
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New 19F NMR methodology reveals structures of molecules in complex mixtures of fluorinated compounds†
Alan J. R.
Smith
,
Richard
York
,
Dušan
Uhrín
 and
Nicholle G. A.
Bell
*
and
Nicholle G. A.
Bell
*
EaStCHEM School of Chemistry, University of Edinburgh, David Brewster Rd, Edinburgh, EH9 3FJ, UK. E-mail: Nicholle.Bell@ed.ac.uk
First published on 25th February 2022
Abstract
Although the number of natural fluorinated compounds is very small, fluorinated pharmaceuticals and agrochemicals are numerous. 19F NMR spectroscopy has a great potential for the structure elucidation of fluorinated organic molecules, starting with their production by chemical or chemoenzymatic reactions, through monitoring their structural integrity, to their biotic and abiotic transformation and ultimate degradation in the environment. Additionally, choosing to incorporate 19F into any organic molecule opens a convenient route to study reaction mechanisms and kinetics. Addressing limitations of the existing 19F NMR techniques, we have developed methodology that uses 19F as a powerful spectroscopic spy to study mixtures of fluorinated molecules. The proposed 19F-centred NMR analysis utilises the substantial resolution and sensitivity of 19F to obtain a large number of NMR parameters, which enable structure determination of fluorinated compounds without the need for their separation or the use of standards. Here we illustrate the 19F-centred structure determination process and demonstrate its power by successfully elucidating the structures of chloramination disinfectant by-products of a single mono-fluorinated phenolic compound, which would have been impossible otherwise. This novel NMR approach for the structure elucidation of molecules in complex mixtures represents a major contribution towards the analysis of chemical and biological processes involving fluorinated compounds.
Introduction
While fluorine-containing compounds are the least abundant natural organohalides,1 modern society has become dependent on numerous man-made fluorinated organic molecules such as pharmaceuticals and agrochemicals.Presently, about 20% of the commercial pharmaceuticals contain fluorine and the proportion of newly approved fluoro-pharmaceuticals is rising steadily.2–4 Similarly, fluoro-agrochemicals have become indispensable for crop production and protecting public health from parasitically transmitted infectious diseases;5 53% of all active agrochemicals registered during 1998–2020 are classed as fluoro-agrochemicals.6 New fragrance and semiochemical molecules can also benefit from fluorination.7 In addition, 18F is the most frequently used radioisotope in positron emission tomography radiopharmaceuticals for both clinical and preclinical research, and the search for simple and efficient 18F-labeling procedures is an active research area.8
Reflecting such interest in fluorinated molecules, design of efficient and environmentally safe fluorination methods9–11 and scaled up manufacture of fluorinated molecules12 are among the most active fields of organic chemistry. Enzymatic13 and chemoenzymatic14–16 platforms for the preparation of fluorinated compounds are also emerging. To support these developments, there is a need to characterise fluorinated molecules using efficient analytical methods, amongst which 19F NMR spectroscopy plays a prominent role. What makes 19F the ideal NMR nucleus is its high sensitivity, 100% natural abundance, large chemical shift dispersion and strong and far-reaching spin–spin interactions.
An important advantage of 19F over other nuclei is the absence of the background signal, reflecting the lack of fluorinated endogenous compounds. 19F NMR has the ability to study fluorinated molecules in the presence of other CHN-containing molecules and mixtures of fluorinated compounds produced by chemical or chemoenzymatic reactions could in principle be analysed with minimal clean-up steps or compound separation.
In its simplest form, 1D 19F NMR has been widely used in studies of biodegradation and biotransformation of fluorinated compounds17–19 and has helped to characterise their catabolic pathways20–24 and identify cryptic liabilities and features with potentially problematic structural arrangements,25 which can lead to recalcitrance and/or toxicity.26 Nevertheless, studying biodegradation pathways still typically requires isolation of metabolites and their identification using known standards;17 both of these steps could be problematic. Another frequent application of 19F NMR comes from using a fluorinated molecule as one of the reactants in studies of mechanisms and kinetics of chemical reactions.27,28
The methodology presented here aims to make the process of structure elucidation of fluorine-containing molecules contained in (complex) mixtures more efficient. It follows the “NMR spies” approach, where 13C labelled tags provide information about the nuclei in their vicinity,29,30 leading to structural characterisation of molecules. In a recent example, introduction of -O13CH3 groups to a subset of molecules as NMR tags led to structural characterisation of 32 phenolic molecules, or their fragments, in a complex matrix of peat fulvic acid.31
In the case of fluorinated organic compounds, 19F atoms provide a 100% NMR active tags already present in molecules, enabling 19F-centred NMR structure determination. An example of this approach includes the FESTA family of NMR experiments32–34 that provide 1H–19F chemical shift correlation and 1H–19F coupling constants. The FESTA experiments require selective manipulation of individual 1H and 19F resonances, which is neither achievable (in particular for 1H resonances) nor practical for very complex mixtures, such as investigated here.
We have designed a set of nonselective 2D NMR experiments that use far reaching 1H–19F and 19F–13C couplings to obtain 1H and 13C chemical shifts of nuclei multiple bonds away from the fluorine atom. The same experiments also yield accurate values of 1H–19F, 19F–13C and 1H–1H coupling constants and 13C-induced 19F isotopic shifts. Put together, the obtained information allows elucidation of fluorine-containing molecular moieties and in favourable cases complete structure determination of small fluorinated molecules.
We have chosen to illustrate this approach on a study of disinfection by-products (DBPs) produced during water treatment. DBPs are formed when disinfectants react with naturally dissolved organic matter (DOM), anthropogenic contaminants, bromide, and iodide during the production of potable water. Approximately 600–700 DBPs have been reported in the literature so far,35 some of which exhibit severe health effects.36,37 Amongst halogenated DBPs, the focus so far has been on the quantification of trihalomethanes (THMs), haloacetic acids (HAAs) and total organic halides (TOXs).38–41 As the known compounds constitute less than 50% of TOXs produced by chlorination and less than 20% by chloramination,38 new generations of DBPs are being continually identified and classified for high priority toxicity studies.35,42 The commonly used alternative disinfectants to chlorine (ozone, chloramines, and chlorine dioxide) produce lower levels of the four regulated THMs and most HAAs as well as TOXs, however, they increase the concentration of some other priority DBPs.35,38,43 Chloramination also incorporates nitrogen into DOM molecules44 generating N-containing DBPs,39,45 which can be even more toxic than those currently regulated.37,46 Chloramination was therefore chosen for this study and 15N labelled NH4Cl was used in all experiments to prepare 15N-containing compounds amenable to NMR studies.
Analytical techniques for the structure determination of DBPs play an important role in this process. Traditional methods, such as liquid/liquid extraction, GC, GC/MS, and solid-phase extraction/MS,47 often produce only tentative structures that need validation through the use of authentic chemical standards.35 Specialised MS48,49 and MS/MS50,51 techniques are also being used in this field. Ultrahigh resolution Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR-MS) is making contributions to the characterisation of DBPs at the level of molecular formulae, compound class and functional group classification, including identification of compound classes with the highest DBP formation potential.52–58 When ion fragmentation is used more definite structural information can be obtained by MS.49,51,59
On the other hand, the use of NMR spectroscopy in the structure determination of DBPs is rare and usually requires some form of compound separation.60–63 Here we illustrate the power of 19F-centred NMR structure elucidation of fluorinated molecules using a complex mixture of DBPs produced by chloramination of a single fluorine-containing molecule.
Experimental methodology
Chloramination
A 500 ml sample was prepared with LC-MS grade water and 50 mg L−1 of 3-fluoro-4-hydroxybenzoic acid (1). The solution was buffered to pH 7.2 with phosphate buffer. A 15N-monochloramine solution was prepared by slow addition of sodium hypochlorite solution to 15NH4Cl in a chlorine-to-ammonia ratio of 0.8 mol mol−1 and added to the sample in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 mass ratio of carbon: disinfectant, as described previously.64 All samples were kept in the dark at 20 °C for 5 days before the addition of excess Na2S2O3 to stop the reaction. The reaction mixture was adjusted to pH 2.0 using HCl before being pumped through PPL SPE cartridges (1 g, 6 ml, Agilent) at a flow rate of ∼5 ml min−1. Each cartridge was conditioned using methanol followed by acidified Milli-Q water (pH 2). After adsorption of the sample, the column was washed with acidified water in order to minimise the retention of inorganic species. The cartridge was then allowed to dry before being eluted with methanol. The eluent was rotary evaporated to dryness.
