
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sulfonium cations as versatile strongly π-acidic ligands†
Ruiping
Li
 ,
Nitsan
Barel
,
Vasudevan
Subramaniyan
,
Orit
Cohen
,
Françoise
Tibika
and
Yuri
Tulchinsky
*
,
Nitsan
Barel
,
Vasudevan
Subramaniyan
,
Orit
Cohen
,
Françoise
Tibika
and
Yuri
Tulchinsky
*
Institute of Chemistry, The Hebrew University of Jerusalem, Jerusalem, 9190401, Israel. E-mail: yuri.tulchinsky@mail.huji.ac.il
First published on 14th March 2022
Abstract
More than a century old, sulfonium cations are still intriguing species in the landscape of organic chemistry. On one hand they have found broad applications in organic synthesis and materials science, but on the other hand, while isoelectronic to the ubiquitous tertiary phosphine ligands, their own coordination chemistry has been neglected for the last three decades. Here we report the synthesis and full characterization of the first Rh(I) and Pt(II) complexes of sulfonium. Moreover, for the first time, coordination of an aromatic sulfonium has been established. A thorough computational analysis of the exceptionally short S–Rh bonds obtained attests to the strongly π-accepting nature of sulfonium cations and places them among the best π-acceptor ligands available today. Our calculations also show that embedding within a pincer framework enhances their π-acidity even further. Therefore, in addition to the stability and modularity that these frameworks offer, our pincer complexes might open the way for sulfonium cations to become powerful tools in π-acid catalysis.
Introduction
Rethinking the coordination chemistry of main group elements has often led to breakthroughs in metal-based homogeneous catalysis. For instance, extending the chemistry of B, Al, Ga, Sn, and Bi gave birth to the concept of σ-acceptor (aka Z-type) ligands.1 Peters,2 Lu3 and others4 have used complexes of these ligands for such fundamentally important processes as N2 fixation, CO2 reduction, and H2 activation.The electron-withdrawing nature of Z-type ligands also offered new opportunities for π-acid catalysis, as demonstrated by Inagaki with borane-based pincer ligands,5 and Gabbai with ligands based on antimony,6 and carbenium cations.7 On the other hand, a significant advance in π-acid catalysis was achieved by Alcarazo by stretching the π-acceptor properties of phosphine8 and arsine9 to the extreme through the introduction of positively charged substituents.
While seeking to unravel new facets of main group chemistry, the coordination properties of another main-group species, sulfonium cations, have been greatly overlooked. Yet, sulfonium salts are at the forefront of fundamental and applied research due to their countless applications as precursors for sulfur ylides,10 alkyl and aryl group sources in cross-coupling reactions,11 photoacids,12 and many others.13
Compared to isoelectronic and isostructural tertiary phosphines, sulfonium cations have their lone pair stabilized by their positive charge, while their low-lying S–C σ*-orbitals become available for accepting electron density. Therefore, together with sulfoxonium, they have attracted attention as non-metal Lewis acids14 and have been utilized as such for catalysis and anion sensing.15 However, while tertiary phosphines are perhaps the most iconic family of ligands, only three crystallographically characterized sulfonium complexes of Mo(0) and Mn(I) were reported decades ago (Chart 1a), where these ligands exhibited strongly π-acidic character.16 Yet, no sulfonium complexes relevant to catalysis have ever been reported, even though formation of transient metal-coordinated sulfonium intermediates during Pd catalyzed cross-coupling reactions of sulfonium salts has been suggested.11a
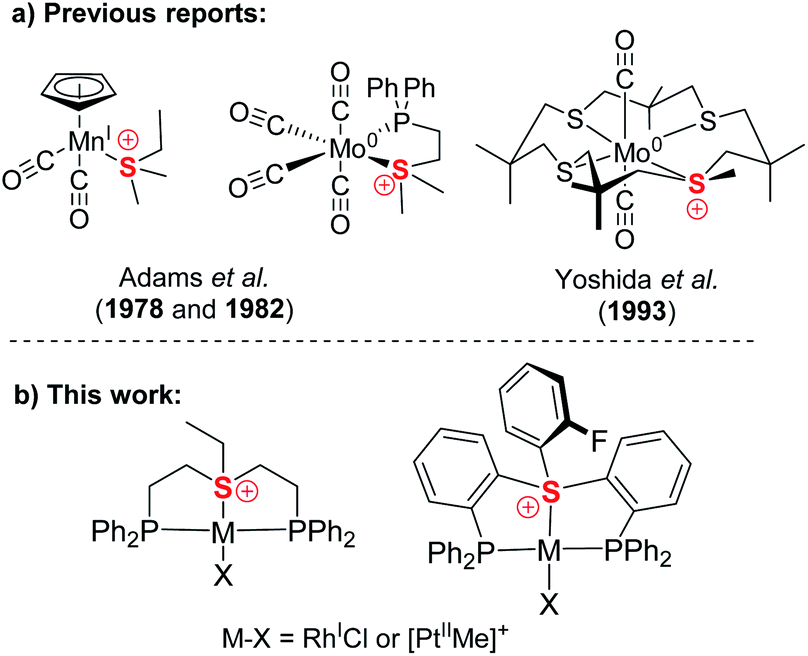 | ||
| Chart 1 Previously reported sulfonium complexes (a) compared to the pincer type sulfonium complexes presented in the work (b). | ||
Here we report the first synthesis and characterization of a series of complexes of both aliphatic and aromatic sulfonium cations with Rh(I) and Pt(II), two representatives of the Pt metal group,17 which lies at the core of today's homogeneous catalysis (Chart 1b). Our in-depth theoretical analysis of sulfonium–metal interaction demonstrated it to be dominated by π-back bonding. This strongly π-acidic character is further enhanced by the pincer frameworks, which also provide our complexes with structural robustness and modularity, both properties of pivotal importance in catalysis.18
Results and discussion
Ligand design and synthesis
Obviously, coordination of the sulfonium cation is hindered by an electrostatic repulsion between its positive charge and that of a metal center (even if partial). So far, the preparation of sulfonium complexes has been achieved by alkylation of the corresponding sulfide complexes. We adopted here a more systematic approach, where the aliphatic or aromatic sulfonium moieties were incorporated within pincer frameworks (I and II, respectively in Chart 2), bearing chelating phosphine arms. A similar strategy was used earlier by Gandelman to achieve coordination of the nitrenium cation.19We designed aliphatic and aromatic sulfonium ligands with NMR active nuclei in the vicinity of sulfur, namely methylene protons in I and a fluorine atom in II (Chart 2), that would allow detecting the formation of an S–M bond in solution, by tracing their chemical shifts and magnetic coupling to NMR-active metal centers, 103Rh and 195Pt.
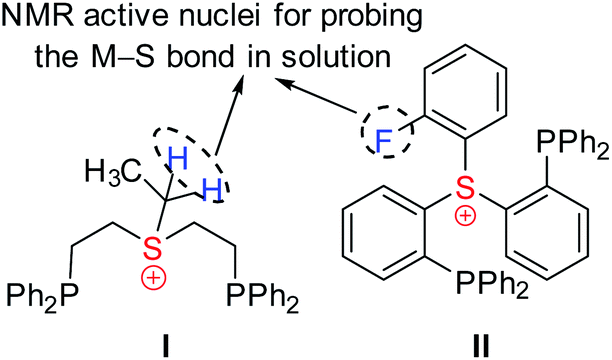 | ||
| Chart 2 Design of sulfonium-based pincer ligands. | ||
Both sulfonium pincer ligands were prepared by alkylation or arylation of the corresponding bis-phosphine sulfide ligands20 with the phosphines protected as borane adducts or phosphine oxides in aliphatic and aromatic systems, respectively (Scheme 1), resulting after deprotection in ligands 4a[OTf] and 4b[OTf]. To obtain XRD structures of sulfonium ligands (Fig. 1) or their complexes (Fig. 3 and 4, vide infra) the triflate counterions were in some cases exchanged for tetraphenylborate or hexafluorophosphate.
