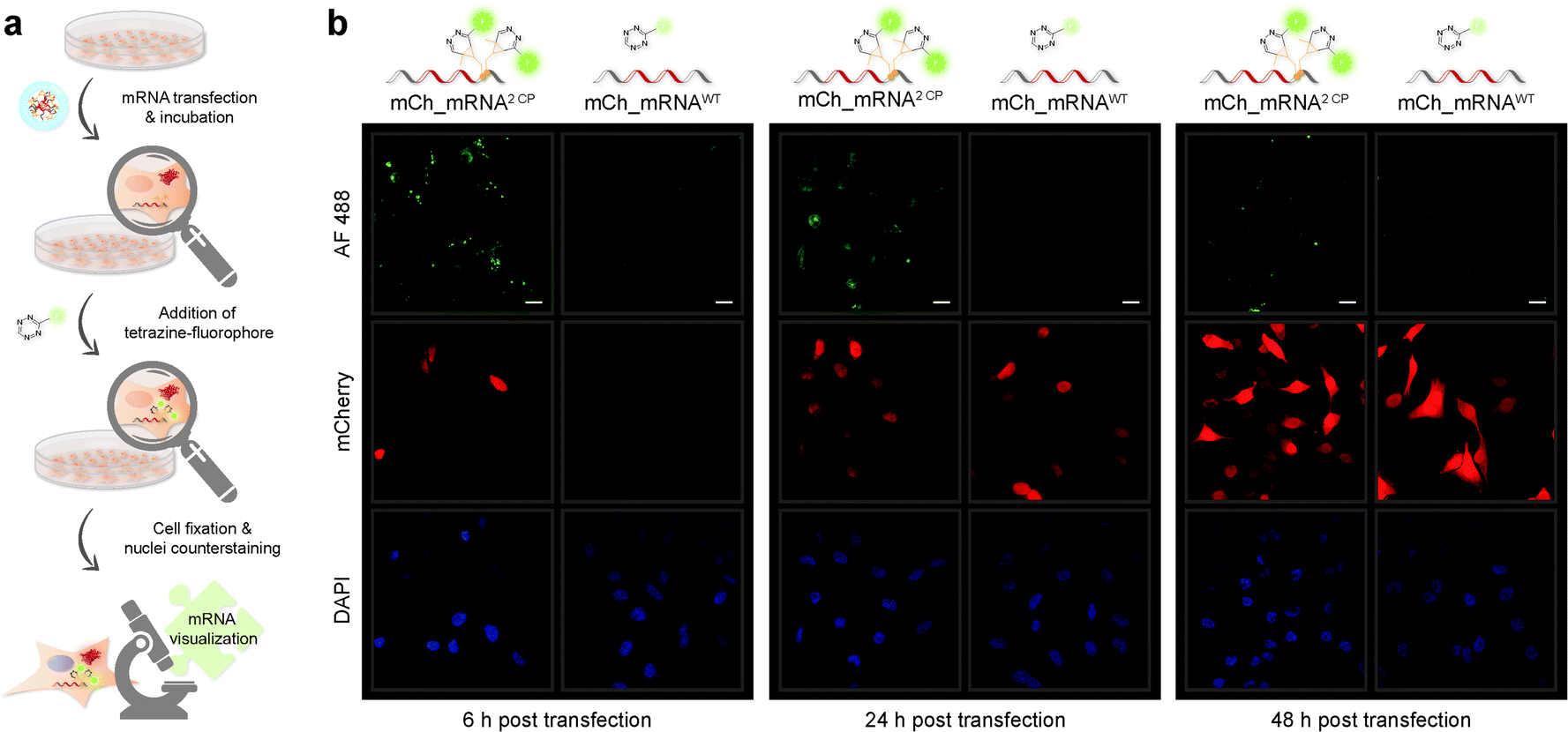 | ||
| Fig. 3 Visualization of cyclopropene modified IVT mRNA. (a) Schematic illustration of the workflow for CP modified mCh_mRNA. HeLa cells were transfected with mCh_mRNA and incubated to allow translation to proceed. Upon addition of TET-FL, the iEDDA click reaction yields fluorescently labeled mRNA. Subsequently, cells were fixed, and cell nuclei were counterstained. (b) HeLa cells transfected with either double CP modified mCh_mRNA2 CP (on the left in each panel) or unmodified mCh_mRNAWT (on the right in each panel). 6 h (left panel), 24 h (middle panel) or 48 h (right panel) post transfection cells were fixed prior to microscopy. Fluorescently labeled mCh_mRNA2 CP shows green fluorescent signals in the AF 488 channel (on the top of each panel). mCherry reporter protein fluorescence is visible with red fluorescence in the mCherry channel (in the middle of each panel). Counterstained cell nuclei show blue fluorescent signals in the DAPI channel (at the bottom of each panel). Scale bars correspond to 20 μm. | ||
Encapsulated, our method facilitates for spatiotemporal mRNA visualization in live cells with a steady green fluorescence and an excellent signal-to-noise ratio for at least 24 h post transfection. Due to advantages of the cell compatible iEDDA18,40 click chemistry, no washing steps are required prior to visualization or fixation.
