
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCovalently linked benzothiadiazole-fullerene adducts for organic optoelectronic devices: synthesis and characterization†
Divambal Appavooab,
Komal Bhardwajcd,
Samarendra P. Singhe,
Emmanuel N. Koukaras f,
Rachana Kumarcd and
Bimlesh Lochab*a
f,
Rachana Kumarcd and
Bimlesh Lochab*a
aMaterials Chemistry Laboratory, Department of Chemistry, Shiv Nadar Institute of Eminence, Delhi-NCR, Gautam Buddha Nagar, Uttar Pradesh 201314, India. E-mail: bimlesh.lochab@snu.edu.in
bDepartment of Chemistry, University of Central Florida, Orlando, FL 32816, USA
cPhotovoltaic Metrology Group, Advanced Materials and Devices Metrology Division, CSIR-National Physical Laboratory, Dr. K. S. Krishnan Marg, New Delhi, India-110012
dAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad-201002, Uttar Pradesh, India
eSemiconductor Physics Laboratory, Department of Physics, School of Natural Sciences, Shiv Nadar Institute of Eminence, India
fLaboratory of Quantum and Computational Chemistry, Department of Chemistry, Aristotle University of Thessaloniki, GR-54124 Thessaloniki, Greece
First published on 15th December 2022
Abstract
Fullerene adducts have attracted attention in a variety of applications including organic optoelectronic devices. In this regard, we have designed a covalently linked donor–acceptor dyad comprising a fluorobenzothiadiazole-thiophene (BTF2-Th) unit with the electron acceptor fullerene in an Acceptor–Donor–Acceptor (A–D–A) molecular arrangement. We synthesized and characterized two new covalently bonded benzothiadiazole-based fullerene molecules, mono-adduct, 7 (benzothiadiazole![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PC61BM = 1:1, anchored terminally via esterification reaction) and multi-adduct, 10-I (benzothiadiazole:PC61BM = n:1, where n ≥ 1, attached directly to the fullerene core via the Prato reaction) using different synthetic strategies. A broadening of the UV-visible spectra of the modified fullerene derivative with strong absorption from 350 to 500 nm and at low wavelengths is observed as compared to PC61BM. A suitable bandgap, good electronic conductivity, and appreciable solubility in solvents suggest their utility in optoelectronic devices. The mono-adduct 7 showed two-order higher electron mobility as compared to bis-adduct 10-I due to retention of extended conjugation in fullerene, as in the case of PC61BM. Experimentally determined optical properties and energy levels of the fullerene adducts were found to be in good agreement and supported by theoretical calculations. The presence of BTF2 affects the ground state dipole moments as well as the absorption strengths, most noticeable in the case of two attached BTF2 moieties. The HOMO and LUMO levels are found to be localized on the fullerene cage with the extension of the HOMO to the BTF2 unit more and the same is noticed in ground state dipole moment in the side-chain functionalized structure. Such structural arrangement provides easy charge transfer between acceptor and donor units to allow a concomitant effect of favorable optoelectronic properties, energy levels of the frontier orbitals, effective exciton dissociation, and charge transport which may reduce processing complexity to advance single material-based future optoelectronic devices.
:PC61BM = 1:1, anchored terminally via esterification reaction) and multi-adduct, 10-I (benzothiadiazole:PC61BM = n:1, where n ≥ 1, attached directly to the fullerene core via the Prato reaction) using different synthetic strategies. A broadening of the UV-visible spectra of the modified fullerene derivative with strong absorption from 350 to 500 nm and at low wavelengths is observed as compared to PC61BM. A suitable bandgap, good electronic conductivity, and appreciable solubility in solvents suggest their utility in optoelectronic devices. The mono-adduct 7 showed two-order higher electron mobility as compared to bis-adduct 10-I due to retention of extended conjugation in fullerene, as in the case of PC61BM. Experimentally determined optical properties and energy levels of the fullerene adducts were found to be in good agreement and supported by theoretical calculations. The presence of BTF2 affects the ground state dipole moments as well as the absorption strengths, most noticeable in the case of two attached BTF2 moieties. The HOMO and LUMO levels are found to be localized on the fullerene cage with the extension of the HOMO to the BTF2 unit more and the same is noticed in ground state dipole moment in the side-chain functionalized structure. Such structural arrangement provides easy charge transfer between acceptor and donor units to allow a concomitant effect of favorable optoelectronic properties, energy levels of the frontier orbitals, effective exciton dissociation, and charge transport which may reduce processing complexity to advance single material-based future optoelectronic devices.
Introduction
With the world moving towards sustainable energy organic solar cells (OSCs) have become very popular and a promising method of harvesting solar energy in an affordable, scalable and efficient manner.1 OSCs utilize either polymer or small molecule organic semiconductors owing to their unique advantages, such as ease of device fabrication by efficient solution-based roll-to-roll printing, light-weight, and mechanical flexibility. Moreover, designing materials at the molecular level allows bandgap tunability, and high optical absorptivity which is desired to achieve high power conversion efficiency (PCE) of the devices. One approach to improve PCE involves optimization of the energy gap between the highest occupied molecular orbital (HOMO) of the donor and the lowest unoccupied molecular orbital (LUMO) of the acceptor to allow maximum energy conversion and to affect open-circuit voltage (Voc). The second approach demands the exploration of strategies to maximize the intermolecular interactions between the donor and acceptor units.1c,2Bulk heterojunction (BHJ) photovoltaic devices revealed high PCE and are vastly based on the blending of a semiconducting polymer/organic small molecule donor with a soluble fullerene derivative as an acceptor, to improve the charge-carrier mobility and exciton separation.3 The fullerene derivatives constitute so far as one of the most explored class of acceptors for BHJ. The parent fullerenes C60/70 rings are tethered usually with organic functionalities to improve the solubility and processability. Such acceptors have vastly improved the device efficacy due to their advantageous properties such as (i) the strong electron affinity to accept and transport electrons in three dimensions, (ii) high electron mobilities, (iii) multiple reversible electrochemical reductions, and (iv) the ability to aggregate in bulk heterojunctions to form both pure and mixed domains of the appropriate length scale to enable charge separation.4 Among the fullerene derivatives, [6]-phenyl-C61 butyric acid methyl ester (PC61BM) acceptor is widely used due to its good solubility, well structurally characterized, further structurally tailored and revealed appreciable compatibility with many conjugated donors.5 Padinger et al.6 reported BHJ device based on a blend of poly(3-hexylthiophene) donor and PC61BM, P3HT:PC61BM, with a PCE of >3.5%, and it has been used as the reference cell in this field.1c,3b,7 Fullerene derivatives have been widely used in optoelectronic device applications. Recently, Karunakaran et al. reported D–A–D type molecules with PCBM as acceptor material for bulk heterojunction-based optoelectronic devices.8 Anthopoulos and others demonstrated PC61BM based ultraviolet phototransistors with balanced electron and hole transport characteristics.9 Field-effect transistors were also reported by Dong et al. using fullerene derivatives having ethylene glycol side chains with different lengths.10 Self-assembled monolayer based on fullerene were also investigated by Lee et al.11 that improved electron transfer at the organic–ZnO interface, without impacting the morphology of active layer. Higher fullerene analogues, like PC71BM, have also been studied extensively as acceptors in OSCs, and recently, Hu and co-workers reported a record 15.3% PCE for a PC71BM based-all small molecule OSCs.12 Functionalization of fullerene raises the LUMO level13 and thus reduces electrochemical losses and simultaneous increase in Voc.14 Multi-adducts of fullerene, more specifically, the bis-adducts have shown better performance than their mono-adducts.15 In general, the higher-adducts than bis-adducts of fullerene show a lower light-harvesting to power conversion performance due to decrease in electron mobility, the occurrence of electronic traps, and a minimal effect on LUMO level.15b,16 Recently multiple functionalized fullerene adducts gained interest for advanced materials and biological applications and thus interesting for exploration.17
Another electron-deficient unit of growing interest is 2,1,3-benzothiadiazole (BT), which exhibits low-lying energy levels that make it an important acceptor building block in materials design to modulate electron transport properties.18 An ease to introduce various functionalities in the benzothiadiazole unit offers rich structural designing flexibility to tune the molecule properties. The most acceptable and preferable methods involve the introduction of electron-withdrawing groups on the aryl ring of BT that allows structural modification using a wide range of reactions.19 Fluorination of BT ring was found to improve the solar cell performance as it lowers the energy of the HOMO level. Moreover, fluorine functionality improves interactions between the donor and acceptor units, thereby enhancing the diffusive properties of the compound.20 Another way of altering electronic properties of BT derivative is through the fusion of the benzothiadiazole unit with widely explored donor units, like thiophene, which has been reported to generate structures with enhanced light-harvesting efficiency.21 Molecules involving fluorobenzothiadiazole (BTF2) and thiophene (Th) have been studied and the proximity of the two moieties allows tunability of electronic properties. The F⋯S interaction between the fluorobenzothiadiazole and thiophene induces planarity and such conformation of the molecule in the solid-state favors intermolecular stacking to form an array of units and thus assists better charge transportation.22 Such intra- and intermolecular interactions (e.g., F⋯H, F⋯S, and F⋯π), favor the crystalline morphology at a nanoscale.23
Use of variation in the arrangement of the acceptor (A)–donor (D) units became popular in light-harvesting materials as they allow better intramolecular charge transfer from the acceptor to the donor units, leading to improved performance of the device.24 Alternating arrangement of units as A–D–A was first introduced by Schulze et al.25 and found that the acceptor/donor interface assists the separation of excitons as electrons in the acceptor and holes in the donor. More recently, Chen et al.26 reported that A–D–A type acceptors exhibit outstanding performance with a PCE of 17.3% in tandem geometry. Such structural arrangements of the acceptor/donor units result in enhanced charge transfer with a simultaneous broadening of the optical absorption range.27
In the current work, we aim to modify the PC61BM acceptor molecule with a covalently linked BTF2-Th unit to obtain an A–D–A type arrangement. Three aspects are considered in the tuning of the acceptor molecule: (i) lowering of bandgap, (ii) efficient inter- and intra-molecular electron transport (iii) to improve optical absorptivity of fullerenes. As a design strategy to attain the former objective, fluorine atoms are incorporated in the acceptor molecule while for the latter objective, benzothiadiazole moiety was covalently tethered to PC61BM to allow directionality for efficient electron transport and improve light absorbance characteristics. Fig. 1 shows the structures of two new covalently bonded benzothiadiazole-based fullerene molecules, mono-adduct (benzothiadiazole:PC61BM = 1:1) and multi-adduct (benzothiadiazole:PC61BM = n:1, where n ≥ 1) which were synthesized and characterized. The thiophene π-bridge (D1, Th) linked difluoro-2,1,3-benzothiadiazole (A1, BTF2) unit is anchored to the fullerene (A2) moiety via a phenyl linker (D2) to design acceptor molecules in D1–A1–D2–A2 configuration. The fullerene moiety was introduced using PC61BM as it is among the most widely used electron acceptor in BHJ devices and offers good solvent solubility and electronic properties.28 The terminal thiophene donor (D1) unit contained a C6-long alkyl chain which not only assists solvent processability of the final molecule but is also known to affect inter- and intramolecular interactions.23a The structural arrangement of the two-hybrid π-bridge units sandwiching A1 unit may favor structural planarity to affect molecular interactions. Furthermore, variation in electron-donating power as provided by the difference in aromaticity between BTF2 and fullerene ring may provide electron conduction gradient to ease the movement of electrons. Backbone modulation of D1–A1–D2 is achieved by altering the mode of its linkage to the fullerene unit, one at the distal end (Acceptor 7) and the other attached to the core (Acceptor 10). The energy levels of the molecules are evaluated theoretically and experimentally.
