
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of distibiranes and azadistibiranes by cycloaddition reactions of distibenes with diazomethanes and azides†
Hanns M.
Weinert
 a,
Christoph
Wölper
a and
Stephan
Schulz
*ab
a,
Christoph
Wölper
a and
Stephan
Schulz
*ab
aInstitute for Inorganic Chemistry, University of Duisburg-Essen, Universitätsstraße 5–7, 45117 Essen, Germany. E-mail: stephan.schulz@uni-due.de
bCenter for Nanointegration Duisburg-Essen (Cenide), University of Duisburg-Essen, Carl-Benz-Straße 199, 47057 Duisburg, Germany
First published on 3rd March 2022
Abstract
Cycloaddition reactions of distibene [L(Me2N)GaSb]2 (L = HC[C(Me)NDipp]2; Dipp = 2,6-i-Pr2C6H3)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) with a series of organoazides RN3 (R = Ph, p-CF3Ph, 1-adamantyl (ada)) yielded azadistibiranes [L(Me2N)GaSb]2NR (R = Ph 1, p-CF3Ph 2, ada 3), whereas Me3SiN3 reacted with insertion into one Ga–Sb bond and formation of L(Me2N)GaSbSb(NSiMe3)Ga(NMe2)L (4). Analogous compounds 5 and 6 formed after heating of 1 and 2 above 60 °C. Prolonged heating of 5 resulted in a [2 + 2] cycloaddition accompanied by elimination of LGa(NMe2)2 and formation of tetrastibacyclobutane 7, while the reaction of 5 with a second equivalent of PhN3 gave heteroleptic azadistibirane 9, which isomerized at elevated temperature to distibene 10. Cycloaddition also occurred in reactions of [L(X)GaSb]2 (X = NMe2, OEt, Cl) with Me3Si(H)CN2, yielding distibiranes [L(X)GaSb]2C(H)SiMe3 (X = NMe211, OEt 12, Cl 13). Compounds 1–13 were characterized by IR, UV-Vis and NMR spectroscopy and sc-XRD. The mechanism of the reaction of [L(Me2N)GaSb]2 with PhN3 and Me3SiN3 and the electronic nature of the resulting compounds were studied by DFT calculations.
with a series of organoazides RN3 (R = Ph, p-CF3Ph, 1-adamantyl (ada)) yielded azadistibiranes [L(Me2N)GaSb]2NR (R = Ph 1, p-CF3Ph 2, ada 3), whereas Me3SiN3 reacted with insertion into one Ga–Sb bond and formation of L(Me2N)GaSbSb(NSiMe3)Ga(NMe2)L (4). Analogous compounds 5 and 6 formed after heating of 1 and 2 above 60 °C. Prolonged heating of 5 resulted in a [2 + 2] cycloaddition accompanied by elimination of LGa(NMe2)2 and formation of tetrastibacyclobutane 7, while the reaction of 5 with a second equivalent of PhN3 gave heteroleptic azadistibirane 9, which isomerized at elevated temperature to distibene 10. Cycloaddition also occurred in reactions of [L(X)GaSb]2 (X = NMe2, OEt, Cl) with Me3Si(H)CN2, yielding distibiranes [L(X)GaSb]2C(H)SiMe3 (X = NMe211, OEt 12, Cl 13). Compounds 1–13 were characterized by IR, UV-Vis and NMR spectroscopy and sc-XRD. The mechanism of the reaction of [L(Me2N)GaSb]2 with PhN3 and Me3SiN3 and the electronic nature of the resulting compounds were studied by DFT calculations.
Introduction
Homonuclear main group element compounds with π bonding contribution have received increasing interest in the last decade due to their promising reactivity in small molecule activation reactions including catalytic processes.1 Double-bonded species in principle should undergo cycloaddition reactions, which belong to the most versatile synthetic tools in organic chemistry (i.e. Diels–Alder reaction and Huisgen cycloaddition).2 Heavier homologues of alkenes, disilenes R2SiSiR2 and digermenes R2GeGeR2,3 as well as diphosphenes RPPR were successfully applied in a variety of [2 + n] (n = 1–4) cycloaddition reactions,4 and diphosphenes reacted with formation of diphosphiranes and azadiphosphiranes (Scheme 1), respectively. In remarkable contrast, virtually no reactions of heavier group 15 dipnictenes RPnPnR containing Pn–Pn double bonds (Pn = As, Sb, Bi) are known, most likely originating from the use of sterically demanding organic substituents, which are applied to avoid the thermodynamically favored dimerization of heavier dipnictenes to cyclotetrapnictanes (kinetic stabilization), which were obtained from reduction reactions of RPnX2 (X = halide).5 Their formation can be rationalized by [2 + 2] cycloaddition of transient dipnictenes, however, the reaction of isolated dipnictene to cyclotetrapnictanes was not yet reported. Diels–Alder-type cycloaddition reactions were only reported for trapped (transient) diarsenes containing sterically less demanding substituents,6e.g. 2,3-bistrifluoromethyl-2,3-arsabicyclo[2.2.2]oct-5-ene was formed in a trapping reaction of in situ formed [CF3As]2 with cyclohexadiene.6a The 1,3 dipolar cycloaddition of nitrile oxides (tmp)CNO (tmp = 2,4,6-(MeO)3C6H2, 2,4,6-Me3C6H2) reported by Ohashi et al. to the best of our knowledge therefore represents the only cycloaddition reaction of a distibene (I).7 This group also reported on the formation of a single product in the reaction of [(Bbt)Sb]2 (Bbt = 2,6-[CH(SiMe3)2]-4-[C(SiMe3)3]-C6H2) with Me3SiN3, but the as-formed compound could not be isolated due to its thermal instability. Apart from these studies, only reactions of distibenes with chalcogens were reported,5e,f,7a,8 yielding three-membered seleno- and tellurodistibirane rings. | ||
| Scheme 1 Heterocycles formed in cycloaddition reactions of dipnictenes. | ||
We are generally interested in the reactivity of electron-rich L(X)Ga-substituted dipnictenes of the type [L(X)GaE]2 (E = As, Sb, Bi),9 and recently reported on cyclic voltammetry (CV) studies and single electron reduction reactions, yielding distibene and dibismuthene radical anions.10 We now extended our studies to cycloaddition reactions and herein report on reactions of distibenes [L(X)GaSb]2 (X = Cl, OEt, NMe2) with diazomethane Me3Si(H)CN2 and a series of organoazides RN3, yielding distibiranes (III) and azadistibiranes (IV), which were found to rearrange upon heating with re-formation of the Sb–Sb double bond and finally to tetrastibacyclobutane (II).
