DOI:
10.1039/D2RA05990H
(Paper)
RSC Adv., 2022,
12, 35989-36001
Synthesis of novel hybrid mesoporous gold iron oxide nanoconstructs for enhanced catalytic reduction and remediation of toxic organic pollutants†
Received
22nd September 2022
, Accepted 2nd November 2022
First published on 15th December 2022
Abstract
The development of highly efficient, rapid, and recyclable nanocatalysts for effective elimination of toxic environmental contaminants remains a high priority in various industrial applications. Herein, we report the preparation of hybrid mesoporous gold–iron oxide nanoparticles (Au–IO NPs) via the nanocasting “inverse hard-templated replication” approach. Dispersed Au NPs were anchored on amine-functionalized iron oxide incorporated APMS (IO@APMS-amine), followed by etching of the silica template to afford hybrid mesoporous Au–IO NPs. The obtained nanoconstructs were fully characterized using electron microscopy, N2 physisorption, and various spectroscopic techniques. Owing to their magnetic properties, high surface areas, large pore volumes, and mesoporous nature (SBET = 124 m2 g−1, Vpore = 0.33 cm3 g−1, and dpore = 4.5 nm), the resulting Au–IO mesostructures were employed for catalytic reduction of nitroarenes (i.e. nitrophenol and nitroaniline), two of the most common toxic organic pollutants. It was found that these Au–IO NPs act as highly efficient nanocatalysts showing exceptional stabilities (>3 months), enhanced catalytic efficiencies in very short times (∼100% conversions within only 25–60 s), and excellent recyclabilities (up to 8 cycles). The kinetic pseudo-first-order apparent reaction rate constants (kapp) were calculated to be equal to 8.8 × 10−3 and 23.5 × 10−3 s−1 for 2-nitrophenol and 2-nitroaniline reduction, respectively. To our knowledge, this is considered one of the best and fastest Au-based nanocatalysts reported for the catalytic reduction of nitroarenes, promoted mainly by the synergistic cooperation of their high surface area, large pore volume, mesoporous nature, and enhanced Au-NP dispersions. The unique mesoporous hybrid Au–IO nanoconstructs synthesized here make them novel, stable, and approachable nanocatalyst platform for various catalytic industrial processes.
1. Introduction
Hybrid mesoporous gold iron oxides have attracted much attention in the fields of catalysis, magnetism, optoelectronics, and theranostic biomedical platforms.1–3 This is mainly due to their porous nature, large surface area and pore volume, as well as their unique physiochemical and magnetic properties. It is well established that at the nanoscale regime, noble metals (Au, Ag, Pd, Pt etc.) are much effective for catalytic conversions,4 especially for the removal of nitro- and/or phenolic-organic pollutants, and other hazardous environmental contaminants.5–8 Gold nanoparticles (Au-NPs), in particular, can effectively catalyze many important chemical transformations such as oxidation reactions (oxidation of cyclohexane, CO, and NH3),9–12 reduction reactions (hydrogenation), and other redox and coupling reactions.13 Furthermore, they are well known to be extremely useful for applications in optics, electronics, and H2O2 sensing,14 as well as for photothermal therapy and theranostic platforms.15 For heterogeneous catalyzed reaction systems, Au-NPs have been typically immobilized on supports such as silica, carbon, organic frameworks, and metal oxides showing in most cases enhanced activities.7,13,16–19 Among those, iron oxide (IO) is considered as one of the most promising support materials for the loading of noble metal NPs due to their abundance, ease of synthesis, low cost, high chemical and thermal stabilities, as well as low toxicity and environmental friendliness.20–22 Moreover, the presence of IO-NPs in metal supported catalytic systems is by itself advantageous, as it allows magnetic separability from the reaction medium (i.e. ease of separation by external magnet in comparison to traditional filtration or centrifugation), avoiding loss of the catalyst and, hence, possible reuse. Structurally, other than the common core–shell nanovehicles,8,17,23 porous materials (mainly mesoporous silica) were employed as a support for dispersing Au-NPs,10,24,25 rendering them highly efficient and recoverable nanocatalysts. For instance, Au-NPs supported on SiO2-coated Fe3O4 materials have been prepared and applied toward catalytic reduction of nitro-aromatic compounds.17 Au-NPs supported on iron oxides (Au/FexOy) have been also synthesized and tested for CO oxidation reactions, showing much higher catalytic activities compared to other Au, iron oxide, or commercial Au/Fe2O3 catalysts.11,26 Recently, Au-NPs loaded on mesoporous Fe2O3 supports have shown to exhibit even better specific activities compared to those supported on nanostructured nonporous Fe2O3 surfaces.27,28 The enhancement in catalytic performance has been mainly attributed to the enhanced diffusivity of the reactant molecules due to the mesopores, increased number of active interface sites between the Au-NPs and the porous iron oxide support, improved dispersion of Au-NPs, and stronger noble metal–support interactions. The mesostructured nature of the support is anticipated to increase the number of Au-support perimeter sites (i.e. more exposed active sites), thus increasing achieved catalytic activities. Moreover, it has been proven that mesoporous IO supports can be loaded with much higher amounts of Au-NPs, which is almost 10 times higher than the Au loading on commercial iron oxides.27 In another report, it was found that the presence of confined Au-NPs on nanocasted Fe2O3 increased the catalytic performance due to both the presence of a microporous/mesoporous structure in the IO support as well as a mixed surface phase of Si and Fe oxides, where Au-NPs are deposited.29 This particular feature is quite interesting along with its effects on the provision of more available active sites, higher Au-support contact surface area, and enhanced reactant diffusivity. All these established results confirm the crucial role of porosity in nanostructured supports, which facilitates a more intimate contact between the confined noble metal NPs and the support. By accommodating Au-NPs both within the large interior pores and/or anchoring them at the exterior surface of the porous material, superior Au-support contact area with improved Au dispersions is attained. The catalytic activities have also been shown to be dependent, albeit to less extent, on Au-NP sizes with 10–20 nm sizes being more active. Consequently, the size, structure, porosity, and stability of the support materials can significantly influence the dispersion of Au-NPs, and, hence, the overall catalytic activities. Thus, the utilization of mesoporous silica and/or iron oxide as a support for the loading of noble metal NPs is particularly advantageous for various catalytic applications.
Several methods have been utilized to synthesize porous noble metal–iron oxide hybrid materials, including chemical reduction of metal ion salts in a specific polymeric matrix, porous support matrix, self-assembly, surfactant-assisted processes, and dispersion or blending of metal NPs with iron oxide.30,31 Nonetheless, the fabrication of well-dispersed and stable Au-NPs with controllable sizes and shapes on porous metal matrices having high surface area and large pore volumes is quite challenging. While the mesopores dictate the size and morphology of the loaded Au-NPs as they are formed in the confined matrix, the high surface area and large pore volume aid in the formation of homogeneously dispersed noble metal NPs on the surface of the support. Consequently, urgent development of robust and reliable procedures to synthesize efficient oxide-supported Au-based nanocatalysts are constantly needed, particularly for chemical and environmental industries worldwide. Herein, we demonstrate, for the first time, the fabrication of hybrid mesoporous Au–IO nanostructures, anchored with highly dispersed Au-NPs (sizes ∼ 15 nm) based on our previously reported “inverse hard-templated replication” of an amino-functionalized IO-incorporated acid-prepared mesoporous silica (IO@APMS-amine) support, which acts as metal stabilizing and size-controlling agent. When tested for the catalytic reduction of nitro aromatic compounds (i.e. nitrophenol and nitroaniline), the as-prepared mesoporous Au–IO NPs were found to be highly efficient with very fast conversion rates, enhanced catalytic efficiencies, and excellent stabilities and recyclabilities. To the best of our knowledge, the mesoporous Au-based IO-supported nanocatalyst developed here is among the most efficient and fastest catalysts reported for the catalytic reduction of nitroarenes. Thanks to the synergistic cooperation of their high surface area, large pore volume, mesoporous nature, and dispersed Au-NPs anchored on the nanocasted support.