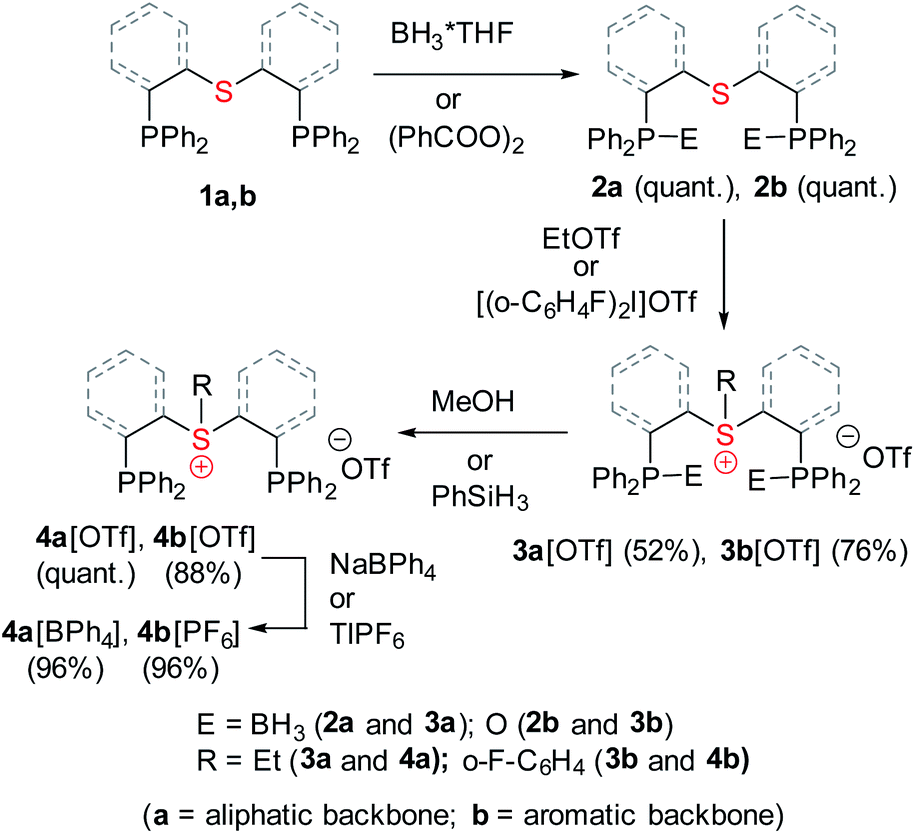 | ||
| Scheme 1 Synthesis of aliphatic and aromatic sulfonium pincer ligands. | ||
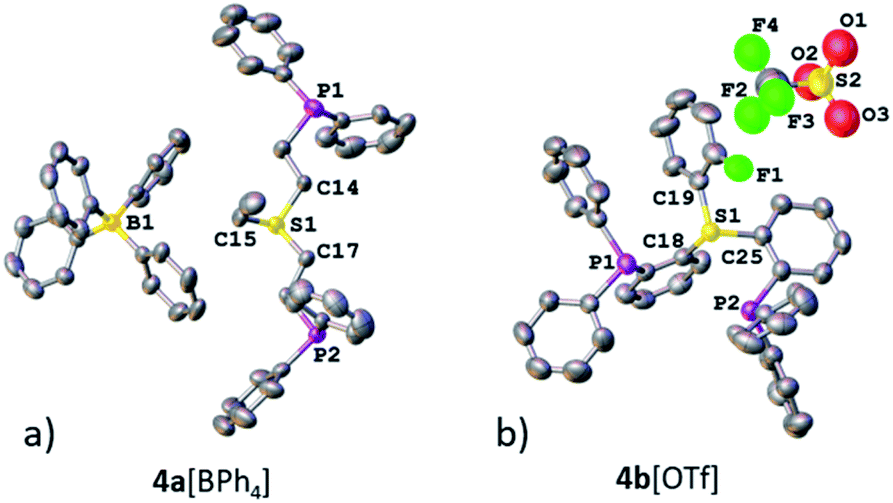 | ||
| Fig. 1 XRD structures of ligands 4a[BPh4] (a) and 4b[OTf] (b). Co-crystallized solvent molecules and hydrogen atoms are omitted for clarity. | ||
Synthesis and characterization of the Rh(I)–sulfonium complexes
The coordinative behavior of the aliphatic sulfonium ligand 4a[OTf] towards Rh(I) was tested by reacting it with [RhCl(COE)2]2 (Scheme 2). A full conversion to a symmetric Rh(I) complex was evident by 31P NMR, as the chemical shift moved from a singlet at −18.2 ppm to a doublet at +46.6 ppm (1JRh–P = 127.8 Hz).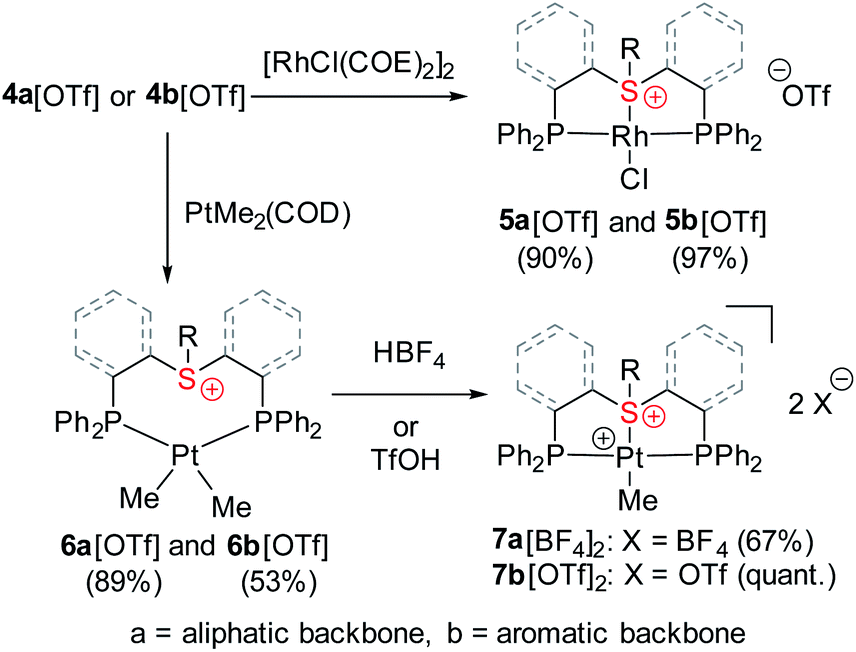 | ||
| Scheme 2 Synthesis of the Rh(I) and Pt(II)–sulfonium complexes. | ||
In the 1H NMR spectrum, significant downfield shifts of all aliphatic signals are observed (Fig. 2). Each of the methylene protons signals a and b divides upon coordination into two (a* and b* pairs, respectively), indicating the formation of a rigid structure with no rotation around C–C bonds. Furthermore, an additional splitting of 1.3 Hz appears in the quartet assigned to the ethyl tail methylene protons (c*). By means of 1H–103Rh HMBC (Fig. S3†), this splitting has been attributed to a through-bond 3JRh–H interaction. The latter is only possible if sulfonium is coordinated to the Rh center.
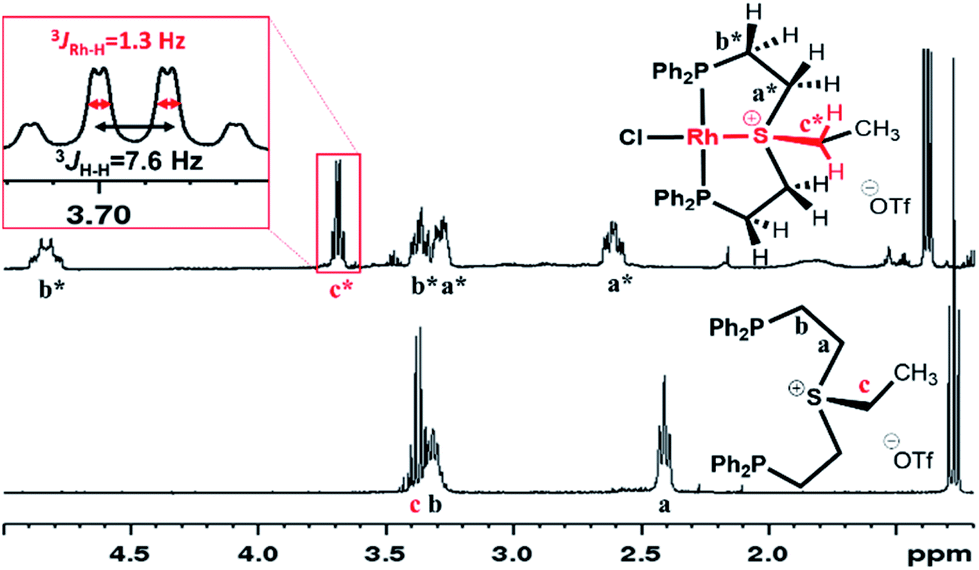 | ||
| Fig. 2 Aliphatic region of 1H NMR spectra of 4a[OTf] and 5a[OTf] in CD2Cl2. | ||
Encouraged by these results, we then turned to the aromatic ligand 4b[OTf] (Scheme 2). Here also, a full conversion of the ligand to a symmetric Rh(I) complex 5b[OTf] was evident from the 31P NMR spectrum, where the chemical shift changed from a singlet at −13.0 ppm to a doublet of doublets at +48.7 ppm (1JRh–P = 126.0 Hz; 5JF–P = 6.0 Hz). Interestingly, the 31P–19F interaction unobservable in the spectrum of the free ligand became noticeable after coordination, perhaps due to the additional rigidity of the formed complex.
