
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of Ca(OH)2 and Na2CO3 through anion exchange between CaCO3 and NaOH: effect of reaction temperature†
Marco Simoni *a,
Theodore Hanein*a,
Chun Long Wooa,
Magnus Nybergb,
Mark Tyrerc,
John L. Provisa and
Hajime Kinoshita*a
*a,
Theodore Hanein*a,
Chun Long Wooa,
Magnus Nybergb,
Mark Tyrerc,
John L. Provisa and
Hajime Kinoshita*a
aUniversity of Sheffield, Department of Materials Science & Engineering, Sheffield, S1 3JD, UK. E-mail: marco.simoni.w@gmail.com; t.hanein@sheffield.ac.uk; h.kinoshita@sheffield.ac.uk
bCEMEX Asia Research AG, Römerstrasse 13, Brügg 2555, Switzerland
cCollegium Basilea, Hochstrasse 51, Basel CH-4053, Switzerland
First published on 11th November 2022
Abstract
The CO2 released upon calcination of limestone accounts for the largest portion of the emissions from the cement, lime, and slaked lime manufacturing industries. Our previous works highlighted the possibility for a no-combustion decarbonisation of CaCO3 through reaction with NaOH solutions to produce Ca(OH)2 at ambient conditions, while sequestrating the process CO2 in a stable mineral Na2CO3·H2O/Na2CO3. In this study, the effect of temperature was assessed within the range of 45–80 °C, suggesting that the process is robust and only slightly sensitive to temperature fluctuations. The proportioning of the precipitated phases Na2CO3·H2O/Na2CO3 was also assessed at increasing NaOH molalities and temperatures, with the activity of water playing a crucial role in phase equilibrium. The activation energy (Ea) of different CaCO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NaOH:H2O systems was assessed between 7.8 kJ·mol−1 and 32.1 kJ·mol−1, which is much lower than the conventional calcination route. A preliminary energy balance revealed that the chemical decarbonisation route might be ∼4 times less intensive with respect to the thermal one. The present work offers a further understanding of the effect of temperature on the process with the potential to minimise the emissions from several energy-intensive manufacturing processes, and correctly assess eventual industrial applicability.
:NaOH:H2O systems was assessed between 7.8 kJ·mol−1 and 32.1 kJ·mol−1, which is much lower than the conventional calcination route. A preliminary energy balance revealed that the chemical decarbonisation route might be ∼4 times less intensive with respect to the thermal one. The present work offers a further understanding of the effect of temperature on the process with the potential to minimise the emissions from several energy-intensive manufacturing processes, and correctly assess eventual industrial applicability.
1 Introduction
The calcination of limestone to give lime (CaCO3 → CaO + CO2) is a crucial step for the production of Portland cement (PC), lime (CaO), slaked lime (Ca(OH)2), and soda ash (Na2CO3), whose worldwide markets exceed 4 Gt,1 ∼55 Mt, ∼15 Mt,2 and 50 Mt3 per year, respectively. Given the heavy carbon footprint of such calcination step,4 these industrial realities are responsible for a significant portion of the total CO2 emissions worldwide.1,5,6 Depending on the targeted product, the very energy-intensive7 calcination of limestone might occur in different calcination designs, with varying efficiencies and carbon emissions;8 specifically, 0.8, 1.0–1.8, and 0.4 t CO2 are emitted to produce 1 t of cement, lime/slaked lime, and soda ash, respectively. However, independently from the calcination unit considered, the emissions arising from this step arise from both the fuel-derived and process CO2. While the process CO2 results from the stoichiometry of CaCO3 decomposition, the fuel-derived emissions arise from combustion of fuels (primarily fossil fuels) necessary to achieve the calcination temperature (above 900 °C).9 The achievement of those environmental goals set by the UN in 2015 (at least 80% of CO2 emission cut from the industry sector by 205010) requires a significant contribution from the cement, lime, and soda ash industries. To achieve this, the application of carbon capture and storage (CCS) technologies11 and the use of alternative fuels12 are expected to be the best candidates; however, both these solutions assume that the thermal calcination of limestone (and the process CO2) is unavoidable. In contrast to this, recent investigations13–16 highlighted the possibility for alternative decarbonisation routes which apply no-combustion approaches for the synthesis of the essential chemical CaO17 from CaCO3. As far as we are aware, the chemical decarbonisation route here characterised represents the only alternative for the sustainable co-synthesis of Ca(OH)2 and Na2CO3. The stoichiometry of the key-reaction is reported in eqn (1); it occurs at ambient conditions and implies the reaction between CaCO3 and aqueous NaOH solutions.| CaCO3 + 2NaOH + xH2O → Ca(OH)2 + Na2CO3·xH2O | (1) |
The process is relatively simple; however, the full fundamental understanding of the system is necessary to determine the feasibility of any scaled-up industrial process, and the reaction rate is assessed in this work. The reaction rate may be defined through eqn (2),18 where the variables t, T, and α (with 0 < α < 1) represent the reaction time, temperature, and extent of reaction, respectively.18 The function f(α) represents the kinetic reaction model depending on the mechanism assessed,19 while f(T) reflects the Arrhenius equation (eqn (3)), depending on the temperature (T), the activation energy (Ea), and the pre-exponential factor (A).
| ∂α/∂t = f(T) × f(α) | (2) |
 | (3) |
Despite the mild temperature range here considered (45–80 °C), it is crucial to assess the response of a given process to eventual fluctuations; ideally, a robust route would not be significantly affected by these variations in the processing parameters. The title study aims to gain a deeper insight into the behaviour of the system considered at increasing temperatures, therefore providing essential information for any eventual scale-up.
2 Materials and methods
2.1 Experimental procedure
The materials used in this work are reagent grade chemicals: Sigma-Aldrich CaCO3 (≥99%), Honeywell Fluka NaOH (≥97%), and distilled water. The ternary composition (wt%) 26.9 (CaCO3):32.7 (NaOH):40.4 (H2O) was first investigated, allowing for a 0.86 conversion efficiency (α) in a previous study;16 therefore, a 20 M NaOH (mol kgH2O−1) was first considered. The relatively high water to solids ratio (1.5 g g−1) ensured homogeneous mixing of the solid powders even at milder stirring conditions, as described below, with respect to a previous characterisation.14
The experimental set up is illustrated in Fig. 1. The 20 m NaOH solutions were prepared in a polypropylene plastic vessel, and placed in the pre-heated water bath for 30 min at the target temperatures Tk of 45 °C, 60 °C, or 80 °C.
 | ||
| Fig. 1 Illustration of the experimental set up discussed above, highlighting the indirect temperature control on the reaction system by setting the surrounding water environment at a Tk temperature through external thermocouple. | ||
Direct contact between the hot plate and the bottom of the reaction vessel was avoided through the placement of an insulating glass disk. The CaCO3 powders were also pre-heated at 80 °C for 2 hours to remove excess water. The CaCO3 was then quickly added to the NaOH solutions and manually mixed, at first, to prevent the agglomeration of the solids. Subsequently, the vessel was covered with a lid to minimise the loss of water and heat throughout the reaction; a magnetic stirring of 300 rpm was set up at increasing residence times of 1, 2, 3, 4 and 5 min. Longer residence times were not considered, since our previous study14 highlighted that negligible reaction progression would occur beyond 5 min. The reaction was also performed in non-isothermal conditions, upon initial heating of the NaOH solutions at the targeted temperatures Ti of 45 °C, 60 °C, and 80 °C (to be referred as 45 °C_5 min_RT, 60 °C_5 min_RT, and 80 °C_5 min_RT, respectively). A residence time of 5 min was solely considered to produce these samples.