Quantification of mRNA and protein levels in cells
In addition to microscopic analysis, we quantified cellular mCh_mRNA levels by RT-qPCR. Concurrently, a quantitative analysis of mCherry reporter protein expression in transfected cells was carried out.For analysis of mCh_mRNA levels by RT-qPCR, we designed two different sets of qPCR primers and hydrolysis probes. By this, we were able to compare the relative quantification results of the variant mCh_mRNA sequences at the mRNA 3′-end (Fig. 4b) and at an internal position within the mCherry protein coding region (Fig. 4c). UB modification sites were excluded for the 3′-end RT-qPCR sequence section, preventing defective reverse transcription caused by the incorporated UBs. Further, we intended to compare the 3′-end and internal RT-qPCR results, enabling an evaluation of mRNA decay for different mCh_mRNA variants.
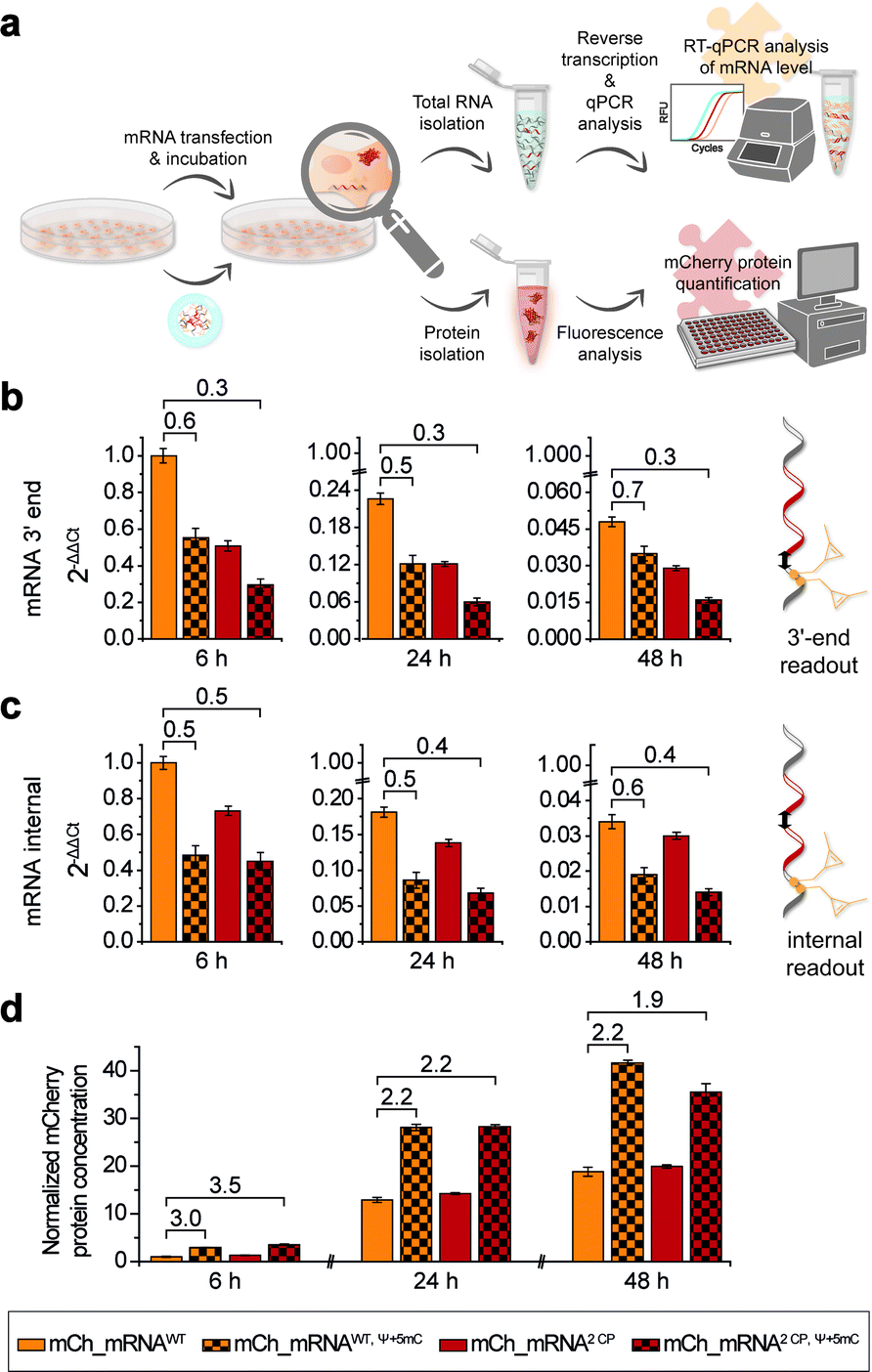 | ||
| Fig. 4 RT-qPCR analysis of IVT mRNA levels after transfection and mCherry protein quantification. (a) Schematic illustration of the workflow for CP modified mCh_mRNA. Seeded cells were transfected with mCh_mRNA, and then incubated and lysed at different time points to either isolate cellular total RNA for subsequent reverse transcription and qPCR analysis or to isolate cellular proteins and quantify mCherry protein concentrations via fluorescence analysis. (b) Fold change of mCh_mRNA amounts by means of 2ΔΔCt values calculated by RT-qPCR at the mCh_mRNA 3′-end using the mCh_qPCR3′-end primers and hydrolysis probe set. The 2ΔΔCt value of unmodified mCh_mRNAWT at 6 h post transfection was set to 1 (n = 4 for mCh_mRNAWT and mCh_mRNA2 CP, n = 2 for mCh_mRNAWT, Ψ+5mC and mCh_mRNA2 CP, Ψ+5mC, technical triplicates for each condition, and mean ± s.d.). (c) Fold change of mCh_mRNA amounts by means of 2ΔΔCt values calculated by RT-qPCR at an internal position within the protein-coding region of mCh_mRNA using the mCh_qPCRinternal primers and hydrolysis probe set. The 2ΔΔCt value of unmodified mCh_mRNAWT at 6 h post transfection was set to 1 (n = 4 for mCh_mRNAWT and mCh_mRNA2 CP, n = 2 for mCh_mRNAWT, Ψ+5mC and mCh_mRNA2 CP, Ψ+5mC, technical triplicates for each condition, mean ± s.d.). (d) Normalized mCherry protein concentrations. The concentration of unmodified mCh_mRNAWT 6 h post transfection was set to 1 (n = 20 for mCh_mRNAWT and mCh_mRNA2 CP, n = 10 for mCh_mRNAWT, Ψ+5mC and mCh_mRNA2 CP, Ψ+5mC, technical duplicates for each condition, mean ± s.d.). | ||