Results and discussion
Synthesis
[L(Me2N)GaSb]2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9a unselectively reacted with organoazides RN3 at ambient temperature with N2 elimination and formation of several reaction products according to 1H NMR spectroscopic studies (Fig. S46†). In contrast, dropwise addition of toluene solutions of PhN3, p-CF3PhN3 and 1-adaN3 to a cooled (−30 °C) suspension of [L(Me2N)GaSb]2 and slow heating to ambient temperature selectively gave azadistibiranes 1–3 (Scheme 2). The reactions can be classified in a formal sense as [2 + 1] cycloaddition reactions, but an initial [2 + 3] dipolar cycloaddition followed by a rapid N2 elimination is also plausible.2,4a,11 However, temperature-dependent in situ1H NMR spectroscopic studies, which were hampered by the low solubility of [L(Me2N)GaSb]2, did not show any additional (intermediate) resonances others than those of azadistibirane 1, hence providing no hints for a [2 + 3] cycloaddition mechanism (Fig. S47†). In contrast, the analogous reaction with Me3SiN3 occurred with insertion of the nitrene into the Ga–Sb bond and subsequent formation of L(Me2N)GaSbSb(NSiMe3)Ga(NMe2)L (4). Comparable findings were reported by Weigand et al. for reactions of diphosphenes with RN3, yielding azadiphosphiranes with aryl azides but imine-substituted diphosphene with Me3SiN3.12
9a unselectively reacted with organoazides RN3 at ambient temperature with N2 elimination and formation of several reaction products according to 1H NMR spectroscopic studies (Fig. S46†). In contrast, dropwise addition of toluene solutions of PhN3, p-CF3PhN3 and 1-adaN3 to a cooled (−30 °C) suspension of [L(Me2N)GaSb]2 and slow heating to ambient temperature selectively gave azadistibiranes 1–3 (Scheme 2). The reactions can be classified in a formal sense as [2 + 1] cycloaddition reactions, but an initial [2 + 3] dipolar cycloaddition followed by a rapid N2 elimination is also plausible.2,4a,11 However, temperature-dependent in situ1H NMR spectroscopic studies, which were hampered by the low solubility of [L(Me2N)GaSb]2, did not show any additional (intermediate) resonances others than those of azadistibirane 1, hence providing no hints for a [2 + 3] cycloaddition mechanism (Fig. S47†). In contrast, the analogous reaction with Me3SiN3 occurred with insertion of the nitrene into the Ga–Sb bond and subsequent formation of L(Me2N)GaSbSb(NSiMe3)Ga(NMe2)L (4). Comparable findings were reported by Weigand et al. for reactions of diphosphenes with RN3, yielding azadiphosphiranes with aryl azides but imine-substituted diphosphene with Me3SiN3.12
 | ||
| Scheme 2 Azadistibiranes 1–3 and distibenes 4–6 formed by cycloaddition reactions of distibene [L(Me2N)GaSb]2 with organoazides. 5 dimerizes to cyclotetrastibane 7 upon thermal treatment and reacts with a second equivalent of PhN3 to 9, which isomerizes to 10 upon heating. | ||
The yield of azadistibiranes 1–3 significantly dropped with increasing reaction times. At ambient temperature, toluene solutions of 1 and 2 underwent slow rearrangement of the nitrene unit and formation of compounds 5 and 6, while heating above 80 °C greatly increases the reaction speed. Azadistibirane 3 in contrast is thermally more stable and was found to decompose unselectively starting at 80 °C. We failed to isolate compound 5 and 6 in pure form due to their slow decomposition in solution (see 1H-NMR Fig. S15 and S18†). Prolonged heating of 5 at 100 °C for 120 h resulted in complete decomposition with formation of LGa(NMe2)2 and tetrastibacyclobutane [(L(NPh)Ga-κGa,κN)2-(μ,η1:1:1:1-Sb4)] 7, which precipitated due to its low solubility in toluene.
[L(Me2N)GaSb]2 was also reacted with two equivalents of Me3SiN3, yielding a mixture of two species. Only the major species 8, in which in contrast to compound 4 one NMe2 group is additionally replaced by an amide group (N(H)SiMe3), was isolated by precipitation upon slow diffusion of CH3CN into the toluene solution. The formation of 8 is unclear, and both toluene and the methyl group of the β-diketiminate ligand can serve as the proton source.13 In contrast, compound 5 was found to react cleanly with a second equivalent of PhN3 to azadistibirane 9, which isomerized to distibene 10 upon heating to 70 °C as well.
We also reacted distibenes [L(X)GaSb]2 (X = NMe2, OEt, Cl) with Me3Si(H)CN2 at ambient temperature (Scheme 3), yielding distibiranes [L(X)GaSb]2C(H)SiMe3 (X = NMe211, OEt 12, Cl 13) with high selectivity. A low temperature in situ1H NMR study showed that the reaction of [L(Me2N)GaSb]2 is fast and even at −80 °C no reaction intermediates were detected (Fig. S48†). Distibiranes 11–13 did not react with additional equivalents of diazomethane or azides even at elevated temperature (100 °C).
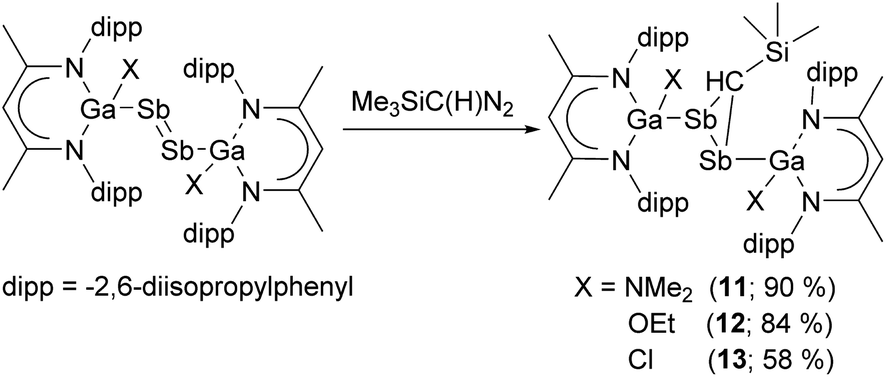 | ||
| Scheme 3 Cycloaddition reactions of [L(X)Ga]2 with Me3Si(H)CN2 to distibiranes 11–13. | ||
Spectroscopic characterization
