
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of a β-cyclodextrin-modified gold nanoparticle-assisted electromembrane extraction method followed by capillary electrophoresis for methadone determination in plasma
Mir Saleh Hoseininezhad-Naminab,
Sibel Aysil Ozkanc,
Elaheh Rahimpour *bd and
Abolghasem Jouybanbe
*bd and
Abolghasem Jouybanbe
aLiver and Gastrointestinal Diseases Research Center, Tabriz University of Medical Sciences, Tabriz, Iran
bPharmaceutical Analysis Research Center and Faculty of Pharmacy, Tabriz University of Medical Sciences, Tabriz, Iran. E-mail: Rahimpour_e@yahoo.com
cAnkara University, Faculty of Pharmacy, Department of Analytical Chemistry, Ankara, Turkey
dInfectious and Tropical Diseases Research Center, Tabriz University of Medical Sciences, Tabriz, Iran
eFaculty of Pharmacy, Near East University, P.O. Box 99138 Nicosia, North Cyprus, Mersin 10, Turkey
First published on 28th November 2022
Abstract
In this study, gold nanoparticles (AuNPs) modified with β-cyclodextrin (β-CD) were used to assist with electromembrane extraction (EME) and were coupled with capillary electrophoresis (CE) and ultraviolet (UV) detection (CE-UV) for the extraction and measurement of methadone from plasma samples. A β-CD-modified AuNP-reinforced hollow fiber (HF) was utilized in this work. The β-CD-modified AuNPs act as an absorbent and provide an extra pathway for the analyte extraction. For obtaining the effect of the presence of β-CD-modified AuNPs in the HF pores, the extraction efficiency of the EME and β-CD-modified AuNPs/EME techniques were compared. Different parameters influencing the extraction efficacy of the EME and β-CD-modified AuNPs/EME methods were optimized. Optimal extractions were performed with 1-octanol as the organic solvent in the supported liquid membrane (SLM), with an applied voltage of 10 V as the driving force across the SLM, and with pH 7.0 in the donor solutions with a stirring speed of 1000 rpm after 20 min and 25 min for the β-CD-modified AuNPs/EME and EME methods, respectively. Under optimal conditions, compared with the EME method, the β-CD-modified AuNPs/EME method exhibited increased extraction efficacy in a short time. The β-CD-modified AuNPs/EME technique demonstrated a lower limit of detection (5.0 ng mL−1), higher extraction recovery (68%), and a more optimal preconcentration factor (135). Furthermore, this method was successfully utilized for measuring methadone in real plasma samples.
1 Introduction
Opioid maintenance treatment (OMT) drugs can be used for the treatment of opioid addiction.1,2 Methadone is an OMT drug used primarily for addiction and pain treatment (Fig. 1). Nevertheless, it is an addictive drug that can be abused. Moreover, the overdose and nonmedical use of methadone can result in death.3,4 Thus, developing a reliable and practical method for measuring its concentration in biological samples is essential.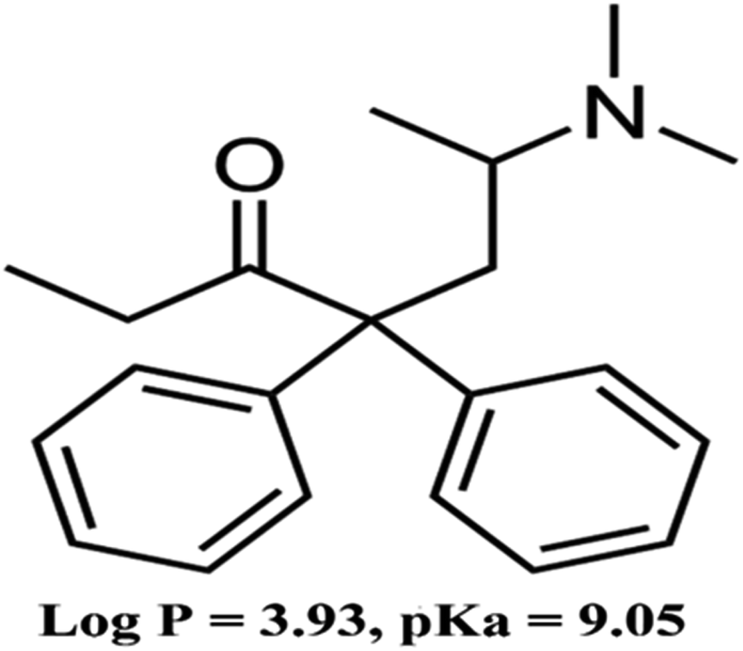 | ||
| Fig. 1 Chemical structure of methadone. | ||
Various techniques, including high-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS),5 gas chromatography-mass spectrometry (GC-MS),6 gas chromatography-flame ionization detector (GC-FID),7 flow-injection analysis,8 capillary electrophoresis (CE),9 and HPLC-UV10 have been used for methadone identification. There are many problems associated with drug analysis of biological samples due to the complexities of biological matrices. They contain salts, different proteins, and other components that can interfere with the analytical method.11 Thus, sample pre-treatment before analysis is essential in bioanalytical chemistry.