Upon reaction, all the pastes were mixed with methanol (30 mL) for further 5 min, ensuring the removal of the unreacted NaOH, whose solubility in methanol is 238 g L−1;20 whereas, the negligible solubility of CaCO3,21 Ca(OH)2,22 Na2CO3 (ref. 23) and Na2CO3·H2O23 in methanol would ensure an unmodified solid phase assemblage upon washing. The solid products and the liquid streams could then be collected separately using Buchner filtration. The solids were dried at 80 °C for 2 h, before being manually ground and sieved below 63 μm for subsequent characterisation though thermogravimetry and X-ray diffraction.
An additional set of experiments was conducted to estimate the Ea of the decarbonisation reaction at different ternary compositions (Table 1), selected from our previous study.16 The same procedure described above was applied here, with the solids being reacted for 1 min at constant temperatures Tk of 30 °C, 45 °C, 60 °C and 70 °C. The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
| ID | NaOH (wt%) | CaCO3 (wt%) | H2O (wt%) | NaOH/CaCO3 (mol mol−1) | H2O/CaCO3 (g g−1) | H2O/NaOH (mol mol−1) | α16 |
|---|---|---|---|---|---|---|---|
| NaOH_10m | 14.3 | 14.3 | 71.4 | 2.5 | 3.0 | 5.5 | 0.07 |
| NaOH_12m | 24.5 | 24.5 | 51.0 | 2.5 | 2.1 | 4.6 | 0.46 |
| NaOH_15m | 30.1 | 20.0 | 49.9 | 3.8 | 2.5 | 3.7 | 0.69 |
| NaOH_17m | 37.2 | 8.1 | 54.7 | 11.5 | 6.7 | 3.3 | 0.96 |
2.2 Characterisation techniques
 | ||
| Fig. 2 Generic temperature-trend of the TG data (wt%) and DTG data (wt%/⁰C), red and black lines, respectively, showing the weight loss events of the sample associated with: Na2CO3·H2O, Ca(OH)2, and CaCO3. | ||
The measurement was repeated 6 times for each sample to estimate the measurement error (±0.16 wt%, ± 0.10 wt% and ± 0.16 wt% for Na2CO3·H2O, Ca(OH)2 and CaCO3, respectively).
 | (5) |
3 Theory
This investigation aims to provide the scientific community with a deeper insight into the chemistry and energetic demand of a novel CaCO3 decarbonisation route,16 with the potential to lead the transition towards sustainable cement, lime, and soda ash industries. To highlight this, an energetic comparison with the state-of-the-art processes is presented in Section 5, highlighting the potential advantages of the alternative one. To do that, the concomitant by-production of soda ash Na2CO3 was also considered, whose conventional manufacturing route through Solvay process requires a high energy supply. This early-stage investigation, together with other ones focusing on the processing conditions14 and the nature of the raw materials used,15 will provide a strong base to the potential scale-up of the process. In these terms, future work will aim to design a laboratory-scale process simulating the ideal industrial one, providing a stronger dataset to assess the effective energy requirements of the route, and therefore allowing for a proper techno-economic analysis.4 Results and discussion
4.1 Effect of temperature and CO2 capture
The XRD analysis of the 45 °C_n, 60 °C_n, and 80 °C_n samples are shown in Fig. 3A–C, respectively. | ||
| Fig. 3 XRD patterns for the samples produced at a constant temperature of 45 °C (A), 60 °C (B), and 80 °C (C), and increasing residence times; the patterns also include those samples produced at ambient conditions and with an initial temperature of 45 °C, 60 °C, and 80 °C. | ||
Upon reaction, only Ca(OH)2 (ICSD collection code: #191851), Na2CO3 (ICSD collection code: #5009), Na2CO3·H2O (ICSD collection code: #1852), and unreacted CaCO3 (ICSD collection code: #80869) could be detected, with respective main reflection angles at 2θ of 29.5°,27 34.1°,28 16.9° (ref. 29) and 30.1°.30 No additional phases were detected, suggesting the absence of secondary and competing reactions. The TG data for the 45 °C_n, 60 °C_n and 80 °C_n series is shown in Fig. 4A–C, respectively.
 | ||
| Fig. 4 TG/dTG analysis for the samples series 45 °C_n (A), 60 °C_n (B), and 80 °C_n (C) showing the weight losses in the temperature ranges of 50–130 °C, 310–470 °C and 560–800 °C corresponding to the presence of Na2CO3·H2O, Ca(OH)2 and CaCO3, respectively. | ||
The phase quantification was performed through the weight losses observed in the TG data for CaCO3 (at 560–800 °C), Ca(OH)2 (at 310–470 °C) and Na2CO3·H2O (at 50–130 °C). The estimated quantities for all the samples discussed are summarised in Table 2, where the processing conditions used are also reported.