All compounds except 7 and 10 are soluble in non-polar solvents such as n-hexane and toluene or solvents of intermediary polarity such as thf, while they are insoluble in polar solvents, i.e., DMF or acetonitrile. 7 and 10 are only slightly soluble in toluene or benzene. They exhibit the typical resonance for the L(X)Ga ligand with characteristic singlets around 5 ppm (γ-H) and 1.5–2 ppm (Me) of the β-diketiminate ligand, as well as sets of doublets (2–0.5 ppm) and septets (4.5–2.5 ppm) for the isopropyl groups. Azadistibiranes 1 and 2 each exhibit a set of resonance for the ortho-protons of the phenyl group, which are shifted to 5.28 and 5.1 ppm, respectively. This shift to higher field is less pronounced for 9 (5.9–6.1 ppm) and likely caused due to the proximity to the Dipp groups, while insertion products 5, 6, and 10 and tetrastibacyclobutane 7 exhibit this resonance in the typical range of aromatic amines. The proton resonances of the Me3SiN units in 4 and 8 are found at 0.4 and 0.5 ppm and the N(H)SiMe3 resonances in 8 at −0.6 ppm. Distibiranes 11–13 are unsymmetric molecules (C1 symmetry) with restricted rotation, hence all carbon atoms (except Me3Si) are magnetically inequivalent, leading to two γ-H, four Me singlets, 16 doublets and 8 septets for the Dipp ligands, while the CHSiMe3 resonances are found at 1.81, 2.13 and 2.18 ppm, respectively. In contrast, azadistibiranes 1–3 each show only a single resonance for both γ-H atoms. This finding either points to trigonal planar-coordinated N atoms (compare R–C/N–Δ angle Table 2), to fast inversion processes or to an equilibrium between the azadistibirane and the distibaimine 15 (see Scheme 4). Since our calculated thermodynamic energies (Scheme 4) don't support the formation of such an equilibrium, we explain these findings by fast inversion processes, since the inversion of ternary amines, in contrast to the heavier pnictogens, is typically very fast at room temperature.14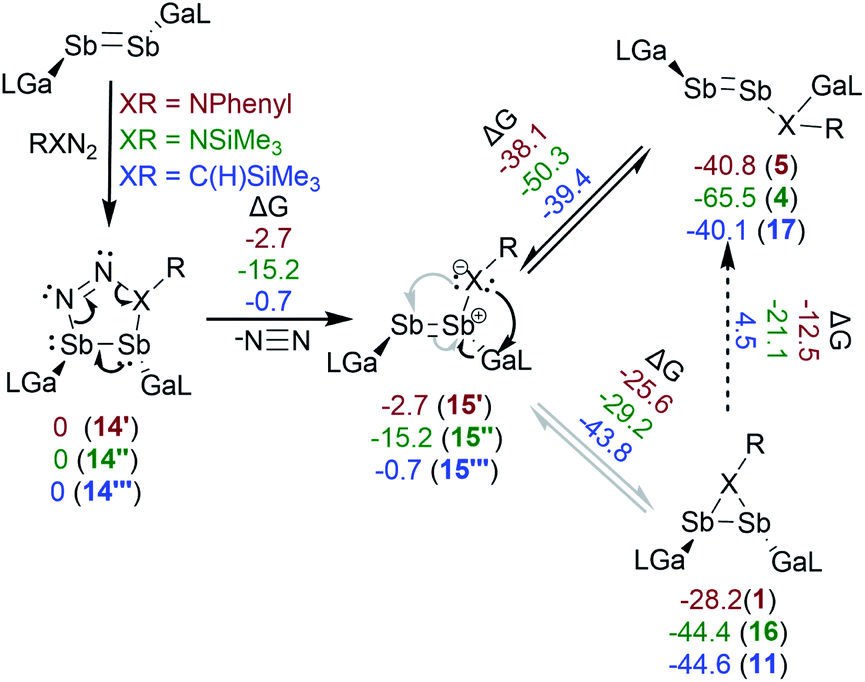 | ||
| Scheme 4 Proposed reaction mechanism for the azadistibiranes and amidodistibenes formation from reactions with PhN3 and Me3SiN3. Free Gibbs energies (normalized to the respective 1,2-distibatriazene in kcal mol−1) of the optimized structures at the PBE0/def2-SVP(D3BJ) level of theory are given. | ||
UV-Vis spectra of toluene solutions of 4–6 and 8 show adsorption bands for the π → π* transition between 494 and 502 nm,15 while 10 shows an absorption maxima at 584 nm (Fig. S50†), which is red shifted compared to [L(Me2N)GaSb]2 (430 nm)10 and to amido-substituted distibenes (510–514 nm).16 The red shift indicates a smaller π–π* energy gap, most likely due to the π-donating NMe2 groups, which increase the energy level of the π-orbitals relative to the π*-orbitals and hence decrease the transition energy (Fig. 2).17 In contrast, toluene solutions of azadistibiranes 1–3 and distibiranes 11–13 are slightly orange and yellow and only show weak adsorption bands at the edge of the visible spectrum (Fig. S49 and S52†), which are partially overlayed by the adsorption of the L(Me2N)Ga ligand.10b
Single crystal X-ray structures
The molecular structures of 1–13 were determined by sc-XRD. Single crystals were obtained from solutions in n-pentane (13, Fig. S58†), n-hexane (4, Fig. S55;†6, Fig. S56†), benzene (11, Fig. S57†) and toluene (1, 7, 9, 10, Fig. 1; 2, Fig. S53†) upon storage at ambient temperature (7, 9, 10), 7 °C (11) or −30 °C (1, 2, 4, 13) for 48 h, respectively. Single crystals of 12 (Fig. 1) were formed upon slow evaporation of a n-hexane solution and those of 3 (Fig. S54†), 5 (Fig. 1) and 8 (Fig. 1) by slow diffusion of CH3CN into toluene solutions. 2 and 9–11 crystallize in the triclinic crystal system (Tables S1a and b†) and all others in the monoclinic crystal system. Selected bond lengths and angles of distibenes 4–6, 8 and 10 are summarized and compared to those of [L(Me2N)GaSb]2 in Table 1. The Sb–Sb bonds in the distibenes (2.6541(4) 4; 2.6750(3) 5; 2.6627(3) 6; 2.6534(5) 8; 2.6688(1) Å 10) are slightly elongated compared to that of [L(Me2N)GaSb]2 (2.6477(3) Å). Remarkably, distibenes 4–6 show short intramolecular N⋯Sb distances (2.472(3) 4; 2.395(1) 5; 2.424(2) Å 6) between the NMe2 group and the neighboring Sb atom and elongated (by 0.1 Å) Ga–NMe2 bonds. This was neither observed for distibene [L(X)GaSb]2 nor 8 and 10. Distibenes 4–6 also exhibit shorter Ga–Sb bond lengths (2.5836(5) 4; 2.5842(3) 5; 2.5834(4) Å 6) compared to that of [L(Me2N)GaSb]2 (2.6200(4) Å). The electronegative imine ligand most likely increases the electrophilicity of the Sb atom, which favors a dative intramolecular interaction of the amine group. | ||
| Fig. 1 Molecular structures of [L(Me2N)GaSb]2NPh (1), [L(Me2N)Ga]SbSb[N(Ph)Ga(NMe2)L] (5), [(L(PhN)Ga-κGa,κN)2-(μ,η1:1:1:1-Sb4)] (7), [L(Me2N)Ga]SbSb[N(SiMe3)Ga(N(H)SiMe3)L] (8), [L(Me2N)GaSb][L(Me2N)GaN(Ph)Sb]NPh (9), [L(Me2N)GaN(Ph)Sb]2 (10), and [L(EtO)GaSb]2C(H)SiMe3 (12). H atoms are omitted for clarity and displacement ellipsoids are drawn at the 50% probability level. For 7 and 10 the symmetry generated part is displayed in pale colors. | ||
9a for comparison
| [L(Me2N)GaSb]2c | 4 | 5 | 6 | 8 | 10 c | |
|---|---|---|---|---|---|---|
| a Values are discussed for the amido substituted part. b Here N(H)SiMe3. c Inversion symmetry. | ||||||
| Sb–Sb | 2.6477(3) | 2.6541(4) | 2.6750(3) | 2.6627(3) | 2.6534(5) | 2.6688(1) |
| Sb–Ga | 2.6200(4) | 2.5836(5) | 2.5842(3) | 2.5834(4) | 2.6241(5) | 3.2907(4) |
