
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFlow cytometry-based rapid detection of Staphylococcus aureus and Pseudomonas aeruginosa using fluorescent antibodies†
Joo-Kyung Kima,
Hanool Yunb,
Chang-Hun Yeomb,
Eun-Jung Kimc,
Wooseong Kimd,
Chang-Soo Lee e,
Byung-Gee Kimac and
Hee-Jin Jeong*b
e,
Byung-Gee Kimac and
Hee-Jin Jeong*b
aInterdisciplinary Program for Biochemical Engineering and Biotechnology, Seoul National University, Seoul, 08826, South Korea
bDepartment of Biological and Chemical Engineering, Hongik University, Sejong, 30016, South Korea. E-mail: heejinjeong@hongik.ac.kr
cBio-MAX/N-Bio, Seoul National University, Seoul, 08826, South Korea
dCollege of Pharmacy and Graduate School of Pharmaceutical Sciences, Ewha Womans University, Seoul 03760, South Korea
eDepartment of Chemical Engineering and Applied Chemistry, Chungnam National University, Daejeon 34134, South Korea
First published on 2nd December 2022
Abstract
Staphylococcus aureus (S. aureus) and Pseudomonas aeruginosa (P. aeruginosa) are major pathogens frequently detected in food and beverage poisoning, and persistent infections. Therefore, the development of a rapid method that can detect these pathogens before serious multiplication is required. In this study, we established a flow cytometry (FCM)-based detection method that allows rapid acquisition of cell populations in fluid samples by using a fluorescent antibody against S. aureus or P. aeruginosa. Using this method, we detected these pathogens with a 103 to 105 CFU order of limit of detection value within 1 hour. The FCM-based method for the detection of S. aureus and P. aeruginosa offers the possibility of high-throughput analysis of pathogens in food, environmental, and clinical sources.
Introduction
Food and beverage poisoning is caused by bacterial pathogens in contaminated foods and drinks that grow in the stomach.1 Staphylococcus aureus (S. aureus) and Pseudomonas aeruginosa (P. aeruginosa) are major Gram-positive and Gram-negative food-borne pathogens, respectively, leading to long-term chronic diseases, typhoid fever, septicaemia, gastroenteritis, and serious infections.2,3 Although antibiotics against these pathogens have been developed, such as oxacillin for S. aureus and imipenem for P. aeruginosa, the evolution of antibiotic-resistant strains is increasing.4 Moreover, S. aureus and P. aeruginosa cause infections even at low concentrations. Therefore, rapid detection of these pathogens is crucial for the timely testing of food and beverage poisonings.Traditional methods for detecting these pathogens are based on bacterial culture- and colony-counting methods.5,6 However, these procedures require 3–5 d, which is time-consuming and impractical for real-time application. Polymerase chain reaction (PCR)-based detection7–11 is faster than conventional culture- and colony-counting-based methods, but still requires 4–5 h for repeated amplification cycles. Our group recently developed an enzyme-linked immunosorbent assay (ELISA) systems for detecting S. aureus12 and P. aeruginosa13 by using commercially available antibodies and recombinant antibodies. We produced recombinant antibodies via a mammalian cell culture system and revealed that these antibodies had high antigen-binding efficiency and a low detection limit (LOD), which enabled the detection of S. aureus or P. aeruginosa under 104 CFU. However, ELISA requires several steps, including immobilising, blocking, washing, and incubating antigens or antibodies in the wells of a 96-well plate. Thus, the ELISA-based detection system requires 20 h to conduct the whole process, which is faster than conventional plating detection methods but remains limited to one-pot detection. Therefore, developing a novel method for the rapid detection of pathogens that can be applied to point-of-care diagnostic tests is necessary.
Flow cytometry (FCM) is a high-throughput analysis method that enables the measurement of the properties of individual cells in a flow.14 FCM using fluorescent antibodies is one of the reagentless immunoassays that require no immobilisation steps. Namely, by reacting the fluorescent antibodies to the antigens on the cells and then applying them to the fluid system, the positive signal from an antigen-bound antibody can be separated from the extra signal from a free antibody, which does not bind to the antigen. FCM is highly automated because cells pass in a flow and are focused by laser beam, then each cell is characterised by its scattering and fluorescence properties.15 Moreover, FCM allows the analysis of cells at a rate of over 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 per minute.16 This high speed of FCM is an advantage over culture-based methods, which requires time-consuming dilution procedures and a waiting period until the formation of colonies on a selective agar plate. FCM provides information regarding the cells present in the culture, which can be collected within several minutes.17
000 per minute.16 This high speed of FCM is an advantage over culture-based methods, which requires time-consuming dilution procedures and a waiting period until the formation of colonies on a selective agar plate. FCM provides information regarding the cells present in the culture, which can be collected within several minutes.17
Bacterial cells are more difficult to analyse using FCM than eukaryotic cells because the volume of bacteria are 1000-fold smaller than those of eukaryotes.15 Moreover, because prokaryotes do not have organelles, their cellular complexity is lower than that of eukaryotes, resulting in the former having weaker scattering signals than the latter.18 Therefore, FCM is typically used in immunoassays for eukaryotic cells, such as cancer and immune cells, but its use for the detection and analysis of pathogens has limitations. Despite this difficulty, rapid FCM-based methods, which can detect pathogens in water, food, and clinical samples, have been developed.19–22 Some methods using fluorescent antibodies combined with FCM have been reported to detect S. aureus or P. aeruginosa.23–26 However, these methods were not optimised to detect low concentrations of pathogens, because the studies have focused on the application of pathogens in real samples. For example, Kennedy et al. detected the food-borne pathogen S. aureus by cultivating cultures in broth until the exponential phase, followed by mixing the cultures with fluorescent antibodies.7 However, the cells were grown until their optical density at 600 nm reached 0.2, with a cell population of 1.0 × 107 CFU mL−1, and a high concentration of cells was used to evaluate the detection method.
In this study, we generated fluorescent antibodies against S. aureus and P. aeruginosa by conjugating a fluorescent dye to commercially available or recombinant antibodies. We then established an FCM-based assay method using these fluorescent antibodies for the rapid detection of these pathogens (Fig. 1A).