In the last few decades, scientists focused on miniaturization, efficiency, sensitivity, and environmentally friendly pre-treatment techniques such as solid-phase extraction (SPE), solvent bar microextraction, headspace solid-phase microextraction, liquid-phase microextraction, and electromembrane extraction (EME).12,13 Among these emerging techniques, fast, excellent sample clean-up and highly selective extraction can be obtained with EME.14 With this method, charged analytes are extracted across a supported liquid membrane (SLM) on a microporous polypropylene hollow fiber (HF) based on electrokinetic migration. The use of an electrical field over the SLM increases the extraction efficacy.15 In the EME method, the flux of analytes across an SLM is described by the modified Nernst–Planck equation. Based on this equation, the magnitude of the applied potential influences the flux of the analyte.14 Using EME, clean extracts have been obtained from biological samples such as whole blood,16 saliva,17 urine,18 breast milk,19 and plasma.18 This method has been developed for different types of acidic and basic drug extraction.20–22
Recently, nanostructures such as carbon nanotubes, silver nanoparticles, maghemite nanoparticles, copper nanoparticles, and titanium dioxide nanoparticles have been utilized in the SLM to increase the efficacy of the EME method.23–26 Gold nanoparticles (AuNPs) are used widely in diagnosis, medical investigations, and analytical chemistry.27,28 Functionalization can be utilized to produce stable, smaller, efficient, and highly active nanoparticles. Thiols, mercaptans, citrates, polymers, and amines are some capping agents that can increase the efficacy of nanoparticles and prevent their aggregation.29,30 β-Cyclodextrin (β-CD) is a known functionalizing agent that is utilized for self-assembly and surface modification of AuNPs.31,32 Modification with β-CD enhances the chemical and physical stability of AuNPs and prevents their aggregation. The functionalization of nanoparticles leads to ease in biofunctionalization.33,34
In the current work, a novel strategy was developed to promote the EME method for the extraction of methadone by inserting β-CD-modified AuNPs into the pores of a HF. The presence of β-CD-modified AuNPs in the HF wall can enhance the surface area and increase the analyte migration. Herein, the β-CD-modified AuNP-assisted EME method coupled with capillary electrophoresis was fully validated for the determination of methadone in a plasma sample, and its efficiency was compared with an unmodified EME method for methadone extraction.
2 Materials and methods
2.1 Chemicals and reagents
Methadone was kindly supplied by Tofigh Daru Pharmaceutical Company (Tehran, Iran). All chemicals utilized in this study were of analytical grade. HCl, NaOH, and potassium dihydrogen phosphate (KH2PO4) were supplied from Merck (Darmstadt, Germany). 1-Heptanol, nitrobenzene, 1-octanol, and dihexyl ether were purchased from Fluka (Buchs, Switzerland). Deionized (DI) water was supplied from Shahid Ghazi Pharmaceutical Company (Tabriz, Iran).2.2 Stock solution
The methadone stock solution was prepared at 5000 ng mL−1 in methanol. The stock solution was stored at 4 °C and protected from light. Sample solutions were freshly prepared from the stock solution every day.2.3 Capillary electrophoresis conditions
An Agilent Technologies 7100 (Waldbronn, Germany) capillary electrophoresis system equipped with diode-array detection (190–600 nm) was utilized for analyses. The separation was performed on a 48.5 cm bare fused silica capillary with a 40 cm effective length and an internal diameter of 50 μm (Agilent). The background electrolyte (BGE) solution was a 100 mmol L−1 borate buffer adjusted to pH 3.0. Before using the CE device, the capillary was washed for 30 min with 1 mol L−1 sodium hydroxide. Then, DI water was used for 30 min, and after that, the capillary was washed for 30 min with BGE solution.35Before beginning electrophoresis every day, the capillary was flushed with 0.1 mol L−1 sodium hydroxide (10 min), DI water (10 min), and BGE solution (10 min). Between analyses, the capillary was washed using 0.1 mol L−1 sodium hydroxide for 3 min, DI water for 3 min, and BGE for 5 min. All samples and BGE were filtered with a polytetrafluoroethylene (PTFE) syringe filter (0.20 μm pore size). Hydrodynamic injection at a pressure of 0.5 psi for 10 s was used for all samples. The constant voltage and capillary temperature were set at 20 kV and 25 °C, respectively. The detection of methadone was accomplished at 195 nm.
2.4 Equipment for EME
A screw-capped homemade glass sample compartment was used with a volume of 7 mL (internal diameter 1 cm and height 7 cm). A porous HF with 0.2 mm wall thickness, 0.6 mm internal diameter, and a pore size of 0.2 μm was utilized for the acceptor solution (PP Q3/2 polypropylene HF (Membrana, Wuppertal, Germany)). For two donor and acceptor solution electrodes, platinum wires were used with a 3 mm interelectrode distance and a diameter of 0.2 mm.36 These electrodes were coupled to a power supply with a programmable voltage range of 0 to 300 V and current range of 0 to 1000 mA (model PS858, S.KAR, Tabriz, Iran). A multimeter was applied to monitor the current during extraction (Zhangzhou Weihua Electronic Co., Ltd. China, Model M890C+). The EME system was stirred using a magnetic bar with a heater-magnetic stirrer during the extraction (model RCT basic from IKA Company, Staufen im Breisgau, Germany).2.5 Synthesis of β-CD-modified AuNPs
β-CD-modified Au NPs were synthesized by adding 0.023 g of β-CD and 200 μL of HAuCl4 50 mmol L−1 to a beaker containing 30 mL deionized water. The mixture was stirred until complete dissolution of reagents occurred. Then, the solution was heated to 100 °C, and 2–3 drops of NaOH (0.07 g mL−1) solution was added. Approximately 8 minutes later, a cherry red solution was formed, confirming the synthesis of AuNPs.37 The synthesized nanoparticles were stored in darkness at 4 °C.2.6 Procedure for β-CD-modified AuNPs/EME
The HF was cut into 4 cm parts to prepare the nanoparticle-immobilized membrane. β-CD-modified AuNPs were dispersed in the 1-octanol organic solvent. Then, the contents of the HPLC syringe were gently injected into the HF. The SLM (HF filled with the β-CD-modified AuNPs and 1-octanol mixture) was sonicated until the mixture spread and dispersed within the HF pores. The excess solvent in the HF was removed by blowing with the medical syringe. Then, utilizing the micro-syringe, 20 μL of HCl with pH of 1 (acceptor solution) was added to the lumen of the HF. After that, the end of the HF was closed by nonconductive plastic. Then, 1 mL of plasma sample diluted with 3 mL of water with a pH of 7.2 was utilized as a donor solution. Positive and negative electrodes were inserted into the donor and acceptor solution. The positive and negative electrodes were connected to a DC power supply, and the EME system was placed on the stirrer.After the extraction process, with the micro-syringe, the acceptor phase was collected and transferred to a micro-vial, followed by CE. The principle of the EME method is shown in Fig. 2. The desired compounds were extracted from a donor, using an SLM, into an acceptor phase. This mass transfer is affected by electrokinetic migration through a stable external electric field in the SLM. The SLM includes an organic solvent (1-octanol and β-CD-modified AuNPs) fixed on the pores of a porous polymeric membrane.