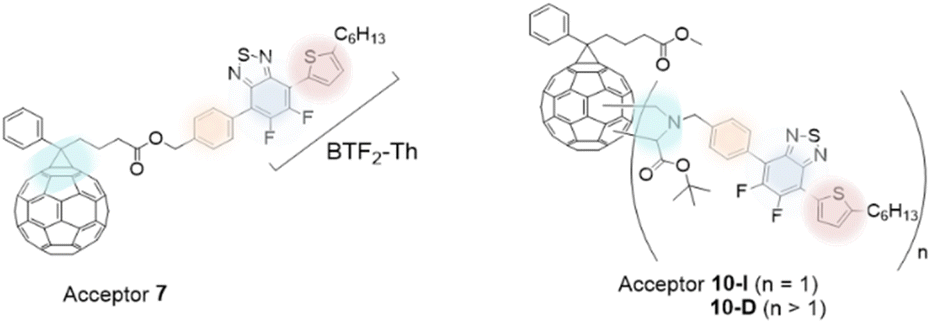 | ||
| Fig. 1 The mono- and multi-adduct acceptors (7: PC61BTF2, 10-I: PC61BM-BTF2 and 10-D: PC61BM-nBTF2). | ||
Results and discussion
Synthesis and characterization of benzothiadiazole derivatives
A multi-step synthesis was adopted to obtain the thiophene-benzothiadiazole-fullerene compounds (7 and 10) as shown in Scheme 1. The synthesis of compound 5 is shown in Scheme S1 (ESI†). The nitro group of commercially available 4,5-difluoro-2-nitroaniline was reduced to an amino group using in situ generated hydrogen by reaction of HCOONH4 in the presence of Pd/C to form compound 2. Intramolecular cyclization of 2 with SOCl2 yielded difluorobenzothiadiazole, 3. Bromination of 3 with bromine and iron powder afforded dibromo-difluorobenzothiadiazole, 4.29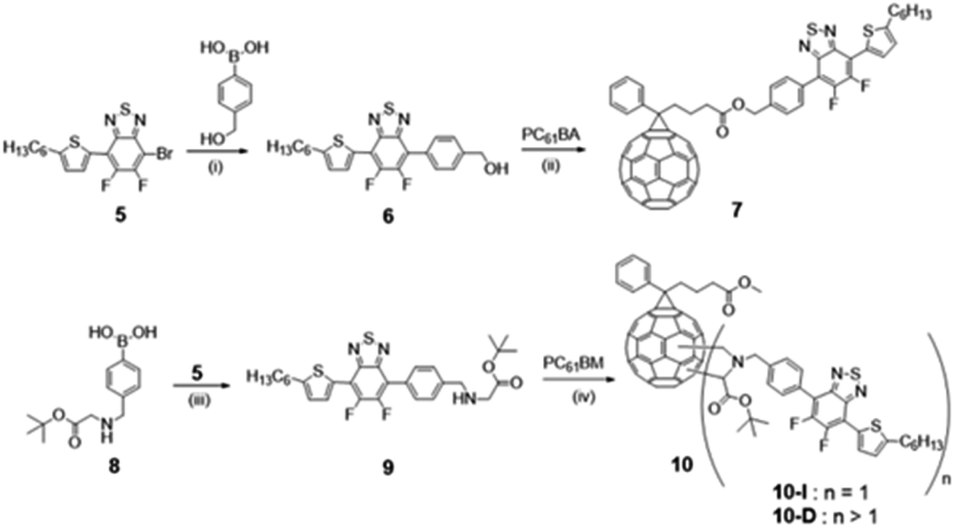 | ||
| Scheme 1 Synthesis of 7 PC61BTF2, 10 PC61BM-nBTF2 where n ≥ 1, reagents and conditions: (i) Pd(PPh3)4, Na2CO3, THF, 66 °C, 18 h, 47% (ii) EDC·HCl, DMAP, ODCB, 0 °C to rt, 2 d, 69% (iii) Pd(PPh3)4, K2CO3, THF, 66 °C, 18 h, 83% (iv) CH2O, PhMe, 110 °C, 18 h, 42% (for 10-I). | ||
Compound 4 underwent Stille coupling reaction with (5-hexylthiophen-2-yl)trimethylstannane, 1, in the presence of Pd2(dba)3 pre-catalyst and tri(o-tolyl)phosphine ligand to generate 5 as a monosubstituted product.30 Due to the formation of unwanted disubstituted analogue, 5 is formed in a moderate yield.31 Structures of the compounds 1–5, synthetic and characterization details (Fig. S1–S9†) are provided in the ESI.† Suzuki coupling reaction between 5 and 4-(hydroxymethyl)phenylboronic acid and 4-aminomethylphenylboronic acid, 8, using Pd(PPh3)4 as catalyst and sodium carbonate as base produced compounds 6 and 9, respectively (Scheme 1).32 The structure of PC61BM was modified using two different pathways, one as on the side chain other on to the fullerene ring. In the former case, benzothiadiazole derivative 6 was attached to the PC61BM molecule via the ester group, with the replacement of methyl group in PC61BM with the BTF2 to form fullerene adduct 7, PC61BTF2. Deprotection of the ester group was first carried out to form PC61BA (synthetic and characterization details are provided in the ESI†), followed by a Steglich esterification reaction between 6 and PC61BA in the presence of EDC·HCl and DMAP to form 7 as a brown solid in a yield 69%.33
To understand the structure–activity relationship i.e., attaching the Ph-BTF2-Th to PC61BM at the fullerene core, in the latter case, a bis-adduct fullerene derivative (compound 10, Scheme 1) was also synthesized. Functionalization of the C60 frame can be achieved by several reactions to form different fullerene derivatives.34 Prato reaction is one of the most widely used reactions to functionalize fullerene. It proceeds via a 1,3-dipolar cycloaddition of the intermediate ylide, formed by the decarboxylation of the iminium salt, onto a [6,6] double bond of the fullerene ring to form fulleropyrrolidines.35 Prato-type reaction between 9 and PC61BM, using paraformaldehyde yielded compound 10. Different bis-adduct isomers, as well as multi-adduct fullerenes, were obtained. From the purification by column chromatography, two fractions were collected, 10-D and 10-I, the latter being the major fraction. Fraction 10-I is most likely the bis-adduct regio-isomers (abbreviated as PC61BM-BTF2) and 10-D (abbreviated as PC61BM-nBTF2, where n > 1), the multi-adduct derivatives. The regio-isomers involved the addition of BTF2 at the fullerene core. The bis-adduct isomers were not separated since it has been reported that the LUMO energy levels of the different isomers are comparable enough that their photovoltaic properties are not affected.15a,b
The formation of the different compounds was followed by 1H, 13C, and 19F NMR. The synthesis of compound 5 was confirmed by comparing the 1H NMR spectrum of product 5 and that of 1 (see Fig. S1 and S8†). In CDCl3, a significant downfield shift of the characteristic thiophene “a” proton from 7.04 ppm (in compound 1) to 8.07 ppm (in compound 5) is observed due to the replacement of the trimethylstannane moiety by fluorinated benzothiadiazole group. The highly electronegative fluorine atoms in the molecule led to the deshielding of the “a” proton and thus account for a downfield shift of the corresponding signals. From Fig. S10,† 1H NMR spectrum of the desired Suzuki product 6, obtained by coupling reaction of 5 with (hydroxymethyl)phenylboronic acid, is evident from the formation of new signals in the aromatic region at 7.80 ppm (“b” proton) and 7.57 ppm (“c” proton) and the benzylic (Ph-CH2) “e” protons at 4.80 ppm. The successful esterification reaction between the alcohol, 6 and the acid, PC61BA (NMR is provided in Fig. S12†), and formation of modified PC61BM with the replacement of methyl ester with benzyl ester i.e., fullerene linked with Ph-BTF2-Th (mono-adduct 7) is also suggested by the prominent chemical shift of benzylic (Ph-CH2) protons from 4.80 ppm (“*” proton) to 5.22 ppm (“#” proton), as shown in Fig. 2. This resonance towards the downfield value is associated with the deshielding effect induced by the adjacent ester group. Detailed structure characterization to confirm the formation of 7 is provided in Fig. S13.†
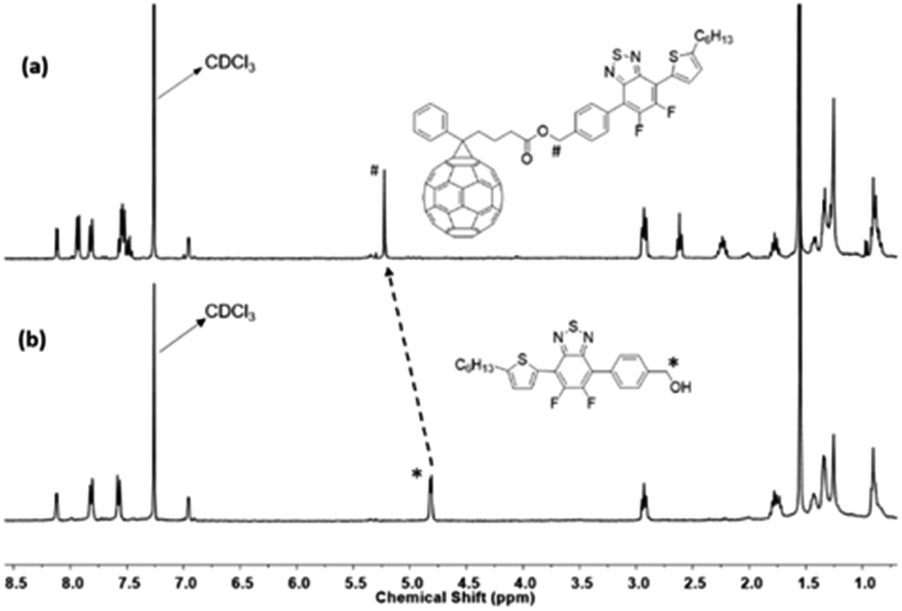 | ||
| Fig. 2 1H NMR spectra of (a) 7 and (b) 6 in CDCl3. | ||
Likewise, the formation of bis- and multi-adducts of PC61BM with Ph-BTF2-Th was studied with NMR. The structures of precursor compound 8–9 were confirmed by NMR (Fig. S14–S17†). 1H NMR confirms the formation of bis- and multi-adducts of PC61BM. Fig. S18† shows the stacked 1H NMR spectra of PC61BM, 9, 10-D, and 10-I and it can be seen that the aromatic region changes from well-defined signals in PC61BM and 9, to broad multiplets in 10-D and 10-I. Similarly, the aliphatic region shows additional signals, which may correspond to the splitting of signals due to the fullerene ring, as reported in the literature.36
Further structural confirmation of successful synthesis of the compound was confirmed by 13C NMR spectroscopy. The 13C NMR spectrum of the Stille coupling product 5, Fig. S9,† showed a shift in the carbon signal of C–Br after replacement of bromide with thiophene, from 99.39 ppm (Fig. S7†) in the dibromo compound 4 to 96.34 ppm in 5. This carbon signal bound to bromine i.e., C–Br at 96.34 ppm also confirms the Suzuki coupling reaction product, 6, as indicated by the disappearance of resonance at 96.34 ppm in the 13C NMR spectrum of 6 (Fig. S11†) due to bromine substitution. 13C NMR spectrum in Fig. 3 supports the formation of the benzyl ester of fullerene linked with Ph-BTF2-Th to form PC61BTF2, 7. A new signal at 172.97 ppm corresponds to the carbonyl carbon as well as several other resonances in the region 125 and 148 ppm due to the fullerene ring were observed. Additionally, a shift in the benzylCH2 signal from 65.21 ppm in alcohol, 6 (Fig. S11†) to 66.08 ppm in the ester product, 7 was recorded (Fig. 3). In the case of the bis- and multi-adducts, the 13C NMR spectra show multiple complex signals, which remained inconclusive to confirm the structures of products, 10-I, and 10-D, as shown in Fig. S19 and S20,† respectively.