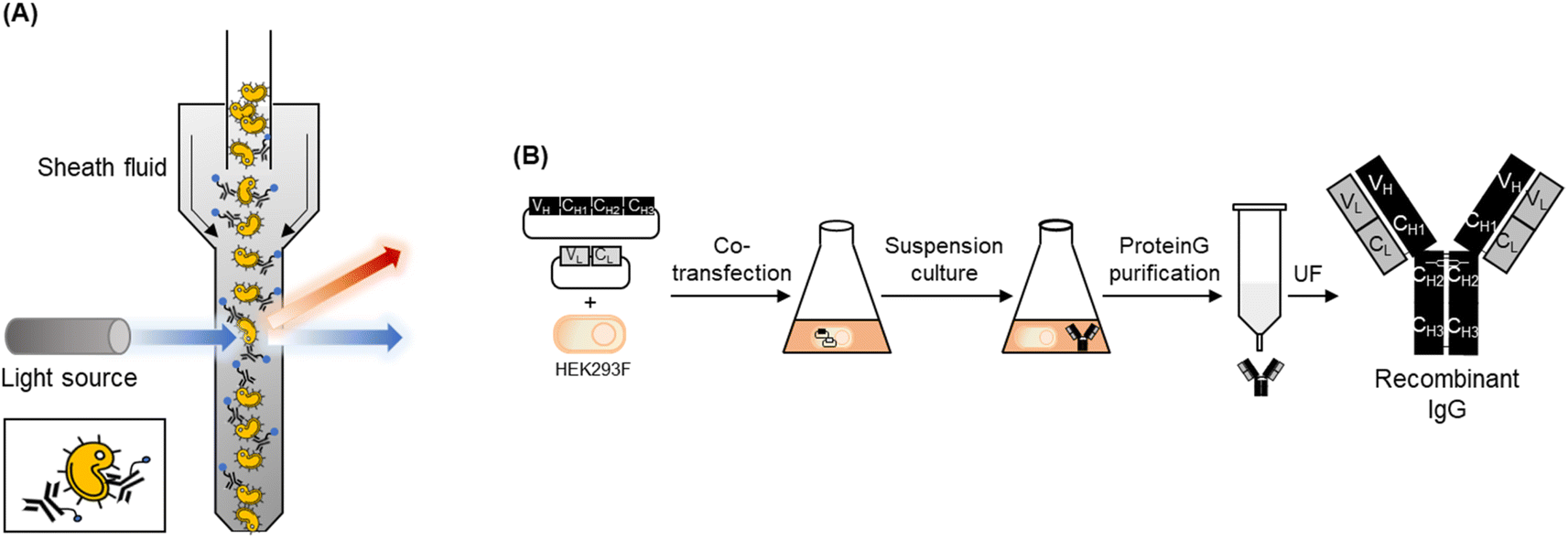 | ||
| Fig. 1 (A) Schematic image of flow cytometry analysis using a fluorescent antibody to detect pathogens. (B) Schematic representation of an entire step for producing a recombinant full-sized antibody using HEK293F cells. UF indicates ultra-filtration. | ||
Results and discussion
Production of recombinant antibodies
In our previous study, we generated recombinant full-sized IgGs against S. aureus12 and P. aeruginosa13 using mammalian cells. To produce recombinant anti-S. aureus antibodies, we constructed three mammalian cell-expressing plasmids with the insertion of the variable domain of three antibodies against wall teichoic acid on the surface of S. aureus: 6DWI, 6DW2, and 6DWC, whose amino acid sequences and CDR regions differed.27 When we expressed the vectors using HEK293F cells and confirmed their antigen-binding activities against WKZ-1, an MSSA strain, and WKZ-2, an MRSA strain, by using indirect ELISA, 6DW2 showed the highest response among the three antibodies. The EC50 and LOD values of 6DW2 were superior to those of commercially available antibodies. When we constructed a mammalian cell-expressing plasmid with the insertion of a variable domain of an antibody against PcrV, a needle-tip protein of the type III secretion system of P. aeruginosa, and expressed the plasmid using HEK293F cells, the antibody detected two antibiotic-susceptible P. aeruginosa strains, UCBPP-PA14 and ATCC 27853, and one antibiotic-resistant P. aeruginosa strain, ATCC BAA-2108, with higher or similar sensitivity than commercial anti-P. aeruginosa antibodies. Based on these results, we expressed 6DW2 and 6CYF using our previously established mammalian cell culture method and compared the responses to FCM analysis to those of the commercial antibodies (Fig. 1B). According to the method described in our previous paper,12,13 we expressed 6DW2 and 6CYF and purified each protein by using protein A affinity beads. We used these proteins for fluorescent labelling.Fluorescent labelling
We used an N-hydroxysuccinimide (NHS)-based labelling method to conjugate the fluorescent dye to the antibody. NHS coupling is a highly effective and stable cross-linking method that can label the amine groups of proteins by using an NHS ester-conjugated fluorescent dye. Carbonyl carbons of NHS ester react with primary amines in the N-terminal of the protein and/or to the side chain of a lysine (Lys) residue(s) of the protein to form amide crosslinks. When the pH of the buffer in the sample reaches 7.2–9.0, primary amines formed nucleophiles and reacted with carbonyl carbons, which are electrophiles. Subsequently, NHS acted as the leaving group and was substituted with primary amines (Fig. 2A).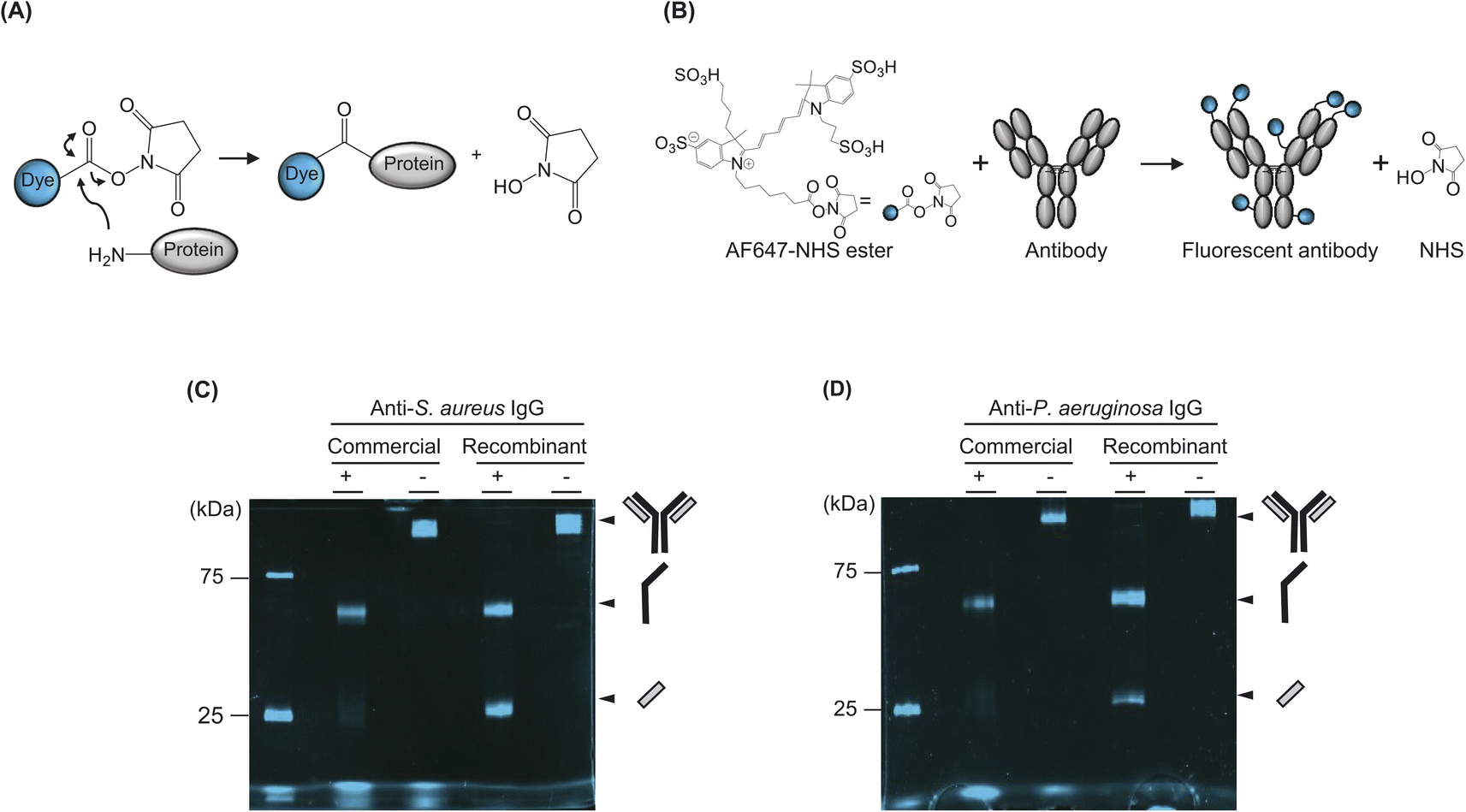 | ||
| Fig. 2 (A) Schematic representation of NHS coupling that leads to conjugation between fluorescent dye and protein. (B) Schematic representation of NHS coupling-based antibody labelling using Alexa647–NHS ester dye. The number and position of dyes on the antibody were schematically imaged (C) SDS–PAGE analysis of fluorescent commercial or recombinant antibody against S. aureus. (D) SDS–PAGE analysis of fluorescent commercial or recombinant antibody against P. aeruginosa. + and − indicates a sample reduced by adding 100 mM DTT and heating, and a non-reduced sample, respectively. | ||
In this study, we used NHS-ester-conjugated Alexa647, a commercially available organic dye with emission and excitation wavelengths of 651 nm and 672 nm, respectively (Fig. 2B). Specifically, we used an Alexa647 dye because its wavelength is suitable for the FCM filter. We mixed commercial or recombinant antibodies with the dye at 1:20 molar ratio and loaded it to a sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE) gel with or without denaturation, which boiled the sample in the dithiothreitol-containing loading buffer. We did not purify the samples after labelling, because a purification process leads to the loss of the sample even though the free dyes can be eliminated. Because the size of the unconjugated free dye was sufficiently small to be separated from the conjugated dye of fluorescent antibody attached to the large size of cells, the free dye was not detected via FCM analysis. Instead, when we analysed the samples using FCM, we eliminated the free dye by a centrifuge process after reacting the fluorescent dye-labelled antibody to the cell. Namely, we mixed the fluorescent antibody and cells for 30 min and centrifuged the samples. Subsequently, the supernatant was washed by pipetting, and the sample was loaded into the FCM equipment.
We observed the SDS–PAGE gel by using a fluorescence imaging system and confirmed that the samples were successfully labelled (Fig. 2C and D). Notably, when we loaded the denatured commercial antibody to separately confirm the labelling of the H and L chains of the antibody, the L chain was almost unlabelled, whereas the H chain was relatively well labelled. Although confirming the exact number of Lys residues on the surface of the antibody was not possible because the three-dimensional structures of these antibodies had not been revealed, we observed that the L chains of both commercial antibodies were not labelled. By contrast, the recombinant antibody, produced from HEK293F cells showed a higher labelling efficiency than commercial antibodies.
Flow cytometry analysis
The fluorescent antibody was reacted with S. aureus or P. aeruginosa cells and analysed using FCM with a laser capable of exciting Alexa647 dye. To acquire a sufficient cell population to obtain a stable event parameter of the instrument, we had to adjust the minimum total cell number to 3 × 106 CFU. Therefore, we incubated fluorescent antibodies with various concentrations of the pathogen, ranging from 1 × 104 CFU to 3 × 106 CFU, and adjusted the total cell number by adding non-pathogenic bacteria, BL21, up to 3 × 106 CFU. After mixing, we eliminated the free dye and unbound fluorescent antibodies by removing the supernatant after centrifugation and adding PBS and then conducted FCM analysis. The histogram of the sample in the presence of 3 × 106 CFU of BL21, which was used as a negative control, was on the left side, and the histograms of the sample in the presence of pathogens were shifted to the right (Fig. 3A and B and S1†). Clear differences in the fluorescent patterns were observed among individual samples in the presence of different numbers of cells that were distinguishable from the fluorescent patterns of 3 × 106 CFU of BL21 cells. We gated the peak range in the presence of 3 × 106 CFU of the pathogen, which is the maximum signal, and determined the positive signal ratio of the samples by calculating the ratio between the fluorescence intensity of the BL21-only sample in the gated range and the fluorescence intensity of the pathogen-including sample in the gated range.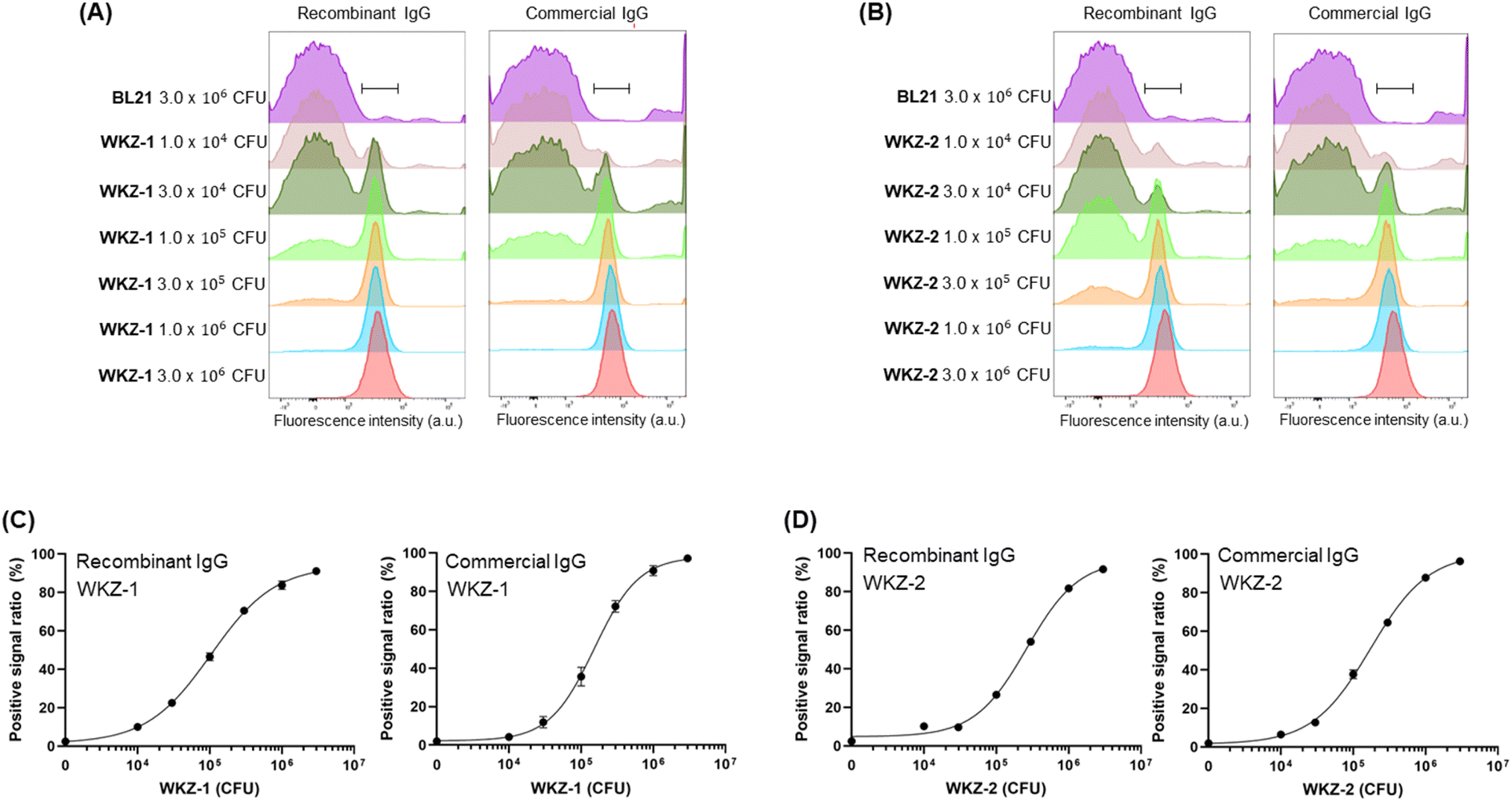 | ||
| Fig. 3 (A) Overlaying histograms obtained by flow cytometry analysis after incubation of fluorescent recombinant or commercial antibody with different numbers of WKZ-1 and BL21. The total cell numbers of each assay were adjusted to 3 × 106 CFU by adding BL21 cells to the different numbers of pathogen cells. (B) Overlaying histograms obtained by flow cytometry analysis after incubation of fluorescent recombinant or commercial antibody with different numbers of WKZ-2 and BL21. The total cell numbers of each assay were adjusted to 3 × 106 CFU by adding BL21 cells to the different numbers of pathogen cells. (C and D) Positive signal ratio of the mean of fluorescence intensity of range-gated in each histogram. Error bars represent ±1 SD (n = 3). | ||
The fluorescent dye-labelled antibody against S. aureus showed a dose-dependent binding efficiency to both WKZ-1 and WKZ-2 (Fig. 3C and D). The LOD values of the recombinant antibody against WKZ-1 and WKZ-2 were 5.17 × 103 CFU and 2.73 × 103 CFU, respectively, which were lower than the value in the use of a commercial antibody, 1.59 × 104 CFU and 9.65 × 103 CFU for WKZ-1 and WKZ-2, respectively (Table 2). When we used these recombinant and commercial antibodies for indirect ELISA in our previous study,12 the LOD values of the recombinant antibody against WKZ-1 and WKZ-2 were 1.5 × 103 CFU and 3.6 × 102 CFU, respectively, which were lower than the values for the commercial antibody, 1.0 × 104 CFU and 7.5 × 103 CFU, respectively. The ELISA results corresponded to the results of the FCM analysis, indicating the usefulness of the recombinant antibody for more sensitive detection of S. aureus than can be performed by a commercial antibody. Notably, a sample could be analysed within 5 min of assay time without the preincubation of cells by using the FCM method described in this study. Thirty minutes of hands-on time for reacting fluorescent antibody and cells followed by 5 min for washing were only necessary for preparing the samples. The LOD values of FCM were estimated as 103 to 104 CFU order, which are similar to those of ELISA and considerably lower than those of bacterial detection methods. Because the assay time for FCM is much faster than that of other conventional methods such as ELISA, which requires 1–2 d to obtain results, a few hours of preincubation for cultivating the cells to reach a cell number over the LOD value can be additionally performed before the FCM analysis.
| Nucleotide | Amino acid |
|---|---|
| atgggcggtggaggtggttataccatacgatgttccagattacgctgaagtccagctggtcgagtcaggtggcggtctggtgcagcccggtgggtctcttcggctgagctgtgccgcgagcgggtttacctttagtagttacgcgatggattgggttcgccaggcccccgggaaaggcttagaatgggtttccgcaatcacgatgagtggcattaccgcgtattatacagacgatgtcaagggccggtttaccatctctcgggacaatagcaaaaatactctgtatttgcagatgaactccctccgggcggaagatacagccgtgtattactgtgcgaaagaggaattcctcccgggcacacactacttttatggtatggatgtatggggccaggggaccaccgtgaccgtgggtggtggcgggagcggtggtggcgggtccggaggaggtggctccgctattcagatgacccagagcccgtcatctttaagtgcaagtgttggcgaccgcgtgaccattacgtgccgggcaagtcaggggattcggaacgatttaggctggtatcagcagaaaccggggaaagccccgaagttattaatctacagtgcaagtacgctgcagagcggggtgccgtcccgctttagcggctcgggttctgggacggattttacacttacgattagttcattgcagccagaagattttgcgacctactactgtttgcaggattacaactacccatggactttcggccagggcaccaaggttgagattggggggggttctcatcatcatcatcatcatggcggatccgactacaaggacgacgatgacaaataa | MGGGGGYPYDVPDYAEVQLVESGGGLVQPGGSLRLSCAASGFTFSSYAMDWVRQAPGKGLEWVSAITMSGITAYYTDDVKGRFTISRDNSKNTLYLQMNSLRAEDTAVYYCAKEEFLPGTHYFYGMDVWGQGTTVTVGGGGSGGGGSGGGGSAIQMTQSPSSLSASVGDRVTITCRASQGIRNDLGWYQQKPGKAPKLLIYSASTLQSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCLQDYNYPWTFGQGTKVEIGGGSHHHHHHGGSDYKDDDDK |
| Strain | Antibody | EC50 (CFU) | LOD (CFU) | |||
|---|---|---|---|---|---|---|
| 1st trial | 2nd trial | 3rd trial | Average | |||
| WKZ-1 | Commercial Ab | 1.45 × 105 | 1.51 × 105 | 1.60 × 105 | 1.52 ± 0.08 × 105 | 1.59 × 104 |
| rAb (HEK293) | 1.01 × 105 | 1.06 × 105 | 1.10 × 105 | 1.06 ± 0.05 × 105 | 5.17 × 103 | |
| WKZ-2 | Commercial Ab | 1.79 × 105 | 1.71 × 105 | 1.77 × 105 | 1.76 ± 0.04 × 105 | 9.65 × 103 |
| rAb (HEK293) | 2.73 × 105 | 2.55 × 105 | 2.66 × 105 | 2.65 ± 0.09 × 105 | 2.73 × 103 | |
| UCBPP PA14 | Commercial Ab | 6.30 × 105 | 4.43 × 105 | 4.61 × 105 | 5.12 ± 1.03 × 105 | 6.61 × 104 |
| rAb (HEK293) | 2.06 × 106 | n.d. | n.d. | n.d. | n.d. | |
| rAb (E. coli) | 2.26 × 106 | 9.73 × 105 | 1.65 × 106 | 1.63 ± 0.64 × 106 | 3.16 × 105 | |
| ATCC 27853 | Commercial Ab | 1.49 × 106 | 1.40 × 106 | n.d. | n.d. | 1.41 × 105 |
| rAb (HEK239) | 1.97 × 106 | n.d. | n.d. | n.d. | n.d. | |
| rAb (E. coli) | 4.81 × 106 | 4.07 × 106 | 7.28 × 105 | 3.20 ± 2.18 × 106 | 1.37 × 105 | |
| ATCC BAA-2108 | Commercial Ab | 8.69 × 105 | 2.17 × 106 | 1.25 × 106 | 1.43 ± 0.67 × 106 | 5.16 × 104 |
| rAb (HEK293) | 4.25 × 106 | n.d. | 2.24 × 106 | n.d. | 8.48 × 104 | |
| rAb (E. coli) | n.d. | 5.13 × 106 | 5.13 × 106 | n.d. | n.d. | |
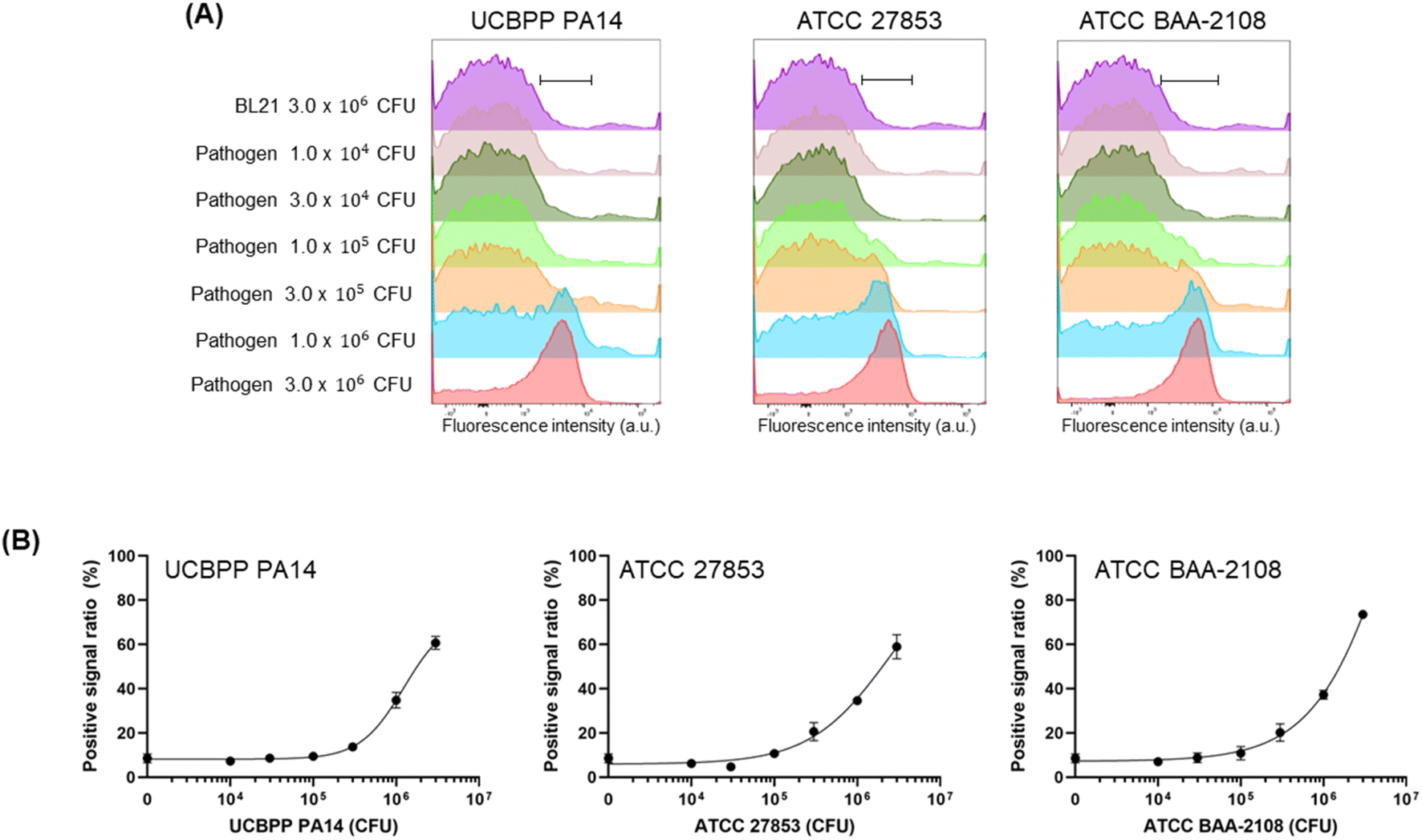
Next, we prepared a fluorescent dye-labelled antibody against P. aeruginosa and mixed it with various numbers of three types of P. aeruginosa strains and BL21 up to 3 × 106 CFU to adjust the entire cell number to be equal to 3 × 106 CFU in the individual assay. As a result, the signal increased in a pathogen cell-number-dependent manner (Fig. 4, S2 and S3†). The LOD value of the recombinant antibody and commercial antibody against ATCC BAA-2108 was 8.48 × 104 CFU and 5.16 × 104 CFU, respectively, indicating that the recombinant antibody can detect the pathogen slightly more sensitively than the commercial antibody can. The exact LOD value of the recombinant antibody against UCBPP PA14 and ATCC 27853 could not be calculated, because the signal on the titration curve was not fully saturated; thus, the equation of the curve was not estimated. However, when we compared the curve patterns and EC50 values of recombinant antibody and commercial antibody, we estimated that the LOD value for detecting the UCBPP PA14 of the recombinant antibody might be higher than that of the commercial antibody, and the value for detecting the ATCC 27853 of recombinant antibody might be similar to that of the commercial antibody. The lowest LOD value of the ELISA assay using a commercial antibody for UCBPP PA14, ATCC 27853, and ATCC BAA-2108 was 105 CFU order, 101 CFU order, and 104 CFU order, respectively, and the LOD values using a recombinant antibody for ATCC27853 and ATCC BAA-2108 was 104 CFU order (the LOD of PA14 was not calculated).13 These results demonstrated that the FCM analysis described herein can detect P. aeruginosa, and fluorescent recombinant or commercial antibodies can be used for the assay. The fact that a recombinant antibody can be produced in a more cost-effective manner than the price of a commercial antibody can compensate for the higher LOD value against ATCC 27853 of the recombinant antibody than the commercial antibody.