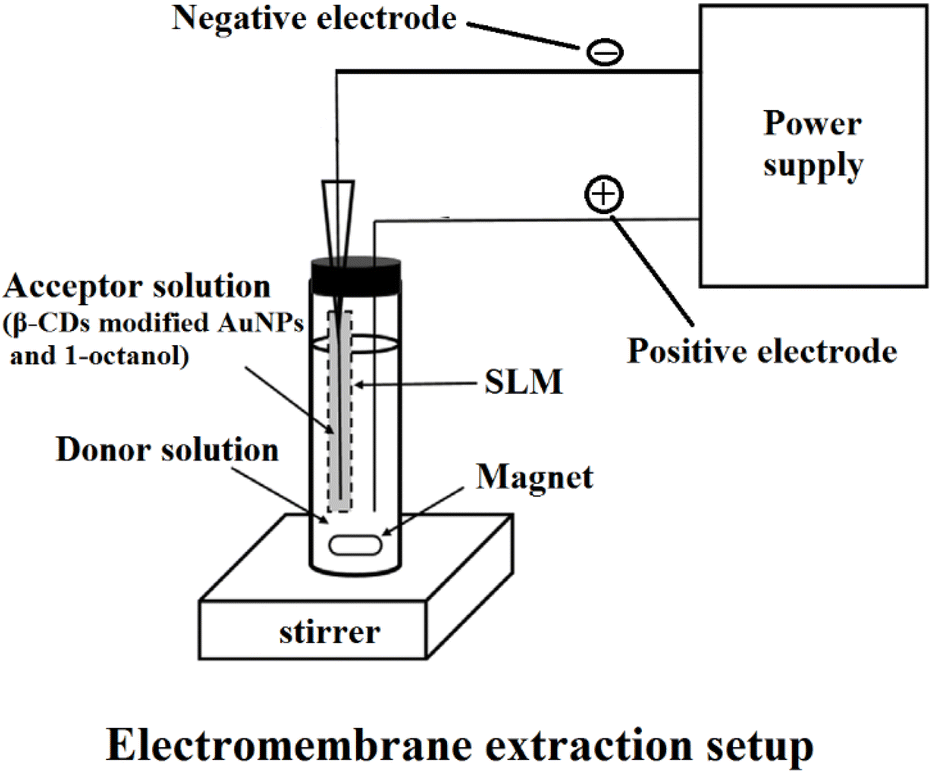 | ||
| Fig. 2 Principle of EME. | ||
2.7 Calculation of extraction recovery, preconcentration factor, and relative recovery
The extraction recovery (ER%) of methadone from the plasma samples was calculated according to eqn (1):
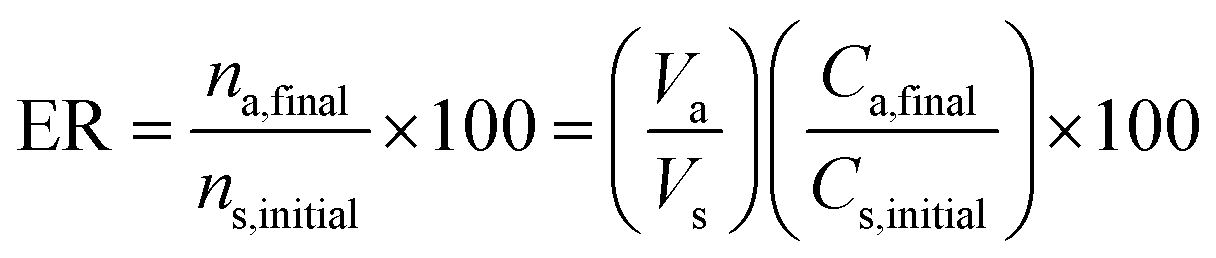 | (1) |
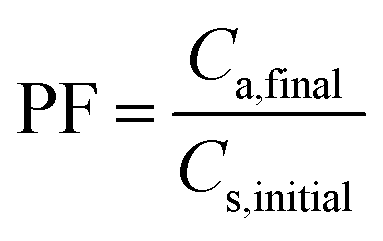 | (2) |
Percent relative recovery (% RR) was calculated with eqn (3):
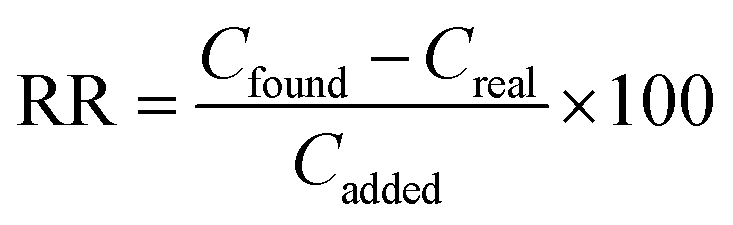 | (3) |
2.8 Ethical considerations
The sample donors signed a consent form approved by the Ethics Committee at Tabriz University of Medical Sciences (code of ethics committee: IR.TBZMED.REC.1400.426).3 Results and discussion
3.1 Characterization of β-CD-modified Au NPs
TEM analysis was used to investigate the size and morphology of the prepared nanoparticles. Fig. 3A shows that the β-CD-modified Au NPs possess a spherical morphology with an average size of <30 nm that was confirmed by dynamic light scattering (DLS) measurement (Fig. 3B).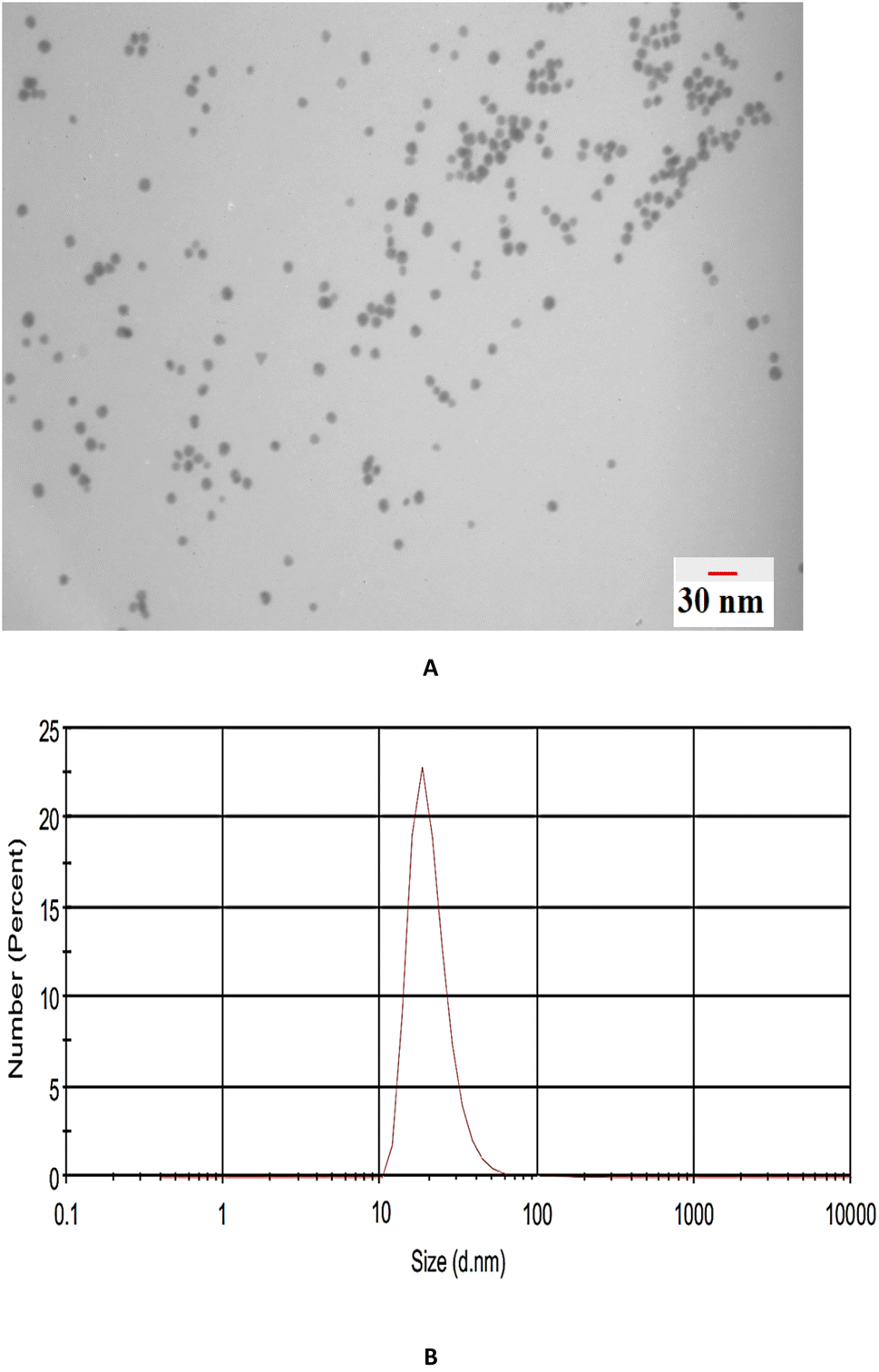 | ||
| Fig. 3 (A) TEM image and (B) DLS analysis of β-CD-modified AuNPs. | ||
3.2 Optimization of EME and β-CD-modified AuNPs/EME conditions
Different parameters affecting the extraction process were optimized to establish excellent extraction. The initial experimental conditions during the optimization were (1) 100 ng mL−1 of methadone, 750 rpm as the stirring rate, voltage of 15 V, 15 min extraction time, donor solution pH of 7.0, and 300 μL of β-CD-modified AuNPs for effect of the SLM composition on the EME and β-CD-modified AuNPs/EME