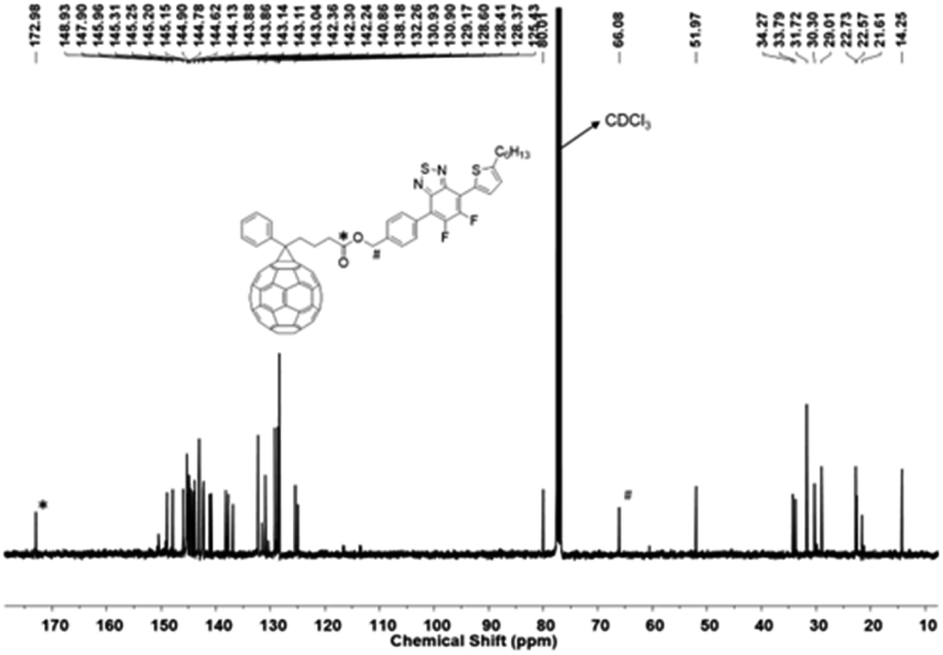 | ||
| Fig. 3 13C NMR spectrum of 7 in CDCl3. | ||
19F NMR of the fluorine-containing compounds was recorded and each reaction undergone by the compounds was reflected by the shifts of the corresponding F signals. Fig. 4 shows the stacked 19F NMR spectra of 4, 5, 6, and 7 (PC61BTF2). Transformation of symmetrical structure dibromo-difluorobenzothiadiazole, 4 to asymmetrical structure bromothiophenyl-linked benzothiadiazole, 5 is confirmed by the appearance of initial one F signal at −118.59 ppm transformed to two doublets (−120.37 and −127.72 ppm). The substitution of the second bromine atom in 5 by the benzyl alcohol group is apparent by the shift in F resonance value from −127.72 and −120.36 ppm in 5 to −128.89 and −133.36 ppm in 6, respectively. The esterification reaction of 6 did not significantly affect the magnetic environment of fluorine in 7. As a result, no major change in the 19F NMR was noticed, with the appearance of two doublets at a similar position (−128.88 and −133.12 ppm) in the PC61BTF2 structure.
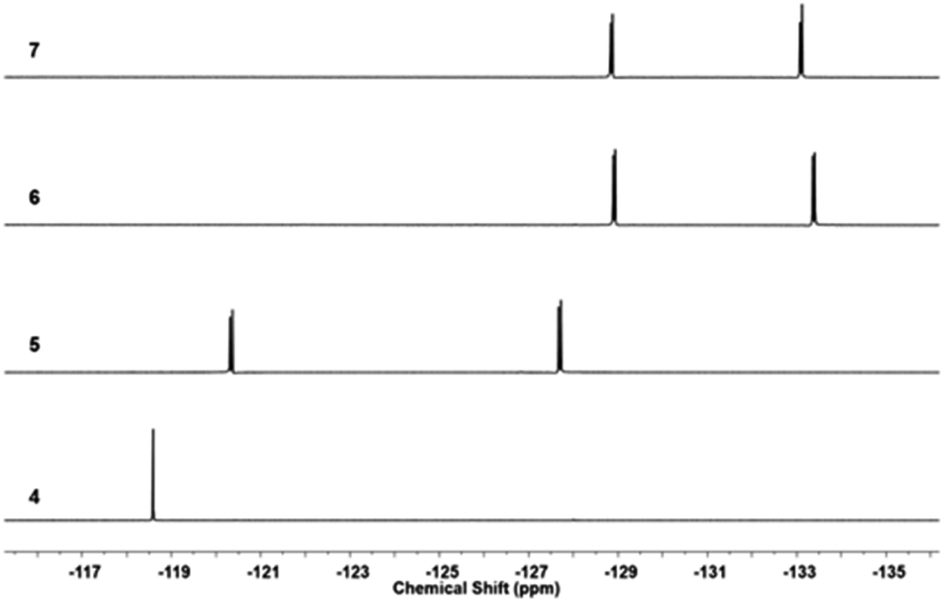 | ||
| Fig. 4 Stacked 19F NMR spectra of 4, 5, 6, and 7 in CDCl3. | ||
19F NMR was also helpful to confirm the successful linking of compound 9, Ph-BTF2-Th anchoring on to PC61BM (Fig. 5) to form 10. Similar to the mono-adduct fullerene, 7, the 19F NMR spectra of the multi-adduct derivatives only showed a slight shift in the fluorine signals when comparing 9 to the multi-adduct products, 10-I and 10-D. The signal at −128 ppm appeared, as a doublet in all cases, whereas the doublet at −133 ppm in 9 undergoes an upfield shift and now appears as broad peaks in 10-I and 10-D 19F NMR spectra. This change may account for the formation of a mixture of isomers during the Prato reaction and was recently reported via light-controlled regioselective synthesis of C60 adducts.37 In the case of 10-D, an additional signal at −132 ppm is noticed, suggesting a different magnetic environment of the fluorine atoms. This supports the addition of multiple benzothiadiazole units onto the fullerene ring to form multiple adducts, PC61BM-nBTF2.
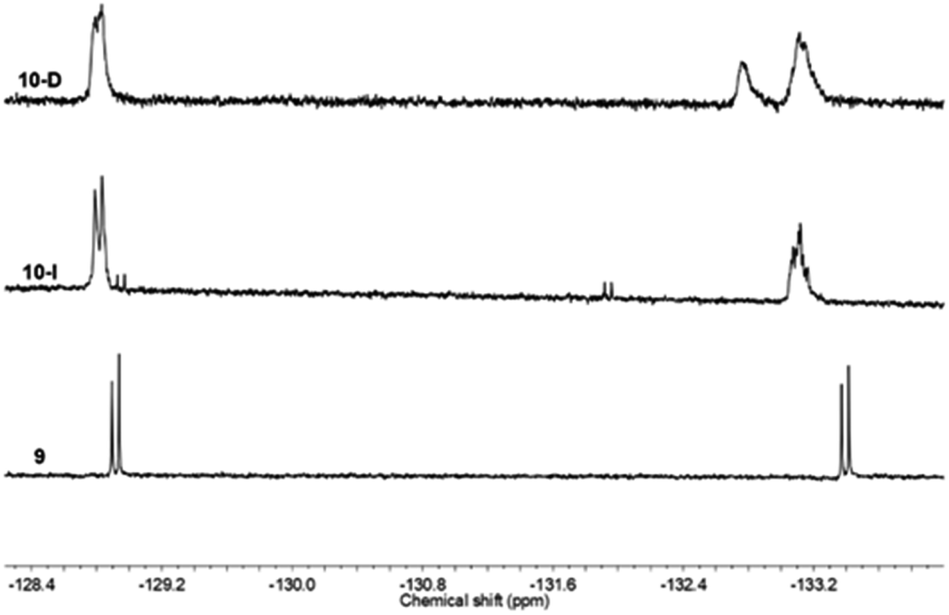 | ||
| Fig. 5 Stacked 19F NMR spectra of 9, 10-D, and 10-I in CDCl3. | ||
FTIR spectroscopy is additionally used to probe the successful esterification reaction between PC61BM and 6. Fig. 6 shows the stacked FTIR spectra, with 7 (PC61BTF2) having characteristic bands of both reactants PC61BA and 6. The O–H stretching peak at 3342 cm−1 in 6 is absent in the spectrum of 7, indicating successful co-reaction. The weak broad O–H stretching in PC61BA at 3270 cm−1 also disappears in 7. Moreover, the carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching undergoes a shift from 1700 cm−1 in PC61BA to 1734 cm−1 in 7 after covalently linking to the benzothiadiazole unit.