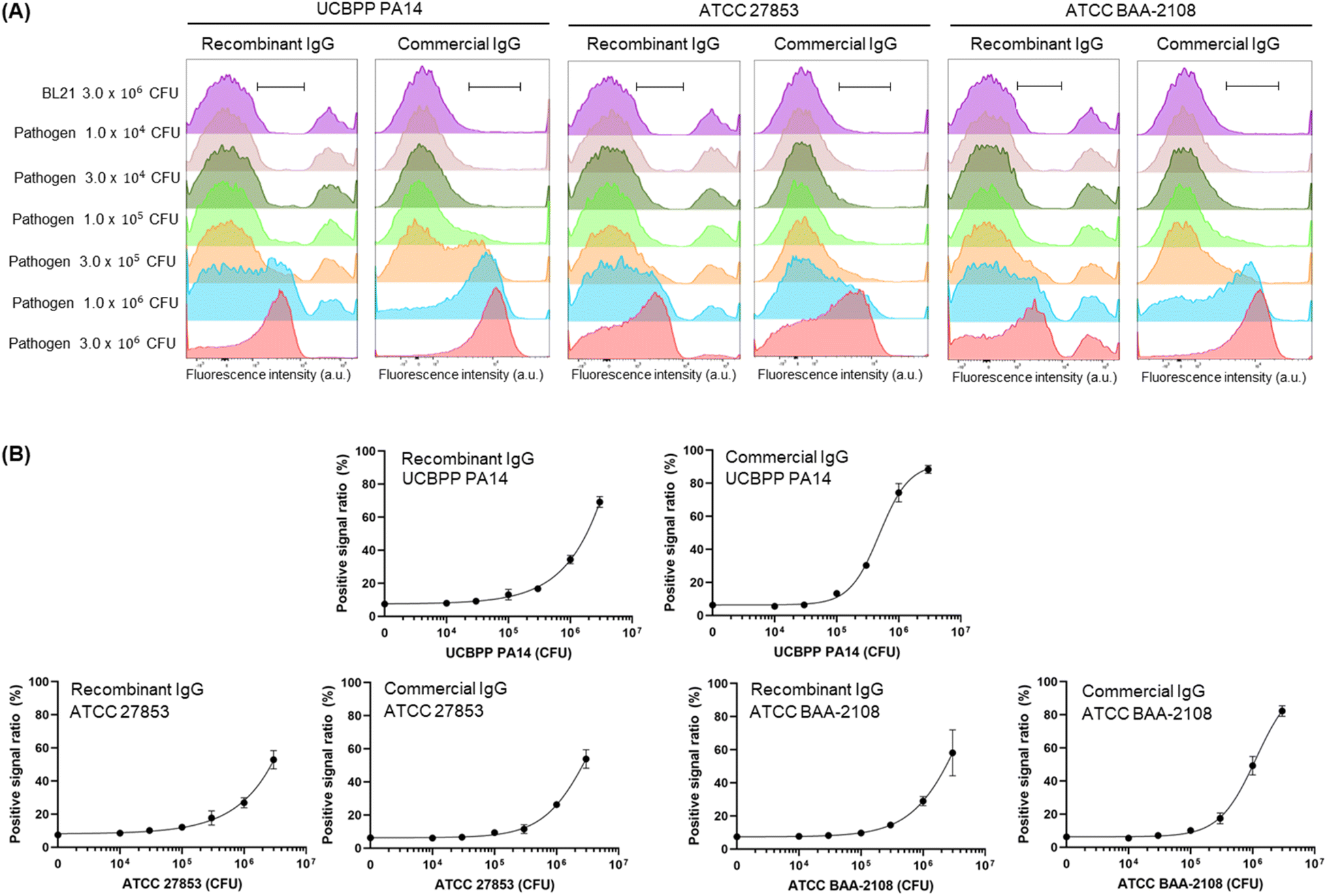 | ||
| Fig. 4 (A) Overlaying histograms obtained by flow cytometry analysis after incubating fluorescent recombinant or commercial antibody with different numbers of P. aeruginosa and BL21. The total cell numbers of each assay were adjusted to 3 × 106 CFU by adding BL21 cells to the different numbers of pathogen cells. (B) Positive signal ratio of the mean of fluorescence intensity of range-gated in each histogram. Error bars represent ±1 SD (n = 3). | ||
Recombinant scFv against P. aeruginosa
Although the FCM analysis of the use of recombinant antibody showed the detection of P. aeruginosa in a range from 104 to 105 CFU within 5 min, we conducted further experiments to generate a single-chain Fv (scFv), the smallest antibody fragment, against P. aeruginosa to extend the candidate of the superior antibody. We previously developed the HEK293F cell-based anti-P. aeruginosa antibody13 and used it for this study as a forementioned. Thus, a possibility was that an scFv that was generated using the Escherichia coli (E. coli) expressing system showed a different response than a full-sized antibody generated using a mammalian cell-expressing system. This phenomenon may be occurred because three-dimensional conformation of the antigen-binding region of antibody can differ despite the amino acid sequences being the same. Moreover, the variable domain of the anti-P. aeruginosa antibody, which we used as an IgG in this study, was derived from the antibody against PcrV, which is located on the outer membrane of P. aeruginosa. Because small antibody fragments generally have higher cell-penetrating efficiency than full-sized antibodies, we estimated that scFvs could reach the PcrVs on the surface of the cells by penetrating highly concentrated cells.We constructed scFv against P. aeruginosa by inserting the VH and VL domains of 6CYF into the E. coli-based scFv-expressing vector. Next, we transformed E. coli with the plasmid and expressed scFv by IPTG induction. Because we added a His-tag at the C-terminus of scFv, we used the tag for Ni-NTA purification (Fig. 5A). When we analysed the protein via SDS–PAGE, we confirmed the expected protein with a molecular weight of 29.6 kDa for the anti-P. aeruginosa scFv was expressed in soluble form with a yield of 172.9 μg in the case of the 100 mL culture (Fig. 5B). We conducted indirect ELISA to confirm the antigen-binding efficiency of the newly generated scFv. We immobilised several concentrations of P. aeruginosa in each well of a 96-well plate and added scFv as a primary antibody, followed by an HRP-conjugated anti-His-tag antibody as a secondary antibody. The signal increased in an antigen-concentration-dependent manner, showing the antigen-binding efficiency of scFv (Fig. S4†). The EC50 values of scFv against UCBPP PA14, ATCC 27853, and ATCC BAA-2108 were 1.63 ± 0.64 × 106, 3.20 ± 2.18 × 106 CFU, and n.d. (not detectable) respectively. We labelled scFv with Alexa647 via NHS coupling and confirmed that the dye was successfully conjugated to scFv (Fig. 5C). As expected, the extra proteins in the sample were also labelled with the fluorescent dye, because the NHS ester-conjugated dye can be labelled to the Lys residues of the proteins. Although the purity of the sample was not high, we moved to the next step using this fluorescent scFv because the extra proteins included in the sample were difficult to bind to a pathogen and were eliminated during the washing step before the analysis and during the flowing step in the fluidic channel of FCM even if some extra fluorescent proteins non-specifically attached to the cells because the binding affinity of such non-specific binding is low. When we used this fluorescent scFv for FCM analysis, we observed that the peak on the histogram was shifted to the right in a pathogen concentration-dependent manner (Fig. 6A and S5†). The LOD values of scFv against UCBPP PA14 and ATCC 27853 were 3.16 × 105 CFU and 1.37 × 104 CFU, respectively. Although the exact LOD value of scFv against ATCC BAA-2108 could not be calculated, it was estimated to be 104 to 105 CFU from the titration curve (Fig. 6B, Table 2). The LOD values of scFv were similar to or slightly higher than those of the commercial antibodies and recombinant IgG. However, the E. coli-based scFv-type recombinant antibody is superior to mammalian cell-based IgG-type antibody production, namely, the former's convenient production, and cost-efficiency. Further, E. coli-based scFv production requires 5 days, whereas mammalian cell-based IgG production requires 2–3 weeks, including several days for inoculating and preculturing the cells and 7 days for the formation of antibodies in the cells. Moreover, the media for mammalian cell culture is more expensive and easier to get contaminated than the E. coli expressing medium. Therefore, the fluorescent anti-P. aeruginosa scFv developed in this study can be used as a probe for FCM analysis, especially when a shorter antibody-producing time and simpler antibody-producing method are required.