methods; (condition), (2) 100 ng mL−1 of methadone, 750 rpm as the stirring rate, 15 min extraction time, donor solution pH of 7.0, and 300 μL of β-CD-modified AuNPs for effect of applied voltage, (3) 100 ng mL−1 of methadone, voltage of 10 V, 15 min extraction time, donor solution pH of 7.0, and 300 μL of β-CD-modified AuNPs for effect of stirring rate, (4) 100 ng mL−1 of methadone, voltage of 10 V, 1000 rpm as the stirring rate, donor solution pH of 7.0, and 300 μL of β-CD-modified AuNPs for effect of extraction time, (5) 100 ng mL−1 of methadone, voltage of 10 V, 1000 rpm as the stirring rate, 20 min extraction time for the β-CD-modified AuNPs/EME method and 25 min extraction time for the EME method, and 300 μL of β-CD-modified AuNPs for effect of donor solution pH, (6) 100 ng mL−1 of methadone, voltage of 10 V, 1000 rpm as the stirring rate, 20 min extraction time for the β-CD-modified AuNPs/EME method and 25 min extraction time for the EME method, and solution pH of 7.0 for effect of β-CD-modified AuNP concentration.In this work, we used solvents tested in previous works for extracting methadone and opioids with the EME method.7,39 To determine the most optimal organic solvent, 1-heptanol, nitrobenzene, 1-octanol, and dihexyl ether were investigated for extraction efficiency using both methods. The results are shown in Fig. 4A, and they indicate that the greatest efficiency was obtained with 1-octanol. 1-Octanol showed the greatest extraction recovery with effective extraction of the analyte. Thus, in this work, 1-octanol was chosen for further experiments.