| Sb–N | — | 2.089(2) | 2.096(1) | 2.111(2) | 2.071(2) | 2.0727(8) |
| Sb–(NMe2) | 3.825(1) | 2.472(3) | 2.395(1) | 2.424(2) | 5.102(3)b | 3.865(1) |
| Ga–N | — | 1.863(2) | 1.865(1) | 1.874(2) | 1.917(2) | 1.8818(8) |
| Ga–(NMe2)a | 1.856(1) | 1.943(2) | 1.955(1) | 1.949(2) | 1.857(2)b | 1.8524(8) |
| N–Sb–Na | — | 74.71(9) | 72.54(5) | 72.53(8) | — | — |
| N–Ga–Na | — | 94.0(1) | 88.57(5) | 89.60(9) | — | — |
| |N–Sb–Ga–N|a | — | 171.2(1) | 152.52(8) | 166.0(2) | — | — |
Selected bond lengths and angles of azadistibiranes 1–3, and 9 and distibiranes 11–13 are summarized in Table 2. Generally, the Sb–Sb bond is significantly elongated compared to [L(Me2N)GaSb]2 (2.6477(3) Å) ranging from 2.7882(2) Å (1) to 2.8291(3) Å (9) and thus lying in the typical range observed for distibanes Sb2R4 (2.77–3.07 Å).18 The Sb–C/N bonds (Sb–C: 2.14–2.29 Å; Sb–N: 2.08–2.11 Å) are in the typical single bond range (Sb–C: 2.04–2.31 Å; Sb–N: 1.95–2.58 Å)19 and the Sb–N–Sb bond angles of the azadistibiranes (83.15(4) 1; 83.0(1) 2; 83.3(1) 3; 87.28(7)° 9) are slightly larger than those of the distibiranes (78.0(1) 11; 78.4(2) 12; 79.2(1)° 13). The angles deviate from the ideal tetrahedral angle for a sp3-hybridized N atom and from the 90° angle expected for an unhybridized Sb atom, most likely resulting from the ring strain. Although the azadistibiranes have formally six electrons (three pnictogen electron lonepairs), Hückel aromaticity can be excluded since both Sb ligands adopt almost perpendicular orientations to the plane in the E configuration (94–115°; Table 2) and the bond length within the three-membered rings are more in line with single bonds.
| Sb–Sb | Sb–C/N | Sb–Ga | Sb–C/N–Sb | C/N–Sb–Sb | Ga–Sb–Δ | R–C/N–Δ | |
|---|---|---|---|---|---|---|---|
| a Δ: centre of the plane span by Sb–Sb–C/N triangle. b Values are discussed for average of the disordered L(Me2N)GaSbN-p-CF3–Ph part. c Values are discussed for the major component of the disordered C(H)SiMe3 part. In both cases the values should be interpreted with caution. d N(Ph) instead of Ga. | |||||||
| 1 | 2.7882(2) | 2.114(1) | 2.6520(2) | 83.15(4) | 48.84(3) | 102.90(2) | 140.2(1) |
| 2.087(1) | 2.6564(2) | 48.01(3) | 104.91(3) | ||||
| 2 | 2.778(3)b | 2.113(3) | 2.6434(3) | 83.0(1)b | 48.98(9)b | 101.82(2)b | 141.9(5)b |
| 2.080(3)b | 2.649(3)b | 48.00(9)b | 103.7(1)b | ||||
| 3 | 2.7987(4) | 2.099(3) | 2.6477(4) | 83.3(1) | 48.56(8) | 104.67(2) | 143.4(3) |
| 2.113(3) | 2.6950(5) | 48.13(8) | 110.71(3) | ||||
| 9 | 2.8291(3) | 2.080(2) | 2.6540(3) | 87.28(7) | 45.48(5) | 102.36(2) | 171.9(2) |
| 2.019(2) | 2.058(2)d | 47.24(5) | 101.03(5)d | ||||
| 11 | 2.7912(5) | 2.139(3)c | 2.6431(6) | 78.0(1)c | 48.55(9)c | 114.56(3)c | 138.4(3)c |
| 2.294(4)c | 2.6400(6) | 53.48(9)c | 100.25(3)c | ||||
| 12 | 2.7912(7) | 2.219(8) | 2.623(1) | 78.4(2) | 51.2(2) | 93.67(6) | 142.3(6) |
| 2.196(8) | 2.627(1) | 50.4(2) | 105.94(5) | ||||
| 13 | 2.7991(3) | 2.225(4)c | 2.6213(4) | 79.2(1)c | 51.3(1)c | 96.70(3)c | 135.9(3)c |
| 2.164(4)c | 2.6521(4) | 49.4(1)c | 107.34(2)c | ||||
Tetrastibacyclobutane 7 shows an inversion center in the Sb4 plane. The Sb–Sb bond lengths (2.8837(2), 2.8449(2) Å) and Sb–Sb–Sb bond angles (94.982(6), 85.018(6)°) within the Sb4 ring slightly deviate from those expected for an ideal square. The Ga–N (1.890(2) Å), Sb–N (2.083(2) Å) and Ga–Sb bond lengths (2.6221(3) Å) are in the typical range of single bonds. The almost flat Ga–Sb–Sb–N unit (torsion angle of −4.13(5)°) resembles a distorted trapezoid, which adopts a perpendicular orientation to the Sb4 plane. The Sb–Sb bond lengths are in the typical range reported for Sb–Sb single bonds,18 whereas the shortest diagonal Sb–Sb distance (4.2230(4) Å) is far longer than that observed in [{L(X)Ga}2-μ,η1:1-Sb4] obtained directly from thermolysis of [L(Me2N)GaSb]2.9a,b
Quantum chemical calculations
The reactions of [L(Me2N)GaSb]2 with PhN3 and Me3SiN3 were investigated by quantum chemical calculations at the PBE0 level of theory.20 Weigand et al. proposed for reactions of the diphosphene cation [(ClImDipp)PP(Dipp)]+ with RN3 (R = Me3Si, Dipp) the initial formation of diphosphatriazoles via 1,3-dipolar cycloaddition,12 which upon elimination of N2 either formed diphospheneimine (Me3SiN3) or rearranged to azadiphosphirane (DippN3). A similar mechanism is likely in our reactions with PhN3 and Me3SiN3, and both the distibatriazole (14) and the distibaimine (15) were found as minima on the energy surface (Scheme 4). The elimination of N2 by a retro [3 + 2] cycloaddition reaction is energetically favored by −15 (Me3SiN3) and −3 kcal mol−1 (PhN3), respectively, which is more than 14 kcal mol−1 less favored compared to the phosphorous species. This might explain why Tokitoh et al. observed no reactivity of [(Bbt)Bi]2 in cycloaddition reaction compared to [(Bbt)Sb]2.7b The formation of both imine structures is thermodynamically unfavored and were not observed by 1H NMR spectroscopy, but they are still likely intermediates in the rearrangement of azadistibiranes to distibenes 4 and 5. The ΔG values of 25–30 kcal mol−1 are in line with slow transformations at 20 °C but fast at 100 °C, but different pathways for the rearrangement reactions can't be excluded.