For RT-qPCR analysis, cells were lysed, and total cellular RNA was isolated at 6, 24 and 48 h post transfection in parallel to protein expression analysis. Reverse transcription from isolated total RNA was performed with a mixture of random primers and oligo(dT) primers. By this, an equal procedure was enabled for all GENAEXT and unmodified mCh_mRNA variants. qPCR was performed in a multiplex assay allowing for simultaneous analysis of mCh_mRNA at the mRNA 3′-end and an internal position together with a GAPDH mRNA sequence as a reference gene. ΔΔCt values were calculated to compare mRNA levels between different mCh_mRNA variants (Fig. 4b, c, ESI Fig. 28 and 29†). Sequence identities of qPCR products were further analyzed by Sanger sequencing (see the ESI†).
For evaluation of transfection efficiencies for different mCh_mRNA variants, we focus on data from the earliest time point after transfection (6 h). Kauffman et al. reported reduced levels for in vitro transfected Ψ-modified mRNA.50 These findings are in accordance with our data for combined Ψ and 5mC modified mCh_mRNA. Similarly, we observe a lower mRNA level for transfected GENAEXT mCh_mRNA containing rTPT3CP in contrast to the unmodified control mRNAs (mCh_mRNA2 CP and mCh_mRNA1 CPvs.mCh_mRNAWT, see Table 1).
For later time points, we accordingly observe reduced mRNA levels for both GENAEXT and Ψ and 5mC modified mCh_mRNA variants in comparison to mCh_mRNAWT (Fig. 4b, c, ESI Fig. 28 and 29†). However, we observe a convergency of GENAEXT and unmodified mCh_mRNA levels over time, if both mRNA variants did not contain additional Ψ and 5mC-modifications (Fig. 4b, c, ESI Fig. 28 and 29†). Thus, we conclude a decreased degradation of GENAEXT mRNA caused by the rTPT3CP modification. Exonucleolytic mRNA decay in the 3′–5′ direction by the exosome51,52 is most likely stalled by the rTPT3CP modification incorporated into GENAEXT mRNA. A similar convergency towards unmodified mCh_mRNA levels is observed for mCh_mRNAWT, Ψ+5mC compared to mCh_mRNAWT, indicating same decelerating effects on mRNA decay for Ψ and 5mC modifications in mCh_mRNA. As Ψ and 5mC modifications are incorporated randomly, structural impacts from chemical base alterations on reverse transcriptase activity can be excluded and considered a systemic factor. Hence, we assume the reported differences in mCh_mRNA amounts to be more likely caused by lipofection and stalled mRNA degradation. Notably, no convergency of mRNA levels is observed for twofold modified mCh_mRNA2 CP, Ψ+5mC in comparison to only Ψ and 5mC modified mCh_mRNAWT, Ψ+5mC. We assume that the effect of stalling mRNA degradation by Ψ and 5mC modifications that are distributed over the entire mRNA sequence predominates and hence overlays the decelerating effect of two site-specific rTPT3CP modifications in the 3′-UTR.