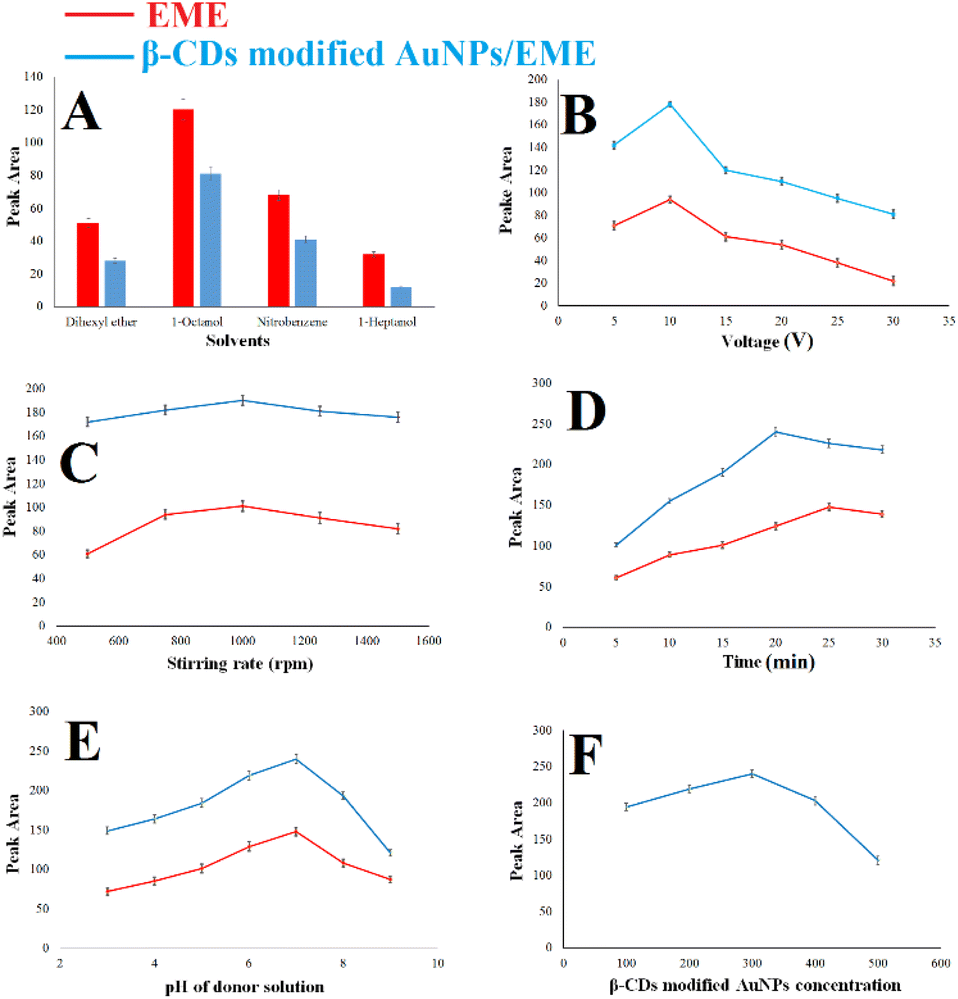 | ||
| Fig. 4 (A) Effect of the SLM composition on the EME and β-CD-modified AuNPs/EME methods; (condition: 100 ng mL−1 of methadone, 750 rpm as the stirring rate, voltage of 15 V, 15 min extraction time, donor solution pH of 7.0, and 300 μL of β-CD-modified AuNPs), (B) effect of applied voltage; (condition: 100 ng mL−1 of methadone, 750 rpm as the stirring rate, 15 min extraction time, donor solution pH of 7.0, and 300 μL of β-CD-modified AuNPs), (C) effect of stirring rate; (condition: 100 ng mL−1 of methadone, voltage of 10 V, 15 min extraction time, donor solution pH of 7.0, and 300 μL of β-CD-modified AuNPs), (D) effect of extraction time; (condition: 100 ng mL−1 of methadone, voltage of 10 V, 1000 rpm as the stirring rate, donor solution pH of 7.0, and 300 μL of β-CD-modified AuNPs), (E) effect of donor solution pH; (condition: 100 ng mL−1 of methadone, voltage of 10 V, 1000 rpm as the stirring rate, 20 min extraction time for the β-CD-modified AuNPs/EME method and 25 min extraction time for the EME method, and 300 μL of β-CD-modified AuNPs). (F) Effect of β-CD-modified AuNP concentration; (condition: 100 ng mL−1 of methadone, voltage of 10 V, 1000 rpm as the stirring rate, 20 min extraction time for the β-CD-modified AuNPs/EME method and 25 min extraction time for the EME method, and solution pH of 7.0). The red color indicates the EME method parameters, and the blue color indicates the β-CD-modified AuNP method. | ||
The pH of the acceptor phase was also optimized by the same method to investigate the effect of pH in the acceptor phase on recovery percentage. The results indicated that extraction performance decreases with an increase in the pH. Thus, a pH value of 1.0 was required for the acceptor phase to obtain effective extraction.