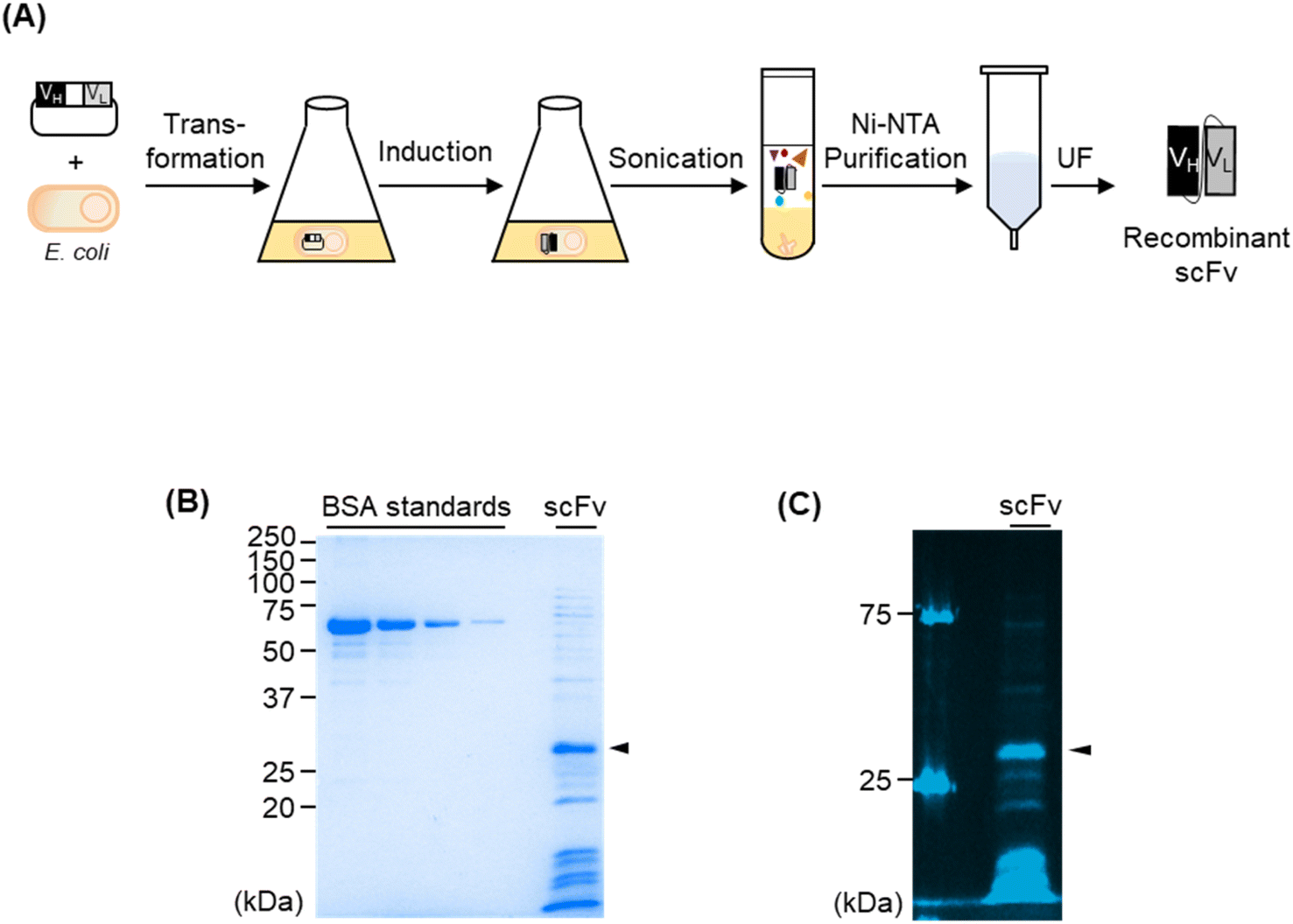 | ||
| Fig. 5 (A) Schematic representation of an entire step for producing a recombinant scFv against P. aeruginosa using E. coli. UF indicates ultra-filtration. (B) SDS–PAGE analysis of scFv. The arrow indicates the band of the target protein. (C) SDS–PAGE analysis of fluorescent scFv. | ||
 | ||
| Fig. 6 (A) Overlaying histograms obtained by flow cytometry analysis after incubation of fluorescent scFv with different numbers of P. aeruginosa strains and BL21. The total cell numbers of each assay were adjusted to 3 × 106 CFU by adding BL21 cells to the different numbers of pathogen cells. (B) Positive signal ratio of the mean of fluorescence intensity of range-gated in each histogram. Error bars represent ±1 SD (n = 3). | ||
Conclusions
In this study, we established an FCM-based method that can detect S. aureus and P. aeruginosa within 1 h, including 1–2 min of mixing fluorescent antibodies and cells, 30 min of reaction, 5 min of washing, and 5 min of analysis. Based on these assays, the minimum number of pathogen for which it was possible to consider a positive signal was 103 to 105 CFU. Because the whole process time for obtaining the assay results is very fast, preincubating the cells before reacting them with fluorescent antibodies can be further performed to increase the concentration of pathogens in the samples over the LOD values. If the bacterial load is less than the LOD for this method, preincubating the cells can increase the number of cells in the sample (i.e. bacteria can be cultured before adding fluorescent antibodies). As the whole process time for this FCM-based method is under 1 h (much faster than other conventional methods such as ELISA that requires approximately 20 h), a pre-incubation step can be added prior to the assay, providing an advantage to this FCM-based rapid detection system. In this study, we focused on evaluating an FCM-based rapid detection method. We thus used commercial or recombinant antibodies that we have used in our previous study. Modifying the antigen-binding sites of antibodies through point mutation would allow screening or selection for reagents with increased sensitivity.We applied our FCM-based method to detect S. aureus in apple juice, which carries a high risk of pathogen contamination. We generated an artificially contaminated beverage sample by adding serially diluted WKZ-2 strain to juice. We then added AF647 dye-labelled 6DW2 to the sample (Fig. S6†). After a 30 min incubation, the sample was centrifuged, washed using PBS, and analysed using FCM. Direct analysis using FCM without centrifugation and washing save 5 min by eliminating pretreatment. However, we washed the sample before loading to FCM because direct injection of commercial juice to FCM can contaminate equipment. We have demonstrated that the FCM-based method described here can detect S. aureus in beverage samples with similar efficacy as detection in PBS. Interestingly, when we centrifuged a mock pathogenic apple juice sample and added fluorescent dye-labelled 6DW2 to the pellet, we observed high background signal, indicating that the order of the pretreatment steps in processing a real sample can considerably affect the outcome.