In case of NSiMe3-substituted species, compound 4 is almost 9 kcal mol−1 more favored than compound 5 with respect to the corresponding azadistibiranes 16 and 1, although this finding is not directly related to the activation barrier. The experimental findings suggest that for 15’’ the [1,2] sigmatropic shift reaction to 4 is both kinetically and thermodynamically favored, whereas compound 1 was isolated in almost quantitative yield upon performing the reaction at low temperature.
A similar mechanism was postulated for reactions of diphosphenes with diazomethanes, and the (intermediary) formation of hydro-diphosphapyrazol and methylene-diphosphene was observed by in situ31P-NMR spectroscopy.21 We identified the corresponding hydrodistibapyrazol (14′′′), methylenedistibene (15′′′) and the insertion product (17) as minima on the energy surface (Scheme 4). In contrast to the azadistibiranes (1, 16), the corresponding distibirane 11 is energetically favored by 4.5 kcal mol−1 compared to the respective insertion product 17, explaining the higher thermal stability of 11. In addition, the energy difference between 15′′′ and 11 (43.8 kcal mol−1) is much larger than those observed for the corresponding imine compounds 15′vs.1 (25.6 kcal mol−1) and 15′′vs.16 (29.2 kcal mol−1), respectively, making its formation upon heating rather unlikely. This perfectly agrees with our experimental findings since no intermediates were observed even at very low reaction temperatures (−80 °C).
To the best of our knowledge, distibenes 4, 5 and 8 represent the only structurally characterized heteroleptic distibenes. The LUMO of distibenes is typically represented by a low-lying π* orbital, whereas the level of the molecular orbitals of the π orbitals are somewhat indistinct since their energy levels are close to those of n+ orbital.5c,15 In contrast, both the π and σ Sb–Sb orbitals of 4 and 5 correspond to the HOMO and HOMO−1, respectively (Fig. S59†), whereas the LUMO is centered at the β-diketiminate ligand and the LUMO+1 is Sb-centred. In addition, these orbitals are significantly polarized towards the Ga-substituted Sb atom. Interestingly for the bisamido-substituted distibene 10 the LUMO is also Sb-centred (Fig. 2).
 | ||
| Fig. 2 Section of the MO diagram of [L(Me2N)GaSbSbN(SiMe3)Ga(NMe2)L] (4) and [L(Me2N)Ga(NPh)Sb]2 (10), isovalue 0.03.23 | ||
The bonding nature was further analysed by natural population analysis (NPA, Table S3†).22 Compared to [L(Me2N)GaSb]2 (−0.16 e) the natural charge on the Ga-substituted Sb atom decreased (−0.69 4, −0.66 e5) while the charge on the amido-substituted Sb atom increased (1.05 4, 1.08 e5), respectively. The charge is even higher than that of distibene 10 (0.63 e). The π-bond is analogously polarized (occupation numbers (ON) π 1.89 e (4), ON π 1.89 e (5) with 71.9% and 71.1% polarization towards the Ga-substituted Sb atom), and the positively polarized Sb atom enables the dative interaction of the NMe2 group. The Wiberg bond order (WBI) of the Sb–NMe2 bond is roughly half of that of the Sb–NR bond (0.22, 0.49 4; 0.21, 0.49 5). Second order perturbation theory analysis revealed that the main interaction is the donation of the N-LP (lone pair) into the Sb–Sb π* orbital (21 kcal mol−14 and 5), which most likely is the main driving force for the ring opening together with the ring strain (Scheme 5).
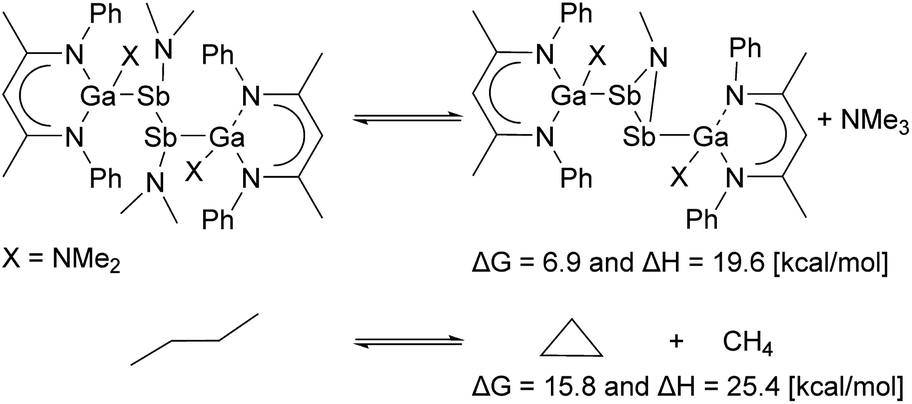 | ||
| Scheme 5 The ring strain was estimated on the PBE0(def2-TZVP/D3BJ) level of theory with the isodesmic model reaction of a bisdimethylamido-substituted distibane to dimethylamine substituted azadistibirane. For comparison the analog method was applied to cyclopropane yielding 25.4 kcal mol−1, which is close to the accepted value of 27.5 kcal mol−1.24 | ||
In addition, TDDFT calculations of 4, 5 and 10 were performed and compared to experimental UV-Vis-data (Table S2†). As was reported for [L(Me2N)GaSb]2,10a one dominant transition at 563 nm was found for 10 (Fig. S62†), which fits nicely to the experimental data. 87% of the transition orbitals comprise of the HOMO and LUMO orbitals, corresponding to known π → π* transition for distibenes.15 Four and two transitions in the visible region of similar intensity and energy were found for 4 (Fig. S60†) and 5 (Fig. S61†), which are blue shifted compared to experimental values. The HOMO and LUMO+1 orbitals are the main contributions to the π → π* transition. Due to the polarized Sb–Sb double bond, the ground state is centred on the L(NMe2)Ga-substituted Sb atom and the excited state on the RN-substituted Sb atom (Fig. 3). The HOMO−1 → LUMO transition contributes to the adsorption band of 4 and 5 in the visible region and can be regarded as metal-to-ligand charge transfer (MLCT) transition as the HOMO−1 (Fig. 2) correlates to the Sb–Sb σ-bond and the LUMO is ligand centred and correlates to C–N π*-bond of the β-diketiminate ligand.
 | ||
| Fig. 3 Depiction of the electron difference density of the ground and excited state of the dominant 4th (left 456 nm) and 3rd (right 463 nm) excitation calculated by tddft [L(Me2N)GaSbSbN(Ph)Ga(NMe2)L] (5), blue hole and red accumulation.23 | ||
The bonding situation of the three-membered rings was also investigated by the atoms in molecules approach (AIM, Tables S4 and S5).†25 The nuclear attractors within the rings are connected to each other via bond paths. The Sb–Sb bond is significantly bent outwards as indicated by ELF distribution (see Fig. 4).26 The ring atoms are connected by polarized σ-bond (NBO) with occupation numbers close to two. NHO directionality and bond bending analysis showed high deviation from the line connecting the nuclear centers (Sb–Sb 33.7°). In contrast, the electron concentration of the C/N–Sb bond is within the spanned triangle and the ring critical point (RCP) is in proximity to the bond critical point (BCP) of the Sb–Sb bond.