O stretching undergoes a shift from 1700 cm−1 in PC61BA to 1734 cm−1 in 7 after covalently linking to the benzothiadiazole unit.
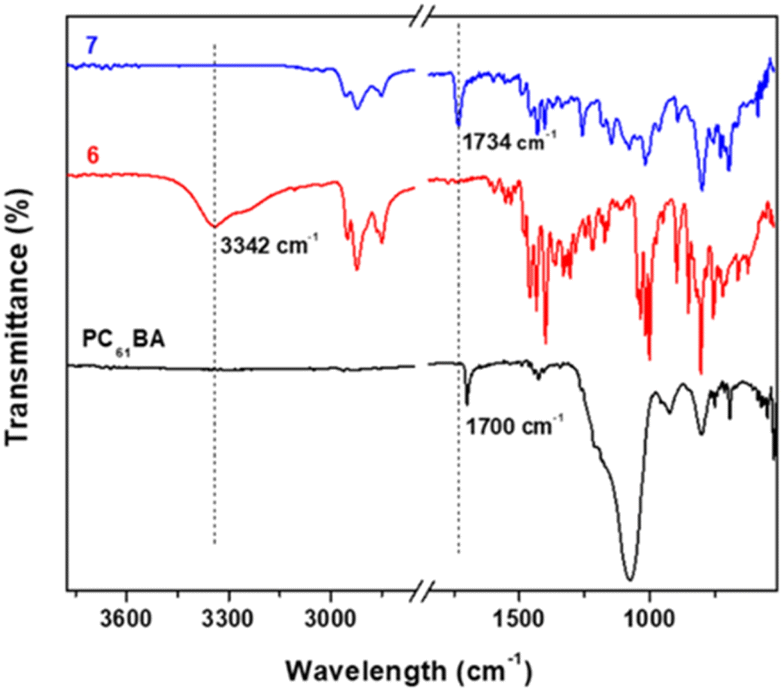 | ||
| Fig. 6 Stacked FTIR spectra of 7, 6, and PC61BA. | ||
Optical and electrochemical properties
UV-visible absorption spectra of PC61BM (instead of PC61BA due to its poor solubility in ODCB), benzothiadiazole alcohol 6, and their corresponding ester, 7 PC61BTF2, in ODCB are represented in Fig. 7A. From the UV/vis spectra, fullerene benzothiadiazole mono-adduct 7 shows the combined absorptions of the alcohol, 6, and PC61BM. At 300 nm, all three compounds show absorption peaks corresponding to the localized π → π* transition in the phenyl rings, similar to that reported in the literature.38 The sharp band at 332 nm in PC61BM, which is associated with the π → π* transition in the fullerene ring, undergoes a slight hypsochromic shift of 6 nm in 7 (λmax = 326 nm). The absorption observed at 420 nm in the spectrum of 6 corresponds to the characteristic intramolecular charge transfer (ICT) transition between electron acceptor (benzothiadiazole moiety) and electron donor (aromatic group) units in 6.38,39 The existence of ICT transition is also present in PC61BTF2 7, along with an additional absorption observed in the 375 to 480 nm region due to the fullerene ring. In the spectrum of 7, a very weak and broad absorption band in the range 445 to 635 nm is also noticed, which is assigned to the first-order forbidden transitions characteristic of fullerenes.40 The UV-visible spectra of compounds 9, 10-D, and 10-I were also recorded in ODCB, as shown in Fig. 7B (Fig. S21,† as recorded). Due to their very close structural similarity, compounds 6 and 9 have similar UV-visible spectra, with absorptions centered at 300 and 420 nm. Along with the retention of absorption features of 9 in PC61BM-nBTF2, the presence of fullerene in 10-D and 10-I is supported by broadening of the bands from 315 to 375 nm due to π → π* transition. Likewise in the case of PC61BTF2 7, a lower intensity absorption at 420 nm is additionally observed in both 10-D and 10-I, which extends to up to ∼600 nm. The optical band gap of monoadduct 7 and bisadduct 10-I was calculated by Tauc plot using the solution data and found to be 2.58 and 2.52 eV respectively (Fig. 8).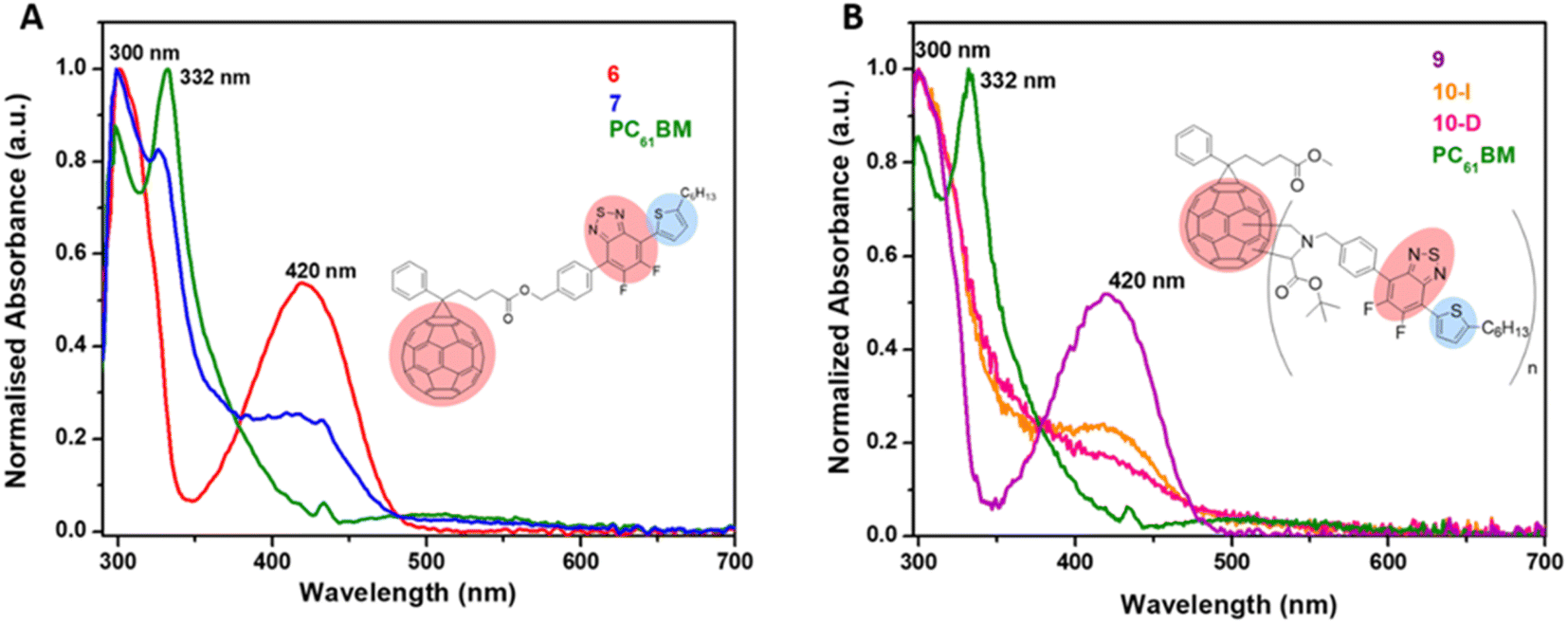 | ||
| Fig. 7 Normalized UV-visible spectra of (A) 6, 7, and PC61BM and (B) 9, 10-I, 10-D, and PC61BM in ODCB (concentrations of 10−5 M). | ||
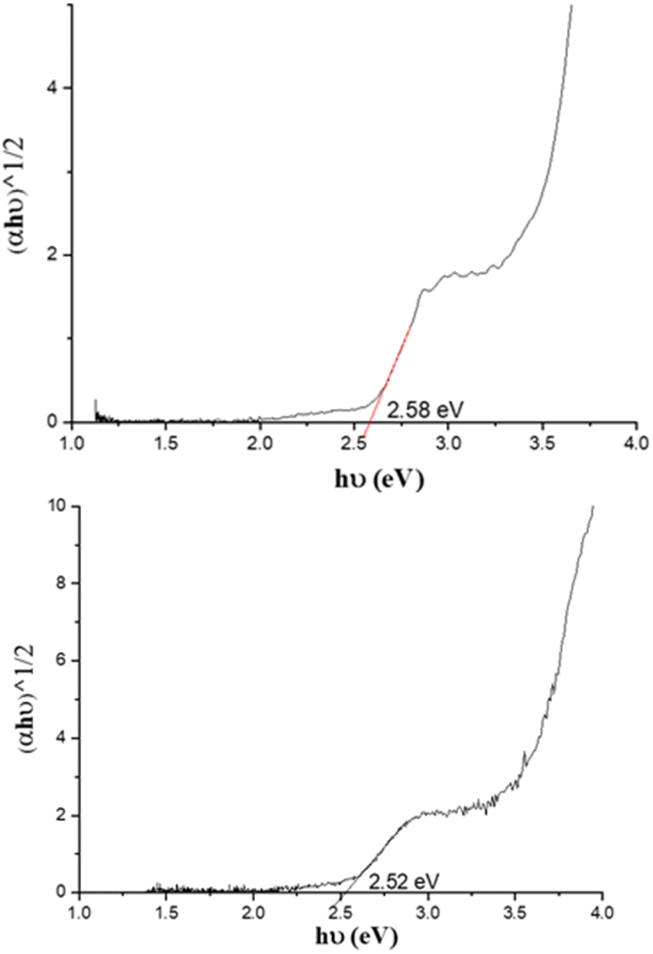 | ||
| Fig. 8 Optical band gap calculated by Tauc plot of monoadduct 7 (top) and bisadduct 10-I (bottom). | ||
To further probe the effect of BTF2 substitution on fullerene at the core (10-D and 10-I) or side chain (7), electrochemical properties of the fullerene derivatives were performed (Fig. 9A and B). Cyclic voltammetry was employed to determine the electrochemical energy gap. The potentials were calibrated by a ferrocene/ferrocenium (Fc/Fc+) redox couple under the assumption that the Fc/Fc+ energy level was −4.8 eV below the vacuum level. The estimated HOMO and LUMO energy levels from the onset oxidation and reduction potentials are determined to study the effect of covalent modification of the fullerene derivatives as compared to PC61BM. Fig. 9 shows a total of four reversible/quasi-reversible reduction potential peaks for both compounds 7 and 10-I. In the negative region, the minima show the reduction events in which electrons are being added to the LUMO of the molecule whereas the maxima in the positive region represent oxidation in which electrons are being removed.