In addition, we constructed a new DNA sequence encoding anti-P. aeruginosa scFv and expressed scFv using a rapid, inexpensive E. coli culture system. The developed scFv showed binding efficiency to three P. aeruginosa strains, which was confirmed by ELISA and FCM. The use of fluorescent IgGs and scFv combined with FCM analysis developed herein can be widely used in applications for detecting S. aureus and P. aeruginosa in samples originating from food, beverages, and clinical sources. We expect that FCM-based rapid and convenience pathogen detection using multicolour fluorescent antibody cocktails, which can be generated by conjugating fluorescent dyes with different emission wavelengths to antibodies that specifically bind each target pathogen will be useful in an extensive array of applications.
Experimental
Chemical gene was synthesized from LncBio (Seoul, Korea). Oligonucleotides and the plasmid miniprep kits were obtained from Bionics (Seoul, Korea). KOD-One DNA polymerase and the In-Fusion HD cloning kit were from Takara-Bio (Tokyo, Japan). E. coli SHuffle T7 Express lysY were obtained from New England Biolabs (Seoul, Korea). Talon beads were obtained from Clontech (Mountain View, CA, USA). The gravity empty column was from Bio-Rad (Daejeon, Korea). Ultrafiltration devices were obtained from Pall (Nanosep Centrifugal-3k; Port Washington, NY, USA). HRP-conjugated anti-DYKDDDDK antibody was obtained from (Biolegend, CA, USA). Spin-type ultrafiltration device was obtained from Millipore (Bedford, MA, USA). Rabbit anti-S. aureus polyclonal IgG and rabbit anti-P. aeruginosa polyclonal IgG were obtained from Thermo (Waltham, Massachusetts, USA). Alexa647–NHS ester was obtained from Broad Pharm (San Diego, USA). Hydroxyl amine-HCl was obtained from Daejung (Cheongju, Korea).Expression and purification of recombinant IgGs
Mammalian cell-based 6DW2 and 6CYF expressions and purifications were performed according to our previous study.12,13 Briefly, HEK293F cells were cultivated in 200 mL of Freestyle 293 medium in a flask at 37 °C in 8.0% CO2. When the density cells reached to 1 × 106 cells per mL, 1.25 μg per mL heavy chain and light chain genes of the anti-6DW2 antibody were co-transfected to the cells using 7.5 μg per mL PEI and incubated for 7 days with 120 rpm at 37 °C in 8.0% CO2. The cultivation was centrifuged (1000 rpm, 5 min, 4 °C) and the antibodies were collected from the cell supernatant through PA purification as follows: 1 mL of the cell supernatant was mixed to 10 μL of PA agarose beads, and incubated for 1 h at RT. The beads were washed three times using 500 μL of PBS, and the antibody was eluted from the beads by adding 100 μL of 0.1 M glycine (pH 2.5). Immediately, 10 μL of PBS was added to the eluent for adjusting the pH of the sample to neutral. The concentration of purified antibody was determined using nanodrop.Gene construction of recombinant scFv
To construct recombinant anti-P. aeruginosa scFv-expressing gene, VH or VL of anti-PcrV antibody expressing genes, 6CYF, were codon-optimized and amplified by polymerase chain reaction (PCR) using primers (5′-catatgggcggtggaggtggtta-3′ and 5′-atgatgatgagaacccccccc-3′), KOD One DNA polymerase, and synthesized DNA (Table 1) as a template DNA. pSrtHA was amplified by PCR using primers (5′-ggggggggttctcatcatca-3′ and 5′-acctccaccgcccatatgtatatc-3′), KOD One DNA polymerase, and pSrtCys::aPDL1scFv.28 Each insert PCR product and vector PCR product was ligated using infusion HD cloning kit, resulting in pSrtHA::a6CYFscFv. The entire coding region sequence of the obtained plasmid was confirmed by sequencing.Expression and purification of scFv
SHuffle T7 Express LysY cells were transformed with pSrtHA::a6CYFscFv cultured at 37 °C for 16 h in LB medium with 100 μg per mL ampicillin and 1.5% agar. A single colony was grown at 37 °C for overnight in 4 mL of 2xYT medium with 100 μg per mL ampicillin and utilized to inoculate 100 mL of 2xYT medium with 100 μg per mL ampicillin and 0.2% glucose. The cells were cultured at 37 °C, and 0.4 mM isopropylthio-β-galactopyranoside was added when an OD600 reached to 0.6. The cells were incubated for 16 h at 16 °C and centrifuged (4000 rpm, 30 min, 4 °C). The pellet was resuspended in 5 mL of binding buffer (50 mM phosphate buffer, 0.3 M sodium chloride (NaCl), 5 mM imidazole, pH 7.4) and sonicated (10 min, pulse time 2 s/2 s). After centrifugation (4000 rpm, 30 min, 4 °C), the supernatant was loaded onto a disposable column containing 0.2 mL of Talon beads. Next, the beads were washed three times with 10 mL of Wash buffer (50 mM phosphate buffer, 0.3 M NaCl, 20 mM imidazole, pH 7.4). After that, 3 mL of elution buffer (50 mM phosphate buffer, 0.3 M NaCl, 150 mM imidazole, pH 7.4) was loaded on the column and the solution passing through was collected. The solution was concentrated then buffer exchanged to PBS buffer (10 mM phosphate buffer, 137 mM NaCl, pH 7.4) using a MWCO 3k ultrafiltration (UF) column. The purity of the target protein was confirmed by sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE), which was loaded with the same volume of loading buffer (8 μL, 250 mM Tris–HCl, 10% SDS, 50% glycerol, 0.05% bromophenol blue, 0.5 M dithiothreitol (DTT), pH 6.8), followed by boiling at 95 °C for 5 min. The protein concentration was determined by SDS–PAGE analysis with BSA as a standard.ELISA
The antigen-binding activity of recombinant anti-P. aeruginosa scFv was tested via indirect ELISA as follows; 100 μL of series diluted P. aeruginosa was immobilized on a 96-well plate (SPL, maxi binding plate) for 16 h at 4 °C. Afterwards, 350 μL blocking buffer (PBS buffer with 3% BSA) was added to the well and incubated 2 h at 25 °C. The well was washed three times with ELISA buffer (PBS buffer with 0.1% BSA). Subsequently, 10 μg mL−1 of purified scFv in 100 μL of ELISA buffer was added to the well and incubated for 1 h at 25 °C. After washing three times using 350 μL of ELISA buffer, 10000-fold diluted HRP-conjugated anti-DYKDDDDK antibody in ELISA buffer was added to the well and incubated for 1 h at 25 °C. The well was washed three times with 350 μL of ELISA buffer and developed with 50 μL of TMB solution for 10 min. The reaction was stopped by adding 50 μL of 1 N H2SO4 to the well, and the absorbance was read using a microplate reader. Dose–response curves were constructed by fitting the absorbance at 450 nm using the GraphPad Prism software (GraphPad Software, San Diego, CA).