3.3 Analytical performance
In the following, the obtained optimization conditions were used to validate the EME and β-CD-modified AuNPs/EME techniques for determining and extracting methadone from plasma samples with CE-diode array detector (DAD). It should be noted that FDA guidelines were utilized to validate the method.It can be concluded that the presence of β-CD-modified AuNPs increased the surface area, increased the analyte migration, and consequently increased the extraction efficiency. Moreover, the β-CD-modified AuNPs/EME method is not very complicated, and the method can be developed by adding a certain amount of nanostructure to the organic solvent. Thus, continuous precision, accuracy, recovery, and stability parameters were investigated for the β-CD-modified AuNPs/EME method.
| Cadded (ng mL−1) | Cfound (ng mL−1) | RR (%) |
|---|---|---|
| 15 | 14.6 | 97.3 |
| 100 | 92.4 | 92.4 |
| 500 | 511.0 | 102.2 |
| Added concentration (ng mL−1) | RE (%) | RSD (%) |
|---|---|---|
| Freeze-thaw stability (cycle 1) | ||
| 15 | −7.67 | 8.30 |
| 500 | −5.90 | 6.11 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| Freeze-thaw stability (cycle 2) | ||
| 15 | −1.72 | 9.19 |
| 500 | 3.02 | 5.58 |
|
||
| Freeze-thaw stability (cycle 3) | ||
| 15 | −4.70 | 11.01 |
| 500 | 7.20 | 5.91 |
|
||
| Stock solution (after 4 weeks at 4 °C) | ||
| 15 | −10.91 | 6.84 |
| 500 | 8.07 | 7.04 |
|
||
| Bench-top stability (after 6 h at room temperature) | ||
| 15 | −7.64 | 5.76 |
| 500 | 5.99 | 5.62 |
3.4 Real sample analysis
To examine the method's efficacy in detecting real samples, the β-CD-modified AuNPs/EME method was used for the extraction and detection of methadone from plasma samples of three patients. First, 1.0 mL of plasma was diluted with 3.0 mL of DI water. For this purpose, β-CD-modified AuNPs/EME was used under optimum conditions. Then, the acceptor solution obtained from the β-CD-modified AuNPs/EME method was injected for CE, and the electropherograms of the results are indicated in Fig. 5. Table 5 provides patient details, the administration of the dose, and the concentration of methadone in the plasma samples. The concentrations of methadone in the three real plasma samples were 164 ± 0.2, 181 ± 0.2, and 144 ± 0.1 ng mL−1. Furthermore, to determine the method's accuracy for the investigation of methadone in real plasma samples with consideration of the matrix effect, the recovery test was performed by the addition of a known amount of methadone (100 ng mL−1). The obtained recovery was between 96.0 to 102.0%, which is in the appropriate range.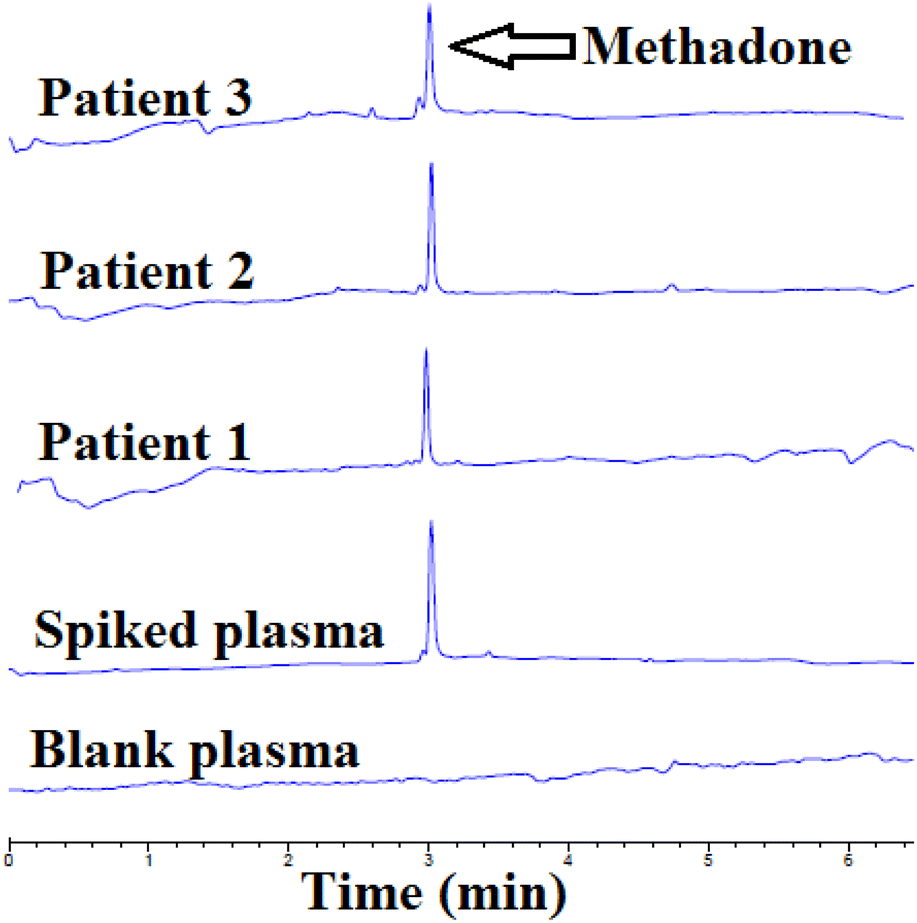 | ||
| Fig. 5 Electropherograms of a blank plasma sample, plasma samples with added amounts of methadone, and real plasma samples of patients after β-CD-modified AuNPs/EME under optimal conditions. | ||
| No. | Gender | Age (year) | Interval between sampling and drug administration (hours) | Daily dose (g) | Methadone in plasma (ng mL−1) |
|---|---|---|---|---|---|
| 1 | Male | 42 | 5 | 0.1 | 164 ± 0.2 |
| 2 | Male | 46 | 4 | 0.2 | 181 ± 0.2 |
| 3 | Male | 44 | 5 | 0.05 | 144 ± 0.1 |
3.5 Comparison with other methods
Based on Table 6, the β-CD-modified AuNPs/EME method was compared with other methods used for the determination of methadone. The results indicated that the current method has lower detection limits than previously used methods, such as EME/CE-UV,45 CNTs-EME/GC-FID,39 EME-SFME/CD-IMS,46 LV-ESSM-LLME/HPLC-UV,47 and LV-EME/CE-UV.48 Furthermore, some instruments used to determine methadone (such as LC-MS and GC-FID) are relatively complicated and expensive. However, the β-CD-modified AuNPs/EME-CE method could provide the same results or better for methadone analysis.| Method | Sample preparation | Sample type | LOD (ng mL−1) | Linear range (ng mL−1) | RSD (%) | PF | % ER | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Surfactant-assisted pulsed two-phase electromembrane extraction.b Corona discharge-ion mobility spectrometry.c Electromembrane extraction and slug flow microextraction.d Low-voltage electrically stimulated stir membrane liquid–liquid microextraction.e Electromembrane extraction at low voltage. | ||||||||
| CE-UV | EME | Urine | — | 15–500 | 7.5 | 130 | — | 45 |