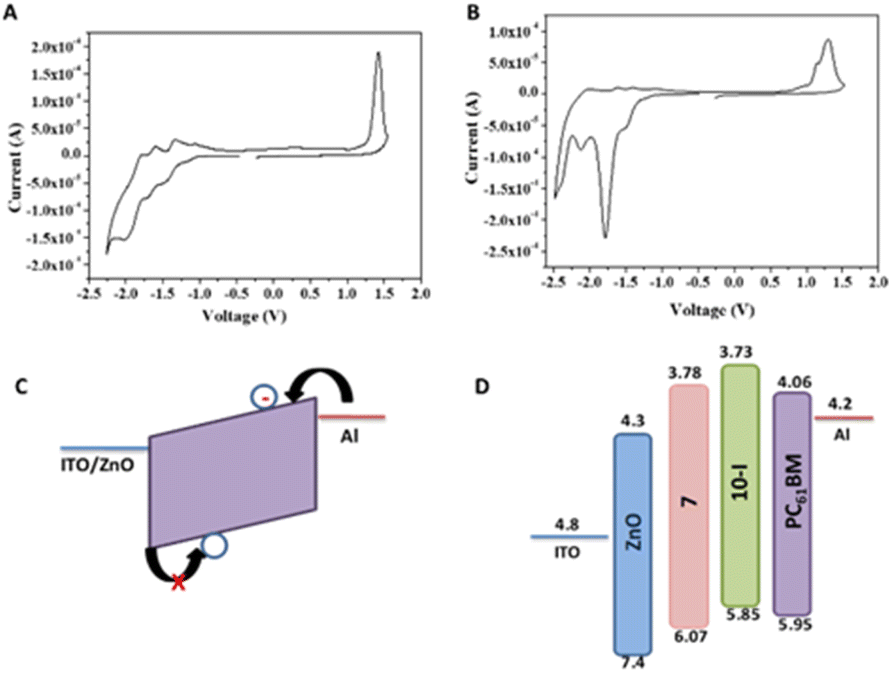 | ||
| Fig. 9 Cyclic voltammetry curves of (A) 7 and (B) 10-I (v/s Fc/Fc+) with 0.1 M n-Bu4NPF6 as a supporting electrolyte and scan rate 100 mV s−1. (C) Schematic representation of fabricated electron-only device and (D) energy level diagram with HOMO and LUMO values. | ||
Details of electrochemical data are summarized in Table 1. Monomer 7 reduces at a lower potential than bis adduct 10-I. Covalent functionalization of PC61BM raises the LUMO levels of both the adducts, however, compound 10-I shows the uplifted LUMO as well HOMO levels compared to 7.41 The uplifted LUMO level of molecule 10-I compared to molecule 7 suggests better electron-accepting properties of the latter which may be prudential for electron mobility. The HOMO energy level of 7 is found to be deeper than both 10-I and PC61BM as can be seen in Table 1. Both optical and electrochemical bandgap values follow the same trend PC61BM < PC61BM-BTF2 < PC61BTF2 range. The observed discrepancy in the calculated value of bandgap from two different techniques, the electrochemical vs. optical measurements, is accounted to the variation in key aspects of the physical process viz. adiabatic or vertical, solvation, exciton binding energy, etc.42
| Entry | λabsmax (nm) | Redonseta (V) | Red−1 (V) | Red−2 (V) | Red−3 (V) | Red−4 (V) | LUMO (eV) | Oxonseta (V) | Ox−1 (V) | HOMO (eV) | Eg(cv) (eV) | Eg(opt)b (eV) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Potentials are measured relative to a Fc/Fc+ redox couple.b Optical bandgap was estimated from Tauc plot; sh stands for the shoulder. | ||||||||||||
| PC61BM | 332 | −0.74 | −0.91 | −1.17 | −1.40 | −1.95 | −4.06 | 1.15 | — | −5.95 | 1.89 | 2.12 |
| 7 | 326, 385 sh | −1.02 | −1.14 | −1.45 | −1.70 | −2.01 | −3.78 | 1.27 | — | −6.07 | 2.29 | 2.58 |
| 10-I | 326, 385 sh | −1.07 | −1.50 | −1.78 | −2.12 | −2.40 | −3.73 | 1.05 | 1.14 | −5.85 | 2.12 | 2.52 |
Charge transport properties
The electron-only devices based on 7 and 10-I were fabricated for studying the charge transport properties using the space-charge-limited current (SCLC) method.43 To investigate detailed electron mobility properties, we fabricated electron only device with configuration: ITO/ZnO/material/Al. SCLC is observed in electron/hole only devices in which using right contact of electrodes either only electrons or hole can be injected. The current so obtained is only space charge limited at higher voltage due to ohmic contact and trap free transport.ITO was deposited on a simple glass slide to form an ITO substrate. On to ITO substrate, the ZnO layer was deposited to block hole injection from ITO. Fullerene adduct (7/10-I) film was spin-coated using a 40 mg mL−1 solution in chlorobenzene. The coated substrate was annealed at ∼120 °C for 15 min. Finally, the Al layer was deposited above the fullerene derivative layer. The n-type semiconductor properties were measured using a Keithley 2420 power source. The electron mobility of the products (7/10-I) was calculated using the Mott–Gurney eqn (1).
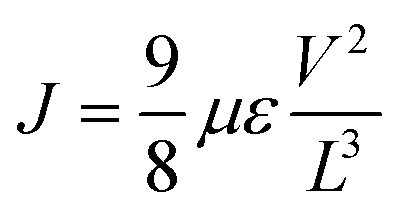 | (1) |
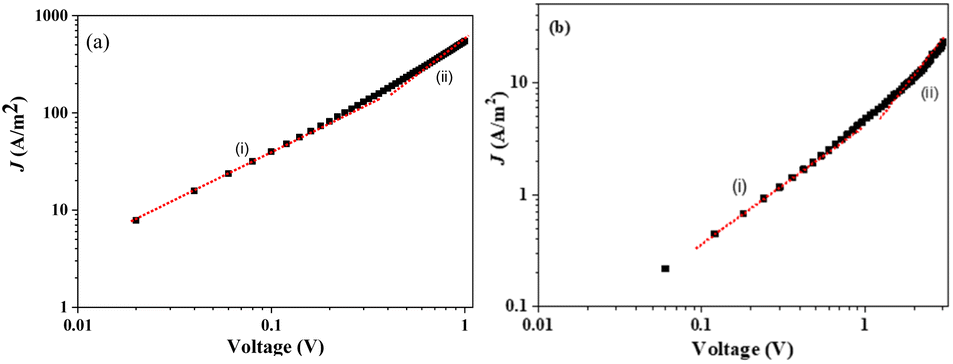 | ||
| Fig. 10 J–V plots for the electron-only devices based on adduct (a) 7 and (b) 10-I. | ||
Theoretical calculations
To probe the effect of functionalization with BTF2 on energy properties theoretical calculations were performed for geometry PC61BM, PC61BTF2 7, PC61BM-nBTF2 10-I (n = 1), and 10-D (n = 2, and 3, shown in the ESI Fig. S22 and S23†). Structures were constructed for PC61BM with phenyl-butyric-acid-methyl groups latched on neighboring carbon atoms. Attempt for latching on next neighboring C60 carbon atoms was also examined for completeness but was not showing an energetically favorable configuration. For adduct 10, functionalization with one (n = 1) BTF2 group was performed on two distinct PC61BM locations, specifically, (a) with the BTF2 group positioned at a para position to phenyl-butyric acid methyl ester (PBM) moiety, and (b) with BTF2 located at a (near meta) position that was found suitable for the later construction of the doubly functionalized PC61BM, to accommodate comparisons. For functionalization with n = 2 BTF2 groups, two near meta locations (analogous to the aforementioned configuration of the n = 1 case) were chosen so that the distance from PBM is the same as the distance between them, specifically, with latching carbon atoms at five hops (PC61BM C atoms) from each other. Finally, for functionalization with n = 3 BTF2 groups, the situation was complicated due to the spatial extent of the BTF2 groups, so latching locations were chosen in a manner to avoid steric effects.The isosurfaces of the frontier orbitals for the PC61BM (reference) and PC61BTF2 structures are shown in Fig. 11a–d.
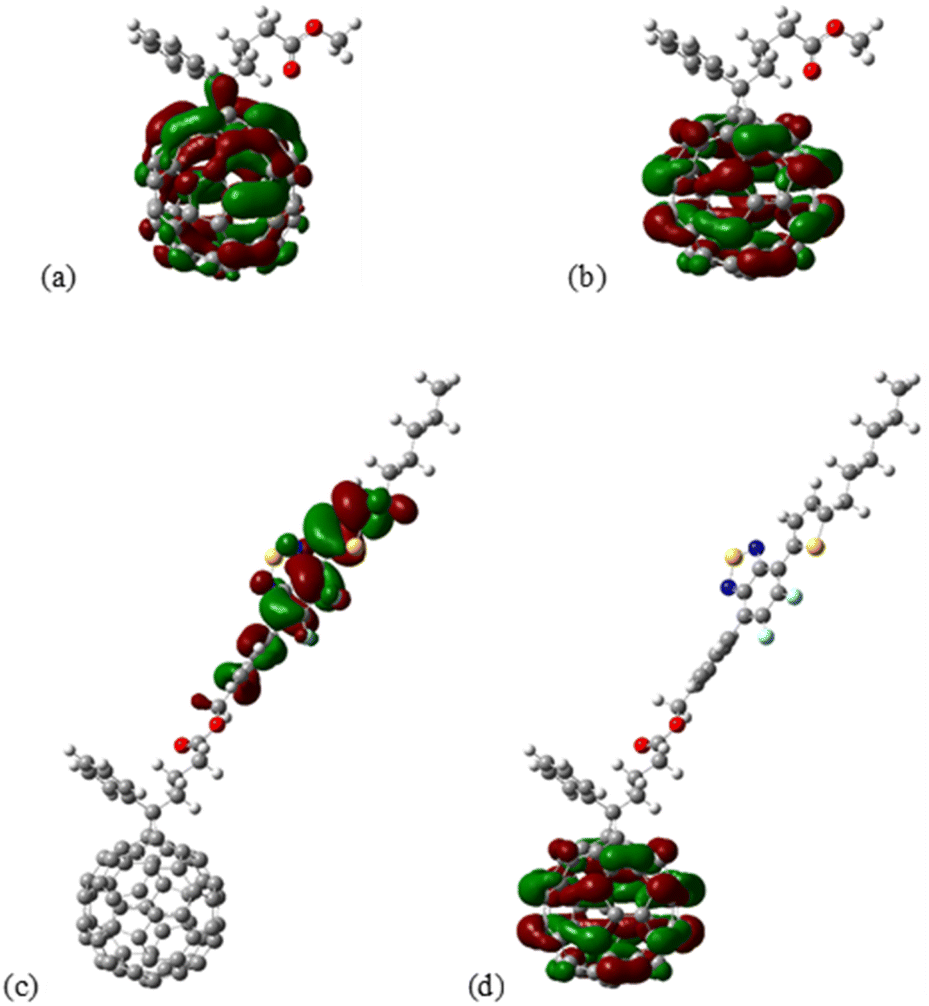 | ||
| Fig. 11 Isosurface of the (a) HOMO and (b) LUMO of PC61BM, as well as the (c) HOMO and (d) LUMO of 7 PC61BTF2 structure. Computed at the M06/def2-SVP level of theory. | ||
From the isosurface plots, the HOMO and LUMO are found to be localized in the fullerene core in the case of PC61BM as can be seen in Fig. 11a and b. Interestingly in the case of PC61-BTF2, the HOMO was significantly localized over the difluorobenzothiadiazole (BTF2) and LUMO extended over the fullerene structure than over the thiadiazole moieties.