:5 mass ratio of carbon: disinfectant, as described previously.64 All samples were kept in the dark at 20 °C for 5 days before the addition of excess Na2S2O3 to stop the reaction. The reaction mixture was adjusted to pH 2.0 using HCl before being pumped through PPL SPE cartridges (1 g, 6 ml, Agilent) at a flow rate of ∼5 ml min−1. Each cartridge was conditioned using methanol followed by acidified Milli-Q water (pH 2). After adsorption of the sample, the column was washed with acidified water in order to minimise the retention of inorganic species. The cartridge was then allowed to dry before being eluted with methanol. The eluent was rotary evaporated to dryness.
NMR experiments and instrumentation
Six new NMR experiments were designed and used in this work: ① 19F-detected variable-time z-filtered 2D 1H, 19F HETCOR (Fig. S7†); ② 19F-detected 2D 1H, 19F TOCSY-HETCOR (Fig. S8†); ③ 1H-detected 2D 19F, 1H CP-DIPSI3-DIPSI2 (Fig. S9†); ④and ④′ 19F-detected 2D 19F, 13C (15N) HMBC optimised for nJFC and 1JFC coupling constants, (Fig. S10† and S11†, respectively), and ⑤ 1H-detected 2D H1CnF (Fig. S12†). Apart from ② all other pulse sequences make use of a double inversion adiabatic sweep;65,66 the pulse sequence ① uses a z-filter to deliver pure phase multiplets;67 the pulse sequence ②, inspired by a 3D TOCSY-HSQC experiment,68 incorporates the 1H chemical shift labelling followed by a spin-lock period before the magnetisation is transferred to 19F for detection; pulse sequence ③ is a simple modification of a 3D 19F–1H heteronuclear TOCSY edited 1H–1H TOCSY69 that removes the 1H chemical shift labelling after the 19F → 1H transfer; the two HMQC based pulse sequences ④ and ④′ use the echo–antiecho quadrature detection as proposed by Bazzo et al.70 but eliminate the 19F chemical shift evolution and yield pure antiphase 13C, 19F doublets; experiment ⑤ is a purposely designed reduced dimensionality71–73 (3,2)D 19F-detected HCF correlation experiment with a simplified polarisation transfer pathway relative to the existing 1H-detected triple-resonance HCF experiment.74 The full analysis of these experiments will be published elsewhere, however, their most relevant aspect for this work, sensitivity, is analysed in the ESI†.The reaction product mixture (30 mg) was dissolved in CD3OH (180 μL) and placed into a 3 mm NMR tube. Spectra involving 19F were acquired on a 500 MHz Bruker Avance III HD NMR spectrometer equipped with a 5 mm QCI-F CryoProbe, while the 1D 1H and a 2D 1H, 15N HSQC spectra were obtained on a 800 MHz AVANCE III NMR spectrometer equipped with a 5 mm TCI cryoprobe. All experiments were performed at 300 K using parameters summarised in Table S1†.
Results and discussion
Hardware requirements and design of 19F-centered experiments
Historically, pulsing on 1H and 19F in one NMR experiment, a requirement for all experiments discussed here, was only possible on a limited number of spectrometers.75 However, this capability is much more common today. When 13C information is sought, three channel NMR spectrometers are required for all but perfluorinated molecules. To boost the sensitivity of such experiments, highly sensitive triple- or quadruple resonance cryoprobes capable of pulsing simultaneously on 1H, 13C and 19F are typically required. Such systems have become more widely available, mainly due to their use in binding studies of biomacromolecules with fluorinated ligands.The chemical shift correlation experiments involving 19F have evolved together with general improvements of liquid-state NMR methodology;75 most notably the use of adiabatic 19F inversion pulses is now widespread.66,76–78 Nevertheless, even some more recent 19F experiments yield magnitude mode spectra,76,78 provide correlation but not the values of coupling constants,76 or contain refocusing periods that generally decrease their sensitivity.77,78 Some phase sensitive experiments yield complicated cross peak structures, thereby lowering their sensitivity.79–81
The new NMR experiments presented here build on these advances, are phase sensitive and produce cross peaks with a simple pattern that allow identification of active coupling constants. They incorporate adiabatic inversion pulses covering a 100 KHz frequency range, ensuring their optimal performance across a range of 19F chemical shifts. The use of a single polarisation transfer interval optimised for nJHF or nJFC coupling constants and the elimination of the effects of passive coupling whenever possible, means that they provide chemical shift correlations mediated by a broad range of coupling constants (4–12 Hz nJHF and 3–26 Hz for nJFC, see Tables S2† and S3). When applicable, they also use 1H or 19F decoupling in the directly detected periods to simplify cross peaks and to boost the sensitivity.
Hundreds of DBPs formed by chloramination of a single molecule
DBPs are typically formed from compounds with activated aromatic rings that react with oxidants to produce modified phenolics and unsaturated aliphatic compounds leading to the generation of trihalomethanes.82 A simple molecule, 3-fluoro-4- hydroxybenzoic acid (1, Fig. 1) was therefore selected as a suitable model compound for chloramination using 15NH4Cl.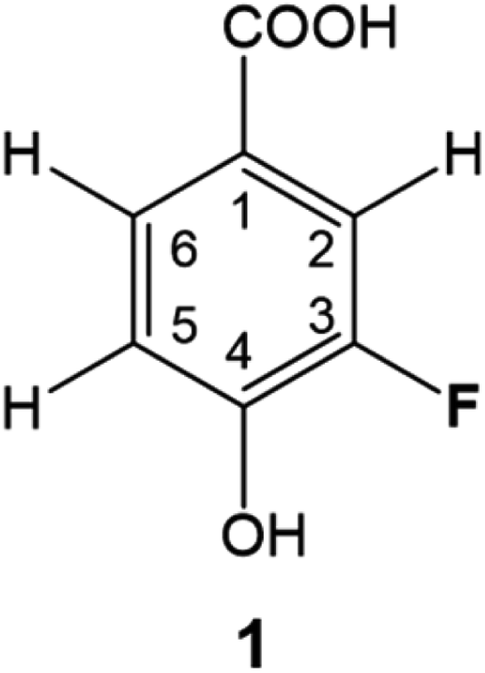 | ||
| Fig. 1 3-Fluoro-4-hydroxybenzoic acid, 1. | ||
A 500 MHz 1H-decoupled 1D 19F spectrum of the reaction mixture produced by chloramination of 1 is very complex; it contains hundreds of peaks of varying intensity spread across a 90 ppm 19F chemical shift range, with the majority and the most intense signals appearing within a 34 ppm range. A partial spectrum is shown in Fig. 2 with thirteen of the most intense resonances numbered. Fig. S1† and S2 present vertical expansions of the full 19F spectrum and the aromatic part of a 1H NMR spectrum of the reaction mixture, respectively.