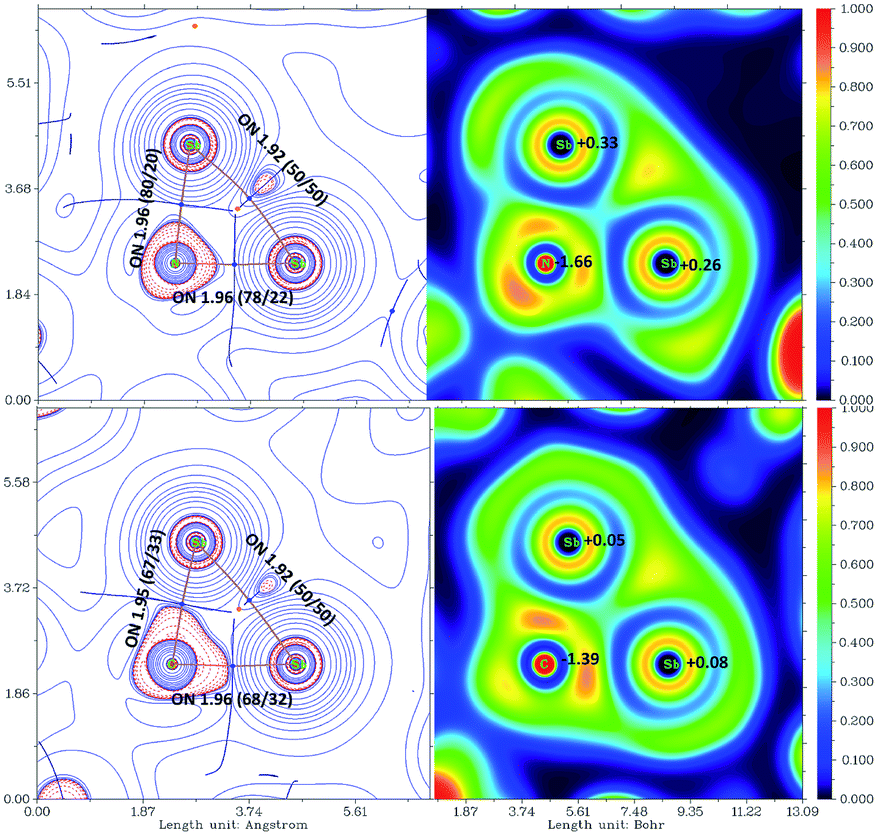 | ||
| Fig. 4 Contour maps of the Laplacian of the electron density (left negative values red, positive blue), bond paths (brown), bcp (light blue), rcp (orange) and basin boundaries (dark blue) are indicated and ELF distribution (right), in the Sb–X–Sb plane of [L(Me2N)GaSb]2NSiMe3 (16) (top) and [L(Me2N)GaSb]2C(H)SiMe3 (11) (bottom). Occupation numbers (ON, |e|) of the σXY bonds according to NBO analysis with squared polarization coefficients and the natural charge is given. | ||
Conclusion
L(X)Ga-substituted distibenes react with organoazides RN3 (R: Ph 1, p-CF3Ph 2, ada 3) and Me3Si(H)CN2 in [2 + 1] cycloaddition reactions to azadistibiranes 1–3 and distibiranes 11–13 (X = NMe211, OEt 12, Cl 13). While cycloaddition reactions are known for diphosphenes, analogous reactions of the heavier group 15 congeners are virtually unknown. Azadistibiranes 1–3 rearrange upon heating to heteroleptic distibenes, formally via insertion of the nitrene unit into the Ga–Sb bond (5, 6), which occurred directly in the reaction with Me3SiN3 (4).5 furthermore reacts with a second equivalent of PhN3 to unsymmetric azadistibirane 9, which isomerizes at elevated temperature to the homoleptic distibene 10. In addition, the [2 + 2] cycloaddition of 5 yields tetrastibacyclobutane 7. Heteroleptic distibenes 4 and 5 showed smaller π–π* gaps compared to the homoleptic distibene [L(Me2N)GaSb]2 according to UV-Vis measurements, and quantum chemical calculations suggest strongly polarized Sb–Sb double bonds. In addition, the π-donating N(R)Ga unit raises the energy of the Sb–Sb bonding orbitals. The HOMO and HOMO−1 correspond to the Sb–Sb bonding π and σ orbitals of 4, 5 and 10, rendering them attractive candidates for the synthesis of unknown distibene radical cations, which is currently under investigation.
Experimental
General procedures
All manipulations were carried out using standard Schlenk and glovebox techniques under argon, which was dried by passing through pre-heated Cu2O pellets and molecular sieve columns. Toluene, n-pentane and n-hexane were dried with a MBraun Solvent Purification System, while benzene and deuterated solvents (toluene-d8, C6D6) were distilled from Na/K alloy, and CH3CN was distilled from CaH2. All solvents were degassed and stored over activated molecular sieves. [L(Cl)GaSb]2,9b [L(EtO)GaSb]2,9d and [L(Me2N)GaSb]29a were prepared according to literature methods. Methyl tert-butyl ether (MTBE) solutions of PhN3 and p-CF3PhN3 were stored over activated molecular sieves (4 Å) at −30 °C for one week prior to use. 1H (300 MHz, 400 MHz, 600 MHz), 13C{1H} (100.7 MHz, 150.9 MHz), 19F (282.3 MHz, 376.5 MHz, 564.6 MHz) and 29Si (79.5 MHz, 119.2 MHz) NMR spectra were recorded using a Bruker Avance DPX-300, a Bruker Avance Neo 400 MHz or a Bruker Avance III HD 600 spectrometer and referenced to internal C6D5H (1H: δ = 7.16; 13C: δ = 128.06) or C6D5CD2H (1H: δ = 2.08; 13C: δ = 20.43). Assignment of the resonances was aided by 2D NMR spectroscopy. IR spectra were recorded in a glovebox using a BRUKER ALPHA-T FT-IR spectrometer equipped with a single reflection ATR sampling module. Microanalyses were performed at the elemental analysis laboratory of the University of Duisburg-Essen. UV-Vis-spectra were recorded on a Shimadzu UV-2600i spectrophotometer in a closed glass cuvette (10 mm) under argon atmosphere.
Caution! Me3SiN3 should be handled with caution under anhydrous conditions in an inert atmosphere or in a well-ventilated fume hood, as hydrolysis can lead to formation of toxic HN3. Azide reactions should generally be performed in small scales.