2. Results and discussion
2.1 Synthesis of mesoporous Au–IO NPs
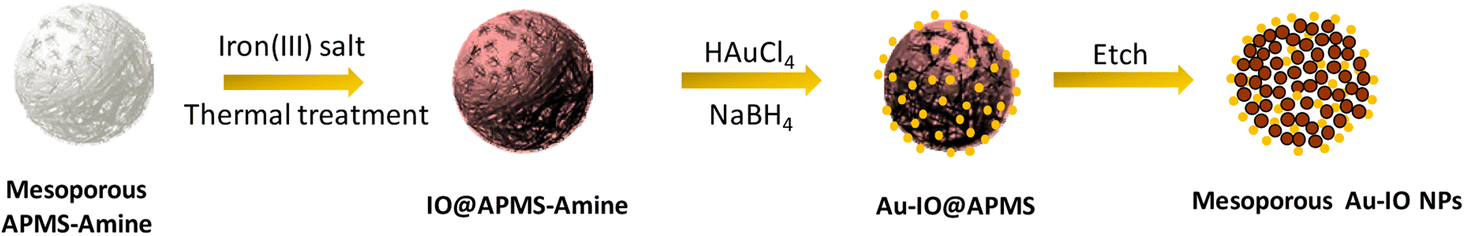
The fabrication of mesoporous Au–IO NPs is illustrated in Fig. 1. The synthesis is based on the nano-casting reverse hard-templated method as in our previous reports,32,33 using APMS as the silica matrix, iron(III) chloride (FeCl3) as the iron precursors, and chlorauric salt (HAuCl4) as the metallic precursor for the AuNP formation. Pore-blocked APMS spheres (∼1.5 μm) were first synthesized,34,35 followed by surface functionalization with –NH2 (amine) groups using 3-aminopropyltriethoxysilane (APTES). Stabilizing aminosilanes were introduced to pore-blocked APMS, which can controllably intercalate Au metal NPs to be anchored on their surfaces.36,37 Repeated impregnation and in situ reduction of FeCl3 salt precursors in a wetting process, followed by subsequent thermal heating afforded IO nanoclusters incorporated within the amino-functionalized mesoporous silica (IO@APMS-amine). AuNPs were then formed on the functionalized amino surface and within the remaining pores of IO@APMS-amine by in situ reduction of adsorbed auric chloride ions with NaBH4.5,25,38 Finally, etching of the silica template with hot NaOH base afforded the final templated mesoporous Au–IO NPs (Fig. 1). Au NPs are most commonly prepared by basic methods, using citrate39 or NaBH4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 40 as reductants producing size-controllable 3–20 nm sized NPs.41 The amine groups (–NH2 and –NH) introduced on the mesoporous walls/surface act as stabilizing centers providing an anchoring surface, which allows formation of dispersed AuNPs with controlled and narrow size distributions. Generally, and in agreement with previous reports, organic functional groups grafted on the surface of porous silica matrices are used to coordinate and stabilize Au precursors for AuNP formation.9,10 This mesopore-directed growth of AuNPs with the help of built-in functionalized amine groups as stabilizing agents is a unique approach, as it is anticipated to aid in the control of the size, shape, and dispersity of the obtained AuNPs. It is to be also noted that although the reduction of chloroauric salt is initiated by the addition of NaBH4, the presence of amine groups also facilitates the reduction process, as has been corroborated earlier.13
40 as reductants producing size-controllable 3–20 nm sized NPs.41 The amine groups (–NH2 and –NH) introduced on the mesoporous walls/surface act as stabilizing centers providing an anchoring surface, which allows formation of dispersed AuNPs with controlled and narrow size distributions. Generally, and in agreement with previous reports, organic functional groups grafted on the surface of porous silica matrices are used to coordinate and stabilize Au precursors for AuNP formation.9,10 This mesopore-directed growth of AuNPs with the help of built-in functionalized amine groups as stabilizing agents is a unique approach, as it is anticipated to aid in the control of the size, shape, and dispersity of the obtained AuNPs. It is to be also noted that although the reduction of chloroauric salt is initiated by the addition of NaBH4, the presence of amine groups also facilitates the reduction process, as has been corroborated earlier.13
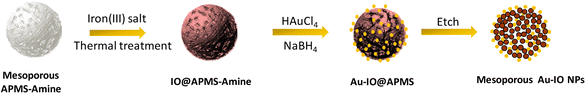 |
| | Fig. 1 Schematic illustration for the preparation of hybrid mesoporous gold iron oxide nanoparticles (Au–IO NPs) via nano-casting reverse methodology using APMS as the silica template. | |
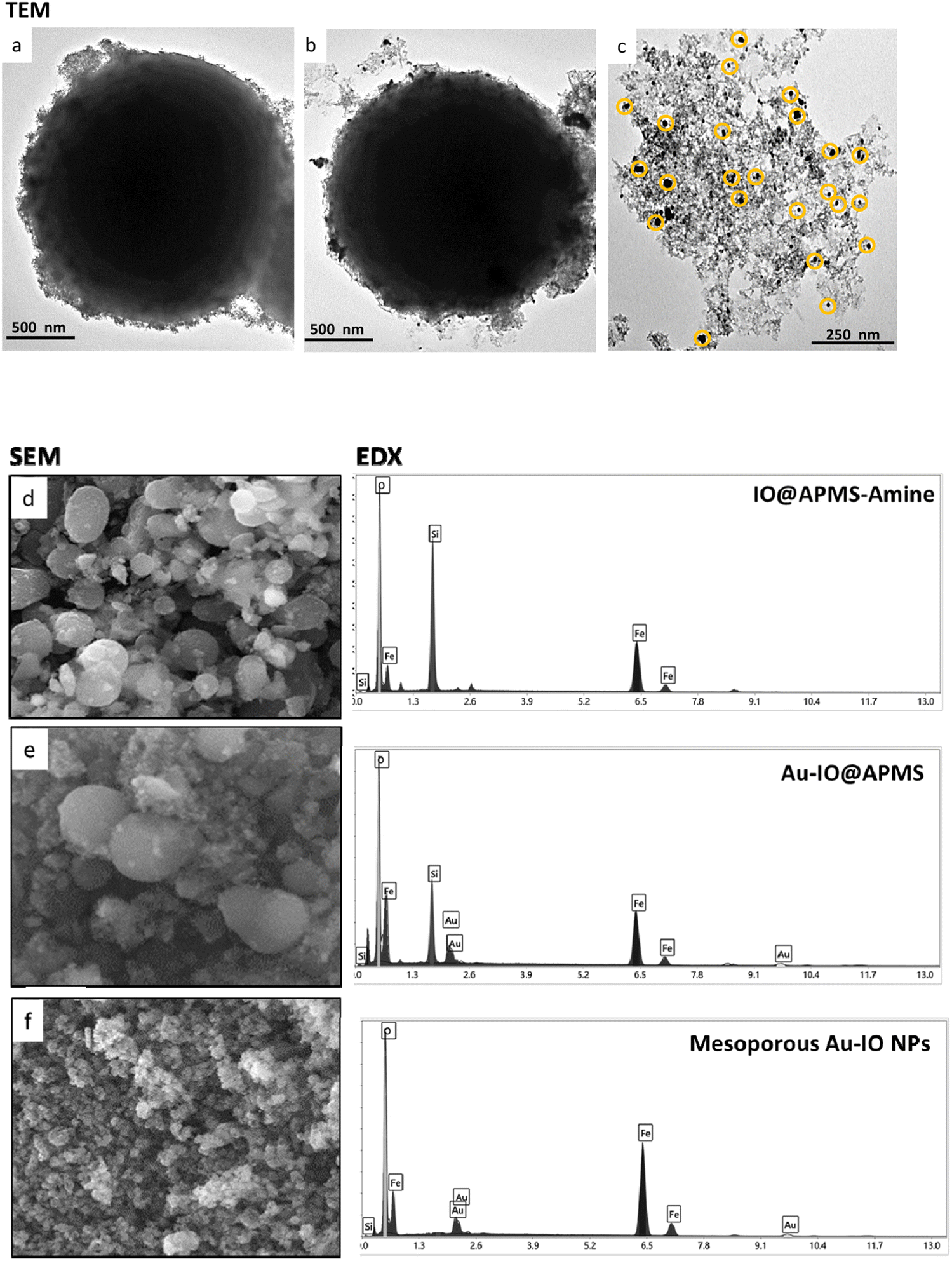
The prepared mesoporous Au–IO NPs were fully characterized by electron microscopy, dynamic light scattering (DLS), Fourier transform infrared (FTIR), X-ray diffraction (XRD), and vibrating-sample magnetometer (VSM). First, the structure, morphology, and composition of mesoporous IO–Au NPs were examined by transmission electron microscopy (TEM) and scanning electron microscopy energy dispersive X-ray (SEM-EDX). Fig. 2a–c and S1† depict TEM images of ∼1.5 μm IO@APMS clearly showing the formation of ultrasmall sized iron oxide nanoclusters (∼1–5 nm) within and surrounding the APMS silica template (Fig. 2a). Once Au NPs were incorporated, a regular arrangement of additional well-dispersed black colored spherical dots (∼15–20 nm) are evident along the surface of the APMS particles (Fig. 2b). No independent agglomeration of the Au NPs was observed along the TEM grid, revealing that the amine moieties controllably anchored the formed Au NPs. Finally, spheroid-like Au–IO nanoparticulate mesostructures are remarkably evident, with mesochannels clearly seen, proving the successful replication of the original mesoporous template. The obtained structures possess mesopores of nanoclustered IO (gray) and the heavier Au (black) NPs depicted in yellow circles (Fig. 2c). SEM-EDX analyses were also conducted to determine the elemental composition of the different prepared samples (Fig. 2d–f). SEM images clearly show the IO- and Au-dotted architectures of both IO@APMS and Au–IO@APMS, along with the formation of smaller-sized mesoporous Au–IO NPs, in agreement with TEM. EDX performed on selected SEM areas confirmed the presence of Fe, O, and Si elements in IO@APMS; Fe, O, Si, and Au in Au–IO@APMS; and only Fe, O, and Au in the final replicated mesoporous Au–IO material. Elemental analysis revealed that the mesoporous support can be incorporated with as high as 14.6 wt% of Au (Table S1†), which is much higher than the loading achieved in previous mesoporous IO-based and nonporous commercial IO supports.27
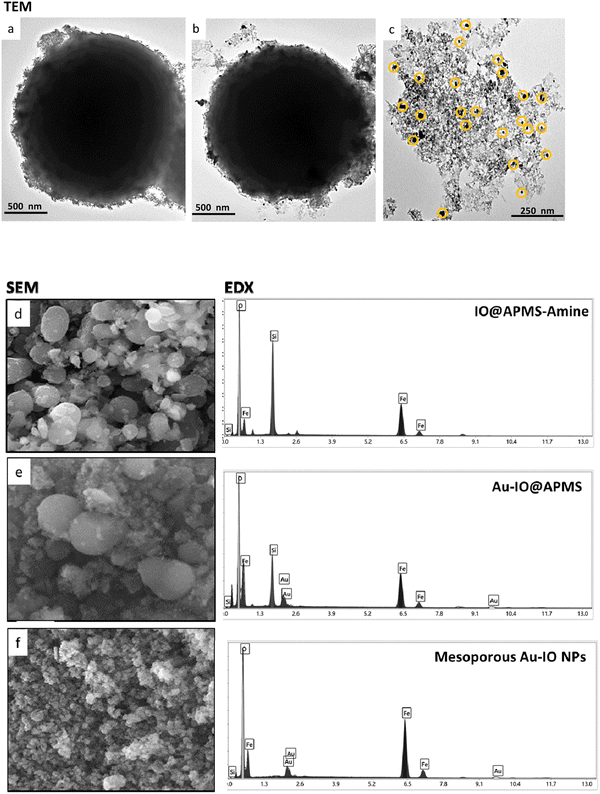 |
| | Fig. 2 Representative TEM images of the different mesoporous Au–IO samples: (a) IO@APMS-amine, (b) Au-grafted IO@APMS, and (c) Au–IO NPs. The images show the successful incorporation of Au onto IO@APMS-amine surface and the clear mesostructure of the final replicated Au–IO NPs. SEM-EDX analyses of the three samples. Left: SEM images of (d) IO@APMS-amine, (e) Au-incorporated IO@APMS, and (f) Au–IO NPs. Right: corresponding EDX spectrum selected from the SEM images of the different samples: (d) IO and silica (Fe, O, Si); (e) IO, silica, and Au (Fe, O, Si, Au); and (f) IO and Au (Fe, O, Au). | |
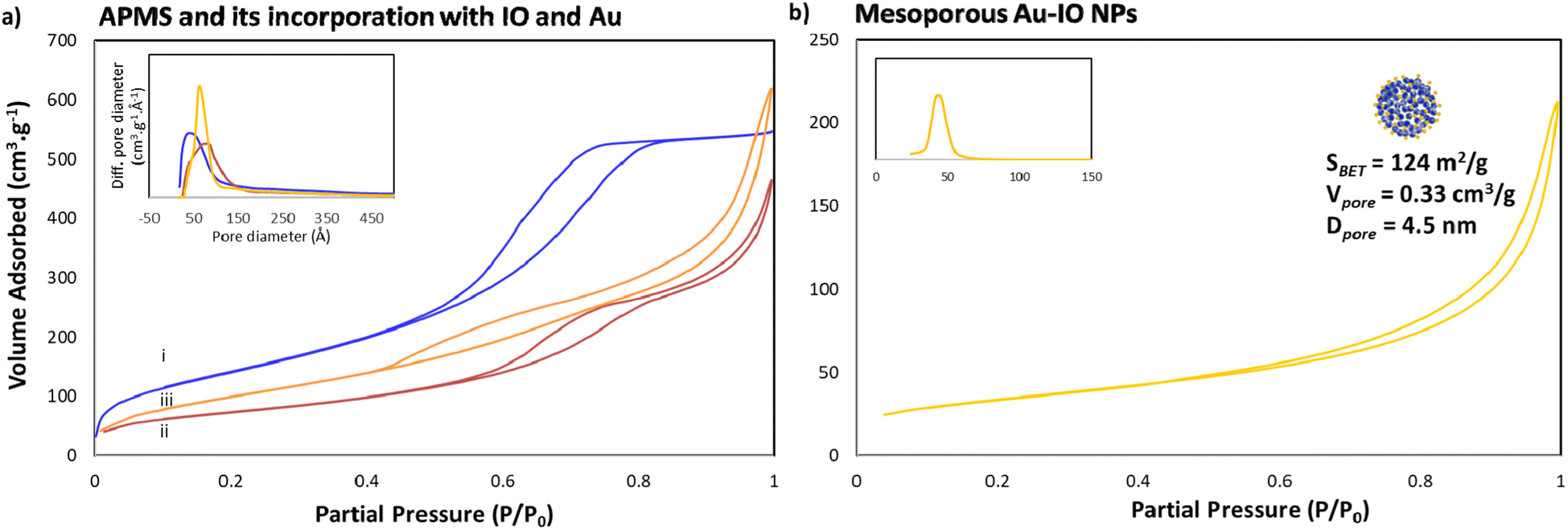
Next, the porous nature of the samples was examined by nitrogen (N2) adsorption/desorption isotherms (Fig. 3). The isotherms clearly demonstrated type IV curves, consistent with reported porosities for similar materials, where the Brunauer–Emmett–Teller (BET) surface area (SBET), Barrett–Joyner–Halenda (BJH) pore volume (Vpore), and BJH average pore size diameter (dpore) were calculated from the obtained desorption graphs. As expected, iron impregnation indicated reduction in the specific surface area and total pore volume of the original APMS-amine (SBET = 534 m2 g−1 and Vpore = 0.96 cm3 g−1) affording IO@APMS with SBET = 265 m2 g−1, Vpore = 0.73 cm3 g−1, and dpore = 9.5 nm (Fig. 3a). Once incorporated with Au NPs, SBET was found to increase to 360 m2 g−1 and Vpore = 1.0 cm3 g−1 for IO–Au@APMS. This result could be mainly due to the formation of Au NPs on the outer surface via the amine moieties coating the APMS, indications of both IO and Au NPs incorporating and coating the surfaces of the silica walls, respectively. Importantly, following the loading of Au NPs, the mesoporous structure of the sample is still well-retained with dpore = 9.0 nm. Notably, N2 isotherms showed that the final etched mesoporous Au–IO NP sample exhibit relatively high SBET = 124 m2 g−1, Vpore = 0.33 cm3 g−1, and major average dpore = 4.5 nm (Fig. 3b). It is worth pinpointing that this procedure was repeated more than once using different APMS samples with various surface areas and pore volumes obtaining mesoporous Au–IO NPs with SBET of 95–125 m2 g−1 and Vpore of 0.15–0.35 cm3 g−1. All the N2 physisorption data are summarized in Table 1.