In order to quantify amounts of in cellulo translated and expressed mCherry mRNA, transfected cells were lysed, and isolated mCherry protein concentrations were calculated with the assistance of a linear mCherry protein standard added to each measurement (Fig. 4d and ESI Fig. 30†). In order to obtain conclusive protein expression data, we performed ten biologically independent experiments, each of two sets of different combinations of mCh_mRNA variants (complete data sets are presented in the ESI†).
mCherry protein quantification is in accordance with our findings during confocal fluorescence microscopy. GENAEXT CP-modified mCh_mRNAs were proven to be translationally active as no loss in the mRNA function with regard to protein expression is observed in comparison to unmodified mCh_mRNA. Instead, we found a slightly improved expression for GENAEXT mCh_mRNA2 CP and a highly improved expression for mCh_mRNA2 CP, Ψ+5mC in comparison to unmodified mCh_mRNAWT (Fig. 4d).
In general, GENAEXT mCh_mRNA and point-mutated canonical mCh_mRNA show notable differences of expressed protein levels in comparison to unmodified mCh_mRNA (ESI Fig. 30†). This context-dependency which means that different effects can be observed for different sequences and modification sites within the same sequence53 was reported for natural modifications before.15,54 However, exchanging the mCh_mRNA's 3′-UTR sequence led to drastic reduction for all mCh_mRNAUTR 2 variants (ESI Fig. 30†). General trends of protein expression levels for the different mCh_mRNAs remain unchanged over time. This is exemplarily shown for mCh_mRNAWT and mCh_mRNA2 CP either with or without Ψ and 5mC modifications in Fig. 4d.
mCherry protein expression levels were found to be highly increased when Ψ and 5mC modifications were included in the mRNA, underlining their importance for mRNA therapeutic efficacy.6,7 Relative mRNA quantification by RT-qPCR shows reduced transfection levels of 55% and less for mCh_mRNAWT, Ψ+5mC and mCh_mRNA2 CP, Ψ+5mC compared to mCh_mRNAWT, respectively. Observed protein amounts 6 h post transfection show a threefold increase in mCherry protein levels for mCh_mRNAWT, Ψ+5mC and mCh_mRNA2 CP, Ψ+5mC in comparison to mCh_mRNAWT. At later timepoints, the increase in mCherry protein levels remains doubled for both Ψ and 5mC modified mCh_mRNAs.
Most astonishingly, all RT-qPCR measurements revealed lower GENAEXT mCh_mRNA2 CP, Ψ+5mC levels compared to mCh_mRNAWT, Ψ+5mC, but mCherry protein expression from GENAEXT mCh_mRNA2 CP, Ψ+5mC was found to be similarly high compared to protein expression from mCh_mRNAWT, Ψ+5mC. This indicates an improved protein expression which counterbalances the reduced amount of mRNA. Hereby, our data can clearly illustrate that GENAEXT modifications can be included into functional RNA without hampering its biological function.
Cell viability after mRNA transfection and live cell click reaction
In addition, and in order to assess future in vivo application in the course of mRNA transfection treatment and in cell click modification, we investigated cell health. Therefore, we used a nonlytic assay constantly measuring the cellular reducing potential to monitor cell viability. Measurements were performed starting from 6 h post mCh_mRNA transfection, covering the incubation time and addition and subsequent influence of TET-FL (Fig. 5 and ESI Fig. 31†). TET-FL was added 24 h post transfection to ensure assay linearity which was determined in the initial experiments and limited to about 30 h (ESI Fig. 32†). As known from the literature, lipofection can negatively affect cell viability and proliferation.55,56 In accordance to this, all cells transfected with mCh_mRNA variants showed a slightly reduced cell viability in comparison to untreated cells and the transfection control with water (Fig. 5b). Additional Ψ and 5mC modifications improved the cell viability noticeably, underlining the importance of these modifications in transfected mRNAs.5,8 Moreover, transfected GENAEXT mRNA showed no reduction in cell viability. Compared to unmodified mCh_mRNAWT or mCh_mRNAWT, Ψ+5mC, all transfected GENAEXT mRNAs are at similar viability levels. The highest viability values are observed for GENAEXT mCh_mRNA with additional Ψ and 5mC modifications. Time dependent cell viability of all transfected mCh_mRNAs is shown in the ESI (Fig. 31†).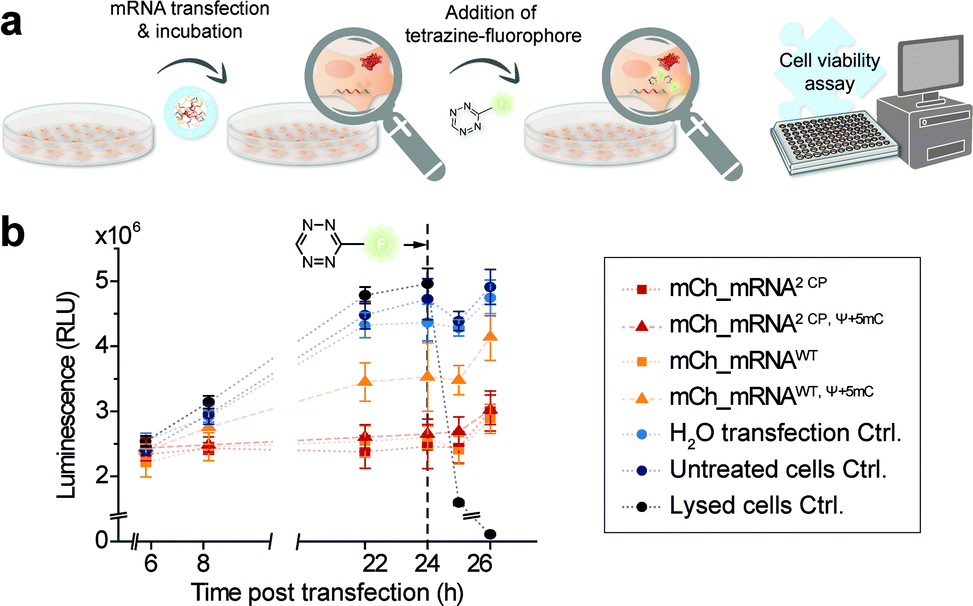 | ||