| GC-FID | CNTs-EME | Plasma | 3 | 10–1000 | 4.4 | 255 | — | 39 |
| LC-MS | EME | Plasma | 0.4 | — | 9.0–10.0 | — | — | 49 |
| GC-FID | SA-PEMEa | Water | 0.6 | 2–1000 | 12.7 | — | — | 7 |
| CD-IMSb | EME-SFMEc | Plasma | 2 | 8–750 | 10.2–10.9 | 406.1 | 63.9 | 46 |
| HPLC-UV | LV-ESSM-LLMEd | Plasma | 2 | 10–800 | 6.5 | 21.5 | 57 | 47 |
| CE-UV | LV-EMEe | Plasma | — | 25–1000 | 2.9–18.7 | 4.1 | 10 | 48 |
| CE-UV | β-CD-modified AuNPs/EME | Plasma | 1.5 | 5–1000 | 1.8–11.2 | 135 | 68 | This study |
4 Conclusions
EME has been introduced as an effective research field since 2006, and its applicability is rapidly increasing due to the numerous superior features of EME. Most significantly, the sensitivity and selectivity can be adjusted by the modified chemical properties of the SLM, for example, by modifying the SLM with nanostructures. The presence of nanostructures in the SLM results in a more effective surface area and enhances the total partition coefficient of the desired analytes.In this work, β-CD-modified AuNPs/EME as a modified approach of the EME method coupled with CE-UV was applied for monitoring the methadone in plasma samples. A comparison of the β-CD-modified AuNPs/EME method with the EME method showed that the former was the more optimal technique for enhancing the methadone extraction efficacy. This technique exhibits excellent outcomes (lower LOD, higher % ER, and higher PF) in a shorter time, and can be considered an alternative to existing methods. A comparison of this method with previous methods showed that the β-CD-modified AuNPs/EME-CE method provided more optimal results. Finally, the validated technique was successfully utilized for monitoring methadone in real plasma samples. This method with different nanostructural modifiers such as CNTs, GO, RGO, ND-G, AgNPs, CuNPs, and maghemite nanoparticles can be employed for other analytes in samples of different matrices (such as urine or exhaled breath condensate) in future studies.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This report is a part of M. S. Hoseininezhad-Namin's PhD thesis submitted to the Faculty of Pharmacy, Tabriz University of Medical Sciences, Tabriz, Iran. The work is financially supported by the Pharmaceutical Analysis Research Center under grant number 67214.References
- T. Clausen, K. Anchersen and H. Waal, Drug Alcohol Depend., 2008, 94, 151–157 CrossRef PubMed.
- M. T. Brugal, A. Domingo-Salvany, R. Puig, G. Barrio, P. De García Olalla and L. De La Fuente, Addiction, 2005, 100, 981–989 CrossRef PubMed.
- K. W. Simonsen, P. Kriikku, G. Thelander, H. M. E. Edvardsen, S. Thordardottir, C. U. Andersen, A. K. Jönsson, J. Frost, D. J. Christoffersen, G. J. M. Delaveris and I. Ojanperä, Forensic Sci. Int., 2020, 313, 110343 CrossRef CAS PubMed.
- A. Casati, R. Sedefov and T. Pfeiffer-Gerschel, Eur. Addict. Res., 2012, 18, 228–245 CrossRef PubMed.
- R. George, M. Lobb, A. Haywood, S. Khan, J. Hardy, P. Good, S. Hennig and R. Norris, Talanta, 2016, 149, 142–148 CrossRef CAS PubMed.
- A. M. Bermejo, A. C. S. Lucas, M. J. Tabernero and P. Fernández, Anal. Lett., 2000, 33, 739–752 CrossRef CAS.
- P. Zahedi, S. S. H. Davarani, H. R. Moazami and S. Nojavan, J. Pharm. Biomed. Anal., 2016, 117, 485–491 CrossRef PubMed.
- R. Montero, M. Gallego and M. Valcárcel, Anal. Chim. Acta, 1990, 234, 433–437 CrossRef.
- S. Hamidi, M. Khoubnasabjafari, K. Ansarin, V. Jouyban-Gharamaleki and A. Jouyban, Anal. Methods, 2017, 9, 2342–2350 RSC.
- S. Taheri, F. Jalali, N. Fattahi, R. Jalili and G. Bahrami, J. Sep. Sci., 2015, 38, 3545–3551 CrossRef PubMed.
- S. Seidi, Y. Yamini, A. Heydari, M. Moradi, A. Esrafili and M. Rezazadeh, Anal. Chim. Acta, 2011, 701, 181–188 CrossRef CAS PubMed.
- M. Havlikova, R. Cabala, V. Pacakova and Z. Bosakova, J. Sep. Sci., 2019, 42, 303–318 CrossRef CAS PubMed.
- M. Havlikova, R. Cabala, V. Pacakova, M. Bursova and Z. Bosakova, J. Sep. Sci., 2019, 42, 273–284 CrossRef CAS PubMed.
- K. S. Hasheminasab and A. R. Fakhari, Anal. Chim. Acta, 2013, 767, 75–80 CrossRef CAS PubMed.
- S. Pedersen-Bjergaard and K. E. Rasmussen, Anal. Bioanal. Chem., 2007, 388, 521–523 CrossRef CAS PubMed.
- A. Gjelstad, K. E. Rasmussen and S. Pedersen-Bjergaard, Anal. Bioanal. Chem., 2009, 393, 921–928 CrossRef CAS PubMed.