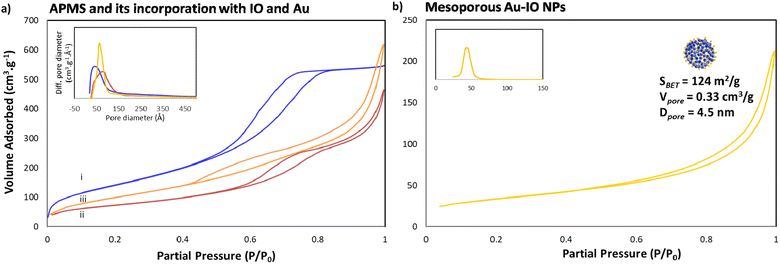 |
| | Fig. 3 N2 physisorption isotherms of (a) (i) APMS-amine, (ii) IO@APMS-amine, and (iii) Au-incorporated IO@APMS; and (b) replicated Au–IO NPs (insets: corresponding pore size distribution plots). The results clearly depict the successful formation of mesoporous Au–IO NPs with high surface area, large pore volumes, and well-controlled pore size distributions. | |
Table 1 N2 physisorption data for the different samples
| Sample |
SBET (m2 g−1) |
Vpore (cm3 g−1) |
dpore (nm) |
| APMS-amine |
534 |
0.99 |
5.9 |
| IO@APMS |
265 |
0.73 |
9.5 |
| Au–IO@APMS |
360 |
1.00 |
9.0 |
| Mesoporous Au–IO NPs |
124 |
0.15 |
4.5 |
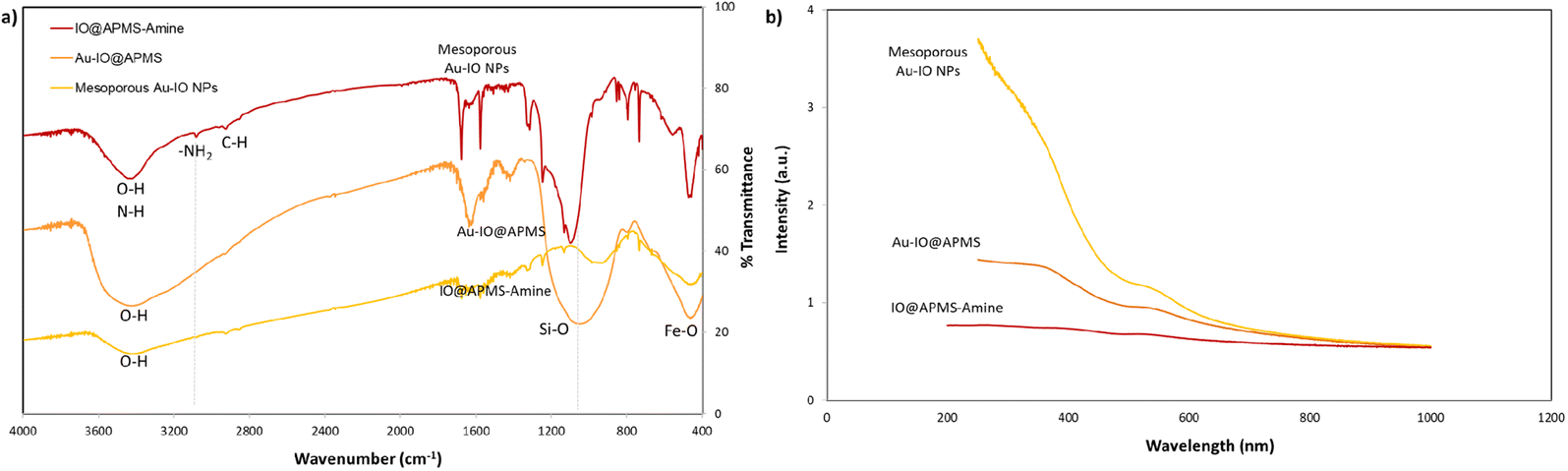
FTIR analysis was then extended to further corroborate the structures of IO@APMS-amine, Au–IO@APMS, and mesoporous Au–IO NP samples (Fig. 4a). In all the samples, the presence of the Fe–O stretching bands at ∼456 to 585 cm−1 confirm the presence of iron oxide. The broad peaks observed at ∼3400 cm−1 depict the O–H and N–H stretching vibration of hydroxyl and amine groups, while the distinctive peaks at 2850 and 2920 cm−1 show the stretching modes of C–H and –CH2– of methyl and methylene groups. In the case of mesoporous Au–IO material, a weaker peak intensity in the 3400 cm−1 region was observed, signifying the interaction of Au NPs with the amine surface moieties. Moreover, the slight decrease of peak at 733 cm−1 ascribed to the Si–O–Si symmetric stretching and Si–O–Fe moiety with Au loading suggests a strong interaction of AuNPs with silica. Importantly, the IR spectrum showed the disappearance of the bands at 1080 cm−1 (corresponding to Si–O–Si) and 960 cm−1 (Si–OH and Si–Oδ−) stretching vibrations and at ∼3080 cm−1 (–NH2 stretching vibrations of the primary amine), indicating the successful incorporation of Au NPs into IO@APMS via the involvement of –NH2 groups. Additionally, the UV-vis absorption spectra of three different samples were also recorded (Fig. 4b). The results confirm the featureless absorption of IO@APMS, with the successful formation of Au NPs displaying a characteristic absorption band centered ∼550 nm due to the Au surface plasmon band (SPB), indicating the presence of well-dispersed nano-sized Au particles. It is well known that with the increase of Au NP sizes and particle agglomeration, the SPB peak shifts to longer wavelength with higher absorbance intensity (∼700 nm). The peak becomes broader and broader due to the coupling of the individual Au NPs when aggregated.42 Thus, the sharp SPB peaks of Au-incorporated samples obtained here indicates a narrow size distribution of well-dispersed Au NPs, corroborating the obtained electron microscopy results. Perimeter sites.
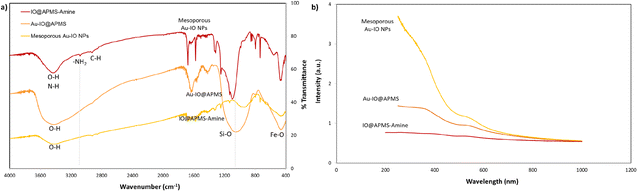 |
| | Fig. 4 (a) FTIR spectra for mesoporous IO@APMS-amine, Au–IO@APMS, and Au–IO NP samples. FTIR clearly shows the disappearance of the 960–1100 cm−1 wide band corresponding to Si–O (1100 cm−1) and Si–O–Fe (960 cm−1) stretching vibrations along with the distinctive iron oxide Fe–O peaks (∼456 and 585 cm−1). (b) Corresponding UV-vis absorption spectra of the three different samples showing the formation of dispersed Au NPs with absorption peak ∼510 nm Au NPs. | |
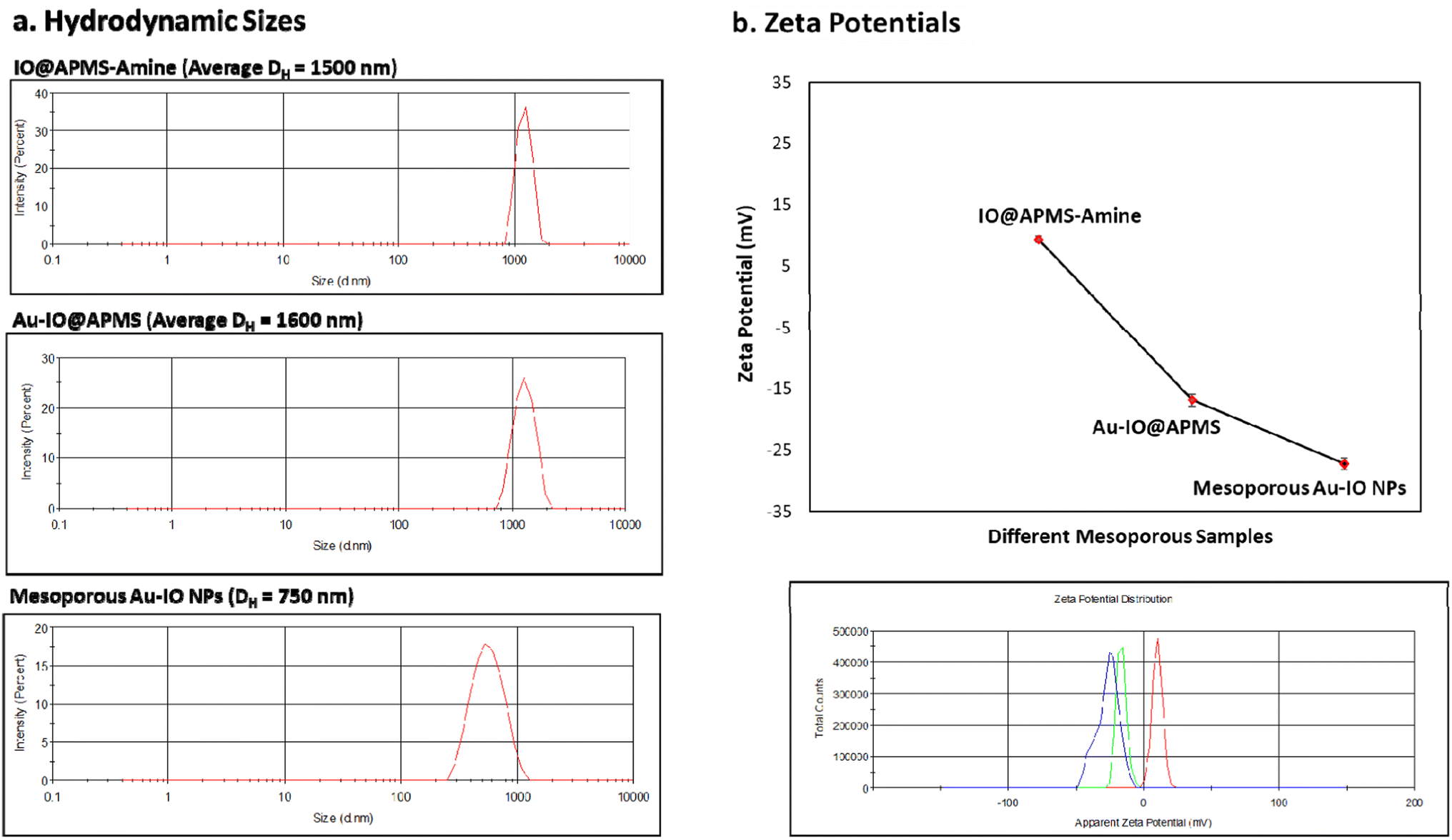
We then assessed the average hydrodynamic sizes (DH) and zeta potentials (ζ) of the different mesoporous constructs in their aqueous dispersions (Fig. 5 and S2†). While the DH sizes for IO@APMS and Au–IO@APMS were found to be similar measuring ∼1500 nm, Au–IO NPs possessed much smaller sizes with average DH ∼ 750 nm (Fig. 5a). This is expected and agrees well with our previous results, mainly due to effective etching of the original mesoporous template. The relatively sharp peaks obtained pinpoint the uniformity and dispersity of the different as-synthesized samples (PDI ∼ 0.5). The surface charges of the different samples were then computed using zeta potentials (ζ) analysis. The results showed average zeta potentials ξ = +9.40 ± 0.56 mV, −16.8 ± 0.50 mV, and −26.4 ± 0.98 mV for IO@APMS-amine, Au–IO@APMS, and mesoporous Au–IO samples, respectively (Fig. 5b). The results indicate a strong interaction of Au NPs with the support enabled by the amine surface modification where positively charged amine-functionalized IO@APMS-amines (ξ-potential = + 9.40 mV) were able to electrostatically attract negatively charged Au NPs (ξ-potential of only Au-NPs = −29.2 mV). This clearly confirm the appropriate anchoring of AuNPs on the aminated surface of IO@APMS as well as their high and good dispersion stability due to electrostatic repulsive forces. It is anticipated that the surface of the IO@APMS microspheres consists mainly of –NH3+ and unmodified Si–OH/Fe–OH groups, where the negatively charged tetrachloroaurate (AuCl4−) ions interact with the ammonium groups through electrostatic interactions resulting in Au dispersed deposition over the whole APMS surface. Both the presence of a mesoporous structure in the support along with a mixed surface phase of Si and Fe oxides, seem to be key parameters, providing improved Au dispersion and higher contact surface area between the confined AuNPs within and/or at the exterior surface of the porous nanostructured support, in accordance with earlier observation.29