| Sample ID | CaCO3 (wt%) | Ca(OH)2 (wt%) | Na2CO3·H2O (wt%) | Na2CO3 (wt%) | LOI (%) | Na2CO3·xH2O/Ca(OH)2 (mol%/mol%) |
|---|---|---|---|---|---|---|
| 45 °C_1 min | 13.7 | 34.5 | 6.6 | 45.2 | 15.4 | 1.1 |
| 45 °C_2 min | 6.4 | 35.6 | 23.6 | 34.4 | 14.9 | 1.1 |
| 45 °C_3 min | 4.2 | 34.3 | 55.9 | 5.6 | 18.3 | 1.1 |
| 45 °C_4 min | 4.8 | 32.9 | 59.6 | 2.7 | 18.8 | 1.1 |
| 45 °C_5 min | 3.2 | 33.5 | 61.6 | 1.7 | 18.5 | 1.1 |
| 45 °C_RT_5 min | 4.6 | 34.7 | 57.0 | 3.7 | 18.7 | 1.1 |
| 60 °C_1 min | 8.9 | 34.8 | 15.1 | 41.2 | 14.6 | 1.1 |
| 60 °C_2 min | 4.5 | 36.8 | 18.0 | 40.7 | 13.5 | 1.1 |
| 60 °C_3 min | 4.2 | 37.7 | 8.6 | 49.5 | 12.2 | 1.1 |
| 60 °C_4 min | 4.3 | 36.7 | 8.9 | 50.1 | 15.0 | 1.1 |
| 60 °C_5 min | 3.1 | 37.8 | 11.1 | 48.0 | 12.2 | 1.1 |
| 60 °C_RT_5 min | 3.2 | 33.5 | 61.6 | 1.7 | 18.5 | 1.1 |
| 80 °C_1 min | 8.5 | 34.7 | 11.1 | 45.7 | 13.8 | 1.2 |
| 80 °C_2 min | 6.1 | 35.9 | 11.0 | 47.0 | 13.0 | 1.2 |
| 80 °C_3 min | 5.2 | 36.1 | 6.6 | 52.1 | 12.0 | 1.2 |
| 80 °C_4 min | 5.5 | 36.1 | 7.9 | 50.5 | 12.3 | 1.2 |
| 80 °C_5 min | 5.2 | 35.8 | 11.2 | 47.8 | 12.6 | 1.2 |
| 80 °C_RT_5 min | 6.0 | 35.2 | 15.0 | 43.8 | 13.4 | 1.2 |
It must be mentioned that the content of Na2CO3 was calculated by subtracting the sum of the other quantified phases Ca(OH)2, CaCO3 and Na2CO3·H2O from the total mass (100%). In fact, since Na2CO3 would start decomposing above 851 °C,31 it could not be directly quantified by TG analysis (up to 800 °C) through the detection of the relevant peak. A higher content of Na2CO3 could also be suggested by the lower LOI registered for samples with similar reaction efficiencies (Table 2). This aspect will be extensively discussed in Section 3.2. However, the quantification was considered reliable since the XRD analysis confirmed the absence of any additional phases in the solid products. Moreover, the ratio between the precipitated Na2CO3·H2O/Na2CO3 and Ca(OH)2 (mol%/mol%) revealed a good accordance to the stoichiometry expressed in eqn (1) (Table 2). In fact, 1 mol of both Ca(OH)2 and Na2CO3·H2O/Na2CO3 should precipitate for each mol of CaCO3 reacted, and the resulting molar ratio between Na2CO3·H2O/Na2CO3 and Ca(OH)2 was expected to be close to unity. Specifically, the ratios were suggesting a slight over-precipitation of Na2CO3·H2O and Na2CO3 with respect to Ca(OH)2 for all the systems studied and that could possibly be reflecting the distribution of the positive (Ca2+) and negative (CO32−) charged sites on the surface of the CaCO3. Statistically, a 27% excess of negatively charged sites may be found on the CaCO3 surface,32 justifying the greater affinity of the CaCO3 to interact with the cationic species Na+ in the liquid bulk to form Na2CO3·H2O/Na2CO3.
Based on the TG data, the extent of reaction (α) was calculated for each system, and the outcomes are reported in Fig. 5.
 | ||
| Fig. 5 Extent of reaction (α) for the samples at different temperatures (45, 60 and 80 °C)and increasing residence times, based on TG analysis; the outcomes are also reported for those samples produced without actively maintaining the temperature throughout the reaction. | ||
The conversion of CaCO3 was high (0.7 < α < 0.8) in the tested reaction conditions, in line with the qualitative XRD data (Fig. 3A–C), showing progressive decrease in the intensity of the CaCO3 main peak at 29.5° 2θ. During the first minute, the system temperature had a significant impact on the extent of the reaction; enhanced conversion efficiencies were gained at higher temperatures, while limited effects were observed at longer residence times. It seems likely that the higher conversion registered at short residence times and higher temperatures could be linked to the lower viscosity of the NaOH solutions, favouring the ionic mobility and the enhanced interaction between the dissolved species and the solid surface and bulk.
The efficiency of the system may also be expressed in terms of CO2 capture, expressed as moles of CO2 precipitated as Na2CO3·xH2O per second of reaction progression. As reported in Fig. 6, the CO2 capture rate was decreasing from ∼4.5 × 10−4 mol sec−1 of CO2 in the first minute of reaction down to two orders of magnitude below (∼10−6 mol sec−1 of CO2) after 5 min of contact time. In other terms, around the 80% of the total process CO2 initially introduced was effectively captured after 1 min of reaction.
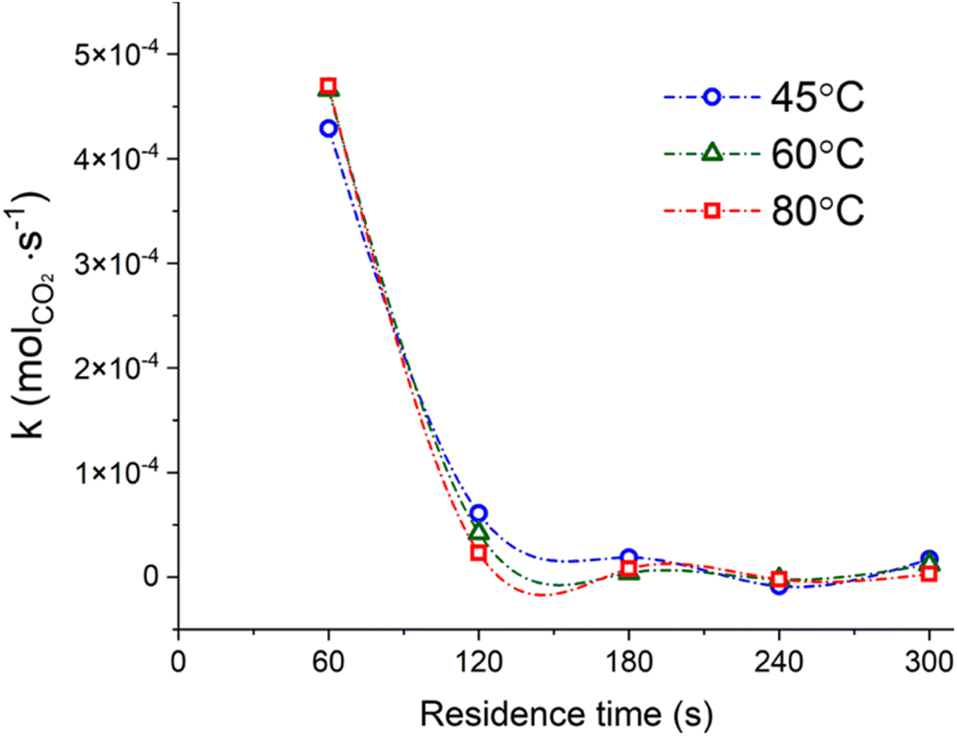 | ||
| Fig. 6 CO2 capture rate, expressed in moles of CO2 precipitated as Na2CO3·xH2O per second of reaction progression, for all the residence times considered at constant temperature; the samples reacted at ambient conditions were not considered for this graph. The lines only work as a guide for the eye. | ||
The samples reacted at ambient conditions (45 °C_5 min_RT, 60 °C_5 min_RT, and 80 °C_5 min_RT) indicated extent of reactions like those reacted at a constant temperature after 5 min (Fig. 5). Decreasing temperature trends could be detected for 45 °C_5 min_RT, 60 °C_5 min_RT, and 80 °C_5 min_RT, with final temperatures of 21.5, 42.1, and 53.6 °C, respectively, after 5 min of residence time. Evidently, the temperature of the system was not significantly influencing the progression of the reaction for a residence time of 5 min.