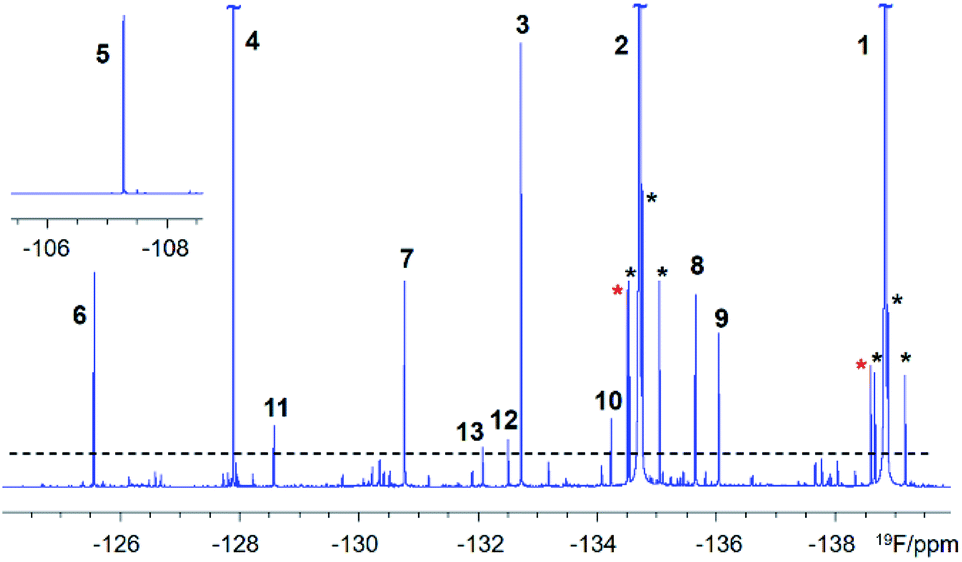 | ||
| Fig. 2 A partial 500 MHz 1H-decoupled 1D 19F spectrum of the chloramination products of 1. Signals above the dashed line are numbered. Black and red asterisks around the two most intense signals, of 1 (the starting material) and 2 (the major product), indicate 13C satellites and their methyl esters as purification by-products, respectively. | ||
Providing fluorine is not removed during the reaction, chloramination products of a fluorinated compound will contain at least one 19F atom. If the reaction causes oligomerisation, molecules with several 19F atoms will also be present. Nevertheless, these will likely be too distant to exhibit 19F–19F couplings and 19F atoms will therefore only couple to protons in 12C molecules and protons and carbons in 13C isotopomers. In molecules that incorporated 15N, couplings of 19F with 15N could arise. The 19F atom thus represents a convenient ‘spy’ that reports on the 19F, 1H, 13C and 15N NMR chemical shifts and numerous coupling constants of fluorinated molecules, underpinning the structural characterisation of DBPs.
19F, 1H and 13C chemical shifts and J couplings determination
Extensive spin–spin interactions involving 19F open numerous magnetisation transfer pathways (Fig. 3) that can be exploited to yield chemical shift correlations of many nuclei.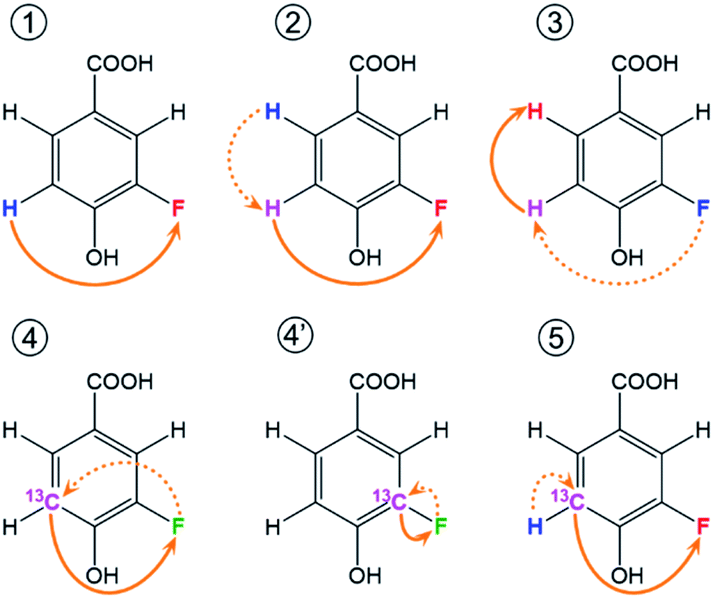 | ||
| Fig. 3 Schematic representation of 19F-centred NMR experiments. The blue, pink and red colours represent the starting, intermediary and the detected nucleus for one example of the magnetization transfer pathways, while green is used when both the starting and detected nucleus are the same. These pathways are used by the following experiments: ① 19F-detected z-filtered 2D 1H, 19F HETCOR; ② 2D 1H, 19F TOCSY-HETCOR; ③ 2D 19F, 1H CP-DIPSI3-DIPSI2; ④ 2D 19F, 13C (15N) HMBC optimised for nJFC (nJFN) coupling constants; ④′ 2D 19F, 13C HMBC optimised for 1JFC coupling constants; ⑤ (3,2)D H1CnF correlation experiment. Dashed and full orange arrows connect the initial and final magnetisation transfer steps, respectively. | ||
A 2D 1H, 19F correlation spectrum (Fig. S3†) illustrates the complexity of the investigated mixture. Zoomed in regions of 19F-centred spectra acquired in this work showing the assignment of signals of compound 9 are presented in Fig. 4.
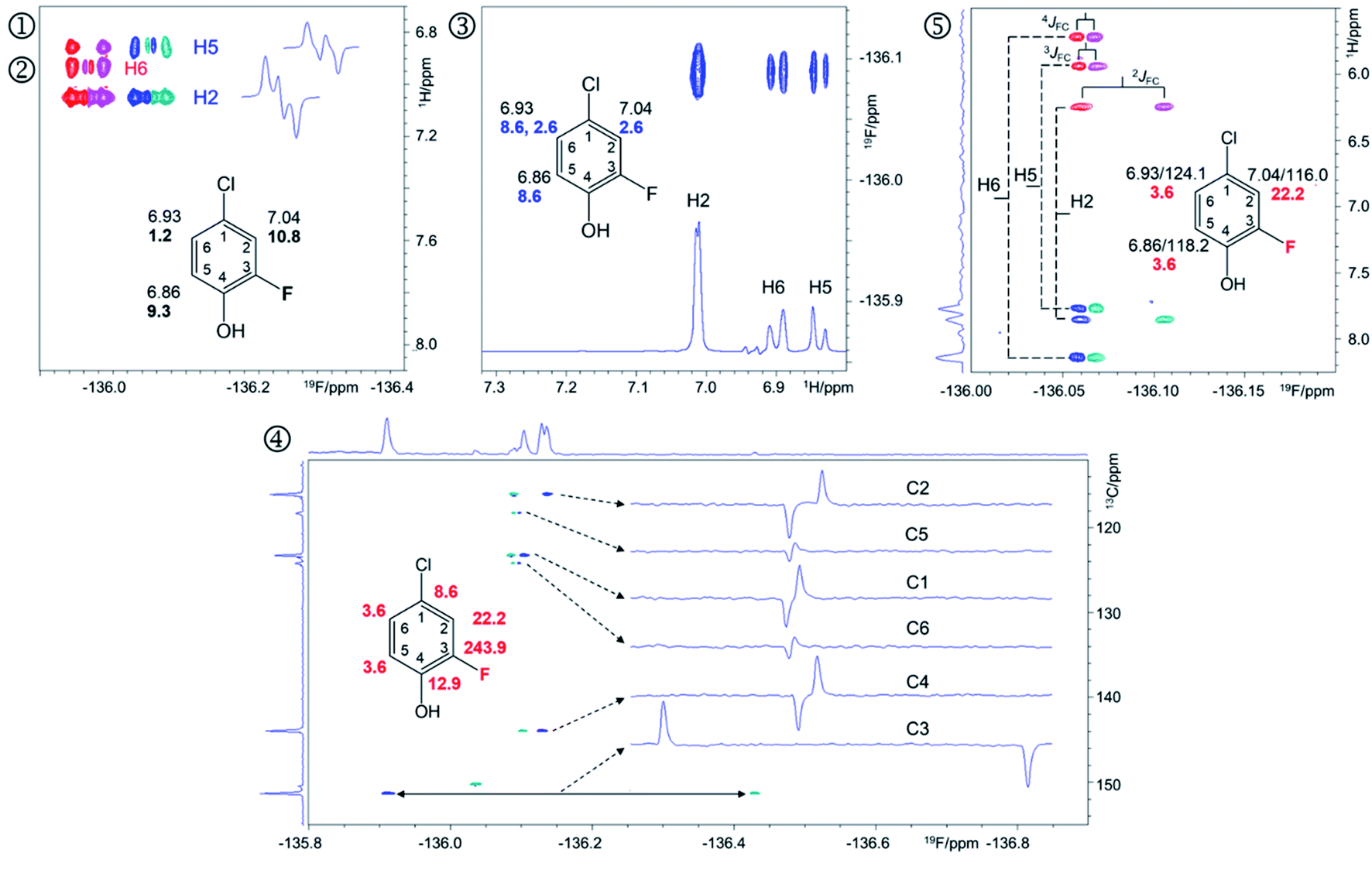 | ||
| Fig. 4 Regions of the 500 MHz NMR spectra acquired with the pulse sequences presented in Fig. S8–S12† showing chemical shift correlations for compound 9. In addition to 2D cross peaks, the figures display the structure of 9 with selected NMR parameters, and where appropriate, F2 traces showing the fine structure of cross peaks. ① Overlay of the 2D 1H, 19F HETCOR (blue/turquoise) and ② 2D 1H, 19F TOCSY-HETCOR (red/magenta) cross peaks. The TOCSY spectrum was left-shifted to facilitate identification of signals. F2 traces through H2 and H5 cross peaks from the HETCOR spectrum are shown. 1H chemical shifts and JHF values (bold) are displayed on the structure; ③ A 2D 19F, 1H CP-DIPSI3-DIPSI2 spectrum; F2 trace at the 19F chemical shift of 9 is shown; 1H chemical shifts and JHH values (blue) are displayed on the structure; ④ A 2D 19F, 13C HMBC spectrum optimised for nJFC coupling constants. Internal F1 and F2 projections and F2 traces at the 13C chemical shifts of 9 are displayed; the JFC values are shown in red; ⑤overlay of two edited 2D(3,2) H1CnF correlation spectra containing individual cross peaks of the F1 doublets that code for 13C chemical shifts. Blue/turquoise and red/magenta colours indicate antiphase JFCF2 doublets in each spectrum. The internal F1 projection of one of the spectra is displayed. Vertical lines connect the corresponding signals with their midpoint marking the 1H chemical shifts. The 1H/13C chemical shifts and JFC coupling constants (red) are indicated. These active coupling constants appear in antiphase, which can cause partial signal cancellation. Thus, to obtain more accurate values it is best to determine them from a 1H coupled 19F spectrum. The H6,F cross peak only appears in the 2D 1H, 19F TOCSY-HETCOR spectrum (②, Fig. 4) because the JH6,F coupling constant is too small to generate a response in the former experiment. A 2D 19F, 1H CP-DIPSI3-DIPSI2 (③, Fig. 4) serves to extend the proton networks beyond the protons coupled to 19F, similarly to 2D 1H, 19F TOCSY-HETCOR experiment. However, as a 1H-detected experiment, it provides values of JHH coupling constants that are beneficial to the structure determination process. A 2D 19F, 13C HMBC spectrum optimised for nJFC coupling constants (④, Fig. 4) provides the chemical shifts and nJFC coupling constants of all 19F-coupled carbons. For one-bond 19F–13C correlations, the sensitivity of the experiment can be enhanced by optimising the polarisation transfer periods for 1JFC coupling constants (pulse sequence of Fig. S11†). If the values of 1JFC coupling are known, the HMBC experiment can be set up to yield the one-bond correlations as well. Finally, the outcome of a simultaneous H1CnF correlation is illustrated in ⑤ (Fig. 4). This intrinsically 3D experiment has been modified using the principles of reduced dimensionality83,84 to produce a (3, 2)D experiment. Here, the 13C chemical shift is coded in the 1H dimension by the width of the F1-doublet. In this experiment two interleaved spectra are acquired, which contain in-phase or antiphase F1 doublets. Editing of these spectra increases the S/N ratio and removes half of the cross peaks in each spectrum, thus reducing spectral overlap. | ||
The 19F-detected z-filtered 2D 1H, 19F HETCOR spectrum (①, Fig. 4) shows HF cross peaks with protons H2 and H5 whose appearance is mediated by large JHF coupling constants.
Sensitivity and resolution limits of 19F-centered NMR
Based on the analysis of signal intensities of the thirteen most intense resonances seen in the 1H-decoupled 1D 19F NMR spectrum of a 30 mg mixture (Fig. 1 and S1†), it can be estimated that compound 11 – the lowest concentration compound that yielded signals in experiments involving 13C – is present at 1 mM (or 30 μg in 180 μL of CD3OH in a 3 mm NMR tube assuming an average molecular weight of 170 g mol−1 for compounds in this mixture). This sensitivity limit applies to an overnight experiment on a 500 MHz NMR spectrometer equipped with a 5 mm QCI-F CryoProbe and a 3 mm sample tube.Exploring a hypothetical scenario, 30 mg of a mixture could contain a 1000 similar size compounds at around 30 μg each. These would be amenable to the structure determination as outlined here, thanks to the remarkable sensitivity of today's NMR spectrometers and the efficiency of the 19F-centered approach. The sensitivity of 1H, 19F correlation experiments is naturally higher with an estimated concentration limit of ∼30 μM (or 1 μg for compounds with Mw = 170 g mol−1 in 180 μl). This statement is supported by the appearance of hundreds of cross peaks in the 2D 1H, 19F HETCOR spectrum (Fig. S3†) associated with 19F signals that are 30 × weaker than the signal of 11. Around 200 spin systems of these minor compounds could be identified in this spectrum. Their cross peaks were resolved due to the exquisite sensitivity of 19F to its chemical environment. The presented analysis thus provides a glimpse into the complexity of mixtures that are amenable to structure elucidation by 19F-centered NMR.
Structure determination process in 19F-centred NMR
In reference (and using symbols to ⑦) to the schematic representation of 19F-centred NMR experiments (Fig. 3) and the example spectra of the chloramination product mixture (Fig. 4), the steps involved in 19F-centred NMR structure determination are discussed below and summarised in a flowchart (Fig. 5).
to ⑦) to the schematic representation of 19F-centred NMR experiments (Fig. 3) and the example spectra of the chloramination product mixture (Fig. 4), the steps involved in 19F-centred NMR structure determination are discussed below and summarised in a flowchart (Fig. 5).
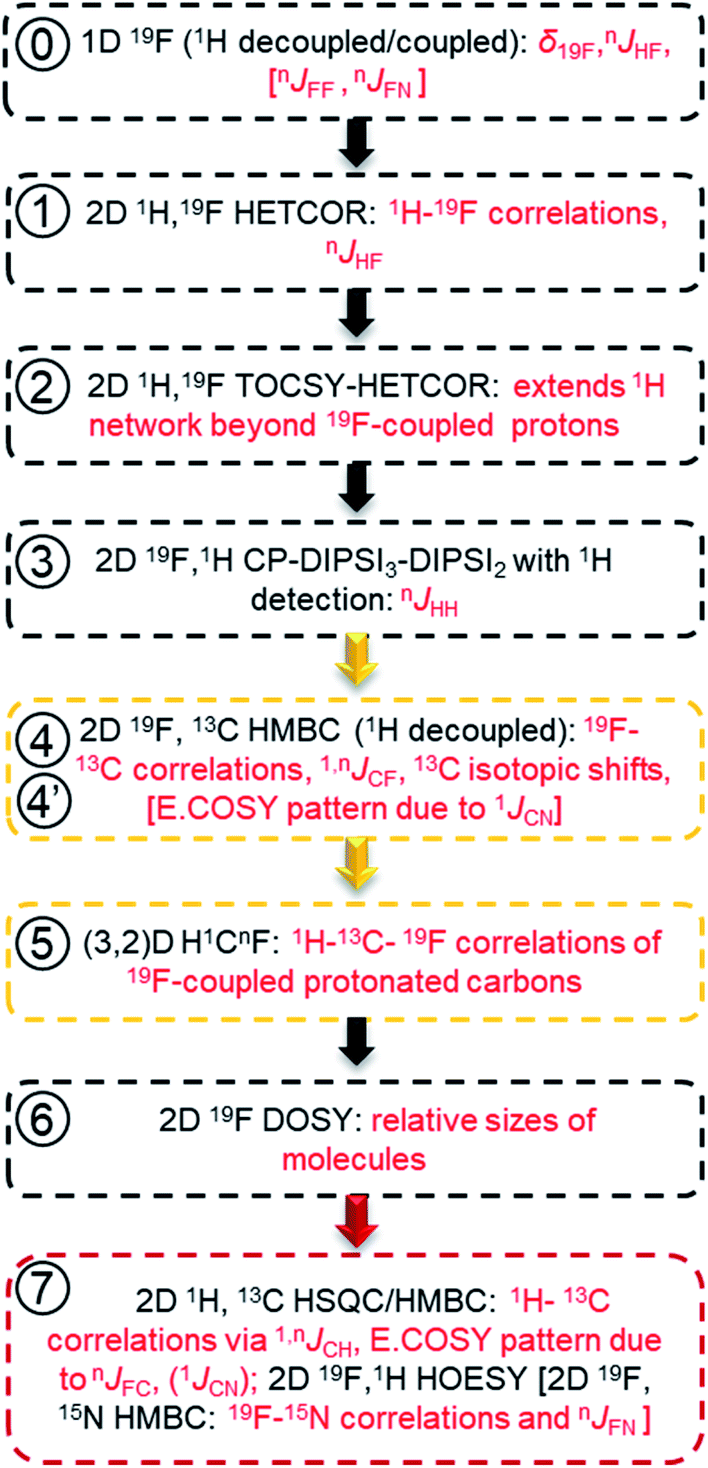 | ||
| Fig. 5 Flow chart for acquiring and working with spectra of mixtures of fluorinated compounds. The information obtained is given in red. Golden and red boxes denote experiments involving 13C, and the experiments for extending the structure beyond the F-containing moieties, respectively. | ||
 The process starts with the acquisition of standard 1D 1H-coupled and 1H-decoupled 19F spectra, which provide 19F chemical shifts and values of nJHF coupling constants.