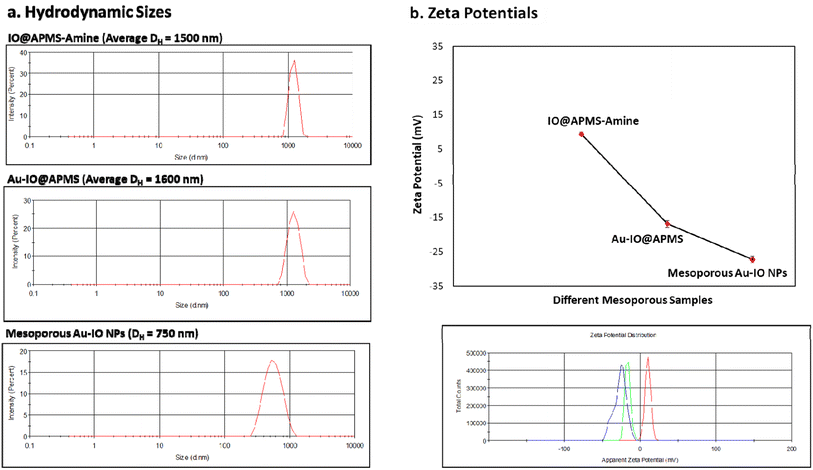 |
| | Fig. 5 DLS and zeta potential measurements. (a) Average hydrodynamic sizes (DH) and (b) average zeta potentials (ξ) of the different mesoconstructs (IO@APMS-amine, Au–IO@APMS, and Au–IO NPs) dispersed in water. The data clearly shows the high uniformity and good size distribution of the different materials, as well as the successful Au grafting on the aminated IO@APMS-amine (ξ = + 9.40 mV) to form mesoporous Au–IO NPs (ξ = −26.4 mV). | |
To determine the magnetic characteristics of the Au–IO samples, field-dependent magnetization (M–H)20–22 of the as-prepared mesostructures at 298 K were recorded (Fig. S3†). The saturation magnetization (Ms) obtained for IO@APMS-amine, Au–IO@APMS, and mesoporous Au–IO NP samples were found to be equal to 1.85 emu g−1, 1.09 emu g−1 and 1.40 emu g−1, respectively (these values are expected to be higher when calculated per gram IO). It is evident that the all-magnetization curves obtained have almost negligible retentivity and coercivity, signifying the superparamagnetic nature of the mesoconstructs. This is advantageous for catalytic reactions, as it provides ease of separation from the reaction medium, enabling regeneration of the catalyst. To further elucidate which phase of magnetic iron oxide we have, XRD of mesoporous Au–IO NPs was conducted. XRD data revealed that the produced iron oxide phase is γ-Fe2O3 (maghemite), with observed diffraction patterns in excellent agreement with previously reported data for maghemite (JCPDS #039-1346).32 Noticeably, the diffraction peaks at 2θ = 38°, 44°, 64°, and 77° indexed to (111), (200), (220), and (311) planes of crystalline Au phases (JCPDS #96-901-3039) are well observed (Fig. S4†). The weight percentage ratio of maghemite to Au phase was found to be 80% γ-Fe2O3: 20% Au. This is in accordance with the EDX results and affirms the very high loading amounts of Au NPs onto the support mainly due to the remarkably high surface area (SBET = 260–360 m2 g−1), large pore volume (Vpore = 0.70–1 cm3 g−1), and appropriate pore diameter (dpore = 9 nm) of the mesostructured support. Moreover, the greater number of surface defects (i.e. steps, edges and kinks) present on mesoporous IO@APMS might be another important factor for the high Au dispersion (Au NPs cannot be easily adsorbed on a flat metal oxide surface nor on nonporous silica-type materials).27
2.2 Catalytic reduction of nitroarenes
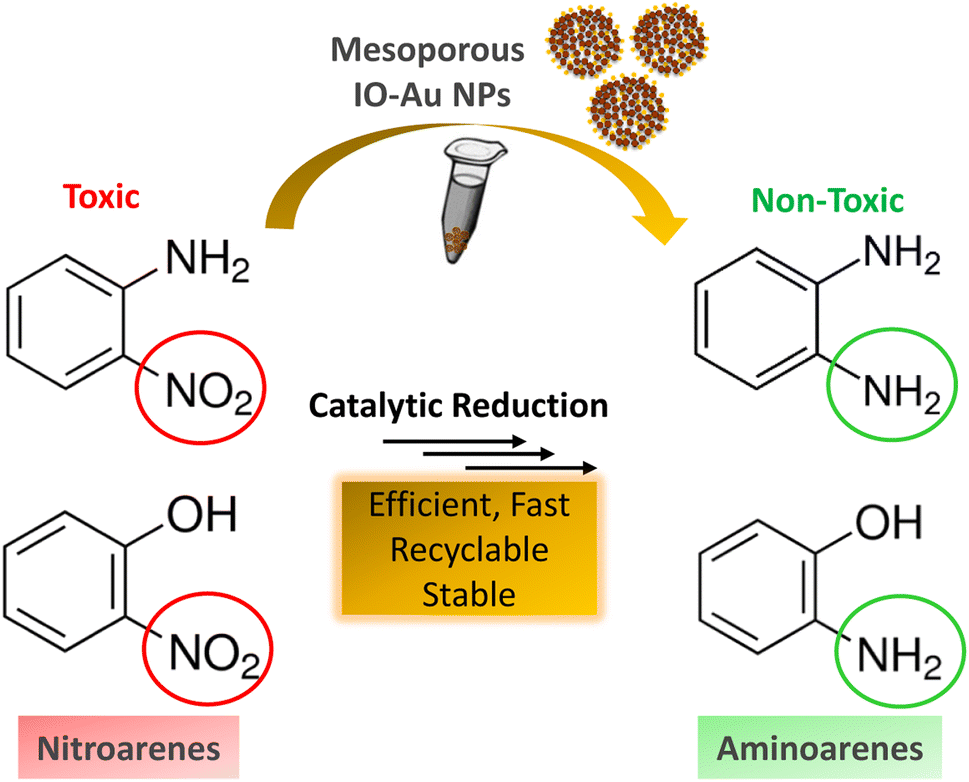
Inspired by their excellent magneto-physiochemical and unique porous properties, the resulting mesoporous Au–IO nanoconstructs were employed for catalytic reduction of 2-nitrophenol and 2-nitroaniline, two of the most common toxic environmental organic pollutants. These nitrobenzene derivatives are known as hazardous and dangerous waste contaminants and are included in the United States Environmental Protection Agency (USEPA) list owing to their potential mutagenic and carcinogenic impacts.43 Short exposure to even low concentrations may cause severe health problems and may lead to cancer and kidney/liver problems. Thus, their catalytic reduction to the non-toxic aminobenzene counterparts is considered to be one of the most promising and sustainable means for their elimination (Fig. 6). Consequently, the development of low-cost, highly stable, ultrafast, and recyclable catalysts for effective elimination of nitroarenes is highly desirable in various industrial applications.44
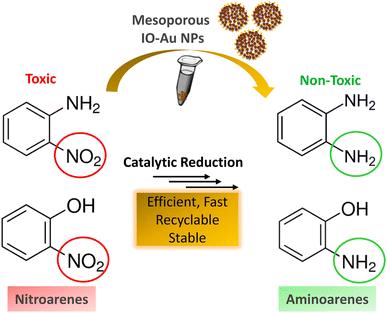 |
| | Fig. 6 Schematic representation for the catalytic reduction of toxic nitroarenes (i.e. 2-nitrophenol and 2-nitroaniline) to their non-toxic amino-counterparts using the hybrid mesoporous Au–IO nanocatalysts. | |
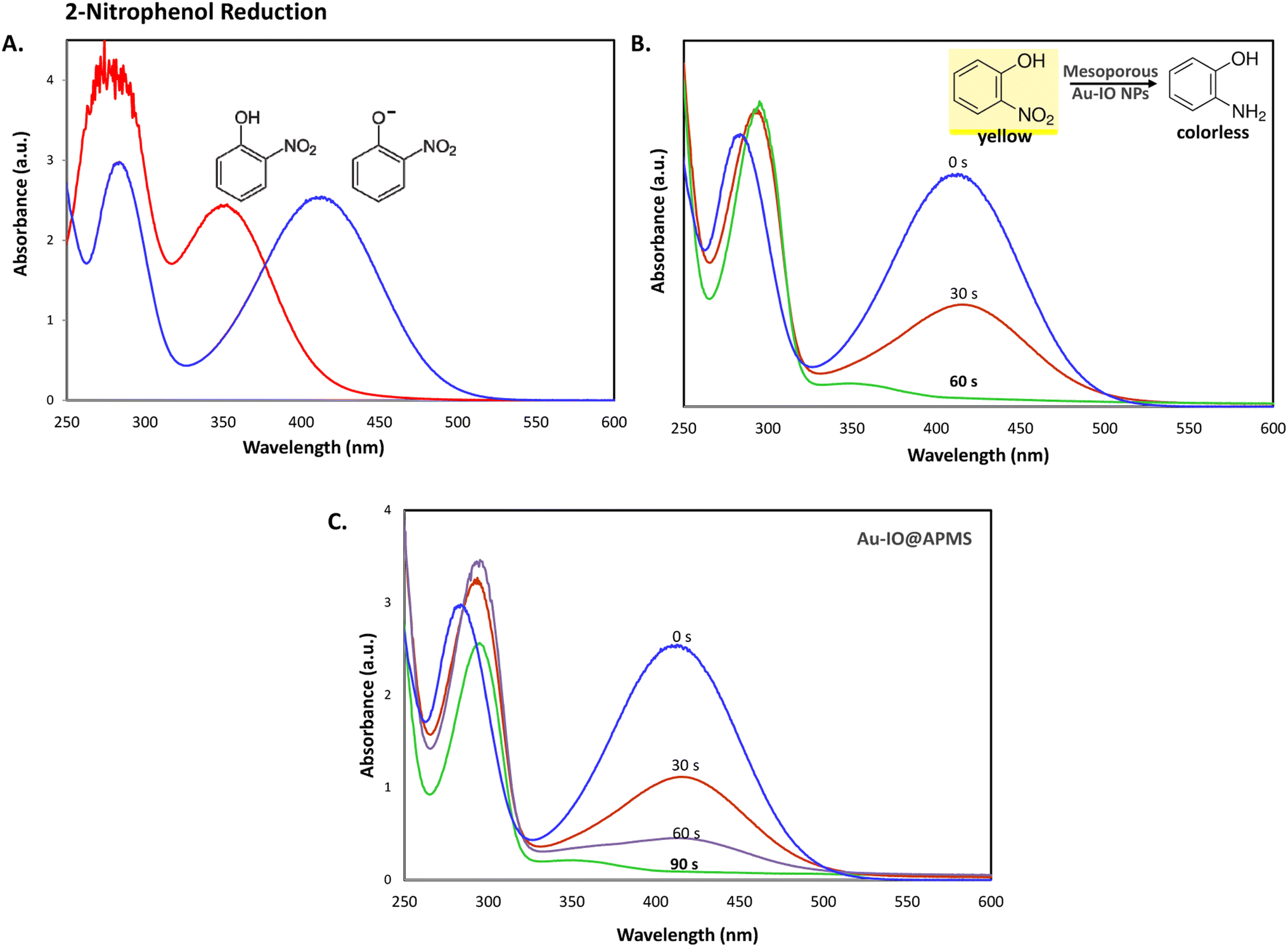
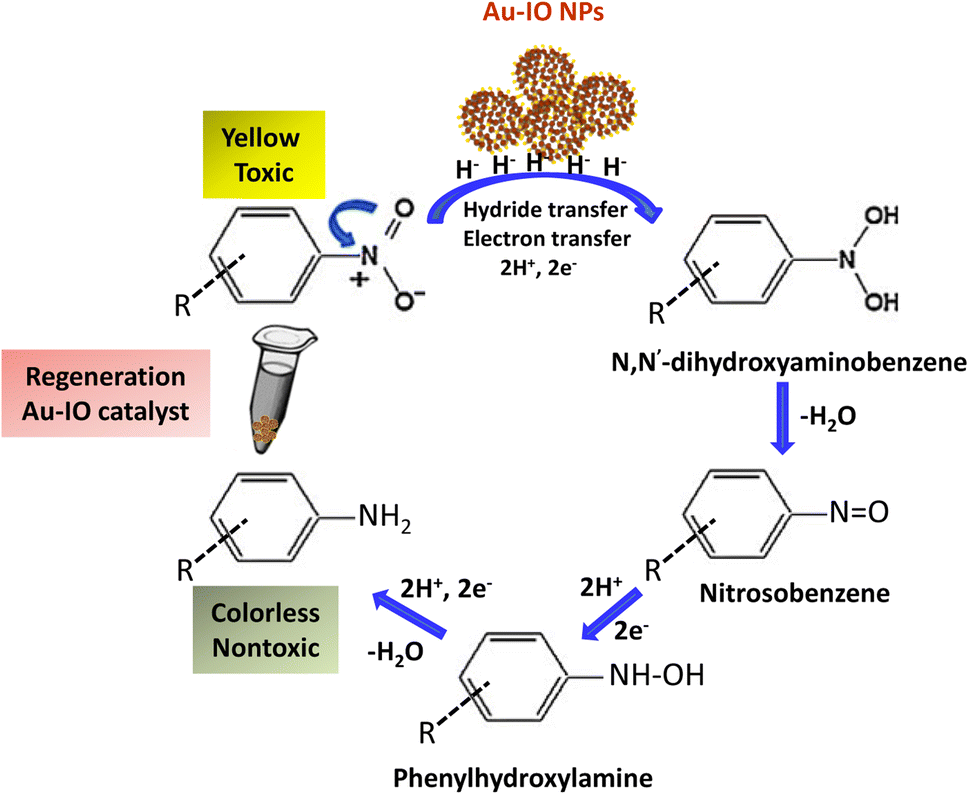
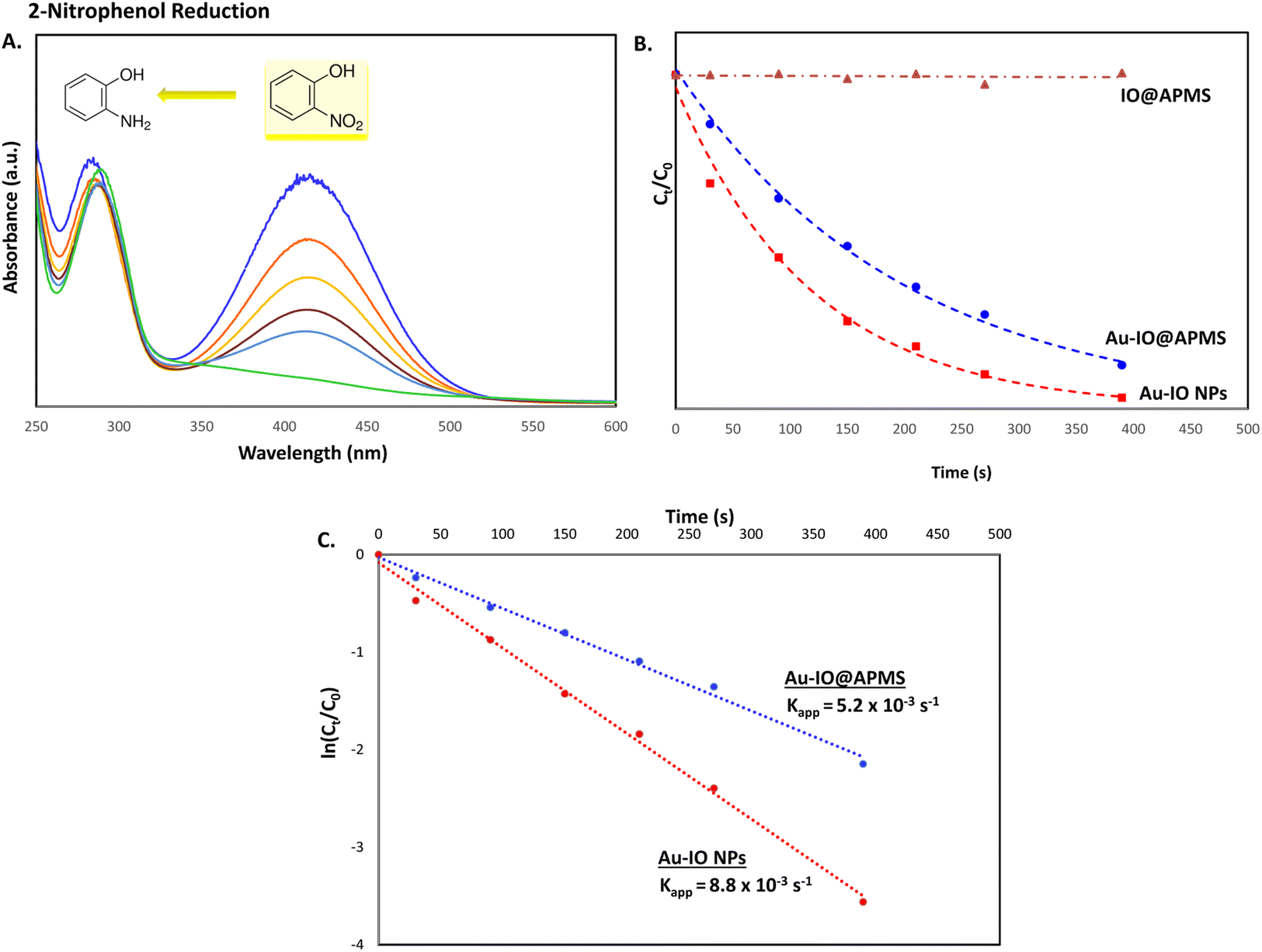
It is well established that at the nano-regime, Au and Au-hybrid NPs are particularly useful for the as-mentioned catalytic conversions. However, bare AuNPs ae usually not that stable, can easily agglomerate through catalytic processes, and are difficult to separate from reaction medium, resulting in clear reduction in catalytic activities. In contrast, the large surface area and pore volume exhibited by the as-prepared Au–IO mesostructures are expected to be beneficial for accommodating substrates, while also providing enhanced diffusion rates of reactant molecules during the catalytic reaction, significantly improving the overall catalytic performance. Furthermore, their intrinsic magnetism aids in their fast separation from the reaction medium by applying external magnet. First, the catalytic performances