The isosurfaces of the frontier orbitals for the three singly functionalized PC61BM structures with BTF2 are shown in Fig. S22a–f.† The HOMO and LUMO of all three structures are localized on the fullerene cage, whereas for all three cases the HOMO extends over the latching carbon of the BTF2 moiety. From Table 2 a clear dependence can be seen of the HOMO eigen energies to the number of attached BTF2 functional groups. This trend is consistent throughout n = 1–3. For the singly functionalized groups, the HOMO of the para and @conf3 functionalized structures are energetically higher when the functional group is closer to the PBM, however, the LUMO has the same energy for both structures.
| Structure | LUMO (eV) | HOMO (eV) | Eg (eV) | Optical gap (eV) | λmax (nm) | f, oscillator strength | |Dipole moment| μg (D) |
|---|---|---|---|---|---|---|---|
| PC61BM | −3.25 | −6.11 | 2.86 | 1.96 | 631 | 0.002 | 5.50 |
| PC61BTF2, 7 | −3.35 | −6.08 | 2.73 | 1.97 | 628 | 0.002 | 5.54 |
| PC61BM-1xBTF2-para, 10-I | −3.14 | −5.88 | 2.75 | 2.71 | 458 | 0.180 | 2.86 |
| PC61BM-1xBTF2, 10-I | −5.97 | −3.14 | 2.83 | 1.93 | 644 | 0.002 | 3.43 |
| PC61BM-1xBTF2@conf3, 10-I | −3.07 | −5.86 | 2.79 | 1.91 | 649 | 0.002 | 5.70 |
| PC61BM-2xBTF2, 10-D | −2.70 | −5.76 | 3.07 | 2.39 | 519 | 0.011 | 1.28 |
| PC61BM-2xBTF2@conf3, 10-D | −3.38 | −5.42 | 2.04 | 1.17 | 1063 | 0.002 | 2.72 |
| PC61BM-3xBTF2, 10-D | −3.21 | −5.32 | 2.11 | 1.20 | 1030 | 0.005 | 2.17 |
The isosurfaces of the frontier orbitals for the doubly functionalized PC61BM structure with BTF2 are shown in Fig. S23a–d.† The HOMO and LUMO are localized on the fullerene cage. As in the singly functionalized cages, the HOMO extends over the latching carbon of the BTF2 moiety for the PC61BM-2xBTF2. Orientation of BTF2 and latching position have an impact on the HOMO and LUMO delocalization, as can be seen in the case of PC61BM-2xBTF2@conf3 where the LUMO is noticeably displaced towards the BTF2 latching positions while remaining over the fullerene cage.
The structure and the frontier orbitals of the triply functionalized structure PC61BM-3xBTF2 are shown in Fig. S24.† To alleviate any steric hindrances the PC61BM-3xBTF2 was designed with the two functional groups that are closest to the PBM with a different (inversed) orientation compared to the structures with n = 1 and 2. The HOMO of n = 3 structure is higher than the others, affirming the trend that the HOMO energy increases with the number of BTF2 functional groups. Interestingly, the effect of the number of attached BTF2 has a stronger effect on (rising) the HOMO level than the effect that the proximity of the BTF2 group to the PBM group. Structure PC61BM-3xBTF2 offers an example of both of these actions and exhibits the highest HOMO eigen energy. Both HOMO and LUMO values determined theoretically and experimentally for various structures are in an appreciable agreement. The theoretical HOMO value was found to decrease with an increase in substitution in the PC61BM core. The frontier orbital values provided in Table 1 suggest that 7, 10-I and 10-D can be used in conjunction with BHJ donors in cases that offer preferred level alignment.
The UV-visible spectra of all the structures under study are provided in Fig. S25.† In analogy with experimental UV-visible spectra, all the structures (7, 10-I, and 10-D) containing BTF2 exhibit significantly strong UV-visible absorbance theoretically too. In the legends of the plots, the relative scale of the absorbance is given to that of the PC61BM spectrum. The strongest absorbance is noted for the PC61BM-2xBTF2 structure that is functionalized with two BTF2 moieties at equal distances from the PC61BM. Regardless of the absorbance magnitude, the main peak for all the functionalized structures is centered at nearly a similar wavelength range (458–460 nm). Of the three singly functionalized structures, the one with BTF2 closer to PBM moiety has a wider and stronger absorbance profile at low wavelengths, near 350 nm. The widest absorbance is noted for the triply functionalized structure that reaches up to large wavelengths, with a low intensity (oscillator strength) onset at λmax = 1030 nm. To examine the contributions to the excitations from specific groups of the structures we have partitioned the structures into groups, specifically the fullerene cage, PBM, and BTF2 (except the first structure). In the ESI,† we have provided the main contributions to the excitations, the charge density as well as the change in charge density on each group (Table S1a–h†). Excitations that result in considerable charge transfer are denoted as bold. We noted that specifically for PC61BM-1xBTF2-para and PC61BM-2xBTF2 the optical gap listed in Table 2 is the 10th and 3rd computed excitation, respectively, that correspond to the first excitations with non-negligible oscillator strengths.
Incidentally, the transition dipole moments for PC61BM-1xBTF2-para listed in Table S2† is comparatively considerably higher and corresponds to the transition with the first non-negligible oscillator strength that is not the first computed excitation. In the ESI,† we provide detailed values for the transition dipole moments for all excitations on all structures. The dipole moment of the adduct in the ground state (μg) was found to slightly increase with the substitution of BTF2 in place of the methyl group in PC61BM, suggesting a marginal increase in polarity of the structure. It is noteworthy that when BTF2 is attached to the core at a para position to the PBM moiety (10-I) the μg value was found to decrease by nearly 2-fold, suggesting a significant cancellation of dipole moments. This alone suggests that the μg value may increase when BTF2 units are attached to the fullerene core. It turns out that this variation is manifested when 2 vs. 3 BTF2 units are attached to the fullerene core. This confirms that the existence of a BTF2 unit in the direction of PBM has a significant controlling effect on the overall dipole moment of the structure. Such variation in dipole moment may induce polarization which is desired to provide directionality to charge carrier mobility and aid charge separation.
Experimental
Materials
PC61BM was purchased from Lumtec, trimethyltin chloride, triethylamine, tri(dibenzylideneacetone)dipalladium (Pd2(dba)3), tri(o-tolyl) phosphine, tetrakis(triphenylphosphine)palladium(0) (Pd(PPh3)4) from Sigma-Aldrich, 2-hexylthiophene, ammonium formate, Pd/C (10%) and 4-(hydroxymethyl)phenylboronic acid from Alfa Aesar, thionyl chloride, bromine, carbon disulfide, and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC·HCl) from Spectrochem, 4,5-difluoro-2-nitroaniline from TCI, iron powder from SRL, hydrochloric acid (35%) from Merck Millipore, glacial acetic acid from Fisher Scientific and 4-(dimethylamino)pyridine (DMAP), n-butyllithium and sodium sulfate from Chemlabs. All the chemicals were used without further purification. All solvents used were AR grade. Tetrahydrofuran (THF) was distilled over sodium using benzoquinone as an indicator under nitrogen atmosphere. Literature procedures were followed in the preparation of compounds 1,45 2,46 3,47 4,29 5,31 8 (ref. 48) and PC61BA49 (see ESI†).Measurements
Proton (1H), carbon (13C), and fluorine (19F) NMR spectra of the compounds were recorded on a Bruker AV400 NMR spectrometer at a proton frequency of 400 MHz and corresponding carbon, fluorine frequencies at room temperature, using CDCl3 and DMSO-d6 as solvents and tetramethylsilane (TMS) as an internal standard and plotted using MestReNova software. Fourier Transform Infrared (FTIR) spectra were recorded on a Nicolet iS5 spectrometer equipped with the iD5-ATR accessory, in the range of 4000 to 400 cm−1. Mass spectrometry analysis was carried out using Agilent HRMS Q-ToF 6540 Series equipped with ESI mode. UV-visible spectra were collected using a Thermo Scientific Evolution 201 spectrophotometer. Samples were dissolved in o-dichlorobenzene (ODCB) and spectra were recorded from 200 to 800 nm with a resolution of 1 nm, using a 1.0 cm UV quartz cuvette. Three electrode system was used while performing cyclic voltammetry (CV) where, the platinum disc was used as a working electrode, silver-wire, and a platinum-wire as reference and counter electrode respectively in a 0.1 M n-Bu4NPF6(tetra-n-butylammonium hexafluorophosphate) supporting electrolyte in anhydrous acetonitrile solution. CV of the prepared fullerene adducts and PC61BM was performed by drop-casting film (from a solution in dichloromethane) on Pt disc working electrode and dried in vacuum for two hours. Current vs. voltage was measured on an Autolab potentiostat. The energy of the highest unoccupied molecular orbital levels (EHOMO) and the lowest unoccupied molecular orbital levels (ELUMO) were calculated according to the following eqn (2).| E(LUMO or HOMO) = −[4.8 + Eonset(red or ox)] eV | (2) |
All of the structures were examined theoretically via computations based on density functional theory (DFT) and time-dependent density functional theory (TD-DFT).50 The M06 (ref. 50d) functional was used throughout for both ground- and excited-state computations. The results reported are all from computations performed using the Gaussian package.51 The ESI† includes further technical details on the computational methods and methodologies employed.
Synthetic procedures
1H NMR (CDCl3, 400 MHz, ppm): 8.12 (d, 1H, J = 3.72 Hz, C–C–CH), 7.81 (d, 2H, J = 7.56 Hz, ArH), 7.56 (d, 2H, J = 8.12 Hz, ArH), 6.95 (d, 1H, J = 2.80 Hz, CH2–C–CH), 4.81 (d, 2H, J = 5.56 Hz, HO–CH2), 2.93 (t, 2H, J = 15.44 Hz, SC-CH2), 1.80–1.33 (m, 8H, AlkH), 0.90 (t, 3H, J = 13.64 Hz, –CH3).
13C NMR (CDCl3, 100 MHz, ppm): 150.65, 150.57, 150.38, 150.32, 149.18, 149.09, 148.23, 148.03, 141.91, 131.45, 131.36, 130.88, 130.85, 129.78, 127.09, 124.92, 65.21, 31.72, 30.29, 29.01, 22.73, 14.24.
19F NMR (CDCl3, 376 MHz, ppm): −128.93 (d), −133.40 (d). MS (ESI(+)): [(M + H)+]: 445.1 (444.1 calculated). FTIR (cm−1) = 3348 (νO–H).
:1 then pure toluene) as a brown solid. Yield: 50 mg (37%). 1H NMR (CDCl3, 400 MHz, ppm): 8.11 (d, 1H, J = 3.64 Hz, C–C–CH), 7.92 (d, 2H, J = 7.16 Hz, ArH), 7.81 (d, 2H, J = 7.52 Hz, ArH), 7.57–7.47 (m, 5H, ArH), 6.95 (d, 1H, J = 3.36 Hz, CH2–C–CH), 5.22 (s, 2H, HO–CH2), 2.93 (t, 4H, J = 14.56 Hz, SC-CH2), 2.62 (t, 2H, J = 14.68 Hz, OOC-CH2), 2.28–2.20 (m, 2H, AlkH), 1.77 (quintet, 2H, J = 30.04 Hz, AlkH), 1.47–1.33 (m, 4H, AlkH), 0.89 (t, 3H, J = 9.64 Hz, –CH3). 13C NMR (CDCl3, 100 MHz, ppm): 172.98, 150.55, 150.50, 149.21, 148.93, 147.90, 145.96, 145.31, 145.20, 145.15, 144.90, 144.81, 144.78, 144.62, 144.54, 144.13, 143.88, 143.86, 143.20, 143.14, 143.11, 143.04, 142.36, 142.30, 142.24, 141.11, 140.86, 138.18, 138.01, 137.70, 136.86, 132.26, 131.55, 131.46, 130.93, 129.17, 128.60, 128.41, 128.37, 125.43, 124.95, 80.01, 66.08, 51.97, 34.27, 33.79, 31.72, 30.30, 29.01, 22.73, 22.57, 21.61, 14.25. 19F NMR (CDCl3, 376 MHz, ppm): −128.88 (d), −133.12 (d). MS (ESI(+)): [(M + H)+]: 1323.1 (1322.1 calculated). FTIR (cm−1): 1735 (νCO).:CH2Cl2 (1:1) eluent. Product 9 was obtained as a yellow solid. Yield: 55 mg (83%).1H NMR (CDCl3, 400 MHz, ppm): 8.11 (d, 1H, J = 3.72 Hz, C–C–CH), 7.77 (d, 2H, J = 7.16 Hz, ArH), 7.52 (d, 2H, J = 812 Hz, ArH), 6.95 (d, 1H, J = 3.56 Hz, CH2-C-CH), 3.90 (s, 2H, Ar-CH2-NH), 3.37 (s, 2H, NH-CH2-CO), 2.93 (t, 2H, J = 15.32 Hz, SC-CH2), 1.77 (quintet, 2H, J = 30.20 Hz, AlkH), 1.48 (s, 9H, C(CH3)3), 1.36–1.25 (m, 8H, AlkH), 0.90 (t, 3H, J = 14.04 Hz, –CH3).
13C NMR (CDCl3, 100 MHz, ppm): 171.79, 150.67, 150.59, 150.31, 150.25, 149.10, 148.28, 140.82, 131.40, 131.31, 130.76, 128.52, 124.91, 81.48, 53.14, 51.10, 31.71, 30.28, 29.01, 28.29, 22.73, 14.24.
19F NMR (CDCl3, 376 MHz, ppm): −128.94 (d), −133.46 (d).
:CH2Cl2 (100:0 to 90:10). Two fractions, compounds 10-D and 10-I were isolated from the column and dried under vacuum to give two shiny brown solids (19 mg for 10-D and 54 mg for 10-I).Conclusions
In conclusion, two new fullerene adducts with A–D–A type arrangement were designed, synthesized, and characterized, where the BTF2-Th unit is covalently anchored to the fullerene either as a terminal group or on to the fullerene core. The molecular structure confirmation of the two fullerene adducts was supported by NMR spectroscopy. The resultant modified structures exhibit wide and strong absorption covering the range from 300 to 600 nm and shown relatively higher absorption in the 350–500 nm region which was nearly absent in PC61BM. Terminal attached BTF2 unit led to a deeper HOMO energy level and higher electron mobility of the adduct than the one with the modified core. Both energy levels and electron mobility are affected due to different placement of BTF2 on fullerene, with HOMO energy is deeper than PC61BM for terminally modified structure. Additionally, the HOMO and LUMO were found to be affected in fullerene adducts with dependence on the location of BTF2 at the periphery vs. core of the fullerene. Computational results show that the presence of BTF2 affects the ground state dipole moments as well as the absorption strengths, most noticeable in the case of n = 2. Current work demonstrates that the fluorinated benzodithiazole unit linked to fullerene has tremendous potential as a building block for high-performance material for optoelectronic applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. A. and B. L. would like to acknowledge the financial support from Shiv Nadar Institute of Eminence. B. L. thanks DST FIST for funding the availability of the MALDI instrument. Computational results presented in this work have been produced using the AUTH Compute Infrastructure and Resources. E. N. K. would like to acknowledge the support provided by the Scientific Computing Office throughout the progress of this research work.References
- (a) M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS PubMed; (b) M. Karakus, D. H. Apaydın, D. E. Yıldız, L. Toppare and A. Cirpan, Polymer, 2012, 53, 1198–1202 CrossRef CAS; (c) H. Hoppe and N. S. Sariciftci, J. Mater. Res., 2004, 19, 1924–1945 CrossRef CAS.
- (a) M. Zhang, Y. Gu, X. Guo, F. Liu, S. Zhang, L. Huo, T. P. Russell and J. Hou, Adv. Mater., 2013, 25, 4944–4949 CrossRef CAS PubMed; (b) H. Medlej, A. Nourdine, H. Awada, M. Abbas, C. Dagron-Lartigau, G. Wantz and L. Flandin, Eur. Polym. J., 2014, 59, 25–35 CrossRef CAS; (c) R. Ganesamoorthy, G. Sathiyan and P. Sakthivel, Sol. Energy Mater. Sol. Cells, 2017, 161, 102–148 CrossRef CAS.
- (a) F. Zhang, O. Inganäs, Y. Zhou and K. Vandewal, Natl. Sci. Rev., 2016, 3, 222–239 CrossRef CAS; (b) H. Kang, G. Kim, J. Kim, S. Kwon, H. Kim and K. Lee, Adv. Mater., 2016, 28, 7821–7861 CrossRef CAS PubMed.
- C. B. Nielsen, S. Holliday, H.-Y. Chen, S. J. Cryer and I. McCulloch, Acc. Chem. Res., 2015, 48, 2803–2812 CrossRef CAS PubMed.
- (a) J. C. Hummelen, B. W. Knight, F. LePeq, F. Wudl, J. Yao and C. L. Wilkins, J. Org. Chem., 1995, 60, 532–538 CrossRef CAS; (b) W. Shi, X. Hou, T. Liu, X. Zhao, A. Sieval, J. Hummelen and T. Dennis, Chem. Commun., 2017, 53, 975–978 RSC; (c) T. Liu, A. J. Misquitta, I. Abrahams and T. J. S. Dennis, Carbon, 2021, 173, 891–900 CrossRef CAS.
- F. Padinger, R. S. Rittberger and N. S. Sariciftci, Adv. Funct. Mater., 2003, 13, 85–88 CrossRef CAS.
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666–12731 CrossRef CAS PubMed.
- G. M. Somashekharappa, M. Paul, C. Govind, R. Mathew and V. Karunakaran, J. Phys. Chem. B, 2022, 126, 4509–4519 CrossRef CAS PubMed.
- W. Huang, Y.-H. Lin and T. D. Anthopoulos, ACS Appl. Mater. Interfaces, 2018, 10, 10202–10210 CrossRef CAS PubMed.
- J. Dong, S. Sami, D. M. Balazs, R. Alessandri, F. Jahani, L. Qiu, S. J. Marrink, R. W. Havenith, J. C. Hummelen and M. A. Loi, J. Mater. Chem. C, 2021, 9, 16217–16225 RSC.
- D. H. Sin, S. H. Kim, J. Lee and H. Lee, Micromachines, 2022, 13, 1613 CrossRef PubMed.
- D. Hu, Q. Yang, H. Chen, F. Wobben, V. M. Le Corre, R. Singh, T. Liu, R. Ma, H. Tang, L. J. A. Koster, T. Duan, H. Yan, Z. Kan, Z. Xiao and S. Lu, Energy Environ. Sci., 2020, 13, 2134–2141 RSC.
- F. Diederich and R. Kessinger, Acc. Chem. Res., 1999, 32, 537–545 CrossRef CAS.
- Y. He, H.-Y. Chen, J. Hou and Y. Li, J. Am. Chem. Soc., 2010, 132, 1377–1382 CrossRef CAS PubMed.
- (a) M. Lenes, G.-J. A. H. Wetzelaer, F. B. Kooistra, S. C. Veenstra, J. C. Hummelen and P. W. M. Blom, Adv. Mater., 2008, 20, 2116–2119 CrossRef CAS; (b) M. Lenes, S. W. Shelton, A. B. Sieval, D. F. Kronholm, J. C. Hummelen and P. W. M. Blom, Adv. Funct. Mater., 2009, 19, 3002–3007 CrossRef CAS; (c) T. E. Kang, H.-H. Cho, C.-H. Cho, K.-H. Kim, H. Kang, M. Lee, S. Lee, B. Kim, C. Im and B. J. Kim, ACS Appl. Mater. Interfaces, 2013, 5, 861–868 CrossRef PubMed.