The process starts with the acquisition of standard 1D 1H-coupled and 1H-decoupled 19F spectra, which provide 19F chemical shifts and values of nJHF coupling constants.
① Chemical shifts of 19F-coupled protons are determined in a 2D 19F, 1H HETCOR experiment; nJHF coupling constants are assigned.
② The 19F-associated proton network is extended by protons not directly coupled to 19F in a 2D 19F, 1H TOCSY-HETCOR experiment.
③ JHH coupling constants are obtained in a 2D 19F, 1H CP-DIPSI3-DIPSI2 experiment; extension of the proton network, established by ①and ②, is possible.
The correlated 19F and 1H chemical shifts and homo- and heteronuclear coupling constants can now be interpreted to propose structural fragments by considering the effect of substituents,85 values of JHF coupling constants86,87 (Table S2†) and JHH coupling constants.
④ 2D 19F, 13C HMBC experiment provides 19F–13C chemical shift correlations, values of 1,nJCF coupling constants and 13C-induced 19F isotopic shifts.
⑤ The 2D(3,2) H1CnF correlation spectra provide a distinction between protonated and non-protonated 19F-coupled carbons and chemical shift correlations of HC pairs.
Experiments involving 19F–13C correlations are very informative and should be performed if sufficient amount of material is available. Considering the effects of substituents,88 the sizes of JFC coupling constants86,87 (Table S3†) and 13C-induced 19F isotopic chemical shifts (Table S4†), structural fragments proposed by the analysis of 1H/19F data can be verified and extended.
⑥ Relative sizes of molecules in a mixture are estimated by a 2D 19F DOSY experiment.
Taking advantage of the large chemical shift dispersion of 19F, interpretation of 19F-detected DOSY spectra89 (Fig. S4†) is straightforward due to minimal signal overlap. A one-shot DOSY experiment90 with rectangular 19F pulses was used here; for spectra covering a wider range of 19F chemical sifts, the use of adiabatic pulses is recommended.91,92 For the studied mixture, the measured diffusion coefficients generally decreased with increasing molecular weight of compounds and their substituents in the order COOH, NO2 and Cl. The contribution from the carboxyl groups was particularly large, presumably because of the formation of hydrogen bonds with the solvent. Assessment of the molecular weight also helps to decide if data beyond the reach of 19F-centred experiments are required.
⑦ 2D 1H, 13C HSQC/HMBC spectra provide one-bond and long-range 1H–13C correlations beyond the reach of the 19F-centered experiment. 2D 19F, 1H HOESY experiments can also help to identify more remote protons.
Using standard 2D 1H, 13C one-bond and long-range correlated experiments alone to analyse complex mixtures is problematic due to the complexity of their spectra. Nevertheless, for larger molecules, which contain spin systems isolated from those containing 19F, protons and carbons identified by 19F-centred experiments can act as starting points for extending the assignments through the analyses of 2D 1H, 13C HSQC/HMBC spectra. Similarly, 2D 19F, 1H HOESY experiments75,93 can reach more remote protons by utilising 19F, 1H NOEs.
Due to use of 15NH4Cl, some fluorinated compounds studied here, contain 15N, which opened another route for obtaining structural information as summarised using square brackets in the flow chart of Fig. 5. The 19F–15N chemical shift correlations can be obtained by a 2D 19F, 15N HMBC experiment (Fig. S10 and S5†). For nitrogen-containing DBPs, carbons directly bonded to 15N are identifiable by the E.COSY pattern of cross peaks in 2D 19F, 13C HMBC spectra caused by relatively large 1JNC (11–13 Hz) coupling constants. The sizes of JFN (or JFF) coupling constants are best determined from 1D 1H-decoupled 19F spectra. A potential presence of 19F–19F interactions can be probed by a 2D 19F, 19F COSY experiment.
Analysis of the chloramination reaction pathways
19F-centred NMR methodology provided a rich set of NMR parameters for the chloramination reaction product mixture (Table S5†), which allowed the structure elucidation of eleven molecules, present in concentrations above the current sensitivity threshold, and partial structures for two additional molecules (Fig. 6). The analysed mixture was prepared in a 5 day experiment, which led to extensive modification of the starting material producing phenolic and likely also non-phenolic compounds, initially via transfer of Cl released from hypochlorous acid, HOCl.50,56 Electrophilic substitution reactions, as the main chlorination mechanism for aromatic substitution,94 resulted in chlorination of 1 producing 2 as the major product. Several DBPs generated by other reactions were also modified in this way–a Cl substitution at the activated ortho position next to an OH group (9→3, 8→13, 4→6). The unexpected appearance of a brominated compound formed from the starting material (1→10) can be explained by the use of NaOCl manufactured by the electrolysis of sodium chloride. Water used in this process contains small amounts of sodium bromide,95 which led to the production of sodium bromate – the source of Br. The presence and the position of Br in compound 10 was established through a comparison of chemical shifts of 2 and 10, which differ only in the nature of the halogen substituent. The experimental differences in the 1H and 13C NMR chemical shifts at corresponding positions agreed perfectly with the values predicted by considering the effects of Cl and Br on the chemical shift of benzene resonance.76,79 In addition, peaks at m/z 232.9255 and 234.9235, corresponding to [C7H3O3F79Br]− and [C7H3O3F81Br]– ions, were detected in FT ICR MS spectra of the product mixture (data not shown), confirming the presence of Br in compound 10.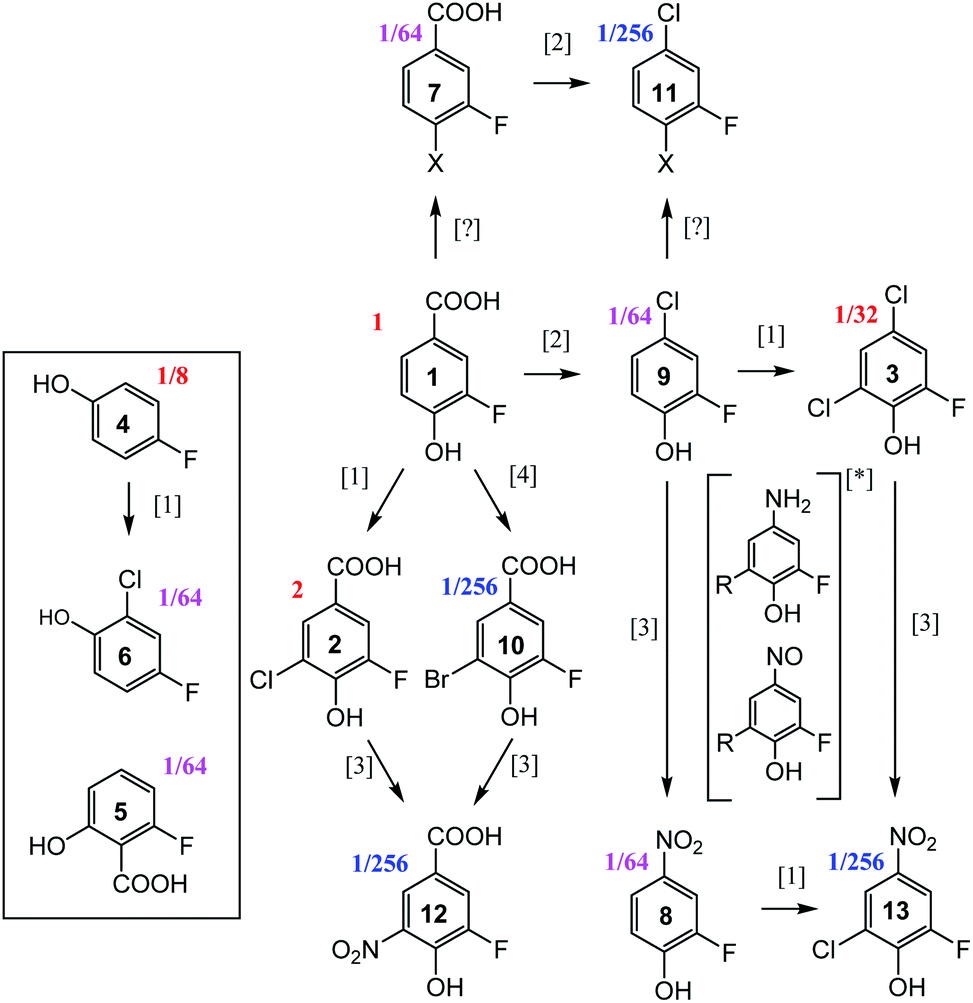 | ||
| Fig. 6 Reaction pathways [1],94 [2],96 [3],50 and [4],95 identified in chloramination of 1. Compounds enclosed in a rectangle fall outside of this classification. Fractions given represent concentrations relative to the starting material, 1, as estimated from the intensity of signals in the 1D 19F NMR spectrum. *unconfirmed intermediates, R = H or Cl. | ||
The second reaction type observed was decarboxylative chlorination96 (1→9 or 7→11). The halogenated sites also continued to react with monochloramine through nucleophilic substitution by H2N in a dechlorinative amination.97 The generated aromatic amines were further oxidised by NH2Cl to form nitroso- and eventually nitro compounds,50 (2→12, 10→12, 9→8, 3→13). An unexpected outcome was the appearance of compounds 4 and 5. These compounds were not part of the starting material, as confirmed by the absence of their signals in the 1H-decoupled 19F spectrum of 1. Their structures were verified by a comparison of NMR parameters with literature data.98,99 Performing such checks is generally recommended, especially in instances where the appearance of the identified compounds is difficult to rationalise. Such comparisons are considered to be reliable due to sensitivity of NMR parameters to molecular structures.
Two additional compounds, containing a tri-substituted benzene ring with a carboxylic group (7) or a chlorine (11) at position C-1, were identified. The differences between the 13C and 1H chemical shifts of the corresponding atoms of these compounds matched the differences observed for an analogous pair of molecules, 1 and 9. A possible mechanism for the formation of compounds 7 and 11 from 1 and 9, respectively, is via resonance stabilised phenoxyl radicals produced by dissociation or abstraction of the phenolic hydrogen.100 This hypothesis is supported by the observed changes of colour of the reaction mixture over the course of 5 days, which could indicate the existence of quinone/semiquinone equilibria. Based on the 19F DOSY spectrum (Fig. S4†), molecules 7 and 11 are the largest, likely dimeric molecules. Attempts to extend their structures using 1H, 13C correlation experiments, as suggested in step ⑦ of Fig. 5, did not yield further information. A 1H, 19F HOESY experiment (not performed here) represents another opportunity for structural characterisation.
The origin of most but not all compounds identified in this study can thus be explained by known reaction mechanisms. It is possible that during the course of chloramination, fluorine radicals were created, further modifying the pool of the produced compounds. This could help to explain the variety of 19F containing compounds (Fig. 1 and ES1†) that are present in concentrations too low to currently allow their structure elucidation. The other source of heterogeneity of the final mixture are the N-containing molecules, as indicated by the richness of its 2D 1H,15N HSQC spectrum (Fig. S6†). None of compounds 2–13 contain a protonated NHx (x = 1, 2) group, indicating that the nitrogenated products of 1 are present at low concentrations.
The number of compounds obtained in our experiments, which admittedly aimed to maximise the production of DBPs, is astounding. Their structural studies will continue to attract attention due to the potential influence of DBPs on human health and the environment.
Conclusions
By analysing a complex mixture of DBPs produced by chloramination of a single fluorine-tagged molecule, we have demonstrated the feasibility of 19F-centred NMR structure determination of small molecules without the need for compound separation. The 19F-centred experiments correlated 19F chemical shifts with those of 1H, 13C and 15N, provided values of JHF, JFC and JNF coupling constants, including 1H–1H chemical shift correlations and JHH coupling constants for a subset of protons. The proposed experiments, which can also be used in their own right, thus collectively represent an efficient NMR approach to the structure determination of mono-fluorinated moieties and small compounds in complex mixtures.Data availability
Data is available on request.Author contributions
NGAB proposed the methodology, designed experiments and performed the structure elucidation. AJRS, DU and RY contributed to the implementation of the experiments and acquisition of spectra. AJRS performed the chloramination reaction. All authors contributed to the analysis of the spectra and writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
NGAB would like to acknowledge NERC soil security programme NE/N020227/1 for funding. AJRS was supported by the Scottish Water and EPSRC grant EP/N509644/1 and RY by the NERC Centre for Doctoral Training, E4 (NE/S007407/1). Instrument support was in part provided by the EPSRC grant EP/R030065/1. The authors would like to thank: Juraj Bella and Dr Lorna Murray at the University of Edinburgh for maintenance of the NMR spectrometers. Dr Dan Fletcher at the University of Dundee for assisting with the acquisition of spectra on the 500 MHz NMR spectrometer. Dr Antonín Lyčka at the University of Hradec Králové for his expert advice during the structure determination of nitrogen containing compounds.References
- D. B. Harper, D. O'Hagan and C. D. Murphy, Fluorinated Natural Products: Occurrence and Biosynthesis, in Natural Production of Organohalogen Compounds, The Handbook of Environmental Chemistry (Vol. 3 Series: Anthropogenic Compounds), ed. G. Gribble, Springer, Berlin, Heidelberg, 2003, vol. 3/3P, DOI:10.1007/b10454.
- M. Inoue, Y. Sumii and N. Shibata, Acs Omega, 2020, 5, 10633–10640 CrossRef CAS PubMed.
- J. L. Han, A. M. Remete, L. S. Dobson, L. Kiss, K. Izawa, H. Moriwaki, V. A. Soloshonok and D. O’Hagan, J. Fluorine Chem., 2020, 239, 109639 CrossRef CAS.
- J. Wang, M. Sanchez-Rosello, J. L. Acena, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed.
- T. Fujiwara and D. O'Hagan, J. Fluorine Chem., 2014, 167, 16–29 CrossRef CAS.
- Y. Ogawa, E. Tokunaga, O. Kobayashi, K. Hirai and N. Shibata, Iscience, 2020, 23, 101467 CrossRef CAS PubMed.
- P. T. Lowe and D. O’Hagan, J. Fluorine Chem., 2020, 230, 109420 CrossRef CAS.
- O. Jacobson, D. O. Kiesewetter and X. Y. Chen, Bioconjugate Chem., 2015, 26, 1–18 CrossRef CAS PubMed.
- T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed.
- T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470–477 CrossRef CAS PubMed.
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed.
- P. T. Nyffeler, S. G. Duron, M. D. Burkart, S. P. Vincent and C. H. Wong, Angew. Chem., Int. Ed., 2005, 44, 192–212 CrossRef CAS PubMed.
- D. O'Hagan and H. Deng, Chem. Rev., 2015, 115, 634–649 CrossRef PubMed.
- J. Fang, D. Hait, M. Head-Gordon and M. C. Y. Chang, Angew. Chem., Int. Ed., 2019, 58, 11841–11845 CrossRef CAS PubMed.
- T. Hayashi, G. Kehr, K. Bergander and R. Gilmour, Angew. Chem., Int. Ed., 2019, 58, 3814–3818 CrossRef CAS PubMed.
- A. Rentmeister, F. H. Arnold and R. Fasan, Nat. Chem. Biol., 2009, 5, 26–28 CrossRef CAS PubMed.
- C. D. Murphy, Biotechnol. Lett., 2010, 32, 351–359 CrossRef CAS PubMed.
- B. D. Key, R. D. Howell and C. S. Criddle, Environ. Sci. Technol., 1997, 31, 2445–2454 CrossRef CAS.
- X. J. Zhang, T. B. Lai and R. Y. C. Kong, in Fluorous Chemistry, ed. I. T. Horvath, 2012, vol. 308, pp. 365–404 Search PubMed.
- M. G. Boersma, T. Y. Dinarieva, W. J. Middelhoven, W. J. H. van Berkel, J. Doran, J. Vervoort and I. Rietjens, Appl. Environ. Microbiol., 1998, 64, 1256–1263 CrossRef CAS PubMed.
- V. S. Bondar, M. G. Boersma, E. L. Golovlev, J. Vervoort, W. J. H. Van Berkel, Z. I. Finkelstein, I. P. Solyanikova, L. A. Golovleva and I. Rietjens, Biodegradation, 1998, 9, 475–486 CrossRef CAS PubMed.
- M. Kiel and K. H. Engesser, Appl. Microbiol. Biotechnol., 2015, 99, 7433–7464 CrossRef CAS PubMed.
- R. Natarajan, R. Azerad, B. Badet and E. Copin, J. Fluorine Chem., 2005, 126, 425–436 CrossRef.
- M. B. Murphy, E. I. H. Loi, K. Y. Kwok and P. K. S. Lam, in Fluorous Chemistry, ed. I. T. Horvath, 2012, vol. 308, pp. 339–363 Search PubMed.
- B. M. Johnson, Y. Z. Shu, X. L. Zhuo and N. A. Meanwell, J. Med. Chem., 2020, 63, 6315–6386 CrossRef CAS PubMed.
- C. D. Murphy, B. R. Clark and J. Amadio, Appl. Microbiol. Biotechnol., 2009, 84, 617–629 CrossRef CAS PubMed.
- E. Nieto-Sepulveda, A. D. Bage, L. A. Evans, T. A. Hunt, A. G. Leach, S. P. Thomas and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2019, 141, 18600–18611 CrossRef CAS PubMed.
- R. Wei, A. M. R. Hall, R. Behrens, M. S. Pritchard, E. J. King and G. C. Lloyd-Jones, Eur. J. Org. Chem., 2021, 2021, 2331–2342 CrossRef CAS.
- N. G. A. Bell, L. Murray, M. C. Graham and D. Uhrin, Chem. Commun., 2014, 50, 1694–1697 RSC.
- N. G. A. Bell, M. C. Graham and D. Uhrin, Analyst, 2016, 141, 4614–4624 RSC.
- G. A. Bell, A. A. L. Michalchuk, J. W. T. Blackburn, M. C. Graham and D. Uhrin, Angew. Chem., Int. Ed., 2015, 54, 8382–8385 CrossRef PubMed.
- L. Castanar, P. Moutzouri, T. M. Barbosa, C. F. Tormena, R. Rittner, A. R. Phillips, S. R. Coombes, M. Nilsson and G. A. Morris, Anal. Chem., 2018, 90, 5445–5450 CrossRef CAS PubMed.
- T. M. Barbosa, L. Castanar, P. Moutzouri, M. Nilsson, G. A. Morris, R. Rittner and C. F. Tormena, Anal. Chem., 2020, 92, 2224–2228 CrossRef CAS PubMed.
- G. Dal Poggetto, J. V. Soares and C. F. Tormena, Anal. Chem., 2020, 92, 14047–14053 CrossRef CAS PubMed.
- S. W. Krasner, H. S. Weinberg, S. D. Richardson, S. J. Pastor, R. Chinn, M. J. Sclimenti, G. D. Onstad and A. D. Thruston, Environ. Sci. Technol., 2006, 40, 7175–7185 CrossRef CAS PubMed.
- S. D. Richardson, M. J. Plewa, E. D. Wagner, R. Schoeny and D. M. DeMarini, Mutat. Res., Rev. Mutat. Res., 2007, 636, 178–242 CrossRef CAS PubMed.
- M. G. Muellner, E. D. Wagner, K. McCalla, S. D. Richardson, Y. T. Woo and M. J. Plewa, Environ. Sci. Technol., 2007, 41, 645–651 CrossRef CAS PubMed.
- G. H. Hua and D. A. Reckhow, Water Res., 2007, 41, 1667–1678 CrossRef CAS PubMed.
- H. Sakai, S. Tokuhara, M. Murakami, K. Kosaka, K. Oguma and S. Takizawa, Water Res., 2016, 88, 661–670 CrossRef CAS PubMed.
- J. F. Lu, T. Zhang, J. Ma and Z. L. Chen, J. Hazard. Mater., 2009, 162, 140–145 CrossRef CAS PubMed.
- H. Gallard and U. von Gunten, Water Res., 2002, 36, 65–74 CrossRef CAS PubMed.
- X. F. Li and W. A. Mitch, Environ. Sci. Technol., 2018, 52, 1681–1689 CrossRef CAS PubMed.
- C. M. M. Bougeard, E. H. Goslan, B. Jefferson and S. A. Parsons, Water Res., 2010, 44, 729–740 CrossRef CAS PubMed.
- A. S. Ginwalla and M. A. Miklta, Environ. Sci. Technol., 1992, 26, 1148–1150 CrossRef CAS.
- T. Bond, M. R. Templeton, N. H. M. Kamal, N. Graham and R. Kanda, Water Res., 2015, 85, 85–94 CrossRef CAS PubMed.
- M. J. Plewa, E. D. Wagner, M. G. Muellner, K. M. Hsu and S. D. Richardson, in Disinfection by-Products in Drinking Water: Occurrence, Formation, Health Effects, and Control, eds. T. Karanfil, S. W. Krasner and Y. Xie, 2008, vol. 995, pp. 36–50 Search PubMed.
- Y. X. Dong, W. Y. Peng, Y. J. Liu and Z. H. Wang, J. Hazard. Mater., 2021, 401, 123884 CrossRef CAS PubMed.
- G. Y. Ding and X. R. Zhang, Environ. Sci. Technol., 2009, 43, 9287–9293 CrossRef CAS PubMed.
- H. Y. Zhai, X. R. Zhang, X. H. Zhu, J. Q. Liu and M. Ji, Environ. Sci. Technol., 2014, 48, 2579–2588 CrossRef CAS PubMed.
- T. T. Gong, Y. X. Tao, X. R. Zhang, S. Y. Hu, J. B. Yin, Q. M. Xian, J. Ma and B. Xu, Environ. Sci. Technol., 2017, 51, 10562–10571 CrossRef CAS PubMed.
- X. R. Zhang, R. A. Minear and S. E. Barrett, Environ. Sci. Technol., 2005, 39, 963–972 CrossRef CAS PubMed.
- A. Andersson, M. Harir, M. Gonsior, N. Hertkorn, P. Schmitt-Kopplin, H. Kylin, S. Karlsson, M. J. Ashiq, E. Lavonen, K. Nilsson, A. Pettersson, H. Stavklint and D. Bastviken, Environ. Sci.: Water Res. Technol., 2019, 5, 861–872 RSC.
- Q. L. Fu, M. Fujii and E. Kwon, Anal. Chem., 2020, 92, 13989–13996 CrossRef CAS PubMed.
- D. M. Bulman and C. K. Remucal, Environ. Sci. Technol., 2020, 54, 9629–9639 CrossRef CAS PubMed.
- H. F. Zhang, Y. H. Zhang, Q. Shi, J. Y. Hu, M. Q. Chu, J. W. Yu and M. Yang, Environ. Sci. Technol., 2012, 46, 4396–4402 CrossRef CAS PubMed.
- E. E. Lavonen, M. Gonsior, L. J. Tranvik, P. Schmitt-Kopplin and S. J. Kohler, Environ. Sci. Technol., 2013, 47, 2264–2271 CrossRef CAS PubMed.
- Z. N. Hao, Y. G. Yin, D. Cao and J. F. Liu, Environ. Sci. Technol., 2017, 51, 5464–5472 CrossRef CAS PubMed.
- M. Gonsior, P. Schmitt-Kopplin, H. Stavklint, S. D. Richardson, N. Hertkorn and D. Bastviken, Environ. Sci. Technol., 2014, 48, 12714–12722 CrossRef CAS PubMed.
- H. F. Zhang, Y. H. Zhang, Q. Shi, H. D. Zheng and M. Yang, Environ. Sci. Technol., 2014, 48, 3112–3119 CrossRef CAS PubMed.
- S. Zheng, J. C. Shi, J. Y. Hu, W. X. Hu, J. Zhang and B. Shao, Water Res., 2016, 107, 1–10 CrossRef CAS PubMed.
- Y. L. Wang, H. J. Liu, G. G. Liu and Y. H. Xie, Sci. Total Environ., 2014, 473, 437–445 CrossRef PubMed.
- S. Zheng, J. C. Shi, J. Zhang, Y. Yang, J. Y. Hu and B. Shao, Water Res., 2018, 132, 167–176 CrossRef CAS PubMed.
- F. M. Wendel, C. L. Eversloh, E. J. Machek, S. E. Duirk, M. J. Plewa, S. D. Richardson and T. A. Ternes, Environ. Sci. Technol., 2014, 48, 12689–12697 CrossRef CAS PubMed.
- X. Wang, J. Wang, Y. H. Zhang, Q. Shi, H. F. Zhang, Y. Zhang and M. Yang, Sci. Total Environ., 2016, 554, 83–88 Search PubMed.
- T. L. Hwang and A. J. Shaka, J. Magn. Reson., Ser. A, 1995, 112, 275–279 CrossRef CAS.
- B. Adams, Magn. Reson. Chem., 2008, 46, 377–380 CrossRef CAS PubMed.
- M. J. Thrippleton and J. Keeler, Angew. Chem., Int. Ed., 2003, 42, 3938–3941 CrossRef CAS PubMed.
- D. Marion, P. C. Driscoll, L. E. Kay, P. T. Wingfield, A. Bax, A. M. Gronenborn and G. M. Clore, Biochemistry, 1989, 28, 6150–6156 CrossRef CAS PubMed.
- H. Hu, P. Kulanthaivel and K. Krishnamurthy, J. Org. Chem., 2007, 72, 6259–6262 CrossRef CAS PubMed.
- D. O. Cicero, G. Barbato and R. Bazzo, J. Magn. Reson., 2001, 148, 209–213 CrossRef CAS PubMed.
- G. Bodenhausen and R. R. Ernst, J. Magn. Reson., 1981, 45, 367–373 CAS.
- Y. Shen, H. S. Atreya, G. H. Liu and T. Szyperski, J. Am. Chem. Soc., 2005, 127, 9085–9099 CrossRef CAS PubMed.
- W. Kozminski and I. Zhukov, J. Biomol. NMR, 2003, 26, 157–166 CrossRef CAS PubMed.
- L. Li and P. L. Rinaldi, Macromolecules, 1997, 30, 520–525 CrossRef CAS.
- J. Battiste and R. A. Newmark, Prog. Nucl. Magn. Reson. Spectrosc., 2006, 48, 1–23 CrossRef CAS.
- R. A. Newmark and R. J. Webb, J. Fluorine Chem., 2005, 126, 355–360 CrossRef CAS.
- A. A. Marchione and B. Conklin, Appl. Magn. Reson., 2017, 48, 485–499 CrossRef CAS.
- A. A. Marchione, R. J. Dooley and B. Conklin, Magn. Reson. Chem., 2014, 52, 183–189 CrossRef CAS PubMed.
- K. A. M. Ampt, R. Aspers, P. Dvortsak, R. M. van der Werf, S. S. Wijmenga and M. Jaeger, J. Magn. Reson., 2012, 215, 27–33 CrossRef CAS PubMed.
- R. Aspers, K. A. M. Ampt, P. Dvortsak, M. Jaeger and S. S. Wijmenga, J. Magn. Reson., 2013, 231, 79–89 CrossRef CAS PubMed.
- K. A. M. Ampt, R. Aspers, M. Jaeger, P. Geutjes, M. Honing and S. S. Wijmenga, Magn. Reson. Chem., 2011, 49, 221–230 CrossRef CAS PubMed.
- J. Sanchis, A. Jaen-Gil, P. Gago-Ferrero, E. Munthali and M. J. Farre, Water Res., 2020, 176, 115743 CrossRef CAS PubMed.
- N. Brodaczewska, Z. Kostalova and D. Uhrin, J. Biomol. NMR, 2018, 70, 115–122 CrossRef CAS PubMed.
- J. Sakas and N. G. A. Bell, Faraday Discuss., 2019, 218, 191–201 RSC.
- Hans Reich's Collection. 1H NMR Spectroscopy, https://organicchemistrydata.org/hansreich/resources/nmr/?index=nmr_index%2F1H_shift#hdata38, (accessed June 2021) Search PubMed.
- B. F. Lutnaes, G. Luthe, U. A. T. Brinkman, J. E. Johansen and J. Krane, Magn. Reson. Chem., 2005, 43, 588–594 CrossRef CAS PubMed.
- F. J. Weigert and J. D. Roberts, J. Am. Chem. Soc., 1971, 93, 2361–2369 CrossRef.
- Hans Reich's Collection, 13C NMR Spectroscopy, https://organicchemistrydata.org/hansreich/resources/nmr/?index=nmr_index%2F13C_shift#cdata-v08, (accessed June 2021) Search PubMed.
- H. Barjat, G. A. Morris, S. Smart, A. G. Swanson and S. C. R. Williams, J. Magn. Reson., Ser. B, 1995, 108, 170–172 CrossRef CAS.
- M. D. Pelta, G. A. Morris, M. J. Stchedroff and S. J. Hammond, Magn. Reson. Chem., 2002, 40, S147–S152 CrossRef CAS.
- J. E. Power, M. Foroozandeh, P. Moutzouri, R. W. Adams, M. Nilsson, S. R. Coombes, A. R. Phillips and G. A. Morris, Chem. Commun., 2016, 52, 6892–6894 RSC.
- C. L. Xu, Y. Wan, D. X. Chen, C. Gao, H. N. Yin, D. Fetherston, E. Kupce, G. Lopez, B. Ameduri, E. B. Twum, F. J. Wyzgoski, X. H. Li, E. F. McCord and P. L. Rinaldi, Magn. Reson. Chem., 2017, 55, 472–484 CrossRef CAS PubMed.
- P. L. Rinaldi, J. Am. Chem. Soc., 1983, 105, 5167–5168 CrossRef CAS.
- M. Deborde and U. von Gunten, Water Res., 2008, 42, 13–51 CrossRef CAS PubMed.
- F. Edition, WHO Chron., 2011, 38, 179 Search PubMed.
- R. A. Larson and A. L. Rockwell, Environ. Sci. Technol., 1979, 13, 325–329 CrossRef CAS.
- J. D. Roberts, H. E. Simmons, L. A. Carlsmith and C. W. Vaughan, J. Am. Chem. Soc., 1953, 75, 3290–3291 CrossRef CAS.
- M. J. Zhang, H. X. Li, H. Y. Li and J. P. Lang, Dalton Trans., 2016, 45, 17759–17769 RSC.
- J. M. Silla, C. J. Duarte, R. Rittner and M. P. Freitas, RSC Adv., 2013, 3, 25765–25768 RSC.
- C. A. McFerrin, R. W. Hall and B. Dellinger, J. Mol. Struct.: THEOCHEM, 2009, 902, 5–14 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc06057k |
| This journal is © The Royal Society of Chemistry 2022 |