of the as-synthesized hybrid mesoporous nanocatalysts IO@APMS, Au–IO@APMS, and Au–IO NPs towards 2-nitrophenol reduction to their non-toxic counterparts in aqueous media is shown in Fig. 7. Time-dependent UV-vis absorption was monitored throughout the reductive catalytic activity of 2-nitrophenol to 2-aminophenol in the presence of NaBH4. As shown in Fig. 7a, the absorption peak at 350 nm corresponds to pure 2-nitrophenol aqueous solution. Upon the addition of freshly prepared NaBH4, the absorption peak shifts to 417 nm, with a color change from pale to dark yellow, ensuring the formation of nitrophenolate solution. Without the addition of the Au catalyst, the reduction reaction cannot proceed even in the presence of NaBH4. Remarkably, when only 1.5 mg of the mesoporous Au–IO NP catalyst was added into the reaction mixture, the reaction rapidly progressed to completion in a very short reaction time of only 60 seconds (s) (Fig. 7b). As can be seen, the absorption peak of 2-nitrophenolate anion dramatically decreased, with a simultaneous new and weak band appearing at 297 nm, confirming the formation of a transparent and colorless non-toxic 2-aminophenol solution. When same amount of Au–IO@APMS was used, the reaction rate was found to be slightly slower with completion of the reaction within 90 s (Fig. 7c). Importantly, same concentration of IO@APMS sample with no incorporated Au exhibited no reduction of 2-nitrophenol even after 24 h of reaction time. When compared to only Au-NPs produced in aqueous basic NaBH4 solution, without the incorporation of IO@APMS support, the catalytic efficiencies were much less taking ∼4 minutes to complete the conversions (Fig. S5†). It is to be noted that when using higher concentrations of mesoporous Au–IO NP or Au–IO@APMS (2 mg mL−1), the reduction reaction was completed extremely fast within only few seconds. All these results strongly confirm that the presence of dispersed Au, but not IO, in addition to its confinement onto and within the mesoporous nanocatalyst support (large surface area and pore volume) are directly responsible for the fast catalytic reduction. Mechanistically, it is believed that upon the addition of Au NPs, the electron donor (BH4−) and electron acceptor (4-nitrophenolate) are both adsorbed on the NP surface where the catalytic reduction of nitrophenolate by hydride ions (H−) inducts via Au–hydride complex.45 Au–H and/or Au–BH4 bond formation with the AuNPs has been suggested.38 Then, interfacial electron transfer occurs from hydrides to nitrobenzene where reduction to corresponding aminobenzene involves two fast intermediate steps via nitrosobenzene and phenylhydroxylamine. Finally, desorption of the product takes place from the nanocatalyst surface to make it free for another cycle. It is to be noted that in the absence of the Au catalyst and the presence of the reducing agent NaBH4, the formation of the 2-nitrophenolate ion is observed, but no further conversion to 2-aminophenol occurs with 417 nm peak remaining unaltered for a long period of time. Likewise, in the absence of NaBH4 and the presence of the Au catalyst, the Au-incorporated IO mesostructures showed no catalytic activities. Thus, both the Au and the hydride species (Au–hydride complex) are crucial to promote the reduction of 2-nitrophenol.46 A plausible mechanism for the reduction of nitroarenes using the Au–IO nanocatalyst is depicted in Fig. 8.
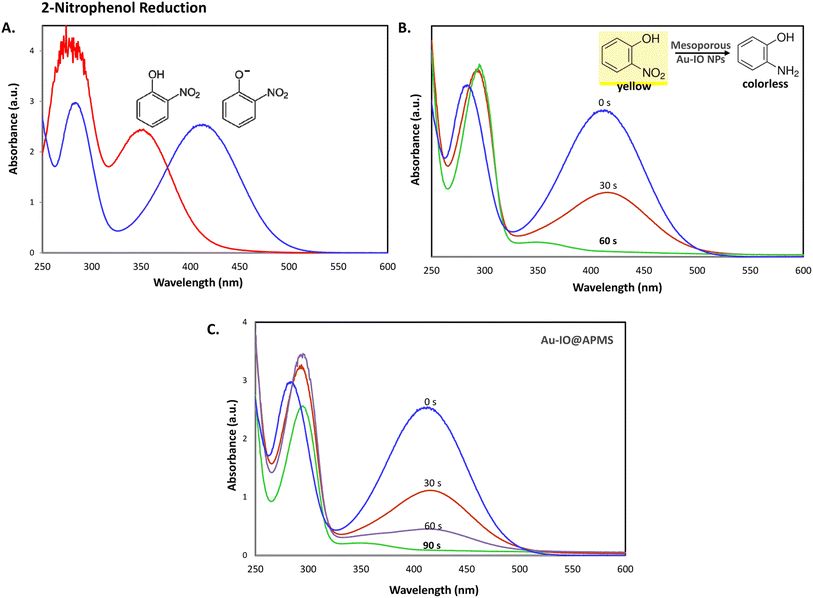 |
| | Fig. 7 (A) UV-vis absorption spectra of 2-nitrophenol before (light yellow) and after (dark yellow) the addition of NaBH4 solution. (B and C) Time progressive UV-vis absorption spectra for 2-nitrophenol reduction using 1.5 mg mL−1 of the mesoporous nanocatalysts (B) Au–IO NP and (C) Au–IO@APMS. Reaction conditions: 2.5 mM of nitroarene solution, 25 mM of freshly prepared NaBH4 (excess), 1.5 mg of catalyst, total volume = 1 mL. | |
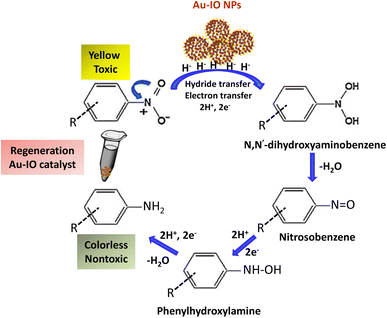 |
| | Fig. 8 A plausible mechanism for catalytic reduction of nitroarenes using mesoporous Au–IO nanocatalysts. | |
The catalytic conversion kinetic rates for reduction of 2-nitrophenol detected using UV-vis were also investigated (Fig. 9a). Since the reducing agent NaBH4 is in high excess relative to 2-nitrophenol, the reaction is expected to be independent of NaBH4 concentration and follow pseudo-first-order kinetics,47 leading to the determination of the rate constant kapp according to the following equation:
where
Ct is the concentration of 2-nitrophenolate at time (
t) and
C0 is the initial concentration of 2-nitrophenolate at
t = 0. The corresponding plots of
Ct/
C0 and ln(
Ct/
C0)
versus reaction time of the various mesostructured-catalyzed reduction reactions are shown in
Fig. 9b and c. From the graphs, it is evident that no reduction was observed for IO@APMS, while Au–IO NP catalyst reduced 2-nitrophenol in slightly quicker times (
kapp = 8.8 × 10
−3 s
−1) compared to mesoporous Au–IO@APMS (
kapp = 5.2 × 10
−3 s
−1).