4.2 Partitioning of Na2CO3·H2O and Na2CO3
The reaction at different conditions resulted in the sequestration of the process CO2 through precipitation of Na2CO3·H2O and Na2CO3 in different proportion, with the x value (eqn (1)) equal to 1 and 0, respectively. Based on the data obtained from TGA (Fig. 4A–C), the molar fraction ν of precipitated Na2CO3, with 0 < ν < 1, was calculated by dividing the moles of Na2CO3 in the samples by the total moles of Na2CO3·H2O and Na2CO3. As introduced in Section 2.1, most samples were produced at a constant temperature (Tk = 45 °C, 60 °C, and 80 °C) whereas others were only initially set up at targeted temperature values (Ti = 45 °C, 60 °C, and 80 °C), following which the system was left at ambient conditions and was allowed to achieve Tf (with Tf < Ti). As reported in Fig. 7, the precipitation of Na2CO3 was dominant at Tk = 45 °C up to 2 min of reaction; beyond that, the precipitation of Na2CO3·H2O was larger despite the constant temperature. Whereas, when considering the systems at Tk = 60 °C and Tk = 80 °C, the precipitation of Na2CO3 was dominant for all the residence times investigated. | ||
| Fig. 7 Molar fraction ν of Na2CO3, expressed as molar ratio between Na2CO3 and Na2CO3·H2O, for the samples produced at 45, 60 and 80 °C at increasing residence times. | ||
Different outcomes were observed when considering the samples reacted at ambient conditions, suggesting a key-role of the temperature on the equilibrium between Na2CO3·H2O and Na2CO3. In fact, while Na2CO3·H2O was dominant at Ti = 45 °C and Ti = 60 °C, with respective Tf values of 21.5 °C and 42.1 °C, Na2CO3 was the main species precipitating at Ti = 80° (Tf = 53.5 °C). Likely, such a different proportion gained at different values of Ti and Tf might be explained by considering the standard enthalpy of reaction ΔHR for x = 0, 1 in eqn (1). The calculation was performed by application of the Hess law and considering standard enthalpies of formation of −1207.4, −426.7, −285.8, −986.1, −1429.7, and −1129.2 kJ mol−1 for CaCO3,33 NaOH,34 H2O,34 Ca(OH)2,34 Na2CO3·H2O,33 and Na2CO3,33 respectively. It appears that the precipitation of Na2CO3·H2O is enhanced at lower temperatures (ΔHR = −69.2 kJ mol−1) than that of Na2CO3 (ΔHR = −54.5 kJ mol−1), justifying the results just discussed. The XRD analysis (Fig. 3A–C) was qualitatively in accordance with the quantification of Na2CO3·H2O and Na2CO3 gained from TGA for both the samples reacted at constant and varying temperatures. In fact, low contents of Na2CO3·H2O (main peak at 16.9° 2θ) were suggested for all the samples reacted at Tk = 80 °C, whereas increased intensities were observed for the Tk = 45 °C series above 2 min of residence time and the sample reacted at ambient conditions with Ti = 60 °C.
The equilibrium between Na2CO3·H2O and Na2CO3 was studied by performing targeted simulations of a simplified system using the PHREEQC software35 with the PITZER(2018) database; an overview of the outcomes is reported in Fig. 8A and B.
 | ||
| Fig. 8 PHREEQC simulation showing the molar ratio between the precipitated Na2CO3 and Na2CaO3·H2O (A) and the activity of water aH2O (B) at 45, 60 and 80 °C and increasing NaOH molalities, up to 20 m; the concentration C0 (B) indicates the initial NaOH concentration considered for the samples here discussed. | ||
The simulations were conducted for a system composed of 1 mole of Na2CO3·H2O and 1 mole of Na2CO3, both dissolved in 0.1 kg of water at Tk = 45 °C, Tk = 60 °C, and Tk = 80 °C, with increasing concentrations of NaOH up to 20 m. The simulated system does not contain Ca2+ ions as in the experimentally tested systems but it is useful to understand the general behaviour of Na2CO3·H2O and Na2CO3 in highly concentrated NaOH solutions. As reported in Fig. 8A, Na2CO3·H2O and Na2CO3 are the dominant species at [NaOH] < 10 m and [NaOH] > 12.5 m, respectively, while the co-precipitation of both species occurs at 10 m < [NaOH] < 12.5 m. Such behaviour might likely depend on the activity of water, which lowers at increasing solute concentrations and system temperature.36 Given the higher proportion of Na2CO3 at Tk = 60 °C and Tk = 80 °C with respect to Tk = 45 °C, it is likely that such phase is favoured at a lower activity of water, i.e. higher temperature. However, an unexpected trend is shown in Fig. 7 for the samples reacted at Tk = 45 °C; in fact, Na2CO3 was the main phase up to 2 min of reaction, whereas the content of Na2CO3·H2O sharply increased beyond that. Likely, the consumption of alkalinity related to the progression of eqn (1) resulted in a lower activity of water, allowing for the precipitation of Na2CO3·H2O rather than Na2CO3. In fact, a higher conversion extent (α = 0.89) was registered after 3 min of reaction, if compared with the outcome after 1 min (α = 0.72) and 2 min (α = 0.85). Most likely, a low activity of water could be maintained at Tk = 60 °C and Tk = 80 °C, even upon enhanced reaction progression, i.e. alkalinity consumption. Whereas the alkalinity drop occurring at an enhanced conversion extent was enough to increase the activity of water in those systems reacted at Tk = 45 °C. This is also confirmed by the experiments conducted at ambient conditions, since the system at Ti = 60 °C mostly formed Na2CO3·H2O at the end of the reaction, when Tf = 42.1 °C was registered.