Fluorescent dye conjugation
Fluorescent antibodies were prepared and confirmed by the following method. 10 μg of proteins in PBS buffer reacted with ×20 mol of Alexa647–NHS ester in the dark at 25 °C for 2 h. 8 μL of fluorescent antibody was mix with 2 μL of transparent loading buffer without bromophenol blue and loaded to the SDS–PAGE gel as a native sample. 8 μL of fluorescent antibody was mix with 2 μL of 0.5 M DTT-containing loading buffer without bromophenol blue, boiled at 95 °C for 5 min, and loaded to the SDS–PAGE gel as a denatured sample. The gel was scanned using a fluorescence scanner (ImageQuant LAS 500, Cytiva, Korea).Cell culture
Two S. aureus strains, WKZ-1 (NR-28984) and WKZ-2 (NR-28985), three P. aeruginosa strains, UCBPP PA14, ATCC 27853, and ATCC BAA-2108, and one E. coli strain, BL21(DE3) were cultured according to our previous study as follows:12,13 the stocks of cultures were inoculated into 3 mL of LB media and incubated overnight at 37 °C with 200 rpm shaking. The sample was centrifuged (3500 rpm, 10 min, 4 °C) and the supernatant was discarded. The diluted cells with PBS reached an OD 600 of 0.5 with a cell density of 108 CFU mL−1.Flow cytometry
Cell cultures were diluted in PBS and transferred into microtubes. 2 μg of fluorescent full-sized antibody and 500 ng recombinant scFv was added to serially diluted pathogen samples in 0.3 mL PBS. After incubation for 1 h at RT, 1% methanol was added. Fluorescent antibody-stained samples were analysed using a FACS CantoII cytometer (BD Biosciences) and data were processed using Flowjo software. Either over 5000 or 10000 events were recorded for each sample. Acquisition was visualized in a cell count/FITC histogram in which regions were created for the blue cell-unit population (FITC). The positive signal population of the samples was obtained by gating based on negative control containing only BL21 and fluorescent antibodies, and the positive ratio for the total number of cells was calculated. Dose–response curves were constructed using the GraphPad Prism software. The EC50 value was calculated from the curve fitting to a 4-parameter logistic equation of Y = bottom + (top − bottom)/(1 + 10((logEC50 − X) × hillslope)) using GraphPad Prism software. The LOD was calculated using following equation: LOD = mean blank + 1.645 × SDblank + 1.645 × SDlow concentration sample.29
Author contributions
Conceptualization, J. K. K. and H. J. J.; methodology and analysis, J. K. K., H. Y., C. H. Y., and E. J. K.; investigation, J. K. K., H. Y., C. H. Y., E. J. K., C. S. L. and W. K.; writing—original draft preparation, J. K. K., H. Y., and C. H. Y.; writing—review and editing, C. S. L., W. K., B. G. K., and H. J. J.; supervision, H. J. J. All authors reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Korea Medical Device Development Fund grant funded by the Korea government (KMDF_PR_20200901_0073), NRF grants funded by the Korean government (2020R1I1A307411712, 2021H1D3A2A02096525), 2022 Hongik University Research Fund, and 2022 Hongik University Innovation Support Program Fund.Notes and references
- E. Abebe, G. Gugsa and M. Ahmed, J. Trop. Med., 2020, 2020, 4674235 CrossRef PubMed.
- Y. Tang, Z. Ali, J. Zou, G. Jin, J. Zhu, J. Yang and J. Dai, RSC Adv., 2017, 7, 51789–51800 RSC.
- S. Y. Tong, J. S. Davis, E. Eichenberger, T. L. Holland and V. G. Fowler Jr, Clin. Microbiol. Rev., 2015, 28, 603–661 CrossRef CAS PubMed.
- S. Affhan, W. Dachang, Y. Xin and D. Shang, Genet. Mol. Res., 2015, 14, 17044–17058 CrossRef CAS PubMed.
- J. Wang, X. Wu, C. Wang, N. Shao, P. Dong, R. Xiao and S. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 20919–20929 CrossRef CAS PubMed.
- X. Zhao, C. Wei, J. Zhong and S. Jin, Biotechnol. Biotechnol. Equip., 2016, 30, 827–833 CrossRef CAS.
- D. Kennedy, U. P. Cronin, A. Piterina and M. G. Wilkinson, Appl. Environ. Microbiol., 2019, 85, e01006 CrossRef CAS PubMed.
- N. M. Kingsford and H. W. Raadsma, Vet. Microbiol., 1995, 47, 61–70 CrossRef CAS PubMed.
- R. Lavenir, D. Jocktane, F. Laurent, S. Nazaret and B. Cournoyer, J. Microbiol. Methods, 2007, 70, 20–29 CrossRef CAS PubMed.
- S. Nakano, T. Kobayashi, K. Funabiki, A. Matsumura, Y. Nagao and T. Yamada, J. Food Prot., 2004, 67, 1271–1277 CrossRef CAS PubMed.
- S. J. Kwon, T. Jeon, D. Seo, M. Na, E. G. Choi, J. W. Son, E. H. Yoo, C. G. Park, H. Y. Lee, J. O. Kim, S. Y. Kim and J. Kang, Tuberc. Respir. Dis., 2012, 72, 293–301 CrossRef PubMed.
- J. K. Kim, G. M. Lim, E. J. Kim, W. Kim, C. S. Lee, B. G. Kim and H. J. Jeong, ACS Omega, 2022, 7, 9690–9700 CrossRef CAS PubMed.
- G. M. Lim, J. K. Kim, E. J. Kim, C. S. Lee, W. Kim, B. G. Kim and H. J. Jeong, BMC Biotechnol., 2022, 22, 21 CrossRef CAS PubMed.
- H. M. Davey, Methods Cell Sci., 2002, 24, 91–97 CrossRef CAS PubMed.
- M. A. Hahn, P. C. Keng and T. D. Krauss, Anal. Chem., 2008, 80, 864–872 CrossRef CAS PubMed.
- D. Kennedy, U. P. Cronin and M. G. Wilkinson, Appl. Environ. Microbiol., 2011, 77, 4657–4668 CrossRef CAS PubMed.
- J. Schellenberg, W. Smoragiewicz and B. Karska-Wysocki, J. Microbiol. Methods, 2006, 65, 1–9 CrossRef CAS PubMed.
- G. M. Cooper, The Cell: A Molecular Approach. The Origin and Evolution of Cells, Sinauer Associates, Sunderland (MA), 2000 Search PubMed.
- B. R. Dixon, J. M. Bussey, L. J. Parrington and M. Parenteau, J. Clin. Microbiol., 2005, 43, 2375–2379 CrossRef PubMed.
- S. B. Doherty, L. Wang, R. P. Ross, C. Stanton, G. F. Fitzgerald and A. Brodkorb, J. Microbiol. Methods, 2010, 82, 301–310 CrossRef CAS PubMed.
- B. C. Ferrari and P. L. Bergquist, Cytometry, Part A, 2007, 71, 265–271 CrossRef CAS PubMed.
- K. Hibi, A. Abe, E. Ohashi, K. Mitsubayashi, H. Ushio, T. Hayashi, H. Ren and H. Endo, Anal. Chim. Acta, 2006, 573–574, 158–163 CrossRef PubMed.
- X. Meng, G. Yang, F. Li, T. Liang, W. Lai and H. Xu, ACS Appl. Mater. Interfaces, 2017, 9, 21464–21472 CrossRef CAS PubMed.
- Y. Shan, C. Xu, M. Wang, Z. Zhu, F.-G. Wu, Z. Shi, Q. Cui and G. M. Arumugam, Talanta, 2019, 192, 154–159 CrossRef CAS PubMed.
- J. P. Diaper and C. Edwards, Microbiology, 1994, 140, 35–42 CrossRef CAS PubMed.
- B. W. S. Liu, Z. Sui, Z. Wang, L. Li, X. Zhen, W. Zhao and G. Zhou, Foodborne Pathog. Dis., 2021, 18, 346–353 CrossRef PubMed.
- R. Fong, K. Kajihara, M. Chen, I. Hotzel, S. Mariathasan, W. L. W. Hazenbos and P. J. Lupardus, mAbs, 2018, 10, 979–991 CAS.
- S. H. Kim and H. J. Jeong, Appl. Sci., 2021, 11, 9149 CrossRef CAS.
- D. A. Armbruster and T. Pry, Clin. Biochem. Rev., 2008, 29(Suppl 1), S49–S52 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra05694a |
| This journal is © The Royal Society of Chemistry 2022 |