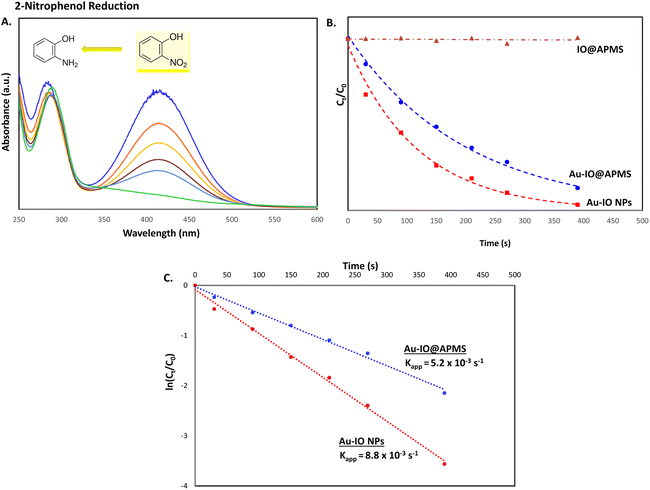 |
| | Fig. 9 (A) Representative time progressive UV-vis absorption spectra for 2-nitrophenol reduction using mesoporous hybrid Au–IO nanocatalysts. (B and C) Kinetic data for 2-nitrophenol reduction using the different hybrid mesoporous IO@APMS, Au–IO@APMS, and Au–IO NP nanocatalysts. (B) Plot of Ct/C0 vs. reaction time; (C) plot of ln(Ct/C0) vs. reaction time. | |
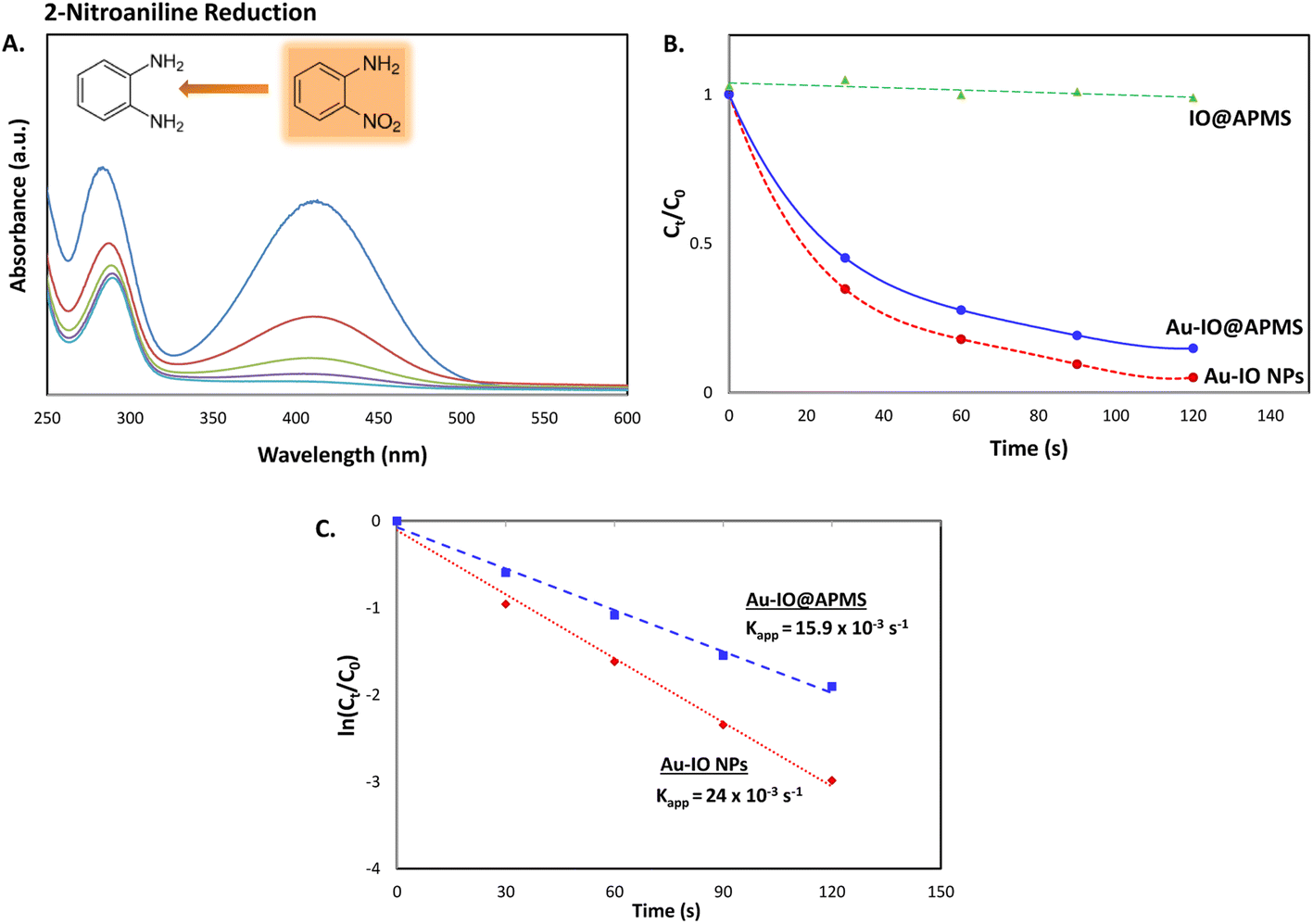
Next, we also studied the use of the hybrid mesoporous Au–IO nanocatalysts for catalytic reduction of 2-nitroaniline monitored by UV-vis spectroscopy (Fig. 10). Although the chemical reduction of nitroaniline,43 a highly toxic pollutant, into the less harmful counterparts is highly needed, its conversion and removal using noble metal-based nanocatalytic systems is studied to much less extent. The 2-nitroaniline orange colored aqueous solution presented two distinguished absorption peaks at 290 and 415 nm (Fig. 10a). In the presence of NaBH4 solution, the intensity of the aforementioned peaks gradually decreased upon addition of mesoporous Au nanocatalysts to 2-nitroaniline/NaBH4 aqueous solution. Similar to 2-nitrophenol reduction reaction, upon adding only NaBH4, no catalytic reduction of 2-nitroaniline to 2-aminoaniline is observed. However, addition of 1.5 mg of mesoporous Au–IO NPs or Au–IO@APMS afforded the non-toxic 2-aminoaniline very fast in 25 and 40 s, respectively (Fig. S6†). The catalytic conversion rates were found to be equal to with kapp = 15.9 × 10−3 s−1 and 23.5 × 10−3 s−1 for the mesoporous Au–IO nanocomposites (Fig. 10b and c). Table 2 shows the computed kinetic catalytic reaction rates of the different mesoporous Au–IO nanocatalysts for reduction of 2-nitrophenol and 2-nitroaniline. Those Au-incorporated mesostructured nanocatalysts were found to have very fast and efficient reduction performance compared to other nanocatalysts reported in the literature, where the hybrid mesoporous Au–IO NPs retaining the fastest catalytic activities. However, extra care should be taken when comparing the efficiencies of different catalysts, as many factors such as nanocatalyst concentrations, amount of the substrate, and overall reaction conditions might be variables. For better quantitative comparison with other systems, the catalytic activity parameter kapp/M (ratio of kapp to the total mass of the catalyst) is typically presented. For instance, the reaction rate constant per unit mass for Au–IO NP towards 2-nitrophenol reduction is calculated to be equal to 17.6 × 10−3 s−1. Table 3 summarizes the catalytic activities of our mesoporous Au–IO NPs compared to other transition metal-based nanoparticulate catalytic platforms used for the reduction of nitroarenes. As depicted, the catalytic activities of the mesoporous Au–IO nanocatalyst developed in this work is considerably greater than those reported in previous works. This affirms the excellent catalytic behavior of our Au-based mesoconstructs due to synergistic factors mainly the dispersed small-sized Au NPs, mesostructures, high surface area, and large pore volumes, facilitating optimum access to Au perimeter active sites, as well as faster transport of the substrates towards the reduction. These interesting results clearly demonstrate that very low concentrations of Au–IO nanocatalysts (only 0.5–1.5 mg) are needed to convert toxic nitroarene solutions to non-toxic counterparts in very short times (only few seconds). This strongly suggests the potential use of the as-prepared hybrid mesoporous Au–IO based nanocatalysts for industrial applications and other catalytic conversion systems.
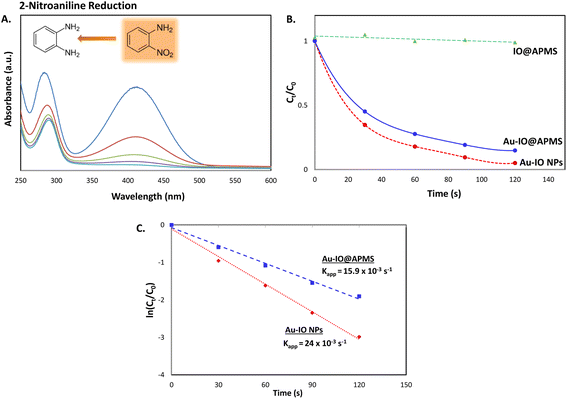 |
| | Fig. 10 (A) Representative time progressive UV-vis absorption spectra for 2-nitroaniline reduction using mesoporous hybrid Au–IO nanocatalysts. (B and C) Kinetic catalytic conversions for 2-nitroaniline reduction using the different mesoporous Au–IO nanocatalysts. (B) Plot of Ct/C0 vs. reaction time; (C) plot of ln(Ct/C0) vs. reaction time. | |
Table 2 Catalytic performance of mesoporous Au nanocatalystsa
| Catalyst |
Substrate |
100% conv. time (sec) |
kapp (s−1) |
kapp/M (s−1 g−1) |
| Reaction conditions: 2.5 mM of nitroarene solution, 25 mM of freshly prepared NaBH4 (excess), 0.5 mg of catalyst, total volume = 1 mL. |
| IO@APMS |
2-Nitrophenol |
No reduction |
N/A |
N/A |
| Au–IO@APMS |
2-Nitrophenol |
60 |
5.2 × 10−3 |
10.4 |
| Mesoporous Au–IO |
2-Nitrophenol |
45 |
8.8 × 10−3 |
17.6 |
| IO@APMS |
2-Nitroaniline |
No reduction |
N/A |
N/A |
| Au–IO@APMS |
2-Nitroaniline |
40 |
15.9 × 10−3 |
31.8 |
| Mesoporous Au–IO |
2-Nitroaniline |
25 |
23.5 × 10−3 |
47 |
Table 3 Catalytic performance of Au nanocatalystsa
| Catalyst |
kapp (s−1) x 10−3 |
kapp/M (s−1 g−1) |
Ref. |
| Reaction conditions: comparison of catalytic activities between mesoporous Au–IO nanocatalyst and other reported Au nanocatalysts for reduction of 4-nitrophenol. Also compare with other state-of-the-art transition metal-based nanocatalysts reported in Table 3.17,49 GO/Fe3O4@PDA/Au stands for Au-NP incorporated polydopamine (PDA)-shelled graphene oxide (GO)/Fe3O4. It is to be noted that reduction of 4-nitrophenol is ∼ 3–4 times faster than that of 2-nitrophenol (i.e. 16 min vs. 60 min for Fe3O4@SiO2–NH2–Au catalyst).49 |
| GO/Fe3O4@PDA/Au |
14.4 |
14.4b |
49 |
| Fe3O4@SiO2–NH2–Au |
7.8 |
2.6b |
17 |
| Fe3O4@SiO2@Aushell |
6.6 |
1.76 |
48 |
| Au@MSNscore–shell |
3 |
N/A |
50 |
| Au@SiO2yolk–shell |
3.9 |
12 |
51 |
| Au@Fe3O4dumbbell |
10.5 |
5.25 |
52 |
| Mesoporous Au–IO |
8.8 |
17.6 |
This workc |
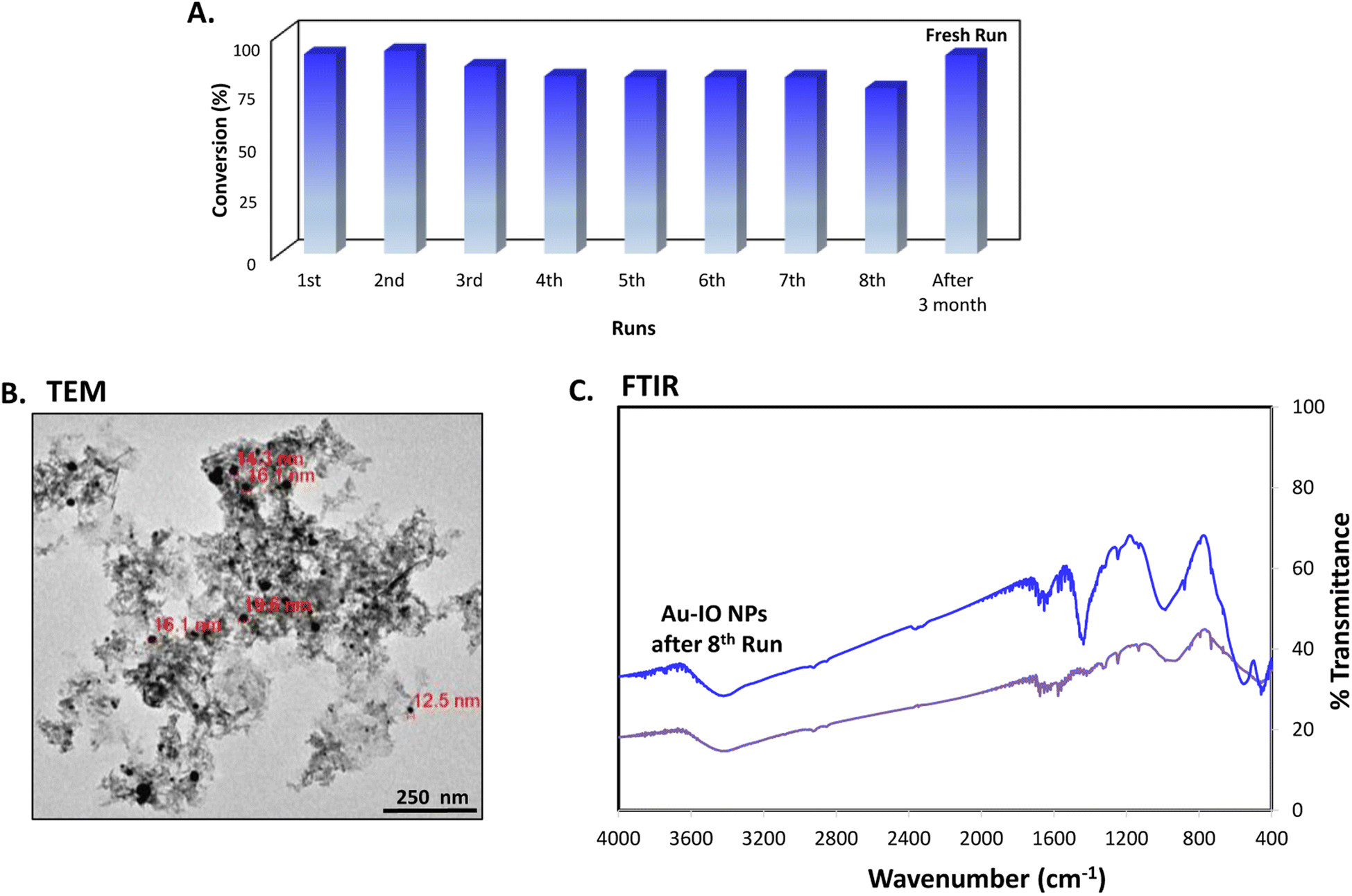
Finally, the stability and recyclability of the mesoporous Au–IO nanocatalyst were also investigated (Fig. 11). It is extremely significant for industrial applications that the utilized nanocatalysts are stable, ready to be used in harsh conditions, and can be recycled many times without the loss in activity. Different strategies have been reported to achieve these goals varying from core-satellite, yolk–shell nanorattle type structures, to magnetic core–gold shells and Au-NPs embedded in core–shell (magnetite-mesoporous silica) microspheres.48 It is therefore imperative to keep enhancing on catalyst stabilities and recyclabilities. Herein, the nanocatalysts were magnetically recovered, washed with water, and then used for the next reduction cycles. As evident, the catalytic conversions didn't stagnate even after the use of the nanocatalyst for 8 consecutive runs (Fig. 11a). The nanocatalyst did not undergo any substantial change in its activity, with only very slight decrease in reaction times noticed during the 8 cycles. This is probably due to adsorption of product (2-aminophenol) over the catalyst surface. To investigate the structural stability of the reused nanocatalyst after the 8th run, TEM and FTIR analysis were conducted. As depicted, the morphology and structural signals were found to be the same as the initial Au–IO NP sample without changes in functionality (Fig. 11b and c). Remarkably, even after 3 months of storage in the fridge at 4 °C, the catalyst efficiently carried out 100% conversion of 2-nitrophenol and 2-nitroaniline within only few seconds, exactly similar to the freshly prepared nanocatalyst samples. This demonstrates the exceptional stability of the mesoporous Au–IO nanocatalyst and that the dispersed AuNPs are still active, validating its outstanding reusability.
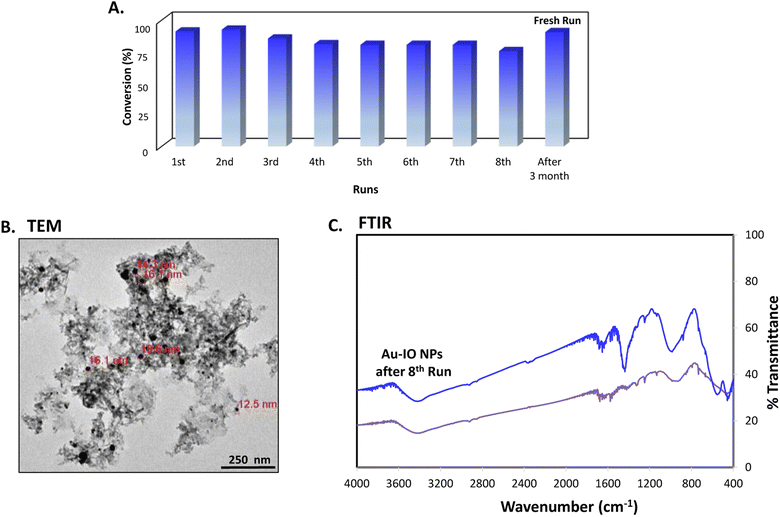 |
| | Fig. 11 (A) Recyclability of the mesoporous Au–IO nanocatalyst against 2-nitrophenol reduction for 8 successive runs along with a fresh run after long storage period (∼3 months). (B) TEM image and (C) FTIR spectra of Au–IO nanocatalysts after the 8th run showing the outstanding stability and reusability of the nanocatalyst. | |
3. Conclusion
In conclusion, we prepared unique hybrid Au–IO mesostructures by nanocasting route using amine-functionalized IO-incorporated APMS (IO@APMS-amine) as the hard silica template. The as-synthesized nanoconstructs were produced with uniform sizes, noticeable aqueous stabilities, good magnetic properties, and excellent porosities. Owing to their high surface areas, large pore volumes, and mesoporous nature (SBET = 124 m2 g−1, Vpore = 0.33 cm3 g−1, and dpore = 4.5 nm), the Au–IO mesoconstructs were evaluated against the catalytic reduction of toxic nitroaromatic compounds. It was found that the mesoporous Au–IO NPs and Au–IO@APMS prepared here act as highly efficient nanocatalysts showing exceptional stabilities (>3 months), high catalytic efficiencies (∼100% conversion within only 60 s for 2-nitrophenol reduction and 25 s for 2-nitroaniline), and excellent recyclability (up to 8 cycles). The catalytic kinetic apparent reaction rate constants (kapp) were calculated to be equal to 8.8 × 10−3 and 23.5 × 10−3 s−1 for 2-nitrophenol and 2-nitroaniline reduction, respectively, faster than any other Au-based nanocatalysts reported earlier. Our results clearly demonstrate that very low concentrations of Au–IO nanocatalysts (only 0.5 mg) are needed to convert toxic nitroarene solutions to non-toxic counterparts in very short times (less than one minute). This is mainly due to synergistic cooperation of the well-dispersed Au, their high surface area, large pore volume, and mesoporous nature allowing substrates to diffuse in and out quickly. This strongly suggests the promising potential for the utilization of the as-prepared hybrid mesoporous Au–IO based nanocatalysts for various industrial catalytic applications.
4. Experimental section
4.1 Materials and methods
Unless otherwise indicated, all chemicals and solvents were obtained from commercial suppliers and used as supplied without further purification. Iron(III) chloride hexahydrate (FeCl3·6H2O), tetrachloroauric acid trihydrate (HAuCl4·3H2O), zinc metal powder, cetyltrimethyl ammonium bromide (CTAB), sodium fluoride (NaF), tetraethyl orthosilicate (TEOS), 3-aminopropyltriethoxysilane (APTES), sodium borohydride (NaBH4), 2-nitrophenol, and 2-nitroaniline were all purchased from UFC Biotechnology and Sigma-Aldrich. Transmission electron microscopy (TEM) were collected on a JEOL-JEM 1400 operating at 120 kV using Gatan camera with Digital Micrograph Imaging software using 400 mesh Formvar/carbon-supported copper grids for sample deposition. SEM images were processed using JSM-7600F JEOL model equipped with EDAX® AMETEK® system. X-ray powder diffraction (XRD) measurements were performed using Rigaku Miniflex 600 model equipped with Cu-Kα radiation (λ = 1.54 Å) and 2θ scan ranging from 0–80°. FTIR spectra (400–4000 cm−1) were recorded as KBr pellet forms using Shimadzu IRAffinity-1. DLS measurements were assessed on Malvern Zetasizer Nano ZS instrument. Magnetic measurements were conducted at room temperature using LakeShore 7404 model VSM having a 1.8 Tesla magnet. Dried samples were exposed to direct current magnetic fields in stepwise increments while swiping the applied field.
4.2 Preparation of mesoporous IO@APMS-amine
APMS silica template was first prepared following our previous work, using CTAB (10 g), H2O (200 mL), conc. HCl (20 mL), TEOS (20 mL), ethanol (70 mL), and NaF (20 mL) heated at 100 °C for 120 min. 1 g of the pore-blocked APMS was then treated with APTES (200 μL) in dry toluene (100 mL) and refluxed while stirring for 4 h. The resulting material was extracted with ethanol 3× in the presence of conc. HCl (1 mL), filtered, and dried under vacuum to yield pore-opened APMS-amine. 300 mg of APMS in 15 mL water was sonicated, followed by the addition of 1 g of FeCl3 aqueous solution (5 mL). Trace amounts of zinc metal was then added and the reaction mixture was vigorously stirred for 24 h at room temperature. A change in color was observed and the resulting material was decanted to separate the zinc, purified by repeated centrifugation/washing steps with water and ethanol (ethanol being the final wash), and finally dried under vacuum. This whole process was repeated using 0.80 g of FeCl3 solution to ensure maximal iron impregnation and filling of the mesopores. The obtained product was then heated slowly to 350 °C at 2 °C min−1 under argon and then left at this temperature for 3 h to afford IO@APMS-amine.