A deeper insight allows for the explanation of the dynamic situation occurring at 10 m < [NaOH] < 12.5 m; in such interval, the system achieves a transitionary aH2O value of 0.603. That corresponds to a chemical potential u of −241.3 kJ mol−1 (eqn (6)), where u0 is the standard chemical potential of formation of pure water,37 R the gas constant, and T the temperature in K.
|
u = u0 + RTlnaw
| (6) |
At this point, the transition Na2CO3·H2O/Na2CO3 is swapped (Fig. 8A), promoting the precipitation of Na2CO3·H2O and Na2CO3 above and below an activity of water of 0.603, respectively. To explain the constant water activity value of 0.603, the gradual formation of Na2CO3 to the detriment of Na2CO3·H2O must be considered; as a result, water is released into the liquid bulk and it counterbalances the further addition of NaOH. In fact, when all the sodium into the system is converted to Na2CO3 and no more H2O is released by the dissolution of Na2CO3·H2O, the activity of water rapidly drops down to just above 0.400 at NaOH 20 m. That corresponds to a higher water activity aH2O within the liquid phase of the system (Fig. 8B), and therefore resulting in a higher proportioning of Na2CO3·H2O with respect to Na2CO3 (Fig. 8A).
4.3 Calculation of Ea
An additional set of experiments was conducted to estimate the activation energy Ea of the decarbonisation reaction at the different starting compositions reported in Table 1, selected from our previous study.16 Based on the amount of CaCO3 and Ca(OH)2 in the reaction products estimated from their TGA data (ESI I–IV†), the extent of reaction (α) was calculated for each reaction. As reported in Fig. 9, beneficial effects of raising temperatures were observed at different levels depending on the starting composition. | ||
| Fig. 9 Conversion extent values (α) of the samples NaOH_10m_n, NaOH_12m_n, NaOH_15m_n and NaOH_17m_n plotted against the temperature initially set up and kept constant throughout the reaction. | ||
In these terms, the effects of temperature (T) is commonly expressed in the reaction rate constant (k) using Arrhenius equation (eqn (7)),38 depending on the pre-exponential factor (A), activation energy (Ea) and gas constant (R).
|
k = Ae(−Ea/RT)
| (7) |
The rate constant k is a change in the extent of reaction (α) per unit time (t). Since the reactions in this sub-set of experiments were all conducted for 1 min, the extent of reaction obtained (Fig. 9) was directly used in eqn (7) as representative of the reaction rate, and thus:
|
α = Ae(−Ea/RT)
| (8) |
Taking the natural logarithm, eqn (8) can also be expressed as:
 | (9) |
By plotting lnα against T−1 (Fig. 10), the activation energy Ea can be calculated from the slope (−Ea/RT); the experimental data for NaOH_10m, NaOH_12m, NaOH_15m, and NaOH_17m led to the fitting eqn (10)–(13), respectively.
| y = 3865.1x + 10.254 R2 = 1.00 | (10) |
| y = 2054.3x + 5.638 R2 = 0.91 | (11) |
| y = 1242.1x + 3.507 R2 = 0.94 | (12) |
| y = 933.3x + 2.591 R2 = 0.93 | (13) |
 | ||
| Fig. 10 Linearized Arrhenius plot showing the correlation between lnk and −1/T (K−1) for the samples series NaOH_10m_n, NaOH_12m_n, NaOH_15m_n and NaOH_17m_n, reflected by the equations, 9, 10, 11, and 12, respectively. | ||
Based on these, the apparent activation energies for NaOH_10m, NaOH_12m, NaOH_15m and NaOH_17m are estimated as 32.1, 17.1, 10.3 and 7.8 kJ mol−1, respectively, and therefore reflecting the limited kinetics at lower NaOH molality. Despite that, lower activation energies were highlighted here, with respect to the conventional calcination route of CaCO3 under inert atmospheres (164–225 kJ mol−1 39) and with varying CO2 partial pressures (213.3–2142.2 kJ mol−1 40). Naturally, this first comparison does not consider the embodied energy required to produce the required NaOH to carry out the chemical calcination of CaCO3. For this reason, the next section will introduce crucial aspects to fully consider when assessing its real feasibility from an industrial point of view.
5 Industry-oriented considerations
The authors understand that a proper life-cycle assessment needs to be done to understand the industrial feasibility of this novel chemical decarbonisation process, but this is not the aim of the present investigation. Nevertheless, some crucial considerations are reported below to better highlight the potential of the decarbonisation route presented here.It is highlighted that eventual fluctuations of temperature would not negatively affect the reaction yield; moreover, the mild processing conditions required suggest convenient operating costs. However, the energy input required for the synthesis of the stoichiometric NaOH would represent the major obstacle in sight of an industrial application. In fact, 7.7 GJ is required to produce 1 t of NaOH, which occurs alongside the synthesis of 0.03 t of hydrogen gas and 1.1 t of chlorine gas, through the chlor-alkali process.41 Such hydrogen is typically wasted and vented to the atmosphere, whereas the in situ recirculation for power production would lower the energy demand by approximately 34%.42 In these conditions, considering that 0.8 t of stoichiometric NaOH are required for each tonne of CaCO3 reacted, about 3.9 GJEquivalent of electricity should be supplied through usage of fresh fuel. Whereas, the conventional cement production route requires an energy input of 2.5–2.9 GJ per tonne of CaCO3 to decarbonise, considering the theoretical heat of dissociation of CaCO3 (1819.4 kJ43) and efficiencies between 41 and 62%.44 At first glance, our route does not appear economically desirable, since it requires 1.4–1.6 times the energy involved in the conventional route. However, it must be considered that the co-production of Na2CO3 occurs whilst synthetising Ca(OH)2, and important energetic considerations would arise from that. In fact, the production of 1 tonne of Na2CO3 requires 13.6 GJ through the conventional Solvay process,45 which would be completely avoided by supplying the product via the chemical route proposed. According to the stoichiometry depicted in eqn (1), a Na2CO3/Ca(OH)2 weight ratio of 1.43 occurs at the end of the reaction, assuming total conversion of CaCO3. The following considerations are done by normalising all the calculations with respect to cement, lime, and slaked lime, treating Na2CO3 as an added-value by-product. For the calculations, a 1:1 weight ratio between the decarbonised CaCO3 and the resulting cement was considered; in fact, the CaO proportion in PC is around 60–70%,46 and a 0.56 weight ratio occurs between the produced CaO (upon dehydration of Ca(OH)2) and the reacted CaCO3. For clarity, these numbers are reported in Table 3, alongside the cases for lime and slaked lime productions.
| Production type | PC | Lime | Slaked lime | CaCO3 reacted | NaOH reacted | Na2CO3 produced |
|---|---|---|---|---|---|---|
| PC | 1 | — | — | 1 | 0.8 | 1.0 |
| Lime | — | 1 | — | 1.6 | 1.3 | 1.8 |
| Slaked lime | — | — | 1 | 1.2 | 1.0 | 1.4 |
All these considerations were used to produce the energetic comparisons displayed in Fig. 11, where the electrical energy input required to produce the stoichiometric NaOH is reported alongside the thermal energy to be supplied to decarbonise CaCO3 plus the one to produce soda ash. The extremes of the energy efficiencies reported for the PRK design are labelled as “Thermal_1” (η = 41%) and “Thermal_2” (η = 62%) for the PC case, whereas the novel decarbonisation route is displayed as “Chemical” both for PC, lime, and slaked lime. It must be mentioned that the energies outlined were normalised with respect to 1 tonne of PC, according to the stoichiometry just discussed, and therefore expressed in GJEquivalent, i.e., the energy required to produce 1 tonne of PC and the stoichiometric amount of Na2CO3 (1.0 t). It must be mentioned that these values solely refer to the decarbonisation step, without considering additional consumptions associated with the further processing of both reactants and products. It is revealed that chemical decarbonisation allows for ∼4 times lower energy consumption with respect to the thermal route, considering the synthesis of 1.0 tonne of PC and 1.0 tonnes of Na2CO3 (Table 3). For the calculation of the energy requirement from the chemical route, a 10% surplus was considered to include the handling and separation of the materials.
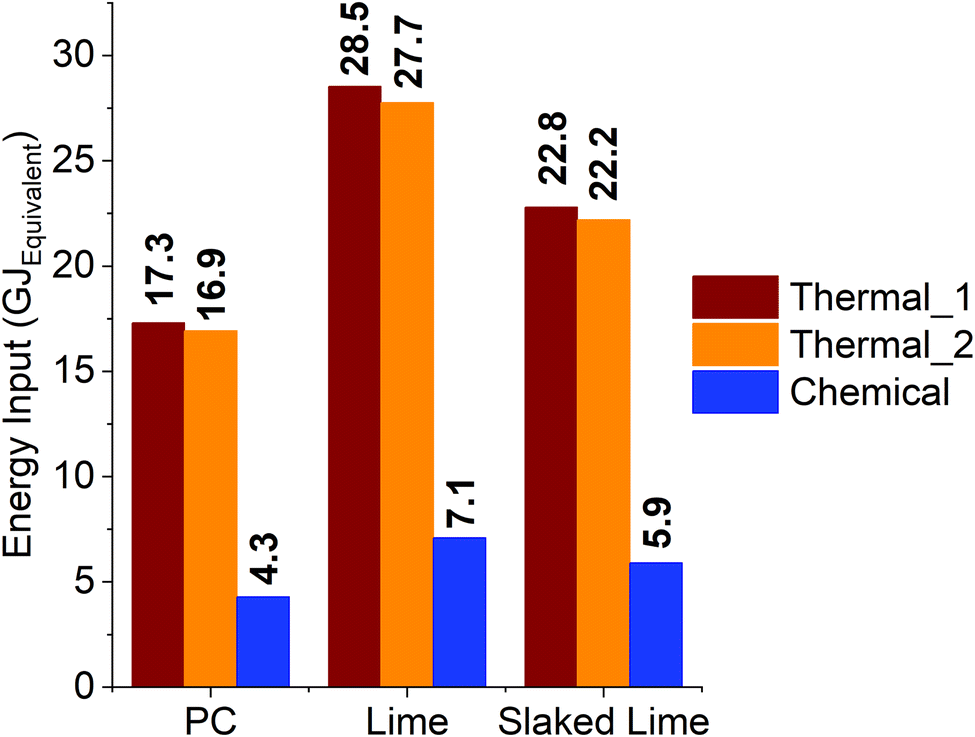 | ||
| Fig. 11 Comparison of the energy consumption between the thermal and chemical approaches to the decarbonisation of CaCO3, referred to the cement, lime and slaked lime industries; the values only depend on the decarbonisation step, considering a PRK design for the thermal calcination in PC production, and PFRK one for lime and slaked lime. | ||
Regarding the production of lime, 1.6 tonnes of initial CaCO3 are required to ensure 1 tonne of product, corresponding to a higher consumption of NaOH with respect to PC. For this reason, the normalisation with respect to lime shows that a higher amount of soda ash is produced, and that is considered for the calculation of the thermal power required from the conventional route. In these terms, the calcination design adopted in the lime/slaked lime industry allows for higher efficiencies (75% < η < 99%44); in Fig. 11, the labels “Thermal_1” and “Thermal_2” refer to the efficiency extremes of 75 and 99%, respectively. The larger consumption of NaOH reflects a higher energy input for the chemical route with respect to PC; despite that, even in this case the thermal route would require ∼4 times the energy employed in the chemical decarbonisation, when 1 and 1.8 tonnes of CaO and Na2CO3 are produced, respectively (Table 3). Finally, the case study for slaked lime Ca(OH)2 highlighted a 3.7–3.8 times lower energy input for the chemical route with respect to the conventional one for the synthesis of 1 and 1.4 tonnes of slaked lime and soda ash, respectively.
The preliminary energetic balance appears promising when sodium carbonate is considered as a co-product (i.e., replacing market soda ash); however, the global market demand of the chemicals involved must be considered. In fact, the current global demand for PC and soda ash is 4 Gt1 and 50 Mt,47 respectively; if the whole global production of PC is performed chemically, an excess production of soda ash would occur. However, the demand for soda ash might significantly grow given the increasing importance of geopolymers in the global perspective, with Na2CO3 used as activator to produce these low-carbon binders.48 Furthermore, in case the excess of soda ash could not be reused, Na2CO3 would represent a safer option for permanent CO2 storage with respect to the current geological disposal of liquid and compressed CO2, whose long-term effects have not been fully understood yet.49
Another drawback of the chemical route is represented by the production of chlorine gas arising from the chlor-alkali process to produce NaOH;41 if the size of the market increases to fulfil the demand for cement production, such emissions might have a heavy impact on the environment. The development of chlorine-based binders, such as alinite,50 could help to mitigate the issue. In fact, despite that the presence of Cl leads to the failure of reinforced concrete through corrosion of the mild steel,51 ∼75% of the worldwide cement is used for unreinforced purposes.52 In addition, an enhancement of the chlor-alkali process with the in situ hydrogen processing and re-use would contribute to the mitigation of the drinkable water crisis expected in the next decades.53 In fact, Na+ and Cl− are removed from the brine (concentrated seawater) fed into the chlor-alkali process, and pure water is a by-product from the reprocessing of hydrogen.54
Alternatively, both the excess production of soda ash and the increasing emissions of chlorine gas might be limited by solely applying the novel route to produce lime and slaked lime, whose markets (50 Mt and 20 Mt, respectively2) are currently closer to that of soda ash (50 Mt47).