- A. A. Guilbert, L. X. Reynolds, A. Bruno, A. MacLachlan, S. P. King, M. A. Faist, E. Pires, J. E. Macdonald, N. Stingelin and S. A. Haque, ACS Nano, 2012, 6, 3868–3875 CrossRef CAS PubMed.
- (a) K.-Q. Liu, J.-J. Wang, X.-X. Yan, C. Niu and G.-W. Wang, Chem. Sci., 2020, 11, 384–388 RSC; (b) C. Fuertes-Espinosa, C. García-Simón, M. Pujals, M. Garcia-Borràs, L. Gómez, T. Parella, J. Juanhuix, I. Imaz, D. Maspoch and M. Costas, Chem, 2020, 6, 169–186 CrossRef CAS.
- J. Yuan, Y. Zhang, L. Zhou, C. Zhang, T.-K. Lau, G. Zhang, X. Lu, H.-L. Yip, S. K. So, S. Beaupré, M. Mainville, P. A. Johnson, M. Leclerc, H. Chen, H. Peng, Y. Li and Y. Zou, Adv. Mater., 2019, 31, 1807577 CrossRef PubMed.
- (a) Y. Zhang, C. Kim, J. Lin and T.-Q. Nguyen, Adv. Funct. Mater., 2012, 22, 97–105 CrossRef CAS; (b) J. Du, M. C. Biewer and M. C. Stefan, J. Mater. Chem. A, 2016, 4, 15771–15787 RSC.
- I.-S. Kim, I.-B. Kim, D.-Y. Kim, S.-H. Kwon and D.-K. Ko, Macromol. Rapid Commun., 2016, 37, 1242–1248 CrossRef CAS PubMed.
- (a) M. Krassas, C. Polyzoidis, P. Tzourmpakis, D. Kosmidis, G. Viskadouros, N. Kornilios, V. Nikolaou, T. Coutsolelos, K. Petridis, M. Stylianakis and E. Kymakis, Energies, 2020, 13, 450 CrossRef CAS; (b) R. Barla, B. Lochab, A. Agrawal, A. Mishra, M. L. Keshtov and G. D. Sharma, Sol. RRL, 2021, 2100402 CrossRef; (c) J. Yuan, Y. Zhang, L. Zhou, C. Zhang, T. K. Lau, G. Zhang, X. Lu, H. L. Yip, S. K. So and S. Beaupré, Adv. Mater., 2019, 31, 1807577 CrossRef PubMed.
- (a) Q. Zhang, M. A. Kelly, N. Bauer and W. You, Acc. Chem. Res., 2017, 50, 2401–2409 CrossRef CAS PubMed; (b) H. Yu, Z. Qi, X. Li, Z. Wang, W. Zhou, H. Ade, H. Yan and K. Chen, Sol. RRL, 2020, 4, 2000421 CrossRef CAS.
- (a) Q. Zhang, L. Yan, X. Jiao, Z. Peng, S. Liu, J. J. Rech, E. Klump, H. Ade, F. So and W. You, Chem. Mater., 2017, 29, 5990–6002 CrossRef CAS; (b) S. Zhang, Y. Qin, M. A. Uddin, B. Jang, W. Zhao, D. Liu, H. Y. Woo and J. Hou, Macromolecules, 2016, 49, 2993–3000 CrossRef CAS.
- (a) T. Qin, W. Zajaczkowski, W. Pisula, M. Baumgarten, M. Chen, M. Gao, G. Wilson, C. D. Easton, K. Müllen and S. E. Watkins, J. Am. Chem. Soc., 2014, 136, 6049–6055 CrossRef CAS PubMed; (b) J.-S. Wu, S.-W. Cheng, Y.-J. Cheng and C.-S. Hsu, Chem. Soc. Rev., 2015, 44, 1113–1154 RSC; (c) X. Wan, C. Li, M. Zhang and Y. Chen, Chem. Soc. Rev., 2020, 49, 2828–2842 RSC; (d) B. Rajkumar, L. Khanam, E. N. Koukaras, G. D. Sharma, S. P. Singh and B. Lochab, ACS Sustainable Chem. Eng., 2020, 8, 5891–5902 CrossRef CAS.
- K. Schulze, C. Uhrich, R. Schüppel, K. Leo, M. Pfeiffer, E. Brier, E. Reinold and P. Bäuerle, Adv. Mater., 2006, 18, 2872–2875 CrossRef CAS.
- L. Meng, Y. Zhang, X. Wan, C. Li, X. Zhang, Y. Wang, X. Ke, Z. Xiao, L. Ding, R. Xia, H.-L. Yip, Y. Cao and Y. Chen, Science, 2018, 361, 1094 CrossRef CAS PubMed.
- Y. Che, Y. Zhang, Y. Yang, C.-H. Liu, R. Izquierdo, S. S. Xiao and D. F. Perepichka, J. Org. Chem., 2020, 85, 52–61 CrossRef CAS PubMed.
- (a) B. C. Thompson and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2008, 47, 58–77 CrossRef CAS PubMed; (b) C. J. Brabec, S. Gowrisanker, J. J. M. Halls, D. Laird, S. Jia and S. P. Williams, Adv. Mater., 2010, 22, 3839–3856 CrossRef CAS PubMed.
- W. Li, C. Du, F. Li, Y. Zhou, M. Fahlman, Z. Bo and F. Zhang, Chem. Mater., 2009, 21, 5327–5334 CrossRef CAS.
- P. Espinet and A. M. Echavarren, Angew. Chem., Int. Ed., 2004, 43, 4704–4734 CAS.
- J.-L. Wang, Z.-F. Chang, X.-X. Song, K.-K. Liu and L.-M. Jing, J. Mater. Chem. C, 2015, 3, 9849–9858 RSC.
- N. Miyaura, T. Yanagi and A. Suzuki, Synth. Commun., 1981, 11, 513–519 CrossRef CAS.
- B. Neises and W. Steglich, Angew. Chem., Int. Ed., 1978, 17, 522–524 CrossRef.
- (a) X. Camps and A. Hirsch, J. Chem. Soc., Perkin Trans. 1, 1997, 1595–1596, 10.1039/a702055d; (b) A. Iwashita, Y. Matsuo and E. Nakamura, Angew. Chem., Int. Ed., 2007, 46, 3513–3516 CrossRef CAS PubMed; (c) S. H. Hoke, J. Molstad, D. Dilettato, M. J. Jay, D. Carlson, B. Kahr and R. G. Cooks, J. Org. Chem., 1992, 57, 5069–5071 CrossRef CAS.
- M. Maggini, G. Scorrano and M. Prato, J. Am. Chem. Soc., 1993, 115, 9798–9799 CrossRef CAS.
- C.-H. Andersson, G. Berggren, S. Ott and H. Grennberg, Eur. J. Inorg. Chem., 2011, 2011, 1744–1749 CrossRef.
- L. Ðorđević, L. Casimiro, N. Demitri, M. Baroncini, S. Silvi, F. Arcudi, A. Credi and M. Prato, Angew. Chem., Int. Ed., 2021, 60, 313–320 CrossRef PubMed.
- I. Bala, R. A. K. Yadav, M. Devi, J. De, N. Singh, K. Kailasam, J. Jayakumar, J.-H. Jou, C.-H. Cheng and S. K. Pal, J. Mater. Chem. C, 2020, 8, 17009–17015 RSC.
- (a) H. Fang, H. Gao, T. Wang, B. Zhang, W. Xing and X. Cheng, Dyes Pigm., 2017, 147, 190–198 CrossRef CAS; (b) Q. Fang, B. Xu, B. Jiang, H. Fu, X. Chen and A. Cao, Chem. Commun., 2005, 1468–1470, 10.1039/b417810f.
- J. P. Hare, H. W. Kroto and R. Taylor, Chem. Phys. Lett., 2013, 589, 57–60 CrossRef CAS.
- R. Kumar, S. Naqvi, N. Gupta and S. Chand, RSC Adv., 2014, 4, 15675–15677 RSC.
- (a) P.-T. Wu, F. S. Kim, R. D. Champion and S. A. Jenekhe, Macromolecules, 2008, 41, 7021–7028 CrossRef CAS; (b) B. C. Thompson, Y.-G. Kim, T. D. McCarley and J. R. Reynolds, J. Am. Chem. Soc., 2006, 128, 12714–12725 CrossRef CAS PubMed; (c) R. E. Aderne, B. G. A. Borges, H. C. Ávila, F. von Kieseritzky, J. Hellberg, M. Koehler, M. Cremona, L. S. Roman, C. M. Araujo and M. L. M. Rocco, Mater. Adv., 2022, 3, 1791–1803 RSC.
- K. Seki, J. Appl. Phys., 2014, 116, 063716 CrossRef.
- S. Naqvi, N. Gupta, N. Kumari, J. Garg and R. Kumar, New J. Chem., 2017, 41, 1933–1939 RSC.
- J. L. Brusso, O. D. Hirst, A. Dadvand, S. Ganesan, F. Cicoira, C. M. Robertson, R. T. Oakley, F. Rosei and D. F. Perepichka, Chem. Mater., 2008, 20, 2484–2494 CrossRef CAS.
- S. Ram and R. E. Ehrenkaufer, Tetrahedron Lett., 1984, 25, 3415–3418 CrossRef CAS.
- H. Zhou, L. Yang, A. C. Stuart, S. C. Price, S. Liu and W. You, Angew. Chem., Int. Ed., 2011, 50, 2995–2998 CrossRef CAS PubMed.
- S. Manku, F. Wang and D. G. Hall, J. Comb. Chem., 2003, 5, 379–391 CrossRef CAS PubMed.
- R. Kumar, S. Khan, N. Gupta, S. Naqvi, K. Gaurav, C. Sharma, M. Kumar, P. Kumar and S. Chand, Carbon, 2016, 107, 765–773 CrossRef CAS.
- (a) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (c) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed; (d) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- R. Gaussian, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennuci, G. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. Hratchian, A. Izmaylov, J. Bloino, G. Zheng, J. Sonnenberg, M. Hada and D. Fox, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- C.-H. Hsieh, Y.-J. Cheng, P.-J. Li, C.-H. Chen, M. Dubosc, R.-M. Liang and C.-S. Hsu, J. Am. Chem. Soc., 2010, 132, 4887–4893 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra06175a |
| This journal is © The Royal Society of Chemistry 2022 |