4.3 Preparation of mesoporous Au–IO@APMS
75 mg of IO@APMS-amine was dispersed in 10 mL water, sonicated, followed by slow addition of 40 mM of HAuCl4·3H2O (16 mg in 1 mL water) in 200 μL portions. 0.4 M of NaBH4 (16 mg in 1 mL water) was subsequently added dropwise, where the color of the solution immediately changed to dark blue. Sonication was continued for 1 h and the suspension was collected by centrifugation, washed several times with water and ethanol, and then dried to afford mesoporous Au-incorporated IO@APMS (Au–IO@APMS). For preparation of only Au-NPs (average sizes ∼20 nm), same procedure with exactly same quantitates and volumes were used but without the addition of IO@APMS-amine.
4.4 Preparation of final mesoporous Au–IO NPs
40 mg of the resulting sample was treated twice with hot 5 M NaOH solution for 24 h to completely remove the silica template, purified by successive centrifugation/washing steps, and dried under vacuum to obtain the final mesoporous Au–IO NPs.
4.5 Reduction of 2-nitrophenol or 2-nitroaniline
In the first step, 25 mM of 2-nitrophenol or 2-nitroaniline aqueous solutions (3.5 mg in 1 mL water) was prepared. Then, 100 μL of the above solution was diluted with 800 μL of water. After that, 100 μL of a freshly prepared aqueous 0.25 M NaBH4 solution (10 mg in 1 mL water) was added where a deep yellow color was obtained. Finally, the above solution was mixed with different mesoporous Au-incorporated samples (0.5–2 mg) and the completion of the reaction was monitored using UV-vis absorption spectroscopy. When the mixture turned colorless, the reaction was complete.
Conflicts of interest
The authors declare no competing financial and/or non-financial interests in relation to the work described.
Acknowledgements
The authors acknowledge funding by the Arab Fund for Economic and Social Development (AFESD) – Arab Fund Fellowship Award Program. The authors thank Drs Nawal Madkhali and Saja Algessair (Department of Physics, IMISU) as well as Dr Rizwan Ali (KAIMRC) for conducting SEM-EDX, XRD and TEM, respectively. Special thanks to the whole staff and faculty members at the nanoBIG group, Faculty of Sciences, Universidad Autónoma de Madrid (UAM) for their support and help.
References
- K. C.-F. Leung and S. Xuan, Chem. Rec., 2016, 16, 458–472 CrossRef CAS PubMed.
- C. Hoskins, Y. Min, M. Gueorguieva, C. McDougall, A. Volovick, P. Prentice, Z. Wang, A. Melzer, A. Cuschieri and L. Wang, J. Nanobiotechnol., 2012, 10, 27 CrossRef CAS PubMed.
- Y. Liu, T.-N. Gao, X. Chen, K. Li, Y. Ma, H. Xiong and Z.-A. Qiao, Inorg. Chem., 2018, 57, 12953–12960 CrossRef CAS PubMed.
- J. M. Walker and J. M. Zaleski, Nanoscale, 2016, 8, 1535–1544 RSC.
- C. Lin, K. Tao, D. Hua, Z. Ma and S. Zhou, Molecules, 2013, 18, 12609–12620 CrossRef CAS PubMed.
- K. S. Shin, Y. K. Cho, J.-Y. Choi and K. Kim, Appl. Catal., A, 2012, 413–414, 170–175 CrossRef CAS.
- P. Pachfule, S. Kandambeth, D. Díaz Díaz and R. Banerjee, Chem. Commun., 2014, 50, 3169–3172 RSC.
- V. K. Gupta, N. Atar, M. L. Yola, Z. Üstündağ and L. Uzun, Water Res., 2014, 48, 210–217 CrossRef CAS PubMed.
- P. Wu, P. Bai, Z. Lei, K. P. Loh and X. S. Zhao, Microporous Mesoporous Mater., 2011, 141, 222–230 CrossRef CAS.
- P. Wu, P. Bai, Z. Yan and G. X. S. Zhao, Sci. Rep., 2016, 6, 18817 CrossRef PubMed.
- S. A. C. Carabineiro, N. Bogdanchikova, P. B. Tavares and J. L. Figueiredo, RSC Adv., 2012, 2, 2957–2965 RSC.
- M. E. Ali, M. M. Rahman, S. M. Sarkar and S. B. A. Hamid, J. Nanomater., 2014, 2014, 192038 Search PubMed.
- K. K. R. Datta, B. V. S. Reddy, K. Ariga and A. Vinu, Angew. Chem., Int. Ed., 2010, 49, 5961–5965 CrossRef CAS PubMed.
- S. K. Maji, S. Sreejith, A. K. Mandal, X. Ma and Y. Zhao, ACS Appl. Mater. Interfaces, 2014, 6, 13648–13656 CrossRef CAS PubMed.
- B. Cheng, H. He, T. Huang, S. S. Berr, J. He, D. Fan, J. Zhang and P. Xu, J. Biomed. Nanotechnol., 2016, 12, 435–449 CrossRef CAS PubMed.
- R. K. P. Purushothaman, J. van Haveren, D. S. van Es, I. Melián-Cabrera, J. D. Meeldijk and H. J. Heeres, Appl. Catal., B, 2014, 147, 92–100 CrossRef CAS.
- K. Bhaduri, B. D. Das, R. Kumar, S. Mondal, S. Chatterjee, S. Shah, J. J. Bravo-Suárez and B. Chowdhury, ACS Omega, 2019, 4, 4071–4081 CrossRef CAS PubMed.
- J. Fang, Y. Zhang, Y. Zhou, S. Zhao, C. Zhang, M. Huang, Y. Gao and C. Yang, New J. Chem., 2017, 41, 4448–4457 RSC.
- M. Sankar, Q. He, R. V. Engel, M. A. Sainna, A. J. Logsdail, A. Roldan, D. J. Willock, N. Agarwal, C. J. Kiely and G. J. Hutchings, Chem. Rev., 2020, 120, 3890–3938 CrossRef CAS PubMed.
- S. Tanaka, Y. V. Kaneti, N. L. W. Septiani, S. X. Dou, Y. Bando, M. S. A. Hossain, J. Kim and Y. Yamauchi, Small Methods, 2019, 3, 1800512 CrossRef.
- Y. Liu, C. J. Jia, J. Yamasaki, O. Terasaki and F. Schüth, Angew. Chem., Int. Ed., 2010, 49, 5771–5775 CrossRef CAS PubMed.
- K. El-Boubbou, Nanomedicine, 2018, 13(8), 929–952 CrossRef CAS PubMed.
- J. Zheng, Y. Dong, W. Wang, Y. Ma, J. Hu, X. Chen and X. Chen, Nanoscale, 2013, 5, 4894–4901 RSC.
- L.-F. Gutiérrez, S. Hamoudi and K. Belkacemi, Catalysts, 2011, 1, 97–154 CrossRef.
- Y. Lin, Z. Li, Z. Chen, J. Ren and X. Qu, Biomaterials, 2013, 34, 2600–2610 CrossRef CAS PubMed.
- Y. Guo, D. Gu, Z. Jin, P.-P. Du, R. Si, J. Tao, W.-Q. Xu, Y.-Y. Huang, S. Senanayake, Q.-S. Song, C.-J. Jia and F. Schüth, Nanoscale, 2015, 7, 4920–4928 RSC.
- S. Tanaka, J. Lin, Y. V. Kaneti, S.-i. Yusa, Y. Jikihara, T. Nakayama, M. B. Zakaria, A. A. Alshehri, J. You, M. S. A. Hossain and Y. Yamauchi, Nanoscale, 2018, 10, 4779–4785 RSC.
- Y. V. Kaneti, S. Tanaka, Y. Jikihara, T. Nakayama, Y. Bando, M. Haruta, M. S. A. Hossain, D. Golberg and Y. Yamauchi, Chem. Commun., 2018, 54, 8514–8517 RSC.
- T. García, J. M. López, B. Solsona, R. Sanchis, D. J. Willock, T. E. Davies, L. Lu, Q. He, C. J. Kiely and S. H. Taylor, ChemCatChem, 2019, 11, 1915–1927 CrossRef.
- B. P. Bastakoti, D. Kuila, C. Salomon, M. Konarova, M. Eguchi, J. Na and Y. Yamauchi, J. Hazard. Mater., 2021, 401, 123348 CrossRef CAS PubMed.
- B. Li, Y. Hao, X. Shao, H. Tang, T. Wang, J. Zhu and S. Yan, J. Catal., 2015, 329, 368–378 CrossRef CAS.
- K. El-Boubbou, R. Ali, H. Al-Zahrani, T. Trivilegio, A. H. Alanazi, A. L. Khan, M. Boudjelal and A. AlKushi, Sci. Rep., 2019, 9, 9481 CrossRef PubMed.
- K. El-Boubbou, R. Ali, S. Al-Humaid, A. Alhallaj, O. M. Lemine, M. Boudjelal and A. AlKushi, Pharmaceutics, 2021, 13, 553 CrossRef CAS PubMed.
- K. El-Boubbou, D. A. Schofield and C. C. Landry, J. Phys. Chem. C, 2012, 116, 17501–17506 CrossRef CAS.
- K. El-Boubbou, D. A. Schofield and C. C. Landry, Adv. Healthcare Mater., 2012, 1, 183–188 CrossRef CAS PubMed.
- J. Kim, S. Park, J. E. Lee, S. M. Jin, J. H. Lee, I. S. Lee, I. Yang, J. S. Kim, S. K. Kim, M. H. Cho and T. Hyeon, Angew. Chem., Int. Ed., 2006, 45, 7754–7758 CrossRef CAS PubMed.
- S. I. Stoeva, F. Huo, J.-S. Lee and C. A. Mirkin, J. Am. Chem. Soc., 2005, 127, 15362–15363 CrossRef CAS PubMed.
- C. Deraedt, L. Salmon, S. Gatard, R. Ciganda, R. Hernandez, J. Ruiz and D. Astruc, Chem. Commun., 2014, 50, 14194–14196 RSC.
- J. Piella, N. G. Bastús and V. Puntes, Chem. Mater., 2016, 28, 1066–1075 CrossRef CAS.
- M. N. Martin, J. I. Basham, P. Chando and S.-K. Eah, Langmuir, 2010, 26, 7410–7417 CrossRef CAS PubMed.
- P. Suchomel, L. Kvitek, R. Prucek, A. Panacek, A. Halder, S. Vajda and R. Zboril, Sci. Rep., 2018, 8, 4589 CrossRef PubMed.
- S. K. Ghosh and T. Pal, Chem. Rev., 2007, 107, 4797–4862 CrossRef CAS PubMed.
- M. I. Din, R. Khalid, Z. Hussain, J. Najeeb, A. Sahrif, A. Intisar and E. Ahmed, RSC Adv., 2020, 10, 19041–19058 RSC.
- A. B. Azzam, R. Djellabi, S. M. Sheta and S. M. El-Sheikh, RSC Adv., 2021, 11, 18797–18808 RSC.
- Y. Chen, L. Feng and S. M. Sadeghzadeh, RSC Adv., 2020, 10, 19553–19561 RSC.
- E. C. B. A. Alegria, A. P. C. Ribeiro, M. Mendes, A. M. Ferraria, A. M. B. Do Rego and A. J. L. Pombeiro, Nanomaterials, 2018, 8, 320 CrossRef PubMed.
- P. Hervés, M. Pérez-Lorenzo, L. M. Liz-Marzán, J. Dzubiella, Y. Lu and M. Ballauff, Chem. Soc. Rev., 2012, 41, 5577–5587 RSC.
- Z. u. Rahman, Y. Ma, J. Hu, Y. Xu, W. Wang and X. Chen, RSC Adv., 2014, 4, 5012–5020 RSC.
- S. Hemmati, M. M. Heravi, B. Karmakar and H. Veisi, Sci. Rep., 2021, 11, 12362 CrossRef PubMed.
- J. Chen, R. Zhang, L. Han, B. Tu and D. Zhao, Nano Res., 2013, 6, 871–879 CrossRef CAS.
- J. Lee, J. C. Park and H. Song, Adv. Mater., 2008, 20, 1523–1528 CrossRef CAS.
- F. h. Lin and R. a. Doong, J. Phys. Chem. C, 2011, 115, 6591–6598 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *abc,
O. M. Lemined and
Daniel Jaque
*abc,
O. M. Lemined and
Daniel Jaque