6 Conclusions
The effects of temperature on the efficiency of a chemical decarbonisation process for CaCO3 was assessed. The reaction appeared effective for the sequestration of the process CO2 partitioned into CaCO3, with over the 80% of it captured within the first minute of reaction. The short residence times and mild heating conditions required look promising in sight of an eventual scale-up. It is supposed that the process might be thermally self-sustained, given (1) the exothermic dissolution of NaOH into water, and (2) the high conversion degrees observed even at ambient conditions. That would reflect a significant advantage from an industrial point of view, suggesting that the process is robust and that no additional costs would be associated with the control of the reaction temperature.The capture of the process CO2 occurs through precipitation of Na2CO3·H2O at milder temperature and pH, whereas Na2CO3 is the dominant Na-based species at higher temperature and alkalinity. Given the need for a final separation of Ca(OH)2 from Na2CO3. xH2O, not discussed here, Na2CO3·H2O might be favourable given the higher solubility with respect to Na2CO3 (330 and 307 g L−1 at 25 °C and 1 atm, respectively). In fact, given the much lower solubility of Ca(OH)2 (1.5 g L−1 at 25 °C and 1 atm), the separation can be carried out in excess of water, and the yield would be maximised when the solubility gap between the phases of interest is large.
The activation energy Ea was determined for different ternary compositions previously tested. Generally, the activation energy barrier was smaller for the system with the higher initial NaOH concentration; moreover, the rate of the chemical decarbonisation appeared much higher with respect to the conventional CaCO3 calcination for all the conditions tested.
Preliminary energetic considerations were also reported, and the comparison between the thermal and chemical route to produce PC, lime, slaked lime, and soda ash was shown. As outlined, the chemical route was ∼4 times more convenient in terms of energy input, if only the decarbonisation step of CaCO3 is considered and when soda ash is considered as a co-product.
Overall, the chemical decarbonisation of CaCO3 may have the potential to drastically reduce the carbon footprint of the related industries, potentially removing the need for high temperature calcination. The main obstacle to overcome is still represented by the general concept that the thermal decarbonisation of CaCO3 is unavoidable, and that a transition would be technically too difficult to operate. However, the current environmental crisis demands for brave and optimistic approaches/decisions backed by the desire for real change towards a sustainable future.
Author contributions
Theodore Hanein and Hajime Kinoshita discovered and conceptualized the technology. Marco Simoni developed the technology and designed the methodology and experiments. Marco Simoni and Chun Long Woo carried out experiments. Marco Simoni drafted the original manuscript. Theodore Hanein, Hajime Kinoshita, and John L. Provis acquired funding, and supervised Marco Simoni and Chun Long Woo. Theodore Hanein, Hajime Kinoshita, John Provis, Mark Tyrer, and Magnus Nyberg reviewed and edited the manuscript.Conflicts of interest
The authors declare that they have no competing interests as defined by RSC Advances, or other interests that might be perceived to influence the interpretation of the article.Acknowledgements
This work was funded by the Engineering and Physical Science Research Council (EPSRC) and CEMEX under grant ID EP/R025959/1. This research utilised the HADES/MIDAS facility at the University of Sheffield established with financial support from EPSRC and BEIS, under grant EP/T011424/1. The group would also express gratitude to Alan Maries, Juan-Carlos Martinez, John Stennet, and Nestor I. Quintero-Mora for the support and knowledge received.References
- R. M. Andrew, Global CO2 emissions from cement production, Earth Syst. Sci. Data, 2018, 10(1), 195–217 CrossRef.
- J.-M. de Vet, et al., Competitiveness of the European Cement and Lime Sectors, WIFO Studies, 2018 Search PubMed.
- W. P. Bolen, Mineral Commodity Summaries–Soda Ash, United States Geological Survey, Reston, VA, 2015 Search PubMed.
- P. D. Warwick, et al., US Geological Survey Carbon Sequestration–Geologic Research and Assessments, Energy Procedia, 2014, 63, 5305–5309 CrossRef CAS.
- E. Gartner and H. Hirao, A review of alternative approaches to the reduction of CO2 emissions associated with the manufacture of the binder phase in concrete, Cem. Concr. Res., 2015, 78, 126–142 CrossRef CAS.
- G. Steinhauser, Cleaner production in the Solvay Process: general strategies and recent developments, J. Cleaner Prod., 2008, 16(7), 833–841 CrossRef.
- E. Worrell, et al., Carbon dioxide emissions from the global cement industry, Annu. Rev. Environ. Resour., 2001, 26(1), 303–329 Search PubMed.
- F. Schorcht, et al., Best available techniques (BAT) reference document for the production of cement, lime and magnesium oxide, European Commission Joint Research Centre Institute for Prospective Technological Studies (Report EUR 26129 EN), Publications Office of the European Union, Luxembourg, 2013 Search PubMed.
- L. Barcelo, et al., Cement and carbon emissions, Mater. Struct., 2014, 47(6), 1055–1065 CrossRef CAS.
- J. Rogelj, et al., Paris Agreement climate proposals need a boost to keep warming well below 2 C, Nature, 2016, 534(7609), 631 CrossRef CAS PubMed.
- D. Leeson, et al., A Techno-economic analysis and systematic review of carbon capture and storage (CCS) applied to the iron and steel, cement, oil refining and pulp and paper industries, as well as other high purity sources, Int. J. Greenhouse Gas Control, 2017, 61, 71–84 CrossRef CAS.
- M. Schneider, et al., Sustainable cement production—present and future, Cem. Concr. Res., 2011, 41(7), 642–650 CrossRef CAS.
- L. D. Ellis, et al., Toward electrochemical synthesis of cement—An electrolyzer-based process for decarbonating CaCO3 while producing useful gas streams, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(23), 12584–12591 CrossRef CAS PubMed.
- M. Simoni, et al., Decarbonisation of calcium carbonate in sodium hydroxide solutions under ambient conditions: effect of residence time and mixing rates, Phys. Chem. Chem. Phys., 2022, 16125–16138 RSC.
- M. Simoni, et al., Effect of Impurities on the Decarbonization of Calcium Carbonate Using Aqueous Sodium Hydroxide, ACS Sustainable Chem. Eng., 2022, 11913–11925 CrossRef CAS PubMed.
- T. Hanein, et al., Decarbonisation of calcium carbonate at atmospheric temperatures and pressures, with simultaneous CO 2 capture, through production of sodium carbonate, Energy Environ. Sci., 2021, 14(12), 6595–6604 RSC.
- A. Dowling, J. O'Dwyer and C. C. Adley, Lime in the limelight, J. Cleaner Prod., 2015, 92, 13–22 CrossRef CAS.
- O. T. Sørensen and J. Rouquerol, Sample controlled thermal analysis: origin, goals, multiple forms, applications and future, Springer Science & Business Media, 2013, vol. 3 Search PubMed.
- O. Levenspiel, Chemical reaction engineering, John Wiley & Sons, 1998 Search PubMed.
- A. Seidell, Solubilities of inorganic and metal organic compounds, 1940 Search PubMed.
- G. W. Morey, The action of water on calcite, magnesite and dolomite, Am. Mineral., 1962, 47(11–12), 1456–1460 CAS.