The 19F NMR spectrum of 5b[OTf] showed only a small downfield shift compared to the free ligand (−104.1 vs. −105.3 ppm, respectively) and no additional splitting by 103Rh could be identified. Likewise, no 19F–103Rh interactions could be detected by HMBC, hence in this case, metal coordination to the aromatic sulfonium moiety could not be validated by NMR alone.
Nevertheless, the irrefutable evidence of sulfonium–Rh bonding in both systems was provided by XRD. Both complexes 5a[BPh4] and 5b[PF6] exhibited a slightly distorted square-planar geometry around the metal (with a τ parameter of 0.1, Table 1), typical of d8 complexes (Fig. 3a and b, respectively). Notably, the sulfonium–Rh(I) bond lengths of 2.126(2) and 2.112(1) Å observed in 5a[BPh4] and 5b[PF6], respectively, are among the shortest reported S–Rh bonds (Table 1). These are significantly shorter than in Rh(I) complexes with sulfides (>2.24 Å) and even with sulfoxides (typically, 2.159–2.291 Å).21 In fact, shorter Rh(I)–S bonds (2.069–2.100 Å) were only observed with the strongest π-acceptor ligands: SO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 22 and the related N-sulfinylaniline.23 These exceptionally short S–Rh bonds in 5a[BPh4] and 5b[PF6] cannot be explained solely by the grip of the pincer framework. Indeed, in both the analogous aliphatic sulfoxide pincer complex 8 that we prepared for comparison (Fig. S17†) and the reported aromatic ones,24 the Rh–S bonds are still longer than in their sulfonium counterparts (2.135 and 2.134 Å, respectively).
22 and the related N-sulfinylaniline.23 These exceptionally short S–Rh bonds in 5a[BPh4] and 5b[PF6] cannot be explained solely by the grip of the pincer framework. Indeed, in both the analogous aliphatic sulfoxide pincer complex 8 that we prepared for comparison (Fig. S17†) and the reported aromatic ones,24 the Rh–S bonds are still longer than in their sulfonium counterparts (2.135 and 2.134 Å, respectively).
| Complex | S–M bond length (Å) | Rh–Cl or Pt–Me bond length (Å) | Average M–P bond length (Å) | S–M–X and P–M–P angles (°) | Geometry index (τ4) | |
|---|---|---|---|---|---|---|
| Rh–Cl | 5a[BPh4] | 2.126(2) | 2.340(2) | 2.296(2) | 179.44(9), 164.80(9) | 0.11 |
| 5b[PF6] | 2.112(1) | 2.324(1) | 2.295(1) | 178.8(1), 166.0(1) | 0.10 | |
| 8 | 2.135(1) | 2.369(1) | 2.313(1) | 172.4(1), 161.2(1) | 0.18 | |
| Pt–Me | 7a[BF4]2 | 2.258(1) | 2.073(5) | 2.304(1) | 177.8(2), 167.3(1) | 0.10 |
| 7b[NTf2]2 | 2.261(1) | 2.060(4) | 2.292(1) | 178.7(2), 165.0(1) | 0.11 | |
| 9[BF4] | 2.336(2) | 2.087(7) | 2.278(2) | 177.6(3), 168.8(1) | 0.10 |
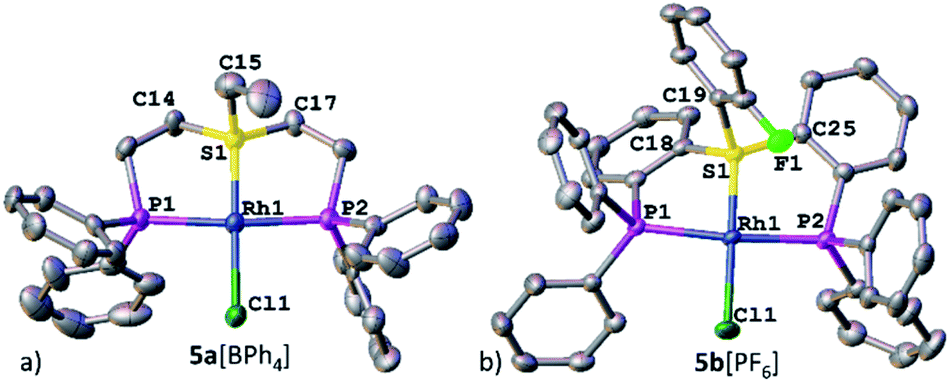 | ||
| Fig. 3 XRD structures of Rh(I)–sulfonium complexes, 5a[BPh4] (a) and 5b[PF6] (b). Co-crystallized solvent molecules, counter anions, and hydrogen atoms are omitted for clarity. | ||
Undoubtedly, these structures not only broaden the very limited pool of known sulfonium complexes but also proved for the first time the coordinating ability of an aromatic sulfonium cation. It is noteworthy, that unlike the α-cationic sulfides, which undergo oxidative addition with electron rich metals,25 the sulfonium complexes 5a[OTf] and 5b[OTf] remained stable as solids and in solutions.
Synthesis and characterization of the Pt(II)–sulfonium complexes
Having shown that stable complexes of sulfonium cations with the neutral RhCl fragment can be obtained, we wondered whether, similarly to cationic nitrenium19b and arenium26 pincer ligands, our frameworks could also induce bonding between these cations and a net positively charged metal fragment, such as [PtMe]+. To achieve that, we first treated ligands 4a[OTf] and 4b[OTf] with Pt(COD)Me2 which resulted in coordination products (Scheme 2), as evident from their 31P NMR spectrum that exhibited downfield shifted peaks at 11.3 or 16.6 ppm with the characteristic 195Pt satellites (1JPt–P = 1813 and 1781 Hz, respectively). The 1H NMR signals at 0.42 and 0.65 ppm were assigned to the methyl protons, confirming the formation of PtMe2 complexes 6a[OTf] and 6b[OTf], respectively. Moreover, these signals appeared as doublets of doublets due to splitting by two magnetically inequivalent P atoms, a configuration only possible when methyl groups are oriented cis to each other (Fig. S1 and S2†). The neutral PtMe2 fragment in 6a[OTf] and 6b[OTf] was then transformed into a cation by protonolysis (by HBF4*OEt2 or HOTf) resulting in the clean formation of complexes 7a[BF4]2 and 7b[OTf]2 (Scheme 2), as attested by new peaks at 42.4 (1JPt–P = 2736 Hz) and 44.3 (1JPt–P = 2768 Hz) ppm, respectively, in 31P NMR. In the aromatic complex 7b[OTf]2, the 31P NMR signals were much sharper than in 6b[OTf], and similarly to the Rh(I) complex 5b[OTf], splitting due to the 31P–19F coupling (5JP–F = 3.3 Hz) became observable.Unlike complexes 6a[OTf] and 6b[OTf], in both 7a[BF4]2 and 7b[OTf]2, the 1H NMR signals at 1.20 and 1.56 ppm, corresponding to single methyls, appeared as triplets indicating magnetic equivalence of the two phosphines, which is only possible in a mutual trans-orientation (Fig. S1 and S2†). Moreover, the signals of the aliphatic protons in 7a[BF4]2 followed a pattern similar to that of 5a[OTf] (Fig. 2), suggesting an analogous structure (Fig. S1†). To further study sulfonium–Pt interaction in solution we applied 1H–195Pt HMBC, once again focusing on magnetic interaction between Pt and the methylene protons of the ethyl tail (Fig. S4†). While in 6a[OTf], this coupling constant is negligible (0.2 Hz, presumably due to 6JPt–H), in 7a[BF4]2 it reaches 7.7 Hz (most likely, due to 3JPt–H), suggesting the presence of a S–Pt bond in 7a[BF4]2, but not in 6a[OTf]. A similar conclusion about S–Pt bonding in 6b[OTf] and 7b[OTf]2 could be drawn by comparing their 19F–195Pt HMBC spectra (Fig. S5†), even though both complexes exhibited nearly identical chemical shifts in 19F NMR (−102.3 and −102.5 ppm, respectively). The former showed no 19F–195Pt correlation, while the latter revealed a prominent cross-peak with a coupling constant of 3.3 Hz, supporting the presence of a sulfonium–Pt bond.