Yield: 18 mg (12.1 μmol, 36%). Anal. calcd for C74H104Ga2N8Sb2: C, 61.54; H, 7.14; N, 7.09%. Found: C, 59.7; H, 7.15; N, 7.27%. ATR-IR: ν 2953, 2920, 2815, 2792, 2750, 1542, 1516, 1457, 1427, 1391, 1313, 1254, 1213, 1172, 1013, 965, 933, 851, 787, 763, 751, 674, 590, 500 cm−1. 1H NMR (400 MHz, C6D6, 25 °C): δ 7.09–7.03 (m, 8H, C6H3-2,6iPr2), 6.84 (dd, 3JHH = 7.2 Hz, 4JHH = 2.0 Hz, 4H, C6H3-2,6iPr2), 6.69 (t, 3J = 7.3 Hz, 4H, C6H3-2,6iPr2), 6.61 (d, 3J = 7.3 Hz, 4H, C6H3-2,6iPr2), 6.50 (t, 3J = 7.2 Hz, 2H, C6H3-2,6iPr2), δ 4.61 (s, 2H, γ-CH), 3.35 (sept, 3JHH = 6.8 Hz, 2H, CH(CH3)2), 3.30 (sept, 3JHH = 6.8 Hz, 2H, CH(CH3)2), 2.88 (s, 6H, N(CH3)2), δ 2.84 (s, 6H, N(CH3)2), 1.48 (s, 12H, ArNCCH3), 1.37 (d, 3JHH = 6.6 Hz, 12H, CH(CH3)2), 1.31 (d, 3JHH = 6.7 Hz, 12H, CH(CH3)2), 1.24 (d, 3JHH = 6.8 Hz, 12H, CH(CH3)2), 1.00 (d, 3JHH = 6.8 Hz, 12H, CH(CH3)2). DEPTQ 13C NMR (150.9 MHz, C6D6): δ 169.2 (ArNCCH3), 168.2 (N–C6H5), 144.3, 143.0, 142.8, 127.1, 124.4, 123.9 (ArC), 128.1, 123.3, 119.3 (N–C6H5), 95.5 (γ-CH), 43.8, 42.9 (N(CH3)2), 29.7, 29.4, 28.1, 27.6 28.7, 27.5 (CH(CH3)2), 25.0, 24.8, 24.4, 24.0 (CH(CH3)2), 23.5 (ArNCCH3).
Data availability
Spectroscopic (NMR, IR, UV-Vis) and crystallographic data (cif file) are provided.Author contributions
M. W. performed the experiments including quantum chemical calculations, C. W. the single crystal X-ray diffraction. The work was supervised by S. S. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful to the Deutsche Forschungsgemeinschaft DFG (St.S., SCHU 1069/22-3) for generous financial support.Notes and references
- (a) C. Weetman, Chem.–Eur. J., 2021, 27, 1941 CrossRef CAS PubMed; (b) C. Weetman and S. Inoue, ChemCatChem, 2018, 10, 4213 CrossRef CAS; (c) T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608 CrossRef CAS PubMed; (d) P. P. Power, Chem. Rec., 2012, 12, 238 CrossRef CAS PubMed; (e) P. P. Power, Nature, 2010, 463, 171 CrossRef CAS PubMed.
- (a) M. Breugst and H.-U. Reissig, Angew. Chem., Int. Ed. Engl., 2020, 59, 12293 CrossRef CAS PubMed; (b) O. Diels and K. Alder, Justus Liebigs Ann. Chem., 1928, 460, 98 CrossRef CAS; (c) R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 633 CrossRef; (d) R. Huisgen, Angew. Chem., 1963, 75, 604 CrossRef CAS.
- F. Hanusch, L. Groll and S. Inoue, Chem. Sci., 2021, 12, 2001 RSC.
- (a) L. Weber, Chem. Rev., 1992, 92, 1839 CrossRef CAS; (b) M. Yoshifuji, Pure Appl. Chem., 2017, 89, 281 CAS; (c) D. Nauroozi and A. Orthaber, Eur. J. Inorg. Chem., 2016, 2016, 709 CrossRef CAS; (d) R. K. Bansal and S. K. Kumawat, Tetrahedron, 2008, 64, 10945 CrossRef CAS; (e) J. D. Protasiewicz, in Comprehensive inorganic chemistry II. From elements to applications, ed. J. Reedijk, Elsevier, Amsterdam, 2nd edn, 2013, pp. 325–348 Search PubMed; (f) M. Yoshifuji, Eur. J. Inorg. Chem., 2016, 607 CrossRef CAS.
- (a) H.-J. Himmel, Eur. J. Inorg. Chem., 2153, 2003, 2003 Search PubMed; (b) H. Voelker, U. Pieper, H. W. Roesky and G. M. Sheldrick, Z. Naturforsch., B: J. Chem. Sci., 1994, 49, 255 CrossRef CAS; (c) T. Sasamori, E. Mieda, N. Takeda and N. Tokitoh, Chem. Lett., 2005, 33, 104 CrossRef; (d) T. Sasamori, E. Mieda, N. Takeda and N. Tokitoh, Angew. Chem., Int. Ed., 2005, 44, 3717 ( Angew. Chem. , 2005 , 117 , 3783 ) CrossRef CAS PubMed; (e) T. Sasamori, E. Mieda and N. Tokitoh, Bull. Chem. Soc. Jpn., 2007, 80, 2425 CrossRef CAS; (f) T. Sasamori, N. Takeda and N. Tokitoh, J. Phys. Org. Chem., 2003, 16, 450 CrossRef CAS; (g) M. Ateş, H. J. Breunig, S. Güleç, W. Offermann, K. Häberle and M. Dräger, Chem. Ber., 1989, 122, 473 CrossRef; (h) L. Dostál, R. Jambor, A. Růžička and J. Holeček, Organometallics, 2008, 27, 2169 CrossRef; (i) S. S. Chitnis, A. P. M. Robertson, N. Burford, J. J. Weigand and R. Fischer, Chem. Sci., 2015, 6, 2559 RSC; (j) S. S. Chitnis, Y.-Y. Carpenter, N. Burford, R. McDonald and M. J. Ferguson, Angew. Chem., Int. Ed., 2013, 52, 4863 CrossRef CAS PubMed; (k) S. S. Chitnis, Y.-Y. Carpenter, N. Burford, R. McDonald and M. J. Ferguson, Angew. Chem., 2013, 125, 4963 CrossRef.
- (a) J. Grobe, A. Karst, B. Krebs, M. Läge and E.-U. Würthwein, Z. Anorg. Allg. Chem., 2006, 632, 599 CrossRef CAS; (b) I. Jibril, L.-R. Frank, L. Zsolnai, K. Evertz and G. Huttner, J. Organomet. Chem., 1990, 393, 213 CrossRef CAS; (c) G. Huttner and I. Jibril, Angew. Chem., 1984, 96, 709 CrossRef CAS; (d) T. Umeyama, K. Naka and Y. Chujo, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 3023 CrossRef CAS; (e) A. Karst, B. Broschk, J. J. Grobe and D. Le Van, Z. Naturforsch., B: J. Chem. Sci., 1995, 50, 189 CrossRef CAS.
- (a) N. Tokitoh, Y. Arai, T. Sasamori, R. Okazaki, S. Nagase, H. Uekusa and Y. Ohashi, J. Am. Chem. Soc., 1998, 120, 433 CrossRef CAS; (b) T. Sasamori, E. Mieda, N. Nagahora and N. Tokitoh, Chem. Heterocycl. Compd., 2006, 42, 1603 CrossRef CAS.
- (a) T. Sasamori, E. Mieda, N. Takeda and N. Tokitoh, Chem. Lett., 2004, 33, 104 CrossRef CAS; (b) T. Sasamori, E. Mieda, A. Tsurusaki, N. Nagahora and N. Tokitoh, Phosphorus, Sulfur Silicon Relat. Elem., 2008, 183, 998 CrossRef CAS; (c) N. Tokitoh, T. Sasamori and R. Okazaki, Chem. Lett., 1998, 27, 725 CrossRef; (d) T. Sasamori and N. Tokitoh, Dalton Trans., 2008, 1395 RSC.
- (a) L. Tuscher, C. Ganesamoorthy, D. Bläser, C. Wölper and S. Schulz, Angew. Chem., Int. Ed., 2015, 54, 10657 ( Angew. Chem. , 2015 , 127 , 10803 ) CrossRef CAS PubMed; (b) L. Tuscher, C. Helling, C. Ganesamoorthy, J. Krüger, C. Wölper, W. Frank, A. S. Nizovtsev and S. Schulz, Chem.–Eur. J., 2017, 23, 12297 CrossRef CAS PubMed; (c) L. Tuscher, C. Helling, C. Wölper, W. Frank, A. S. Nizovtsev and S. Schulz, Chem.–Eur. J., 2018, 24, 3241 CrossRef CAS PubMed; (d) J. Krüger, J. Schoening, C. Ganesamoorthy, L. John, C. Wölper and S. Schulz, Z. Anorg. Allg. Chem., 2018, 644, 1028 CrossRef; (e) L. Song, J. Schoening, C. Wölper, S. Schulz and P. R. Schreiner, Organometallics, 2019, 38, 1640 CrossRef CAS.
- (a) H. M. Weinert, C. Wölper, J. Haak, G. E. Cutsail III and S. Schulz, Chem. Sci., 2021, 12, 14024 RSC; (b) H. M. Weinert, C. Wölper and S. Schulz, Organometallics, 2021, 40, 3486 CrossRef CAS.
- (a) E. Niecke, R. Rüger, M. Lysek and W. W. Schoeller, Phosphorus Sulfur Relat. Elem., 1983, 18, 35 CrossRef CAS; (b) J. Borm, G. Huttner and O. Orama, J. Organomet. Chem., 1986, 306, 29 CrossRef CAS; (c) J. Borm, G. Huttner, O. Orama and L. Zsolnai, J. Organomet. Chem., 1985, 282, 53 CrossRef CAS; (d) W. Güth, PhD thesis, University of Bielefeld, 1987, pp. 12–15.
- K. Schwedtmann, F. Hennersdorf, A. Bauzá, A. Frontera, R. Fischer and J. J. Weigand, Angew. Chem., Int. Ed., 2017, 56, 6218 CrossRef CAS PubMed.
- J. A. B. Abdalla, I. M. Riddlestone, R. Tirfoin and S. Aldridge, Angew. Chem., Int. Ed., 2015, 54, 5098 CrossRef CAS PubMed.
- C. Klmel, C. Ochsenfeld and R. Ahlrichs, Theor. Chim. Acta, 1992, 82, 271 CrossRef.
- P. Vilarrubias, Mol. Phys., 2017, 115, 2597 CrossRef CAS.
- (a) R. J. Schwamm and M. P. Coles, Chem.–Eur. J., 2019, 25, 14183 CrossRef CAS PubMed; (b) D. Dange, A. Davey, J. A. B. Abdalla, S. Aldridge and C. Jones, Chem. Commun., 2015, 51, 7128 RSC.
- M. Miar, A. Shiroudi, K. Pourshamsian, A. R. Oliaey and F. Hatamjafari, J. Chem. Res., 2021, 45, 147 CrossRef CAS.
- Cambridge Structural Database, Version 5.42, see also: F. H. Allen, Acta Crystallogr. Sect. B, 2002, 58, 380–388. 55 hits containing an Sb–Sb bond classified as a single were found of the type R2Sb–SbR2 (R defined as non-antimony, c.n. (Sb) = 3). The Sb–Sb bond lengths range from 2.77–3.07 Å (mean of 2.86(5) Å excluding a single highly ring strain structure 2.64 Å.
- Cambridge Structural Database, Version 5.42, see also: F. H. Allen, Acta Crystallogr. Sect. B, 2002, 58, 380–388. 495 hits containing an Sb–C bond classified as a single were found of the type R2Sb–C (c.n. (Sb) = 3). The Sb-C bond lengths range from 2.04–2.31 Å (mean of 2.17(4)). Analogous, 195 entries for Sb–N were found, 1.95–2.58 Å (mean of 2.06(7))..
- The ORCA quantum chemistry package was used and details regarding the quantum chemical calculations are given in the ESI.† (a) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73 CAS; (b) F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed; (c) F. Neese, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2018, 8, 1 Search PubMed; (d) C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS; (e) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC; (f) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; (g) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed; (h) B. Metz, H. Stoll and M. Dolg, J. Chem. Phys., 2000, 113, 2563 CrossRef CAS; (i) R. Izsák and F. Neese, J. Chem. Phys., 2011, 135, 144105 CrossRef PubMed; (j) F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98 CrossRef CAS; (k) F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC; (l) M. Garcia-Ratés and F. Neese, J. Comput. Chem., 2020, 41, 922 CrossRef PubMed; (m) M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669 CrossRef CAS PubMed; (n) V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995 CrossRef CAS.
- (a) P. Jutzi and S. Opiela, Z. Anorg. Allg. Chem., 1992, 610, 75 CrossRef CAS; (b) G. Etemad-Moghadam, J. Bellan, C. Tachon and M. Koenig, Tetrahedron, 1987, 43, 1793 CrossRef CAS.
- NBO 7.0, E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis, and F. Weinhold, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2018 Search PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed , http://www.ks.uiuc.edu/Research/vmd/..
- (a) R. D. Bach and O. Dmitrenko, J. Am. Chem. Soc., 2004, 126, 4444 CrossRef CAS PubMed; (b) K. B. Wiberg and R. A. Fenoglio, J. Am. Chem. Soc., 1968, 90, 3395 CrossRef CAS.
- (a) P. S. V. Kumar, V. Raghavendra and V. Subramanian, J. Chem. Sci., 2016, 128, 1527 CrossRef CAS; (b) R. F. W. Bader, Chem. Rev., 1991, 91, 893 CrossRef CAS; (c) C. Kalaiarasi, M. S. Pavan and P. Kumaradhas, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 775 CrossRef CAS PubMed; (d) T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
- (a) U. Ekström, L. Visscher, R. Bast, A. J. Thorvaldsen and K. Ruud, J. Chem. Theory Comput., 2010, 6, 1971 CrossRef PubMed; (b) B. Silvi and A. Savin, Nature, 1994, 371, 683 CrossRef CAS; (c) A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures and analytical data, NMR, IR, and UV-Vis spectra of 1–13, computational details and cif files. CCDC 2129215 (1), 2129216 (2), 2129217 (3), 2129218 (4), 2129219, (5), 2129220, (6), 2129221 (7), 2129222 (8), 2129223 (9), 2129211 (10), 2129212 (11), 2129213 (12), and 2129214 (13). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2sc00314g |
| This journal is © The Royal Society of Chemistry 2022 |