- A. Farhad and Z. Mohammadi, Calcium hydroxide: a review, Int. Dent. J., 2005, 55(5), 293–301 CrossRef PubMed.
- J. L. Ellingboe and J. H. Runnels, Solubilities of Sodium Carbonate and Sodium Bicarbonate in Acetone-Water and Methanol-Water Mixtures, J. Chem. Eng. Data, 1966, 11(3), 323–324 CrossRef CAS.
- J. Zelić, D. Rušić and R. Krstulović, Kinetic analysis of thermal decomposition of Ca (OH)2 formed during hydration of commercial Portland cement by DSC, J. Therm. Anal. Calorim., 2002, 67(3), 613–622 CrossRef.
- P. K. Gallagher and D. W. Johnson Jr, The effects of sample size and heating rate on the kinetics of the thermal decomposition of CaCO3, Thermochim. Acta, 1973, 6(1), 67–83 CrossRef CAS.
- M. Hartman, et al., Thermal dehydration of the sodium carbonate hydrates, Chem. Eng. Commun., 2001, 185(1), 1–16 CrossRef CAS.
- S. M. Antao and I. Hassan, Temperature dependence of the structural parameters in the transformation of aragonite to calcite, as determined from in situ synchrotron powder X-ray-diffraction data, Can. Mineral., 2010, 48(5), 1225–1236 CrossRef CAS.
- T. Nagai, et al., Compression mechanism and amorphization of portlandite, Ca (OH)2: structural refinement under pressure, Phys. Chem. Miner., 2000, 27(7), 462–466 CrossRef CAS.
- K. K. Wu and I. D. Brown, A neutron diffraction study of Na2CO3. H2O, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31(3), 890–892 CrossRef.
- A. Arakcheeva, et al., The incommensurately modulated structures of natural natrite at 120 and 293 K from synchrotron X-ray data, Am. Mineral., 2010, 95(4), 574–581 CrossRef CAS.
- K. Motzfeldt, The thermal decomposition. of sodium carbonate by the effusion method, J. Phys. Chem., 1955, 59(2), 139–147 CrossRef CAS.
- S. L. Stipp and M. F. Hochella Jr, Structure and bonding environments at the calcite surface as observed with X-ray photoelectron spectroscopy (XPS) and low energy electron diffraction (LEED), Geochim. Cosmochim. Acta, 1991, 55(6), 1723–1736 CrossRef CAS.
- R. A. Robie and B. S. Hemingway, Thermodynamic properties of minerals and related substances at 298.15 K and 1 bar (105 Pascals) pressure and at higher temperatures, US Government Printing Office, 1995, vol. 2131 Search PubMed.
- M. W. Chase Jr, NIST-JANAF thermochemical tables, J. Phys. Chem., Ref. Data, Monograph, 4th edn, 1998, vol. 9, pp. 1–1951 Search PubMed.
- D. L. Parkhurst and C. A. J. Appelo, Description of input and examples for PHREEQC version 3: a computer program for speciation, batch-reaction, one-dimensional transport, and inverse geochemical calculations, US Geological Survey. 2013 Search PubMed.
- J. Balej, Water vapour partial pressures and water activities in potassium and sodium hydroxide solutions over wide concentration and temperature ranges, Int. J. Hydrogen Energy, 1985, 10(4), 233–243 CrossRef CAS.
- J. G. Speight, Lange's handbook of chemistry, McGraw-Hill, New York, 2005, vol. 1 Search PubMed.
- K. J. Laidler, The development of the Arrhenius equation, J. Chem. Educ., 1984, 61(6), 494 CrossRef CAS.
- L. Fedunik-Hofman, A. Bayon and S. W. Donne, Kinetics of solid-gas reactions and their application to carbonate looping systems, Energies, 2019, 12(15), 2981 CrossRef CAS.
- K. M. Caldwell, P. K. Gallagher and D. W. Johnson Jr, Effect of thermal transport mechanisms on the thermal decomposition of CaCO3, Thermochim. Acta, 1977, 18(1), 15–19 CrossRef CAS.
- S. Lakshmanan and T. Murugesan, The chlor-alkali process: work in progress, Clean Technol. Environ. Policy, 2014, 16(2), 225–234 CrossRef CAS.
- A. Kumar, F. Du and J. H. Lienhard, Caustic Soda Production, Energy Efficiency, and Electrolyzers, ACS Energy Lett., 2021, 6(10), 3563–3566 CrossRef CAS.
- B. R. Stanmore and P. Gilot, Calcination and carbonation of limestone during thermal cycling for CO2 sequestration, Fuel Process. Technol., 2005, 86(16), 1707–1743 CrossRef CAS.
- M. Simoni, et al., Decarbonising the lime industry: State-of-the-art, Renewable Sustainable Energy Rev., 2022, 168, 112765 CrossRef CAS.
- J. T. Houghton, Intergovernmental Panel on Climate Change, Revised 1996 IPCC Guidelines for National Greenhouse Gas Inventories: Greenhouse Gas Inventory Workbook, OECD. 1996 Search PubMed.
- H. F. W. Taylor, Cement Chemistry, 2nd edn, 1997, 0727725920, 9780727725929 Search PubMed.
- S. Jewell and S. Kimball, USGS mineral commodities summaries: 2014, US Geological Survey, p. 2014, http://minerals.usgs.gov/minerals/pubs/mcs/2014/mcs2014.pdf, accessed, 2014 12(12).
- J. L. Provis and S. A. Bernal, Geopolymers and related alkali-activated materials, Annu. Rev. Mater. Res., 2014, 44, 299–327 CrossRef CAS.
- C.-F. Tsang, J. Birkholzer and J. Rutqvist, A comparative review of hydrologic issues involved in geologic storage of CO2 and injection disposal of liquid waste, Environ. Geol., 2008, 54(8), 1723–1737 CrossRef CAS.
- M. Simoni, et al., Producing cement clinker assemblages in the system: CaO-SiO2-Al2O3-SO3-CaCl2-MgO, Cem. Concr. Res., 2021, 144, 106418 CrossRef CAS.
- G. K. Glass and N. R. Buenfeld, The presentation of the chloride threshold level for corrosion of steel in concrete, Corros. Sci., 1997, 39(5), 1001–1013 CrossRef CAS.
- U. N. Environment and K. L. Scrivener, et al., Eco-efficient cements: Potential economically viable solutions for a low-CO2 cement-based materials industry, Cem. Concr. Res., 2018, 114, 2–26 CrossRef.
- W. A. Jury and H. J. Vaux Jr, The emerging global water crisis: managing scarcity and conflict between water users, Adv. Agron., 2007, 95, 1–76 Search PubMed.
- H. Khasawneh, et al., Utilization of hydrogen as clean energy resource in chlor-alkali process, Energy Explor. Exploit., 2019, 37(3), 1053–1072 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra05827h |
| This journal is © The Royal Society of Chemistry 2022 |