Ultimately, the solid-state structures of 6a[BPh4], 7a[BF4]2, and 7b[NTf2]2 (the latter was prepared by treating 6b[OTf] with an excess of bistriflimide) were established by single crystal XRD (Fig. 4c, a and b, respectively). In 6a[BPh4], as expected from the NMR analysis, no Pt–S bond was observed, and the methyl groups indeed exhibited a cis configuration. In contrast, both 7a[BF4]2 and 7b[NTf2]2 exhibited Pt–S bonds of 2.258(1) and 2.261(1) Å, respectively (see Table 1). Surprisingly, despite electrostatic repulsion between the cationic sulfonium and the [PtMe]+ fragment, the Pt–S bond in 7a[BF4]2 is shorter than that in its neutral sulfide analog 9[BF4], 2.336(2) Å, prepared for comparison (Fig. 4d).
 | ||
| Fig. 4 XRD structures of Pt(II) complexes, 7a[BF4]2 (a), 7b[NTf2]2 (b), 6a[BPh4] (c), and 9[BF4] (d). Co-crystallized solvent molecules, counter anions, and hydrogen atoms are omitted for clarity. | ||
Theoretical analysis of metal–sulfonium bonding and the influence of the pincer framework
The exceptionally short metal–sulfonium bonds observed in our Rh complexes prompted us to undertake a computational investigation by DFT. To gain a proper insight, we applied the energy decomposition analysis27 combined with the natural orbitals for chemical valence theory (EDA-NOCV) which provides a quantitative description of L–M bonding in a visual and chemically intuitive manner.28,29 In this method the overall interaction energy (ΔEint) between two molecular fragments (e.g. the sulfonium ligand and the rest of the complex) is assessed by means of EDA; then NOCV is applied to extract the total orbital interaction contribution (ΔEorb) and decompose it into individual constituents (ΔEorb(n)) according to their orbital symmetry. Each such constituent is then represented by a deformation density plot (Δρ(n)) that visualizes the redistribution of charge upon combination of the two molecular fragments.First, we considered the Rh–S bonding interactions in the model monodentate aliphatic and aromatic sulfonium complexes 10a and 10b and compared them with analogous complexes of neutral phosphines, sulfides and sulfoxides, as well as with a few representative cationic ligands. By inspecting the deformation density plots of the most significant orbital interactions (ΔEorb(n)), we could identify a single σ-symmetric interaction that has a clear L → M donation character, and two π-symmetric ones (perpendicular and parallel to the coordination plane) corresponding to the M → L back-donation (see representative deformation density maps of 10a in Fig. 5a and for other maps see Tables S22 and S23†). Interestingly, in the only reported pincer complex of the isoelectronic telluronium cation the σ interaction is in an opposite direction, i.e., it has a M → L character, thus classifying telluronium as a Z-type ligand.30 This difference in σ-bonding characteristics between sulfonium and telluronium can be rationalized by the so-called inert-pair effect,31 which in this case reflects the difference in energy of the 3s electrons of sulfonium compared to the 5s electrons in telluronium. In the latter the energy of this lone pair is too low to play any role in the bonding to the metal; this can only occur thanks to the donation from the metal's d orbital to the σ* orbitals of telluronium. Therefore, while isoelectronic, sulfonium and telluronium systems are not isolobal.
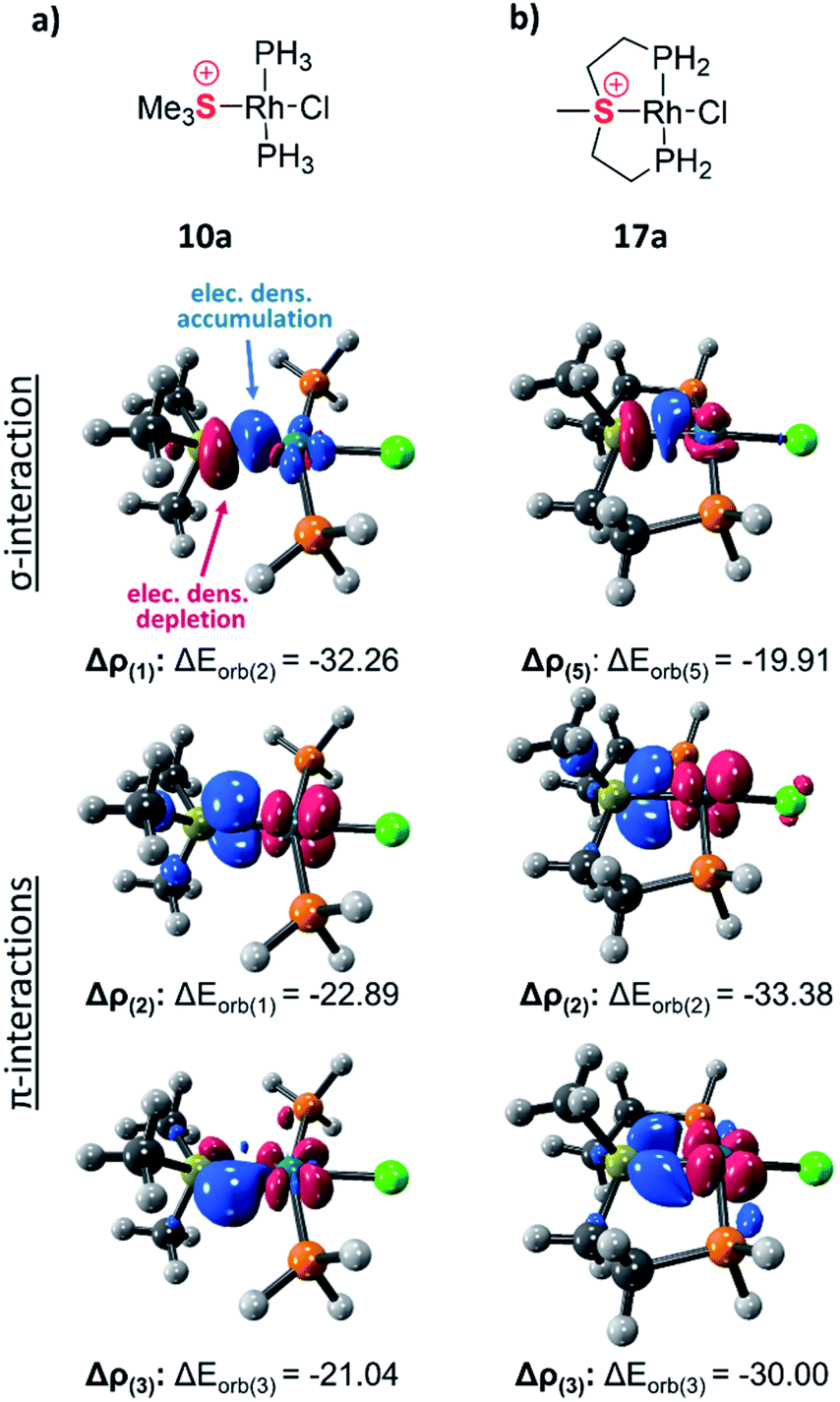 | ||
| Fig. 5 Selected deformation density plots of model complexes 10a (a) and 17a (b) (all energies are given in kcal mol−1). | ||
As evident from Table 2 in terms of their BDEs and σ-donation, sulfonium cations are nearly similar to sulfides and sulfoxides. However, sulfonium cations are significantly stronger π-acceptors, with π-back-bonding interaction being predominant. This is quite unusual and not the case even for the strongly π-acidic perfluorinated phosphines (in complexes 14a–c), where similarly to common phosphines (in 13a and 13b), σ-donation still prevails. This predominance of π-back-donation over σ-donation appears specific only to cationic ligands considered here. Compared to the latter, the π-acidity of sulfonium stands between that of N-heterocyclic nitrenium ([NHN]+, in 15a) and N-heterocyclic phosphenium ([NHP]+, in 15b), and is comparable to Alcarazo's tris-cationic phosphine PR3+ (in 15c).32
| a All energies are given in kcal mol−1. b Sum of the ⊥ and ‖ π-interactions. | ||||||||
|---|---|---|---|---|---|---|---|---|
| MX | RhCl | |||||||
| Ligand types | Sulfonium cations | Sulfides | Sulfoxides | Phosphines | ||||
| Model complexes | 10a | 10b | 11a | 11b | 12a | 12b | 13a | 13b |
| L | SMe3+ | SPh3+ | SMe2 | SPh2 | DMSO | Ph2SO | PMe3 | PPh3 |
| BDE | −38.57 | −36.96 | −36.21 | −38.19 | −39.80 | −44.55 | −70.06 | −57.77 |
| σ-Bonding (% of σ/ΔEorb) | −32.26 (37.3%) | −31.13 (31.0%) | −35.03 (63.5%) | −34.39 (61.0%) | −39.82 (57.1%) | −40.73 (53.5%) | −56.69 (67.3%) | −53.31 (63.3%) |
| π-Backbondinga (% of π/ΔEorb) | −43.93 (50.8%) | −39.40 (39.2%) | −14.89 (27.0%) | −16.10 (28.6%) | −24.42 (35.0%) | −27.55 (38.0%) | −22.06 (26.2%) | −24.19 (28.7%) |
| MX | RhCl | PtMe+ | ||||||
| Ligand type | Perfluorinated phosphines | Cationic ligands | Sulfonium cations | |||||
| Model complex | 14a | 14b | 14c | 15a | 15b | 15c | 16a | 16b |
| L | PF3 | P(CF3)3 | P(C6F5)3 |
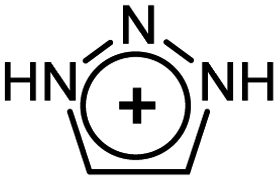 [NHN]+ [NHN]+ |
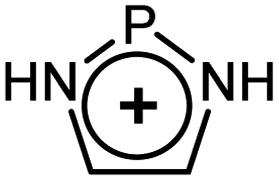 [NHP]+ [NHP]+ |
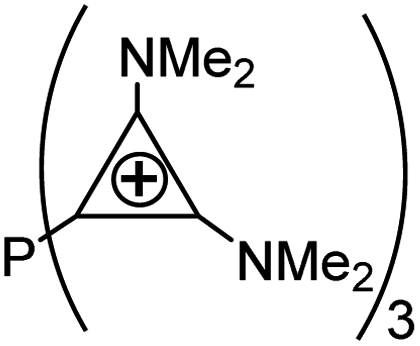 [PR3]3+ [PR3]3+ |
SMe3+ | SPh3+ |
| BDEa | −56.76 | −53.16 | −51.35 | −30.55 | −57.57 | −53.16 | +48.60 | +30.66 |
| σ-Bondinga (% of σ/ΔEorb) | −51.83 (51.4%) | −46.89 (49.7%) | −47.79 (55.1%) | −25.30 (40.3%) | −42.93 (44.3%) | −44.43 (42.6%) | −33.74 (57.3%) | −32.84 (56.9%) |
| π-Backbondinga,b (% of π/ΔEorb) | −44.61 (43.3%) | −40.75 (43.2%) | −32.21 (37.1%) | −29.19 (46.4%) | −62.42 (52.3%) | −46.17 (44.2%) | −17.20 (29.2%) | −13.53 (23.5%) |
With the cationic [PtMe]+ fragment the calculations confirmed that the monodentate sulfonium complexes 16a and 16b (Table 2) are kinetically stable, despite the electrostatic repulsion between the positively charged metal fragment and the sulfonium ligand responsible for the calculated positive BDE values. The obtained density plots of the model Pt complexes 16a and 16b were comparable in shape with those of the Rh complexes 10a and 10b (Fig. S18†), with prominent σ- and π-symmetric interactions. As expected for a positively charged metal center, the contribution of the π back-bonding in these model Pt complexes is significantly weaker than in their RhCl counterparts, yet still not negligible.
The influence of the pincer framework on bonding in both the Rh complexes 17a and 17b and their Pt analogues 18a and 18b is quite pronounced. As evident from Table 3, one can see that in both complexes the geometry deformations imposed by the pincer ligands strengthen the π back-donation within the complexes, so that the overall π/σ ratio significantly increases. Remarkably, in the case of the Pt complexes 18a and 18b π back-bonding even becomes comparable to the σ-donation, in spite of the positive charge on the metal center.
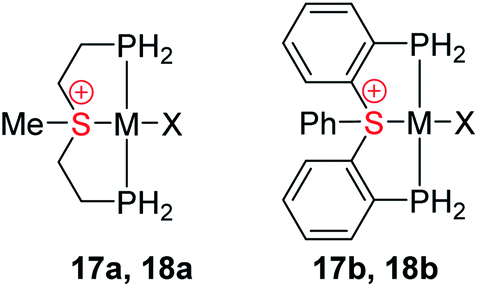
These changes in bonding character can be rationalized by comparing the geometries of the pincer complexes relative to the monodentate ones. The following discussion of the aliphatic and aromatic Rh complexes, as displayed in Fig. 6a, b and S19,† respectively, is also applicable to the Pt systems.
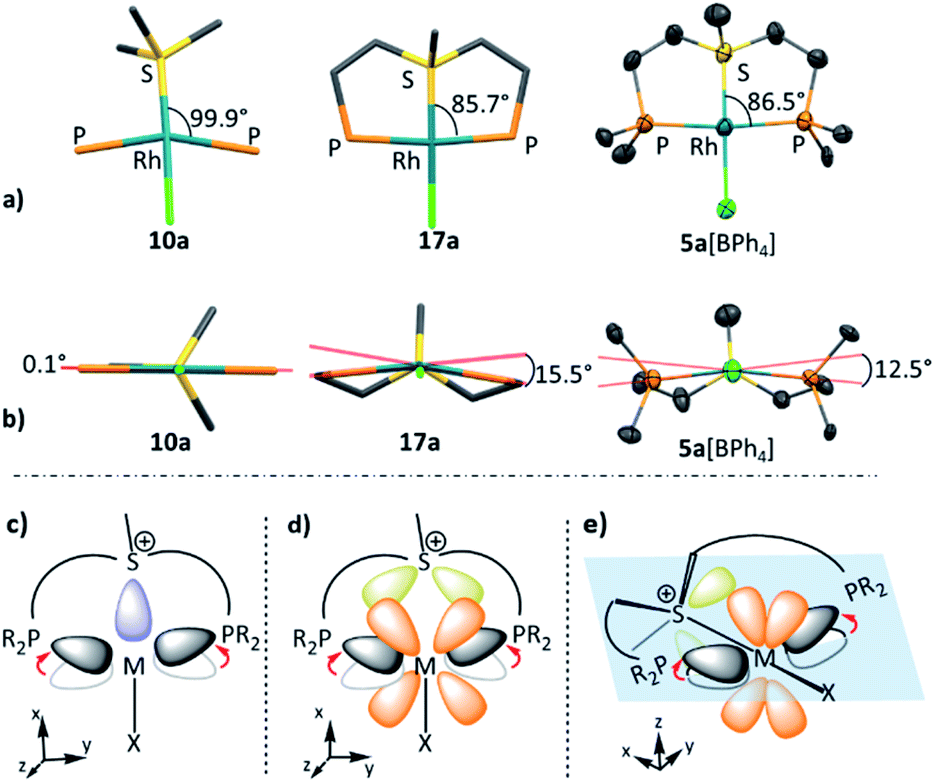 | ||
| Fig. 6 The in-plane (a) and out-of-plane (b) deformations in model complexes 10a, 17a and 5a[BPh4] (hydrogen atoms, BPh4 counter anion, and phenyl rings are omitted for clarity); schematic representation effect of pincer framework induced the in-plane and out-of-plane deformations on the σ (c) and π-interactions (d and e). | ||
In the aliphatic sulfonium pincer systems (both 5a[BPh4] and its model analog 17a), the average P–Rh–S angles are ∼15° smaller than in the optimized monodentate complex 10a (Fig. 6a). Such a decrease essentially pushes the phosphine lone pairs closer to those of sulfonium, increasing repulsive interactions between them. Thus, the sulfonium lone-pair is pushed away from the metal, which results in weakening the σ-donation in pincer complexes (Table 4, column 2). At the same time, this angle reduction also causes a stronger repulsion between the lone pairs of the phosphines and the filled dxy orbital of the metal, shifting electron density closer to the adjacent σ*-orbital of the sulfonium (Fig. 6b). An enhanced in-plane π-back-donation is thus induced (Table 4, column 3).
| Model complex | ΔEorb of L → M | ΔEorb of ‖ M → L | ΔEorb of ⊥ M → L | |
|---|---|---|---|---|
| σ-Donationb | π-Backdonationb | π-Backdonationb | ||
| a For the corresponding deformation density plots, see Tables S22 and S23. b All energies are given in kcal mol−1. | ||||
| Rh–Cl | 10a | −32.26 | −21.04 | −22.89 |
| 10b | −31.13 | −18.10 | −21.30 | |
| 17a | −19.91 | −30.00 | −33.38 | |
| 17b | −18.17 | −30.02 | −29.08 | |
| Pt–Me | 16a | −33.74 | −8.62 | −8.58 |
| 16b | −32.84 | −6.42 | −7.11 | |
| 18a | −47.16 | −17.55 | −22.17 | |
| 18b | −45.02 | −22.70 | −18.87 | |
In addition, the pincer framework also distorts the otherwise nearly planar coordination environment around the metal, pushing the two phosphines out of the coordination plane (Fig. 6b). This in turn results in repulsive interactions with the filled dxz orbital, similarly strengthening the interaction with the perpendicular σ*-orbital of the sulfonium (Fig. 6c). Therefore, π-back-donation in the perpendicular plane increases as well (Table 4, column 4).
Overall, the EDA-NOCV data clearly points out that geometric distortion imposed by the pincer framework not only preserves the unique characteristics of sulfonium cations as weak σ-donors and potent π-acceptors, but also enhances them. For comparison, an analogous attempt to incorporate a phosphenium moiety within a pincer framework resulted in a full charge transfer from the metal to the ligand, transforming it into a phosphide.33
Conclusions
To summarize, in this paper we have consolidated the status of sulfonium cations among the family of rare cationic ligands demonstrating for the first time that their coordination chemistry can be extended to the Pt group metals. We also prepared the very first examples of metal-coordinated aromatic sulfonium cations. These unusual compounds might represent stable analogs of possible transient intermediates forming during Pd-catalyzed cross-coupling of sulfonium salts. Our calculations suggested that sulfonium cations are among the best π-acceptors available. Moreover, the pincer frameworks which offer additional robustness also intensify this propensity. These scaffolds might therefore be the key to transform sulfonium complexes from a chemical curiosity into potential π-acid catalysts, the applications of which are currently studied in our lab.Data availability
Experimental procedures, NMR spectra and computational details are given in ESI.†Author contributions
Y. T. conceived the project, R. L., N. B., and O. C. performed the experiments, V. S. performed the XRD data refinement and DFT calculations, and V. S., Y. T., and F. T. wrote the paper.Conflicts of interest
There are no conflicts of interest.Acknowledgements
We would like to thank Prof. Chris H. Hendon and Mr Min-Chieh Yang from the University of Oregon, USA, and Dr Veniamin Borin from the Hebrew University for their valuable advice in our computational studies. R. L. expresses her gratitude for the CSC-HUJI fellowship. We thank Dr Roy Hoffman for his assistance in performing 2D 103Rh and 195Pt NMR measurements. Our deep gratitude is also given to our colleagues from the Hebrew University, Prof. Dmitri Gelman, Prof. Avi Bino and Prof. Silvio Biali, for their kind help and support throughout our research.Notes and references
- (a) A. F. Hill, G. R. Owen, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 1999, 38, 2759 CrossRef CAS; (b) D. You and F. P. Gabbaï, Trends Chem., 2019, 1, 485–496 CrossRef CAS; (c) A. Amgoune and D. Bourissou, Chem. Commun., 2011, 47, 859–871 RSC.
- (a) J. Fajardo Jr and J. C. Peters, Inorg. Chem., 2021, 60, 1220–1227 CrossRef CAS PubMed; (b) T. J. Del Castillo, N. B. Thompson and J. C. Peters, J. Am. Chem. Soc., 2016, 138, 5341–5350 CrossRef CAS PubMed; (c) J. S. Anderson, J. Rittle and J. C. Peters, Nature, 2013, 501, 84–87 CrossRef CAS PubMed.
- (a) L. J. Clouston, V. Bernales, R. K. Carlson, L. Gagliardi and C. C. Lu, Inorg. Chem., 2015, 54, 9263–9270 CrossRef CAS PubMed; (b) J. Ye, R. C. Cammarota, J. Xie, M. V. Vollmer, D. G. Truhlar, C. J. Cramer, C. C. Lu and L. Gagliardi, ACS Catal., 2018, 8, 4955–4968 CrossRef CAS; (c) B. J. Graziano, M. V. Vollmer and C. C. Lu, Angew. Chem., Int. Ed., 2021, 60, 15087–15094 CrossRef CAS PubMed; (d) M. V. Vollmer, J. Ye, J. C. Linehan, B. J. Graziano, A. Preston, E. S. Wiedner and C. C. Lu, ACS Catal., 2020, 10, 2459–2470 CrossRef CAS; (e) J. T. Moore and C. C. Lu, J. Am. Chem. Soc., 2020, 142, 11641–11646 CrossRef CAS PubMed; (f) R. C. Cammarota, J. Xie, S. A. Burgess, M. V. Vollmer, K. D. Vogiatzis, J. Ye, J. C. Linehan, A. M. Appel, C. Hoffmann, X. Wang, V. G. Young and C. C. Lu, Chem. Sci., 2019, 10, 7029–7042 RSC.
- (a) J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2017, 139, 6074–6077 CrossRef CAS PubMed; (b) M. Devillard, R. Declercq, E. Nicolas, A. W. Ehlers, J. Backs, N. Saffon-Merceron, G. Bouhadir, J. C. Slootweg, W. Uhl and D. Bourissou, J. Am. Chem. Soc., 2016, 138, 4917–4926 CrossRef CAS PubMed; (c) B. R. Barnett, C. E. Moore, A. L. Rheingold and J. S. Figueroa, J. Am. Chem. Soc., 2014, 136, 10262–10265 CrossRef CAS PubMed.
- (a) F. Inagaki, C. Matsumoto, Y. Okada, N. Maruyama and C. Mukai, Angew. Chem., Int. Ed., 2015, 54, 818–822 CrossRef CAS PubMed; (b) A. Ueno, K. Watanabe, C. G. Daniliuc, G. Kehr and G. Erker, Chem. Commun., 2019, 55, 4367–4370 RSC; (c) F. Inagaki, K. Nakazawa, K. Maeda, T. Koseki and C. Mukai, Organometallics, 2017, 36, 3005–3008 CrossRef CAS; (d) R. Murakami, H. Tanishima, D. Naito, H. Kawamitsu, R. Kamo, A. Uchida, K. Kawasaki, C. Kiyohara, M. Matsuo, K. Maeda and F. Inagaki, Tetrahedron Lett., 2021, 78, 153267 CrossRef CAS; (e) R. Murakami and F. Inagaki, Tetrahedron Lett., 2019, 60, 151231 CrossRef; (f) F. Inagaki, K. Maeda, K. Nakazawa and C. Mukai, Eur. J. Org. Chem., 2018, 2018, 2972–2976 CrossRef CAS.
- (a) H. Yang and F. P. Gabbai, J. Am. Chem. Soc., 2015, 137, 13425–13432 CrossRef CAS PubMed; (b) D. You, H. Yang, S. Sen and F. P. Gabbaï, J. Am. Chem. Soc., 2018, 140, 9644–9651 CrossRef CAS PubMed; (c) Y.-H. Lo and F. P. Gabbaï, Angew. Chem., Int. Ed., 2019, 58, 10194–10197 CrossRef CAS PubMed; (d) D. You and F. P. Gabbaï, J. Am. Chem. Soc., 2017, 139, 6843–6846 CrossRef CAS PubMed; (e) S. Sen, I.-S. Ke and F. P. Gabbaï, Organometallics, 2017, 36, 4224–4230 CrossRef CAS; (f) D. You, J. E. Smith, S. Sen and F. P. Gabbaï, Organometallics, 2020, 39, 4169–4173 CrossRef CAS; (g) R. R. Rodrigues and F. P. Gabbaï, Molecules, 2021, 26, 1985 CrossRef CAS PubMed; (h) J. S. Jones and F. P. Gabbai, Chem.–Eur. J., 2017, 23, 1136–1144 CrossRef CAS PubMed.
- (a) L. C. Wilkins, Y. Kim, E. D. Litle and F. P. Gabbaï, Angew. Chem., Int. Ed., 2019, 58, 18266–18270 CrossRef CAS PubMed; (b) E. D. Litle, L. C. Wilkins and F. P. Gabbaï, Chem. Sci., 2021, 12, 3929 RSC.
- (a) P. Redero, T. Hartung, J. Zhang, L. D. M. Nicholls, G. Zichen, M. Simon, C. Golz and M. Alcarazo, Angew. Chem., Int. Ed., 2020, 59, 23527 CrossRef CAS PubMed; (b) T. Johannsen, C. Golz and M. Alcarazo, Angew. Chem., Int. Ed., 2020, 59, 22779–22784 CrossRef CAS PubMed; (c) K. Sprenger, C. Golz and M. Alcarazo, Eur. J. Org. Chem., 2020, 6245–6254 CrossRef CAS; (d) J. Zhang, M. Simon, C. Golz and M. Alcarazo, Angew. Chem., Int. Ed., 2020, 59, 5647–5650 CrossRef CAS PubMed; (e) T. Hartung, R. Machleid, M. Simon, C. Golz and M. Alcarazo, Angew. Chem., Int. Ed., 2020, 59, 5660–5664 CrossRef CAS PubMed; (f) L. D. M. Nicholls and M. Alcarazo, Chem. Lett., 2019, 48, 1 CrossRef CAS; (g) H. Tinnermann, L. D. M. Nicholls, T. Johannsen, C. Wille, C. Golz, R. Goddard and M. Alcarazo, ACS Catal., 2018, 8, 10457–10463 CrossRef CAS; (h) L. D. M. Nicholls, M. Marx, T. Hartung, E. González-Fernández, C. Golz and M. Alcarazo, ACS Catal., 2018, 8, 6079–6085 CrossRef CAS; (i) L. Gu, L. M. Wolf, A. Zieliński, W. Thiel and M. Alcarazo, J. Am. Chem. Soc., 2017, 139, 4948–4953 CrossRef CAS PubMed; (j) E. González-Fernández, L. D. M. Nicholls, L. D. Schaaf, C. Farès, C. W. Lehmann and M. Alcarazo, J. Am. Chem. Soc., 2017, 139, 1428–1431 CrossRef PubMed; (k) G. Mehler, P. Linowski, J. Carreras, A. Zanardi, J. W. Dube and M. Alcarazo, Chem.–Eur. J., 2016, 22, 15320–15327 CrossRef CAS PubMed; (l) M. Alcarazo, Acc. Chem. Res., 2016, 49, 1797–1805 CrossRef CAS PubMed; (m) E. Haldón, A. Kozma, H. Tinnermann, L. Gu, R. Goddard and M. Alcarazo, Dalton Trans., 2016, 45, 1872–1876 RSC; (n) H. Tinnermann, C. Wille and M. Alcarazo, Angew. Chem., Int. Ed., 2014, 53, 8732–8736 CrossRef CAS PubMed; (o) M. Alcarazo, Chem.–Eur. J., 2014, 20, 7868–7877 CrossRef CAS PubMed; (p) A. Kozma, T. Deden, J. Carreras, C. Wille, J. Petuškova, J. Rust and M. Alcarazo, Chem.–Eur. J., 2014, 20, 2208–2214 CrossRef CAS PubMed; (q) J. Carreras, G. Gopakumar, L. Gu, A. Gimeno, P. Linowski, J. Petuškova, W. Thiel and M. Alcarazo, J. Am. Chem. Soc., 2013, 135, 18815–18823 CrossRef CAS PubMed; (r) J. Carreras, M. Patil, W. Thiel and M. Alcarazo, J. Am. Chem. Soc., 2012, 134, 16753–16758 CrossRef CAS PubMed.
- J. W. Dube, Y. Zheng, W. Thiel and M. Alcarazo, J. Am. Chem. Soc., 2016, 138, 6869–6877 CrossRef CAS PubMed.
- (a) S. I. Kozhushkov and M. Alcarazo, Eur. J. Inorg. Chem., 2020, 2020, 2486–2500 CrossRef CAS PubMed; (b) D. Kaiser, I. Klose, R. Oost, J. Neuhaus and N. Maulide, Chem. Rev., 2019, 119, 8701–8780 CrossRef CAS PubMed; (c) V. G. Nenaidenko and E. S. Balenkova, Russ. J. Org. Chem., 2003, 39, 291–330 CrossRef CAS; (d) L.-Q. Lu, T.-R. Li, Q. Wang and W.-J. Xiao, Chem. Soc. Rev., 2017, 46, 4135–4149 RSC; (e) V. K. Aggarwal and C. L. Winn, Acc. Chem. Res., 2004, 37, 611–620 CrossRef CAS PubMed; (f) D. Kaiser, I. Klose, R. Oost, J. Neuhaus and N. Maulide, Chem. Rev., 2019, 119, 8701–8780 CrossRef CAS PubMed; (g) A. C. B. Burtoloso, R. M. P. Dias and I. A. Leonarczyk, Eur. J. Org. Chem., 2013, 2013, 5005–5016 CrossRef CAS.
- (a) J. Srogl, G. D. Allred and L. S. Liebeskind, J. Am. Chem. Soc., 1997, 119, 12376–12377 CrossRef CAS; (b) F. Berger, M. B. Plutschack, J. Riegger, W. Yu, S. Speicher, M. Ho, N. Frank and T. Ritter, Nature, 2019, 567, 223–228 CrossRef CAS PubMed; (c) J. Li, J. Chen, R. Sang, W.-S. Ham, M. B. Plutschack, F. Berger, S. Chabbra, A. Schnegg, C. Genicot and T. Ritter, Nat. Chem., 2020, 12, 56 CrossRef CAS PubMed; (d) P. Cowper, Y. Jin, M. D. Turton, G. Kociok-Köhn and S. E. Lewis, Angew. Chem., Int. Ed., 2016, 55, 2564–2568 CrossRef CAS PubMed; (e) P. S. Engl, A. P. Häring, F. Berger, G. Berger, A. Pérez-Bitrián and T. Ritter, J. Am. Chem. Soc., 2019, 141, 13346–13351 CrossRef CAS PubMed; (f) H. Jia, A. P. Häring, F. Berger, L. Zhang and T. Ritter, J. Am. Chem. Soc., 2021, 143, 7623–7628 CrossRef CAS PubMed; (g) E. M. Alvarez, T. Karl, F. Berger, L. Torkowski and T. Ritter, Angew. Chem., Int. Ed., 2021, 60, 13609–13613 CrossRef CAS PubMed; (h) J. T. Chen, J. K. Li, M. B. Plutschack, F. Berger and T. Ritter, Angew. Chem., Int. Ed., 2020, 59, 5616–5620 CrossRef CAS PubMed; (i) F. Ye, F. Berger, H. Jia, J. Ford, A. Wortman, J. Börgel, C. Genicot and T. Ritter, Angew. Chem., Int. Ed., 2019, 58, 14615–14619 CrossRef CAS PubMed; (j) S.-M. Wang, H.-X. Song, X.-Y. Wang, N. Liu, H.-L. Qin and C.-P. Zhang, Chem. Commun., 2016, 52, 11893–11896 RSC; (k) S.-M. Wang, X.-Y. Wang, H.-L. Qin and C.-P. Zhang, Chem.–Eur. J., 2016, 22, 6542–6546 CrossRef CAS PubMed; (l) Z.-Y. Tian, S.-M. Wang, S.-J. Jia, H.-X. Song and C.-P. Zhang, Org. Lett., 2017, 19, 5454–5457 CrossRef CAS PubMed; (m) Z.-Y. Tian and C.-P. Zhang, Chem. Commun., 2019, 55, 11936–11939 RSC; (n) H. Minami, K. Nogi and H. Yorimitsu, Org. Lett., 2019, 21, 2518–2522 CrossRef CAS PubMed; (o) H. Minami, S. Otsuka, K. Nogi and H. Yorimitsu, ACS Catal., 2018, 8, 579–583 CrossRef CAS; (p) D. C. Simkó, P. Elekes, V. Pázmándi and Z. Novák, Org. Lett., 2018, 20, 676–679 CrossRef PubMed.
- (a) J. V. Crivello, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 4241–4254 CrossRef CAS; (b) Y. Takahashi, S. Kodama and Y. Ishii, Organometallics, 2018, 37, 1649–1651 CrossRef CAS; (c) C. O. Yanez, C. D. Andrade and K. D. Belfield, Chem. Commun., 2009, 827–829 RSC.
- (a) A. Péter, G. J. P. Perry and D. J. Procter, Adv. Synth. Catal., 2020, 362, 2135–2142 CrossRef; (b) X. Wang, L. Truesdale and J. Q. Yu, J. Am. Chem. Soc., 2010, 132, 3648–3649 CrossRef CAS PubMed; (c) X.-G. Zhang, H.-X. Dai, M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2012, 134, 11948–11951 CrossRef CAS PubMed; (d) L.-S. Zhang, K. Chen, G.-H. Chen, B.-J. Li, S. Luo, Q.-Y. Guo, J.-B. Wei and Z.-J. Shi, Org. Lett., 2013, 15, 10–13 CrossRef CAS PubMed.
- (a) Y. Kim, M. Kim and F. P. Gabbaï, Org. Lett., 2010, 12, 600–602 CrossRef CAS PubMed; (b) H. Zhao and F. P. Gabbai, Org. Lett., 2011, 13, 1444 CrossRef CAS PubMed; (c) F. A. Tsao, A. E. Waked, L. Cao, J. Hofmann, L. Liu, S. Grimme and D. W. Stephan, Chem. Commun., 2016, 52, 12418–12421 RSC.
- (a) H. Zhao and F. P. Gabbaï, Nat. Chem., 2010, 2, 984–990 CrossRef CAS PubMed; (b) Y. Kim, H. Zhao and F. P. Gabbaï, Angew. Chem., Int. Ed., 2009, 48, 4957–4960 CrossRef CAS PubMed.
- (a) R. D. Adams and D. F. Chodosh, J. Organomet. Chem., 1976, 120, C39 CrossRef CAS; (b) R. D. Adams and D. F. Chodosh, J. Am. Chem. Soc., 1978, 100, 812–817 CrossRef CAS; (c) R. D. Adams and M. Shiralian, Organometallics, 1982, 1, 883 CrossRef CAS; (d) R. D. Adams, C. Blankenship, B. E. Segmueller and M. Shiralian, J. Am. Chem. Soc., 1983, 105, 4319–4326 CrossRef CAS; (e) T. Yoshida, T. Adachi, K. Sato, K. Baba and T. Kanokogi, J. Chem. Soc., Chem. Commun., 1993, 1511–1513 RSC.
- P. B. Kettler, Org. Process Res. Dev., 2003, 7, 342–354 CrossRef CAS.
- (a) M. Albrecht and G. Van Koten, Angew. Chem., Int. Ed., 2001, 40, 3750–3781 CrossRef CAS; (b) E. Peris and R. H. Crabtree, Chem. Soc. Rev. , 2018, 47, 1959–1968 RSC.
- (a) Y. Tulchinsky, M. A. Iron, M. Botoshansky and M. Gandelman, Nat. Chem., 2011, 3, 525–531 CrossRef CAS PubMed; (b) Y. Tulchinsky, S. Kozuch, P. Saha, M. Botoshansky, L. Shimon and M. Gandelman, Chem. Sci., 2014, 5, 1305 RSC.
- (a) C. Huang, J. Feng, R. Ma, S. Fang, T. Lu, W. Tang, D. Du and J. Gao, Org. Lett., 2019, 21, 9688–9692 CrossRef CAS PubMed; (b) C. O. Yanez, C. D. Andrade and K. D. Belfield, Chem. Commun., 2009, 827–829 RSC.
- Searches were performed on the Cambridge Structural Database on Jan. 2022.
- (a) G. J. Kubas and R. R. Ryan, Inorg. Chim. Acta, 1981, 47, 131 CrossRef CAS; (b) R. R. Ryan, P. G. Eller and G. J. Kubas, Inorg. Chem., 1976, 15, 797–799 CrossRef CAS.
- R. Meij, D. J. Stufkens and K. Vrieze, J. Organomet. Chem., 1979, 164, 353–370 CrossRef CAS.
- (a) D. L. M. Suess and J. C. Peters, Organometallics, 2012, 31, 5213–5222 CrossRef CAS; (b) F. Meyer, E. Hupf, E. Lork, S. Grabowsky, S. Mebs and J. Beckmann, Eur. J. Inorg. Chem., 2020, 3829–3836 CrossRef CAS.
- A. Kozma, J. Petuskova, C. W. Lehmann and M. Alcarazo, Chem. Commun., 2013, 49, 4145 RSC.
- A. Vigalok, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 1998, 120, 477–483 CrossRef CAS.
- (a) T. Ziegler and A. Rauk, Inorg. Chem., 1979, 18, 1755 CrossRef CAS; (b) T. Ziegler and A. Rauk, Theor. Chim. Acta, 1977, 46, 1 CrossRef CAS.
- (a) A. Michalak, M. Mitoraj and T. Ziegler, J. Phys. Chem. A, 2008, 112, 1933–1939 CrossRef CAS PubMed; (b) M. Mitoraj and A. Michalak, Organometallics, 2007, 26, 6576–6580 CrossRef CAS; (c) M. Mitoraj and A. Michalak, J. Mol. Model., 2007, 13, 347–355 CrossRef CAS PubMed; (d) M. Mitoraj and A. Michalak, J. Mol. Model., 2008, 14, 681–687 CrossRef CAS PubMed; (e) M. Mitoraj, H. Zhu, A. Michalak and T. Ziegler, Int. J. Quantum Chem., 2009, 109, 3379–3386 CrossRef CAS.
- (a) M. Mitoraj and A. Michalak, Organometallics, 2007, 26, 6576–6580 CrossRef CAS; (b) M. Mitoraj and A. Michalak, J. Mol. Model., 2007, 13, 347–355 CrossRef CAS PubMed.
- (a) T.-P. Lin and F. P. Gabbaï, J. Am. Chem. Soc., 2012, 134, 12230–12238 CrossRef CAS PubMed; (b) T. P. Lin and F. P. Gabbaï, Angew. Chem., Int. Ed., 2013, 52, 3864–3868 CrossRef CAS PubMed.
- T. Overton, J. Rourke, M. Weller, F. Amstrong and P. Atkins, Inorganic Chemistry, Oxford University Press, 5th edn, New York, 2010, p. 267 Search PubMed.
- J. Petuskova, M. Patil, S. Holle, C. W. Lehmann, W. Thiel and M. Alcarazo, J. Am. Chem. Soc., 2011, 133, 20758 CrossRef CAS PubMed.
- B. Pan, Z. Xu, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Inorg. Chem., 2012, 51, 4170–4179 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2119886–2119894. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2sc00588c |
| This journal is © The Royal Society of Chemistry 2022 |