| Fig. 5 Cell viability assay to evaluate effects of mCh_mRNA transfection and live cell click reaction. (a) Schematic illustration of the workflow for CP modified mCh_mRNA. Seeded cells were transfected with mCh_mRNA variants and incubated and treated with TET-FL 24 h after transfection. The reducing potential of differently treaded cells was measured by a luciferase-coupled, nonlytic assay at several time points. (b) Measured luminescence directly refers to cell viability by means of the luciferase-coupled reaction in order to analyze the reducing potential of viable cells. Instead of TET-FL, the lysed cell controls were treated with Triton X-100 24 h post transfection (n = 1, technical triplicates of each condition, mean ± s.d.). | ||
In the context of our experiments, addition of TET-FL does not affect cell viability either. Only a minor decrease in the measured luminescence is detected one hour after tetrazine addition, and luminescence increases again subsequently. As an additional control for maximal loss of the measured luminescence signal and thus cell viability, former untransfected cells were treated with Triton X-100 simultaneously with tetrazine addition.
Taken together, our data emphasize the beneficial effect on cell viability when natural base modifications Ψ and 5mC are included in the mRNA sequence. A combination of our rTPT3CP modification together with Ψ and 5mC can equally successfully be applied with regard to cell viability.
Discussion
Taken together both the results for relative quantification of mCh_mRNA levels by RT-qPCR and mCherry protein quantification, we demonstrate efficient translation of IVT mRNAs modified with UBs in combination with natural RNA modifications in cells. Similar translation efficiencies for double CP modified and unmodified mCh_mRNA prove no impairment of ribosome activity by GENAEXT mRNA in transfected cells. Reduced mRNA amounts as found by RT-qPCR suggest an even improved protein translation for mCh_mRNA2 CP, Ψ+5mC and mCh_mRNA2 CP in comparison to mCh_mRNAWT, Ψ+5mC and mCh_mRNAWT, respectively. Additional Ψ and 5mC base alterations were shown to strongly increase mRNA translation and protein expression.Elevated protein expression from IVT mRNA bearing natural RNA modifications such as Ψ and 5mC has provisionally been attributed to altered interactions with the ribosome: ribosome-pausing has been shown to substantially affect the folding of nascent polypeptide chains.57 Translation from mRNAs comprising modifications most likely leads to correctly folded proteins by inducing ribosome pausing events alongside the mRNA sequence statistically modified with natural RNA modifications such as Ψ and 5mC in this work. Additional modifications with the CP-modified rTPT3 nucleobase in the mRNA's 3′-UTR further improve protein translation. Differences in mRNA stability by reduced RNA degradation were found for Ψ and 5mC modified mCh_mRNAWT, Ψ+5mC as well as for CP-modified mCh_mRNA2 CP in comparison to unmodified mCh_mRNAWT, but not for twofold modified mCh_mRNA2 CP, Ψ+5mC in comparison to unmodified mCh_mRNAWT, Ψ+5mC. Thus, we concluded a dominant impact reducing mRNA degradation by multifold included modified natural bases Ψ and 5mC in relation to a couple of site-specific rTPT3CP modifications. Surprisingly, twofold modified mCh_mRNA2 CP, Ψ+5mC resulted in the highest protein expression with regard to reduced amounts of mRNA analyzed by RT-qPCR. As we can further rule out improved transfection efficiencies, we propose regulatory mechanisms positively affecting the translational processes, either via activating translation or inhibiting translation repression. RNA-binding proteins (RBPs) bind to 3′-UTR cis-elements both in a sequence and structure dependent manner to recruit effector proteins.58 Interactions of the CP-modified 3′-UTR section with regulatory factors may promote efficient formation of translationally active complexes and thus contribute to an improved translation. Alternatively, microRNAs (miRNAs) have been shown to repress translation as they target complementary sequences in the 3′-UTR.58 Mutations in the 3′-UTR with the CP-modified rTPT3 nucleobase incidentally targeting a yet unknown miRNA recognition site may possibly hinder translational interference. Future studies will be focused on investigating the role of rTPT3CP modifications in various 3′-UTR sequences to answer this question.
In our study, we could show that the combination of both chemical base alterations of Ψ and 5mC together with site-specific introduction of the unnatural rTPT3CP nucleobase in the mRNA's 3′-UTR results in a synergistic effect, notably increasing translational efficiency.
Conclusions
In this study, we demonstrate synthesis and application of a functional and protein-encoding GENAEXT mRNA with site-specific modifications in its 3′-UTR for the first time. This modification, the cyclopropene modified rTPT3, is introduced in a facile and quick manner and does not affect mRNA functionality when incorporated into its 3′-UTR. Live cell iEDDA click reaction of cyclopropene modified GENAEXT mRNA with a tetrazine-fluorophore derivative enabled mRNA visualization after transfection into HeLa cells by confocal fluorescence microscopy. We show that our method is not only capable for in-cell mRNA visualization but also a powerful tool for localization and time-dependent tracking of mRNAs in living cells.mRNA levels after transfection into cells and expression of the fluorescent reporter protein mCherry were studied by RT-qPCR and fluorescence quantification, respectively. The obtained data confirm the benefit of naturally occurring RNA modifications Ψ and 5mC2–4 for protein expression. Transfection proceeds with lower efficiency for modified mRNA, GENAEXT mRNA or with natural base modifications only. This is counterbalanced by an outstanding mRNA translation efficiency. GENAEXT mRNA with additional natural base modifications Ψ and 5mC represents a powerful synergetic tool to monitor, control and consequently improve mRNA translation, mRNA stability, protein expression, and ultimately cell viability.
The GENAEXT approach can be applied to desired mRNA sequences of arbitrary length. Functionalization of the mRNA by iEDDA click reaction with tetrazine derivatives allows a plethora of applications. Besides attachment of various reporter groups for in-cell experiments, this approach could be employed for in vitro pull-down experiments identifying mRNA binding proteins in future.
Further development of this approach will open up new and promising possibilities for fine tuning of mRNA characteristics and efficient packing and delivery of mRNAs into cells by covalent attachment to carrier systems. Moreover, by the introduction of artificial regulatory elements in untranslated regions of mRNAs, potential to control mRNA translation may be unlocked.
Data availability
All raw data have been included in the ESI.†Author contributions
L. B., C. D. and S. K. S. conceived the project; L. B. and S. K. S. designed the experiments; L. B. performed experiments, L. B., C. D. and S. K. S. analysed the data and wrote the manuscript, S. K. S. acquired funding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has received financial support from the Boehringer Ingelheim Stiftung (Plus 3 Grant to S. Kath-Schorr) and the Deutsche Forschungsgemeinschaft (KA 3699/6-1). We thank Dr Frank Eggert for support in chemical synthesis.References
- A. Vitiello and F. Ferrara, Brief review of the mRNA vaccines COVID-19, Inflammopharmacology, 2021, 29, 645–649 CrossRef CAS PubMed.
- K. Karikó, et al., Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability, Mol. Ther., 2008, 16, 1833–1840 CrossRef PubMed.
- K. Karikó, et al., Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA, Nucleic Acids Res., 2011, 39, e142 CrossRef PubMed.
- B. R. Anderson, et al., Incorporation of pseudouridine into mRNA enhances translation by diminishing PKR activation, Nucleic Acids Res., 2010, 38, 5884–5892 CrossRef CAS PubMed.
- K. Karikó, et al., Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA, Immunity, 2005, 23, 165–175 CrossRef PubMed.
- K. D. Nance and J. L. Meier, Modifications in an Emergency: The Role of N1-Methylpseudouridine in COVID-19 Vaccines, ACS Cent. Sci., 2021, 7, 748–756 CrossRef CAS PubMed.
- J. W. Park, et al., mRNA vaccines for COVID-19: what, why and how, Int. J. Biol. Sci., 2021, 17, 1446–1460 CrossRef CAS PubMed.
- L. Warren, et al., Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA, Cell Stem Cell, 2010, 7, 618–630 CrossRef CAS PubMed.
- D. R. Davis, Stabilization of RNA stacking by pseudouridine, Nucleic Acids Res., 1995, 23, 5020–5026 CrossRef CAS PubMed.
- Y.-C. Chang, et al., Synthesis and solution conformation studies of 3-substituted uridine and pseudouridine derivatives, Bioorg. Med. Chem., 2008, 16, 2676–2686 CrossRef CAS PubMed.
- E. Kierzek, et al., The contribution of pseudouridine to stabilities and structure of RNAs, Nucleic Acids Res., 2014, 42, 3492–3501 CrossRef CAS PubMed.
- D. M. Mauger, et al., mRNA structure regulates protein expression through changes in functional half-life, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 24075–24083 CrossRef CAS PubMed.
- Y. V. Svitkin, et al., N1-methyl-pseudouridine in mRNA enhances translation through eIF2α-dependent and independent mechanisms by increasing ribosome density, Nucleic Acids Res., 2017, 45, 6023–6036 CrossRef CAS PubMed.
- P. Dave, E. Griesbach, G. Roth, D. Mateju and J. A. Chao, Single-molecule imaging reveals the coupling of translation and mRNA decay, bioRxiv, 2021 DOI:10.2139/ssrn.3885982.
- I. A. Roundtree, et al., Dynamic RNA Modifications in Gene Expression Regulation, Cell, 2017, 169, 1187–1200 CrossRef CAS PubMed.
- H. Depmeier, et al., Strategies for Covalent Labeling of Long RNAs, ChemBioChem, 2021, 22, 2826–2847 CrossRef CAS PubMed.
- S. Croce, et al., Chemoenzymatic Preparation of Functional Click-Labeled Messenger RNA, ChemBioChem, 2020, 21, 1641–1646 CrossRef CAS PubMed.
- A.-C. Knall and C. Slugovc, Inverse electron demand Diels-Alder (iEDDA)-initiated conjugation: a (high) potential click chemistry scheme, Chem. Soc. Rev., 2013, 42, 5131–5142 RSC.
- J. K. Bashkin and L. A. Jenkins, The Role of Metals in the Hydrolytic Cleavage of DNA and RNA, Comments Inorg. Chem., 1994, 16, 77–93 CrossRef CAS.
- L. Anhäuser, S. Hüwel, T. Zobel and A. Rentmeister, Multiple covalent fluorescence labeling of eukaryotic mRNA at the poly(A) tail enhances translation and can be performed in living cells, Nucleic Acids Res., 2019, 47, e42 CrossRef PubMed.
- D. A. Malyshev and F. E. Romesberg, The expanded genetic alphabet, Angew. Chem., Int. Ed., 2015, 54, 11930–11944 CrossRef CAS PubMed.
- Y. J. Seo, et al., Transcription of an expanded genetic alphabet, J. Am. Chem. Soc., 2009, 131, 5046–5047 CrossRef CAS PubMed.
- D. A. Malyshev, et al., PCR with an expanded genetic alphabet, J. Am. Chem. Soc., 2009, 131, 14620–14621 CrossRef CAS PubMed.
- A. M. Leconte and F. E. Romesberg, Amplify this! DNA and RNA get a third base pair, Nat. Methods, 2006, 3, 667–668 CrossRef CAS PubMed.
- I. Hirao, et al., An efficient unnatural base pair for PCR amplification, J. Am. Chem. Soc., 2007, 129, 15549–15555 CrossRef CAS PubMed.
- T. Mitsui, et al., An efficient unnatural base pair for a base-pair-expanded transcription system, J. Am. Chem. Soc., 2005, 127, 8652–8658 CrossRef CAS PubMed.
- I. Hirao, et al., A two-unnatural-base-pair system toward the expansion of the genetic code, J. Am. Chem. Soc., 2004, 126, 13298–13305 CrossRef CAS PubMed.
- A. W. Feldman and F. E. Romesberg, Expansion of the Genetic Alphabet: A Chemist's Approach to Synthetic Biology, Acc. Chem. Res., 2018, 51, 394–403 CrossRef CAS PubMed.
- M. Kimoto and I. Hirao, Genetic alphabet expansion technology by creating unnatural base pairs, Chem. Soc. Rev., 2020, 49, 7602–7626 RSC.
- C. Domnick, et al., Posttranscriptional spin labeling of RNA by tetrazine-based cycloaddition, Org. Biomol. Chem., 2019, 17, 1805–1808 RSC.
- C. Domnick, et al., Site-specific enzymatic introduction of a norbornene modified unnatural base into RNA and application in post-transcriptional labeling, Chem. Commun., 2015, 51, 8253–8256 RSC.
- F. Eggert and S. Kath-Schorr, A cyclopropene-modified nucleotide for site-specific RNA labeling using genetic alphabet expansion transcription, Chem. Commun., 2016, 52, 7284–7287 RSC.
- F. Eggert, et al., Illuminated by foreign letters – strategies for site-specific cyclopropene modification of large functional RNAs via in vitro transcription, Methods, 2017, 120, 17–27 CrossRef CAS PubMed.
- D. R. Gallie, The cap and poly(A) tail function synergistically to regulate mRNA translational efficiency, Genes Dev., 1991, 5, 2108–2116 CrossRef CAS PubMed.
- L. Li, et al., Natural-like replication of an unnatural base pair for the expansion of the genetic alphabet and biotechnology applications, J. Am. Chem. Soc., 2014, 136, 826–829 CrossRef CAS PubMed.
- N. C. Shaner, et al., Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein, Nat. Biotechnol., 2004, 22, 1567–1572 CrossRef CAS PubMed.
- M. L. Blackman, et al., Tetrazine ligation: fast bioconjugation based on inverse-electron-demand Diels-Alder reactivity, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS PubMed.
- N. K. Devaraj, et al., Tetrazine-based cycloadditions: application to pretargeted live cell imaging, Bioconjugate Chem., 2008, 19, 2297–2299 CrossRef CAS PubMed.
- J. Yang, et al., Synthesis and reactivity comparisons of 1-methyl-3-substituted cyclopropene mini-tags for tetrazine bioorthogonal reactions, Chem.–Eur. J., 2014, 20, 3365–3375 CrossRef CAS PubMed.
- N. Devaraj, Advancing Tetrazine Bioorthogonal Reactions through the Development of New Synthetic Tools, Synlett, 2012, 23, 2147–2152 CrossRef CAS.
- C. Domnick, et al., EPR Distance Measurements on Long Non-coding RNAs Empowered by Genetic Alphabet Expansion Transcription, Angew. Chem., Int. Ed., 2020, 59, 7891–7896 CrossRef CAS PubMed.
- A. X.-Z. Zhou, et al., Progress toward Eukaryotic Semisynthetic Organisms: Translation of Unnatural Codons, J. Am. Chem. Soc., 2019, 141, 20166–20170 CrossRef CAS PubMed.
- A. M. Pyka, et al., Diels-Alder cycloadditions on synthetic RNA in mammalian cells, Bioconjugate Chem., 2014, 25, 1438–1443 CrossRef CAS PubMed.
- J. L. Kirschman, et al., Characterizing exogenous mRNA delivery, trafficking, cytoplasmic release and RNA-protein correlations at the level of single cells, Nucleic Acids Res., 2017, 45, e113 CrossRef CAS PubMed.
- J. Surre, et al., Strong increase in the autofluorescence of cells signals struggle for survival, Sci. Rep., 2018, 8, 12088 CrossRef PubMed.
- O. I. Kolenc and K. P. Quinn, Evaluating Cell Metabolism Through Autofluorescence Imaging of NAD(P)H and FAD, Antioxid. Redox Signaling, 2019, 30, 875–889 CrossRef CAS PubMed.
- A. A. Heikal, Intracellular coenzymes as natural biomarkers for metabolic activities and mitochondrial anomalies, Biomarkers Med., 2010, 4, 241–263 CrossRef CAS PubMed.
- F. Bartolomé and A. Y. Abramov, in Probing mitochondrial function, ed. V. Weissig and M. Edeas, Humana Pr, New York, NY, 2015, pp. 263–270 Search PubMed.
- A. Fiszer-Kierzkowska, et al., Liposome-based DNA carriers may induce cellular stress response and change gene expression pattern in transfected cells, BMC Mol. Biol., 2011, 12, 27 CrossRef CAS PubMed.
- K. J. Kauffman, et al., Efficacy and immunogenicity of unmodified and pseudouridine-modified mRNA delivered systemically with lipid nanoparticles in vivo, Biomaterials, 2016, 109, 78–87 CrossRef CAS PubMed.
- A. Chlebowski, et al., RNA decay machines: the exosome, Biochim. Biophys. Acta, 2013, 1829, 552–560 CrossRef CAS PubMed.
- J. C. Zinder and C. D. Lima, Targeting RNA for processing or destruction by the eukaryotic RNA exosome and its cofactors, Genes Dev., 2017, 31, 88–100 CrossRef CAS PubMed.
- S. H. Boo and Y. K. Kim, The emerging role of RNA modifications in the regulation of mRNA stability, Exp. Mol. Med., 2020, 52, 400–408 CrossRef CAS PubMed.
- H. Shi, et al., Where, When, and How: Context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers, Mol. Cell, 2019, 74, 640–650 CrossRef CAS PubMed.
- Y. Granot and D. Peer, Delivering the right message: challenges and opportunities in lipid nanoparticles-mediated modified mRNA therapeutics-an innate immune system standpoint, Semin. Immunol., 2017, 34, 68–77 CrossRef CAS PubMed.
- A. Wadhwa, et al., Opportunities and Challenges in the Delivery of mRNA-based Vaccines, Pharmaceutics, 2020, 12(2), 102 CrossRef CAS PubMed.
- A. A. Komar, A pause for thought along the co-translational folding pathway, Trends Biochem. Sci., 2009, 34, 16–24 CrossRef CAS PubMed.
- C. Mayr, Regulation by 3'-Untranslated Regions, Annu. Rev. Genet., 2017, 51, 171–194 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2sc00670g |
| This journal is © The Royal Society of Chemistry 2022 |