- S. Seidi, Y. Yamini, A. Saleh and M. Moradi, J. Sep. Sci., 2011, 34, 585–593 CrossRef CAS PubMed.
- S. Pedersen-Bjergaard and K. E. Rasmussen, J. Chromatogr. A, 2006, 1109, 183–190 CrossRef CAS PubMed.
- I. J. Ø. Kjelsen, A. Gjelstad, K. E. Rasmussen and S. Pedersen-Bjergaard, J. Chromatogr. A, 2008, 1180, 1–9 CrossRef CAS PubMed.
- A. Gjelstad, K. E. Rasmussen and S. Pedersen-Bjergaard, J. Chromatogr. A, 2006, 1124, 29–34 CrossRef CAS PubMed.
- M. Balchen, A. Gjelstad, K. E. Rasmussen and S. Pedersen-Bjergaard, J. Chromatogr. A, 2007, 1152, 220–225 CrossRef CAS PubMed.
- M. R. Payán, M. A. B. López, R. F. Torres, M. V. Navarro and M. C. Mochón, Talanta, 2011, 85, 394–399 CrossRef PubMed.
- M. Ramos-Payán, R. Fernández-Torres, J. L. Pérez-Bernal, M. Callejón-Mochón and M. T. Bello-López, Anal. Chim. Acta, 2014, 849, 7–11 CrossRef PubMed.
- M. S. Hoseininezhad-Namin, E. Rahimpour, S. A. Ozkan and A. Jouyban, Anal. Methods, 2022, 14, 212–221 RSC.
- Z. Tahmasebi, S. S. H. Davarani and A. A. Asgharinezhad, Biosens. Bioelectron., 2018, 114, 66–71 CrossRef PubMed.
- M. Khajeh, S. Pedersen-Bjergaard, M. Bohlooli, A. Barkhordar and M. Ghaffari-Moghaddam, J. Sci. Food Agric., 2017, 97, 1517–1523 CrossRef PubMed.
- A. Tomar and G. Garg, Global J. Pharmacol., 2013, 7, 34–38 Search PubMed.
- T. Khan, N. Ullah, M. A. Khan, Z. U. R. Mashwani and A. Nadhman, Adv. Colloid Interface Sci., 2019, 272, 102017 CrossRef CAS PubMed.
- S. Alex and A. Tiwari, J. Nanosci. Nanotechnol., 2015, 15, 1869–1894 CrossRef CAS PubMed.
- T. John, A. Gladytz, C. Kubeil, L. L. Martin, H. J. Risselada and B. Abel, Nanoscale, 2018, 10, 20894–20913 RSC.
- S. Pande, S. K. Ghosh, S. Praharaj, S. Panigrahi, S. Basu, S. Jana, A. Pal, T. Tsukuda and T. Pal, J. Phys. Chem. C, 2007, 111, 10806–10813 CrossRef CAS.
- T. Huang, F. Meng and L. Qi, J. Phys. Chem. C, 2009, 113, 13636–13642 CrossRef.
- M. R. Bindhu, K. Ancy, M. Umadevi, G. A. Esmail, N. A. Al-Dhabi and M. V. Arasu, J. Photochem. Photobiol., B, 2020, 210, 111965 CrossRef PubMed.
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025–1102 CrossRef PubMed.
- B. Seyfinejad, M. Khoubnasabjafari, S. E. Ziaei, S. A. Ozkan and A. Jouyban, Daru, J. Pharm. Sci., 2020, 28, 615–624 CrossRef PubMed.
- B. Seyfinejad, A. Meshkini, P. Habibolahi, S. A. Ozkan and A. Jouyban, Electrophoresis, 2020, 41, 666–677 CrossRef PubMed.
- R. Rajamanikandan, A. D. Lakshmi and M. Ilanchelian, New J. Chem., 2020, 44, 12169–12177 RSC.
- L. Xu, P. C. Hauser and H. K. Lee, J. Chromatogr. A, 2008, 1214, 17–22 CrossRef PubMed.
- K. S. Hasheminasab, A. R. Fakhari and M. H. Koruni, J. Sep. Sci., 2014, 37, 85–91 CrossRef PubMed.
- M. S. Hoseininezhad-Namin, B. Seyfinejad, S. A. Ozkan, J. Soleymani, M. Khoubnasabjafari, V. Jouyban-Gharamaleki, E. Rahimpour and A. Jouyban, J. Pharm. Biomed. Anal., 2022, 219, 114959 CrossRef PubMed.
- M. Balchen, L. Reubsaet and S. Pedersen-Bjergaard, J. Chromatogr. A, 2008, 1194, 143–149 CrossRef CAS PubMed.
- A. Sarafraz-Yazdi, A. H. Amiri and Z. Es'haghi, Chemosphere, 2008, 71, 671–676 CrossRef CAS PubMed.
- S. Asadi and S. Nojavan, Anal. Chim. Acta, 2016, 923, 24–32 CrossRef CAS PubMed.
- S. Seidi, Y. Yamini and M. Rezazadeh, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2013, 913–914, 138–146 CrossRef CAS PubMed.
- M. H. Koruni, H. Tabani, H. Gharari and A. R. Fakhari, J. Chromatogr. A, 2014, 1361, 95–99 CrossRef PubMed.
- M. Behpour, M. Maghsoudi and S. Nojavan, J. Chromatogr. A, 2022, 1678, 463355 CrossRef PubMed.
- K. M. Ara and F. Raofie, Talanta, 2017, 168, 105–112 CrossRef PubMed.
- N. C. Domínguez, A. Gjelstad, A. M. Nadal, H. Jensen, N. J. Petersen, S. H. Hansen, K. E. Rasmussen and S. Pedersen-Bjergaard, J. Chromatogr. A, 2012, 1248, 48–54 CrossRef PubMed.
- C. Huang, L. E. E. Eibak, A. Gjelstad, X. Shen, R. Trones, H. Jensen and S. Pedersen-Bjergaard, J. Chromatogr. A, 2014, 1326, 7–12 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |