
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePd/Cu-catalyzed access to novel 3-(benzofuran-2-ylmethyl) substituted (pyrazolo/benzo)triazinone derivatives: their in silico/in vitro evaluation as inhibitors of chorismate mutase (CM)†
Gangireddy Sujeevan Reddyab,
Sharda Shuklaab,
Harshavardhan Bhuktarab,
Kazi Amirul Hossaina,
Rebecca Kristina Edwina,
Varadaraj Bhat Giliyarub,
Parimal Misraa and
Manojit Pal *a
*a
aDr. Reddy's Institute of Life Sciences, University of Hyderabad Campus, Gachibowli, Hyderabad 500 046, India. E-mail: manojitpal@rediffmail.com
bManipal College of Pharmaceutical Sciences, Manipal Academy of Higher Education, Madhav Nagar, Manipal 576 104, Karnataka, India
First published on 21st September 2022
Abstract
In view of the reported chorismate mutase (CM or MtbCM) inhibitory activities of 3-indolylmethyl substituted (pyrazolo/benzo)triazinone derivatives the structurally similar 3-(benzofuran-2-ylmethyl) substituted (pyrazolo/benzo)triazinones were designed and evaluated in silico against CM. The docking of target molecules was performed at the interface site of MtbCM (PDB: 2FP2). All the best ranked molecules participated in a strong H-bonding with the ILE67 of the B chain at the backbone position in addition to several hydrophobic/van der Waals interactions with the hydrophobic residues. Based on encouraging docking results, the one-pot synthesis of newly designed benzofuran derivatives was carried out using tandem Pd/Cu-catalyzed Sonogashira cross-coupling followed by intramolecular cyclization of 2-iodophenols with appropriate terminal alkynes. A range of novel 3-(benzofuran-2-ylmethyl) substituted (pyrazolo/benzo)triazinone derivatives were prepared in high (>80%) yields. Three molecules i.e. 3h, 3i and 3m that participated in good interaction with CM in silico showed encouraging (64–65%) inhibition at 30 μM in vitro. An SAR within this class of molecules suggested that the benzotriazinone series in general was better than the pyrazolotriazinone series. Based on molecular docking in silico, CM inhibition in vitro and computational ADME prediction the benzofuran derivatives 3i and 3m seemed to be of further medicinal interest in the context of discovery and development of new anti-tubercular agents.
1. Introduction
While COVID-19 has emerged as the leading cause of death worldwide since 2019, tuberculosis (TB) the previous major killer still poses a considerable threat to human health. Indeed, as a single infectious agent Mycobacterium tuberculosis has been the cause of death of 1.4 million people including 0.2 million HIV-positive patients according to the 2020 report of the WHO.1 Besides the co-morbidity with HIV-AIDS the other factors causing TB related death include the long treatment duration e.g. 6–9 months, increased occurrence of multi or extensive drug resistance and declining importance or endeavor in anti-infective drug research. Over the years chorismate mutase or CM (EC 5.4.99.5) or MtbCM (Mycobacterium tuberculosis CM) has been explored as one of the pharmacological targets for the identification of potential anti-tubercular agents.2 Because of its presence in bacteria but not in animals and its key role in the survival of bacteria2–4 the enzyme CM (that catalyzes the Claisen rearrangement of chorismate to prephenate) has emerged as an interesting drug target. Due to our interest in the area of anti-tubercular agents5 we have reported the synthesis and evaluation of a range of small organic molecules as inhibitors of CM.6,7 Recently, we have explored the 3-indolylmethyl substituted (pyrazolo/benzo)triazinone derivatives A (Fig. 1) for the identification of potent inhibitors of CM.8 In further continuation of this research we became interested in assessing a library of compounds represented by B (Fig. 1) that was obtained via replacing the indole moiety of A by a benzofuran ring. The choice of benzofuran framework was mainly prompted by the fact that beside being an integral part of several reagents such as benzbromarone, amiodarone, etc. (that are already in patient use) this framework has been explored previously for the identification of anti-tubercular as well as anti-bacterial agents.9 Further, chemically both indole and the benzofuran ring share some common structural features.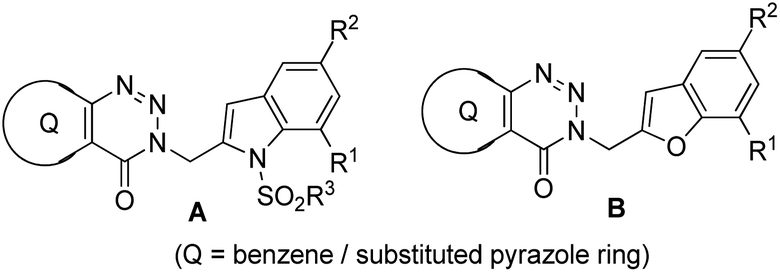 | ||
| Fig. 1 Reported inhibitors of CM based on (pyrazolo/benzo)triazinone derivatives A and the proposed template B. | ||
Moreover, CM inhibitory potential of benzofuran derivatives have been documented recently.10 Herein, we report the in silico studies, chemical synthesis and in vitro CM inhibitory potential of a series compounds based on B (Fig. 1).
2. Results and discussion
Initially, before undertaking the actual synthesis of library of molecules based on B (Fig. 1) the merit and potential of the current designing approach was assessed via the in silico docking study of several 3-(benzofuran-2-ylmethyl) substituted (pyrazolo/benzo)triazinone derivatives derived from B. Due to the beneficial role of allosteric regulation of protein in its function as well as in many biological processes, the allosteric sites are considered as attractive targets for the design of new NCEs (new chemical entities). On the other hand, study has confirmed the allosteric behavior of Saccharomyces cerevisiae chorismate mutase (ScCM) on the basis of conformational dynamics and solvent entropy.11 Further, the allosteric inhibition of MtbCM by all three aromatic amino acids (Trp, Phe or Tyr at 0.5 mM) at interface site has been noted previously.12Since the binding energy of our studied ligands at interface site was lower than the other possible sites of MtbCM, hence we thought that our molecules were preferably stabilized at the interface site of MtbCM (which is a homodimer). Nevertheless, the docking of our molecules at the interface site of MtbCM (PDB: 2FP2) was performed using the open-source tool AutoDock Vina.13 Additionally, the MarvinSketch14 and AutoDock tool15 were also used for conducting the docking studies and the docking score of the best pose of each ligand along with that of the reported compound8 A-1 is presented in Table 1. To check the reproducibility of docking results, the docking of each individual ligand was performed at least for five times when the maximum difference in the scores obtained was found to be within the range of ±0.2. The validation of docking protocol was carried out by re-docking the co-crystal ligand (TSA) and calculating the RMSD difference between co-crystal one and docked one in PyMOL.16 The maximum RMSD being 1.713 (<2) clearly confirmed the correctness of the docking protocol employed.
| a Figure within the bracket represents AutoDock Vina score (kcal mol−1). |
|---|
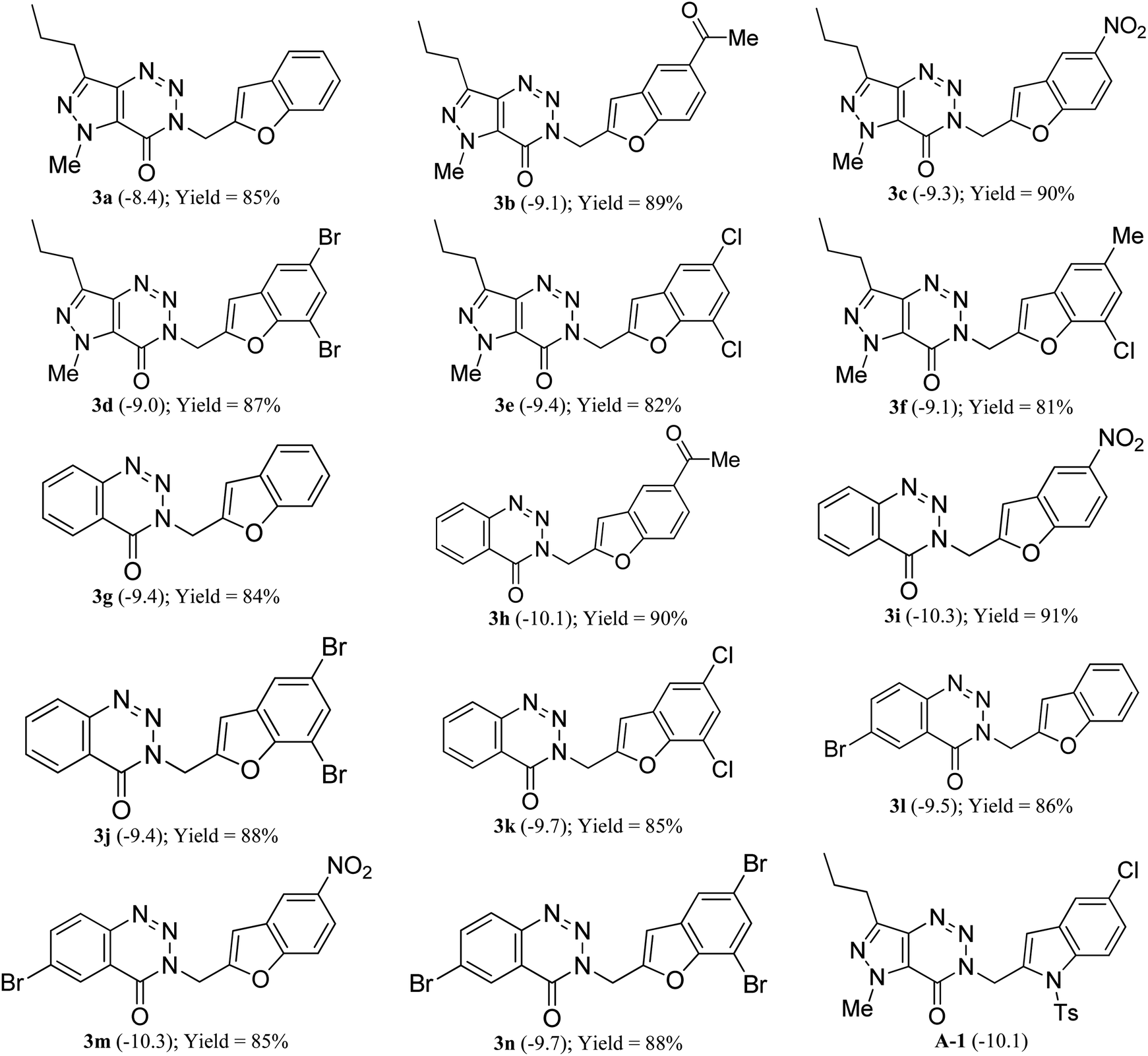 |
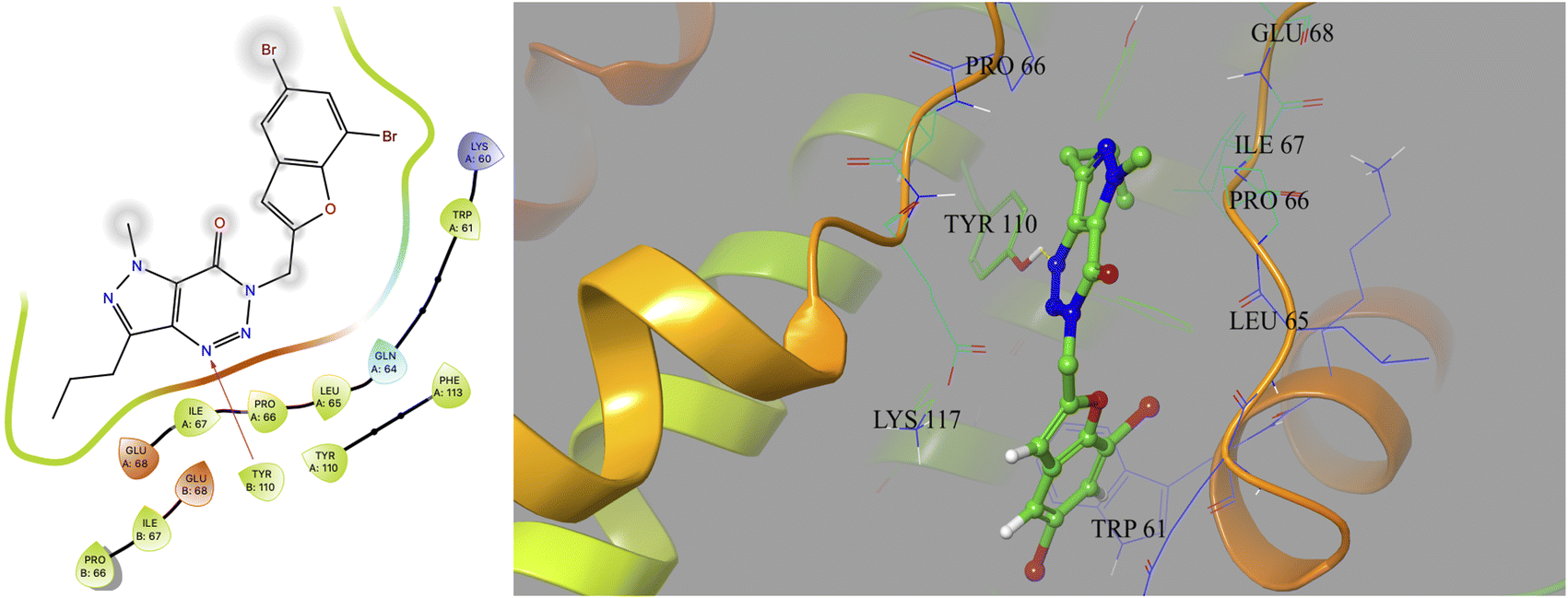
Based on docking scores (Table 1) the compound 3h, 3i and 3m (score ∼ −10 kcal mol−1) appeared as potential hits in the in silico studies and was comparable to the known compound A-1. Indeed, the compound 3h (Fig. 2) formed a strong H-bond through the ketone carbonyl group with the ILE67 of B chain at the backbone position. Additionally, the molecule also participated in the hydrophobic/van der Waals interactions with the hydrophobic residues (LEU65, PRO66, TYR110, PHE113, LEU65 of both A and B chain) and hydrophobic regions (polar and charged residues e.g. GLU68 of both A and B chain and GLN64, LYS117 of A chain). The compound 3i (Fig. 3) showed H-bonding interaction through its nitro group with the backbone atom of ILE67 of B chain in addition to a π–π interaction with TYR110 of B chain (which could be the reason of its slightly better docking score than compound 3h). Similar hydrophobic contacts mainly with LEU56, PRO66, PHE113, GLU68, etc. of both the chain were also found in this case. Like compound 3i, the involvement of nitro group in the H-bond interaction was noted for compound 3m (Fig. 4) in addition to its participation in the usual hydrophobic interactions.
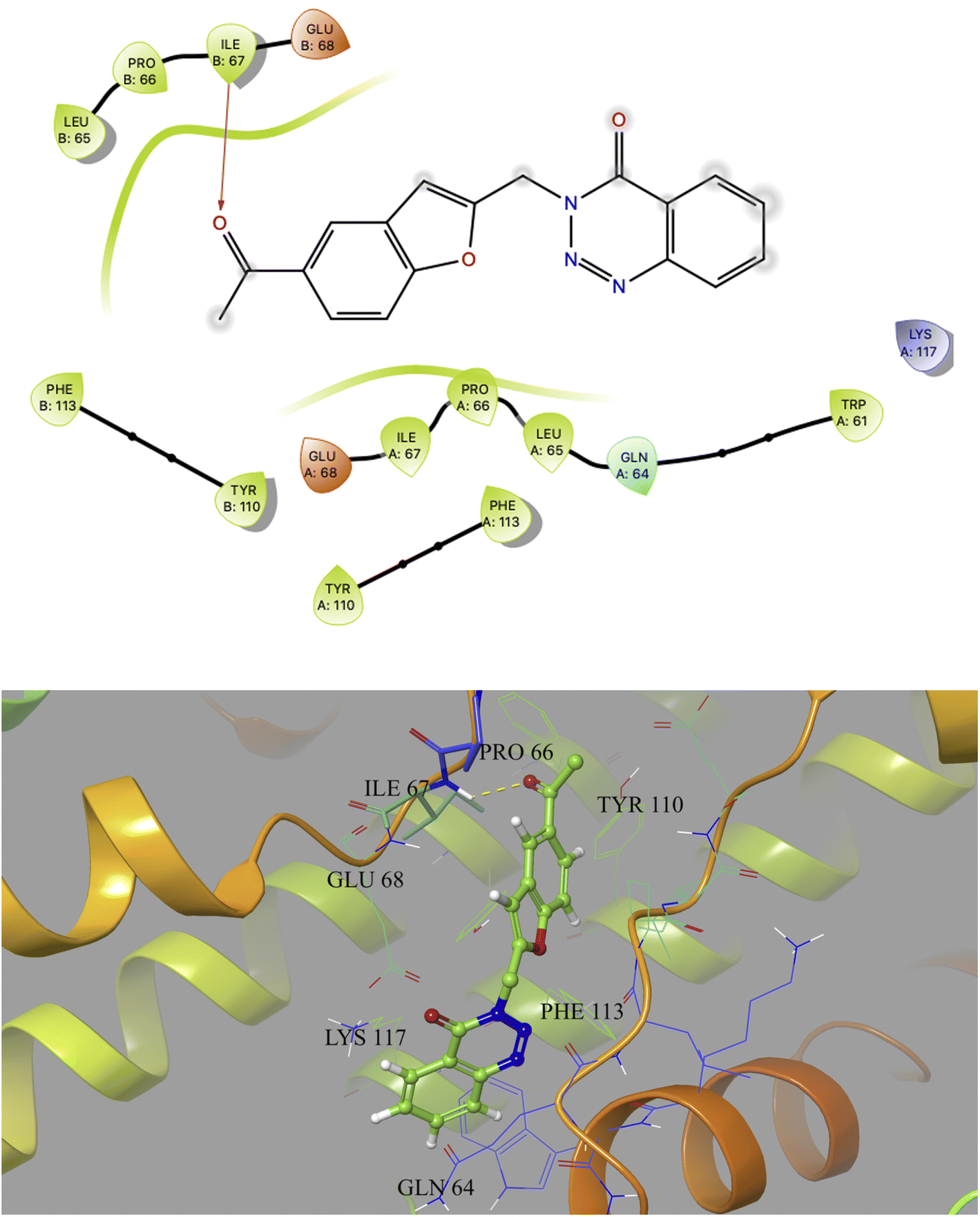 | ||
| Fig. 2 2D and 3D interaction (H-bond is shown in yellow colour) of compound 3h with the interface residues of MtbCM (PDB: 2FP2) [prepared using Maestro visualizer (Schrödinger, LLC)]. | ||
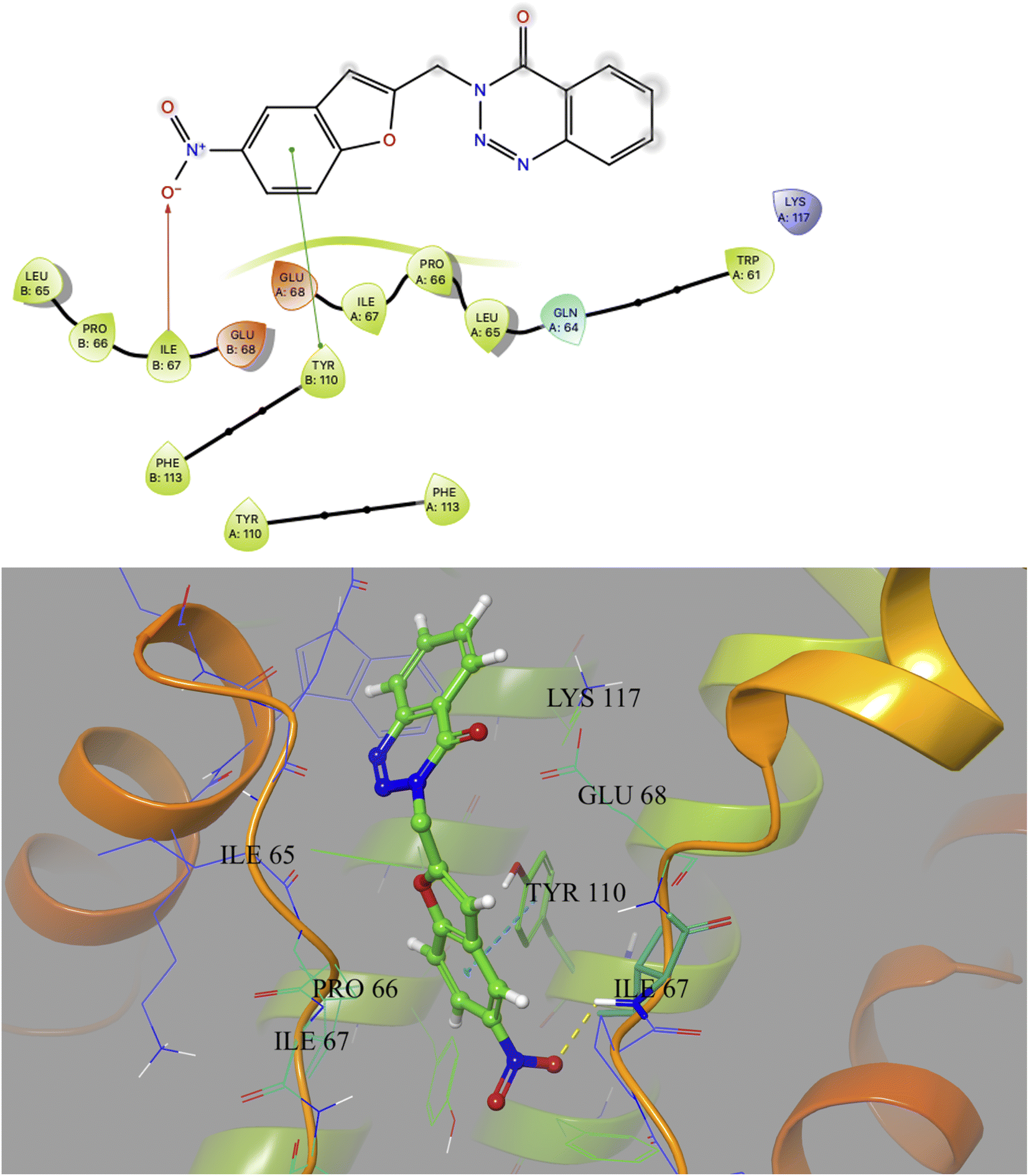 | ||
| Fig. 3 2D and 3D interaction (H-bond and π–π interaction shown in yellow and cyan colour, respectively) of compound 3i with the interface residues of MtbCM (PDB: 2FP2). | ||
 | ||
| Fig. 4 2D and 3D interaction (H-bond and π–π interaction shown in yellow and cyan colour, respectively) of compound 3m with the interface residues of MtbCM (PDB: 2FP2). | ||
Nevertheless, in the light of promising overall outcome of the in silico study it was become necessary to correlate these observations with the data generated through an in vitro assay. Hence, the chemical synthesis as well as some preliminary in vitro evaluation of compounds presented in Table 1 was undertaken and the results are presented in the following sections. Notably, the in silico studies, synthesis and in vitro evaluation of 3-(benzofuran-2-ylmethyl) substituted (pyrazolo/benzo)triazinone derivatives as potential inhibitors of chorismate mutase (CM) was not known previously.
Among the several possible strategies considered for the synthesis of targeted compounds based on B (Fig. 1), we envisioned that the construction of the benzofuran ring could be a convenient as well as effective approach for serving the purpose. While numerous approaches are known for the construction of 2-substituted benzofuran ring,17 the one-pot method18–35 involving the tandem transition-metal-catalyzed Sonogashira cross-coupling36 followed by intramolecular cyclization of 2-halogenated phenols with terminal alkynes gained considerable interest due to its convenience, functional group tolerance and environmental advantages. Indeed, one of us has been involved in exploring this strategy since 1992 (ref. 37) and its subsequent applications in the preparation of potential bioactive compounds.38–41 In further continuation of this efforts we decided to adopt a similar one-pot synthetic strategy in the current study for accessing the targeted 3-(benzofuran-2-ylmethyl) substituted (pyrazolo/benzo)triazinone derivatives. Thus, the Pd/Cu-catalyzed coupling of the required terminal alkyne (1) [e.g. 5-methyl-3-(prop-2-ynyl)-7-propyl-3H-pyrazolo[4,3-d][1,2,3]triazin-4(5H)-one (1a) or 3-(prop-2-yn-1-yl)benzo[d][1,2,3]triazin-4(3H)-one (1b) or 6-bromo-3-(prop-2-yn-1-yl)benzo[d][1,2,3]triazin-4(3H)-one (1c)] obtained via the reported method8 with appropriate 2-iodophenols (2) afforded the expected products (3) in good to excellent yields (Scheme 1).
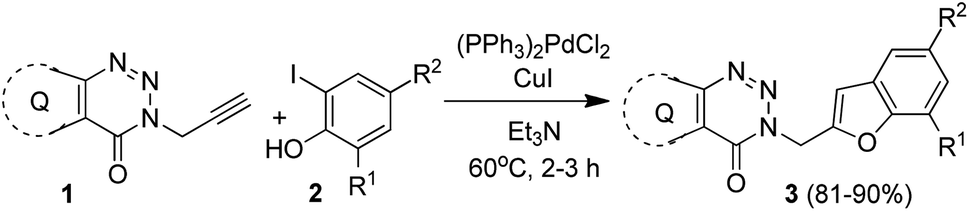 | ||
| Scheme 1 Pd/Cu-catalyzed synthesis of 3-(benzofuran-2-ylmethyl) substituted (pyrazolo/benzo)triazinone derivatives. | ||
We used a similar reaction conditions reported by us previously20,37a with some suitable modifications as the use of terminal alkyne 1 for the coupling with 2-iodophenols was not explored earlier. For example, a longer reaction time i.e. 16–24 h was required in the previously reported method when the reaction was performed using 2–3.5 mol% of (Ph3P)2PdCl2, 3–5 mol% of CuI and 2 equiv. of Et3N in DMF. In the current study, we have observed that the reaction time could be decreased significantly with the marginal increase in the catalyst loading such as the use of 5 mol% of (Ph3P)2PdCl2 and CuI. Nevertheless, a range of reaction conditions by changing the catalyst, base and solvent was examined for the coupling of alkyne 1a with 2-iodophenol 2a (Table 1). Among the combination of various Pd-catalyst, ligand PPh3 and co-catalyst CuI tested (entries 1–7, Table 2), the combination of (Ph3P)2PdCl2 and CuI (entry 7, Table 2) was found to be most effective for affording the desired product 3a in high yield. Further, the use of a solvent such as DMF (entries 1–4, Table 2) or EtOH (entries 5 and 6, Table 2) was counterproductive and was not necessary in the present case because the excess of Et3N could play the role of base as well as solvent (entry 7, Table 2). Indeed, the reaction was completed within 2 h when performed in Et3N alone whereas product yield was not increased when longer reaction time was used (entry 7 vs. 8, Table 2). However, in view of the known toxicity of Et3N an attempt was made to reduce its volume by conducting the reaction using Et3N as a base, and dry DMF as a solvent in the presence of Pd(PPh3)2Cl2 and CuI as catalysts for 4 h. The desired product 3a was obtained in 55% yield in this case that was less than that of entry 7 of Table 2. The reaction did not proceed in the absence of co-catalyst CuI (entry 9, Table 2) or the catalyst Pd(PPh3)2Cl2 (entry 10, Table 2). Thus, the condition of entry 7 of Table 2 was used for the preparation of various other analogues of 3a.
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Base | Solvent | Time (h) | % Yieldb |
| a Reactions were carried out using alkyne 1a (1 mmol), 2a (1.2 mmol), catalyst and base (2.0 mmol) in a solvent (5 mL) under nitrogen at 60 °C.b Isolated yield.c 5 mol% of each catalyst used.d 5 mol% of each catalyst and 10 mol% of PPh3 used. | |||||
| 1 | 10% Pd/C/CuI | K2CO3 | DMF | 6 | 0c |
| 2 | 10% Pd/C/PPh3/CuI | K2CO3 | DMF | 6 | 15d |
| 3 | Pd(OAc)2/PPh3/CuI | K2CO3 | DMF | 6 | 20d |
| 4 | Pd(OAc)2/PPh3/CuI | Et3N | DMF | 6 | 30d |
| 5 | Pd(OAc)2/PPh3/CuI | Et3N | EtOH | 6 | 20d |
| 6 | Pd(PPh3)2Cl2/CuI | Et3N | EtOH | 6 | 40c |
| 7 | Pd(PPh3)2Cl2/CuI | Et3N | Et3N | 2 | 85c |
| 8 | Pd(PPh3)2Cl2/CuI | Et3N | Et3N | 6 | 83c |
| 9 | Pd(PPh3)2Cl2 | Et3N | Et3N | 6 | 0c |
| 10 | CuI | Et3N | Et3N | 6 | 0c |
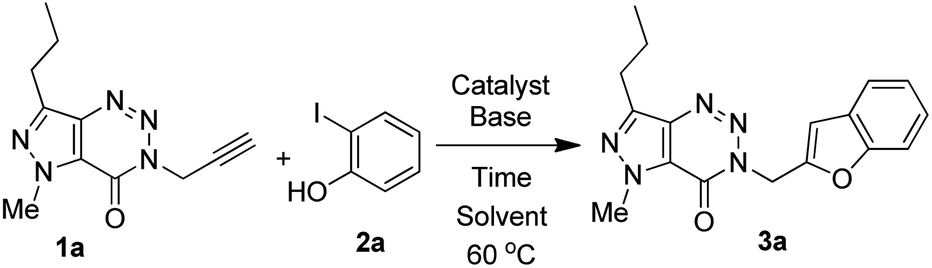
Three different terminal alkynes (1a–c) were employed for the reaction with a variety of 2-iodophenols (2a–f) in the presence of (Ph3P)2PdCl2 and CuI in Et3N alone at 60 °C (Table 1). The 2-iodophenols may contain the electron withdrawing group like CH3CO or NO2, halogen such as Cl or Br and electron donating group like CH3. The reactions proceeded smoothly in all these cases and reached to the completion within 2–3 hours affording a range of desired products (3) in high (>80%) yields. All the 3-(benzofuran-2-ylmethyl) substituted (pyrazolo/benzo)triazinone derivatives (3a-n) obtained were characterized with the help of 1H as well as 13C NMR and MS spectral data. Briefly, the presence of benzofuran ring in these compounds was indicated by the appearance of C-3 proton in 1H NMR spectra as a singlet in the range δ 6.9–7.2 and C-3 carbon in 13C NMR at 106 ppm. The linker “CH2” moiety appeared near δ 4.5 and 45 ppm in the 1H and 13C NMR spectra, respectively. Further the signal near 155 ppm in 13C NMR spectra was due to the “C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O” carbon. These are shown in the selected 1H and 13C NMR data of two representative compounds 3c and 3i (Fig. 5). The compound 3c was further characterized by the presence of NCH3 group at δ 4.17 and the n-propyl protons at δ 2.93 (t, CH2), 1.86–1.73 (m, CH2) and 0.94 (t, CH3) in 1H NMR spectra.
O” carbon. These are shown in the selected 1H and 13C NMR data of two representative compounds 3c and 3i (Fig. 5). The compound 3c was further characterized by the presence of NCH3 group at δ 4.17 and the n-propyl protons at δ 2.93 (t, CH2), 1.86–1.73 (m, CH2) and 0.94 (t, CH3) in 1H NMR spectra.
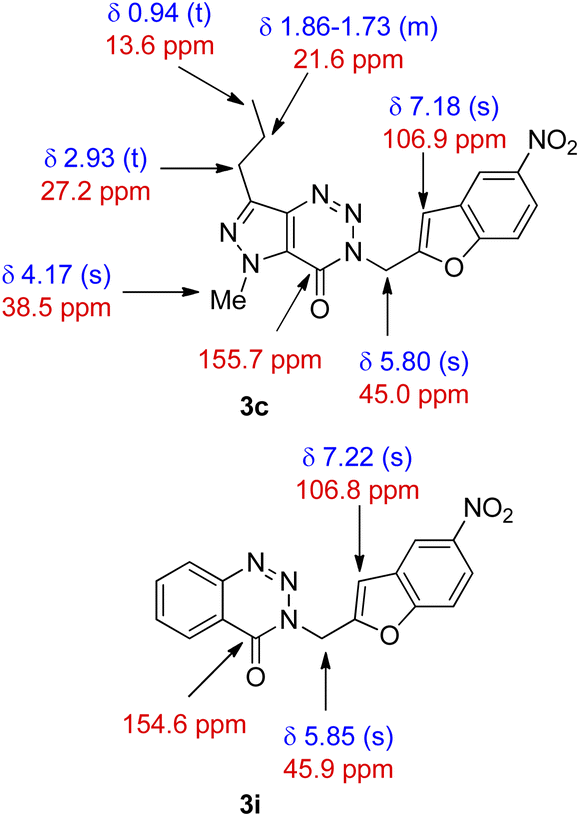 | ||
| Fig. 5 Selected 1H (blue) and 13C NMR (red) spectral data of compound 3c and 3i. | ||
From the view point of reaction mechanism,20,22–24,29,32 the involvement of two key steps e.g. Sonogashira coupling followed by intramolecular cyclization in the current approach are depicted in Scheme 2. Thus, the active Pd(0) species generated from Pd(II) participated in oxidative addition to 2 to afford the intermediate E-1 that on trans-organometallation with copper acetylide (generated in situ from compound 1 and CuI) followed by reductive elimination of Pd(0) furnished the internal alkyne E-3 via E-2 thereby completing the first catalytic cycle. In the second catalytic cycle the interaction of Cu(I) species with the alkyne moiety of E-3 (seemed to be aided by the proximate carbonyl group) formed E-4 that on intramolecular ring closure afforded E-5 and then E-6 and finally the product 3 with the regeneration of CuI to complete the second catalytic cycle.
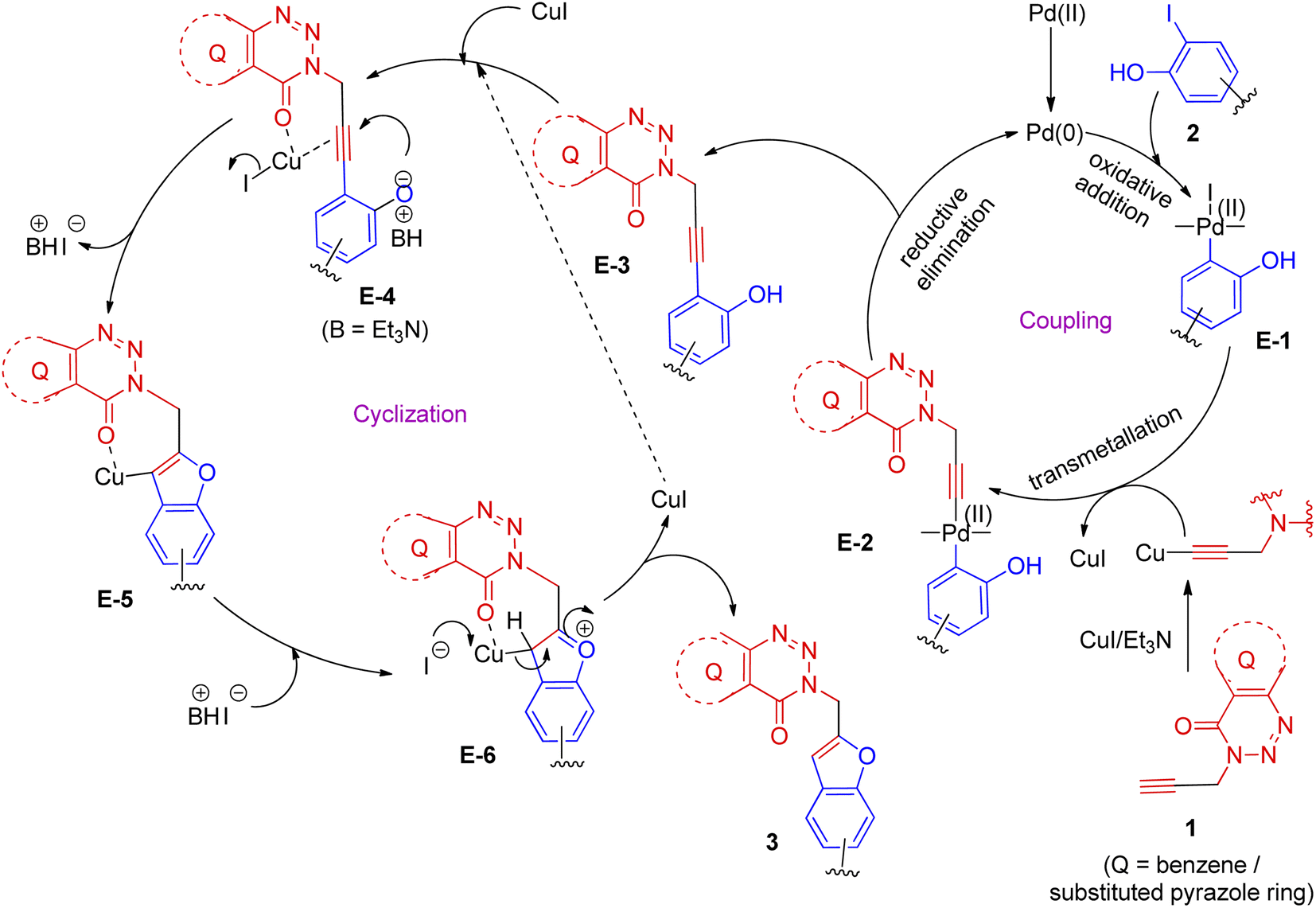 | ||
| Scheme 2 The plausible reaction mechanism for coupling-cyclization of fused 1,2,3-triazin-4-one based alkyne (1) with o-iodophenols (2) under Pd/Cu-catalysis. | ||
Next, the CM inhibitory potential of the synthesized compounds 3 was assessed in vitro42,43 with the help of an assay that measures the catalytic activity of CM during the conversion of substrate chorismate to the product prephenate. The known compound A-1 (Table 1) and a reported inhibitor44 C [i.e. 4-(3,4-dimethoxyphenethylamino)-3-nitro-5-sulfamoylbenzoic acid] was used as the reference compound in this assay. At the concentration of 30 μM the compound 3h, 3i and 3m showed 63.75 ± 1.09, 65.24 ± 0.98 and 64.90 ± 1.05% inhibition respectively that was comparable to 69.67 ± 0.57% inhibition of the reference compound A-1 (53.23 ± 0.13% inhibition at 10 μM was observed for the known inhibitor C). The other compounds that showed significant inhibition of CM included 3k (63.66 ± 2.07), 3n (63.75 ± 1.12) and 3e (61.28 ± 2.03), 3g (60.27 ± 3.22) and 3j (60.09 ± 2.35). Among rest of the compounds 3a and 3d appeared to be the least active (>45% inhibition) that was reflected in the results of docking studies. Indeed, the typical H-bonding interaction (as observed in case of active molecules) was missing in case of compound 3a (Fig. 6) due to the lack of any group at C-5 position of its benzofuran ring. Further, though the molecule participated in the hydrophobic interactions but the number of such contacts was lower than that of active molecules. On the other hand, the compound 3d (Fig. 7) though participated in a H-bond interaction through one of the trazinone nitrogen with the TYR110 of B chain however fewer hydrophobic contacts (with PRO66, ILE67, GLU68 of both A and B chain, and LYS60, TRP61, GLN64, PHE113 of A chain) were noted in this case.
 | ||
| Fig. 6 2D and 3D interaction of compound 3a with the interface residues of MtbCM (PDB: 2FP2). | ||
 | ||
| Fig. 7 2D and 3D interaction of compound 3d with the interface residues of MtbCM (PDB: 2FP2). | ||
Nevertheless, in general both in silico and in vitro evaluation suggested that the compounds belonging to the benzotriazinone series emerged as better active molecules than that from pyrazolotriazinone series. Indeed, all the best active molecules i.e. 3h, 3i and 3m emerged from the benzotriazinone series. Thus, an overview of SAR (Structure–Activity-Relationship) noted for the current series of benzofuran derivatives is presented in Fig. 8. Briefly, the “NO2” or “CH3CO” group at C-5 of the benzofuran ring was favorable for CM inhibition while other substituents like Cl, Br or Me or no substituent at this position reduced the activity. A “H” atom at C-7 position was generally better than the “Cl” or “Br” group at the same position in terms of activity. The inhibitory activity was influenced by the aryl/heteroaryl ring fused with the triazinone moiety and as mentioned above, the benzene ring rather than the pyrazole moiety was found to be beneficial for the CM inhibition of the current series of compounds.
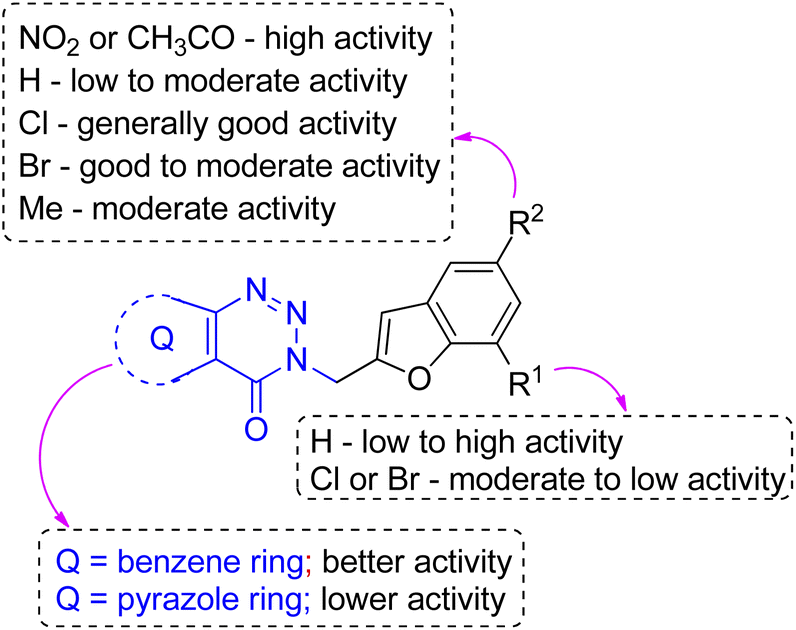 | ||
| Fig. 8 SAR summary for CM inhibitory activities of compound 3. | ||
In view of usefulness of the early detection of ADME (absorption, distribution, metabolism, and excretion) or pharmacokinetic properties of NCEs the computational ADME prediction of compounds 3h, 3i and 3m along with the reference compound A-1 was carried out using SwissADME web-tool.45 It is evident from Table 3 that desirable ADME was predicted for compound 3i and 3m with high GI absorption, no BBB (Blood Brain Barrier) penetration and no P-gp substrate potential. Indeed, the superior bioavailability score over the reference molecule A-1 was predicted for these compounds with no violation of Lipinski's criteria in contrary to A-1. However, BBB penetration was predicted for compound 3h. Thus, the benzofuran derivatives 3i and 3m seemed to be of further medicinal interest for the discovery and development of new and promising inhibitors of CM.
| Properties | Molecules | ||
|---|---|---|---|
| A-1 | 3i | 3m | |
a log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P: lipophilicity.b logS (ESOL): water solubility, calculated by ESOL method which Quantitative Structure–Property Relationship (QSPR) based model.c GI: gastrointestinal.d BBB: blood brain barrier.e P-gp: permeability glycoprotein.f MW: molecular weight. P: lipophilicity.b logS (ESOL): water solubility, calculated by ESOL method which Quantitative Structure–Property Relationship (QSPR) based model.c GI: gastrointestinal.d BBB: blood brain barrier.e P-gp: permeability glycoprotein.f MW: molecular weight. |
|||
| (i) Physicochemical | |||
| Molecular weight (g mol−1) | 511.00 | 322.28 | 401.17 |
| Consensus logPa |
4.41 | 2.34 | 2.88 |
| logS (ESOL)b |
−6.41 (poorly soluble) | −4.32 (moderately soluble) | −5.22 (moderately soluble) |
|
|||
| (ii) Pharmacokinetics | |||
| GIc absorption | High | High | High |
| BBBd permeation | No | No | No |
| P-gpe substrate | No | No | No |
|
|||
| (iii) Druglikenss | |||
| Lipinski rule | 1 violation: MWf > 500 | No violation | No violation |
| Veber rule | No violation | No violation | No violation |
| Bioavailability score | 0.17 | 0.55 | 0.55 |
3. Conclusion
In conclusion, 3-(benzofuran-2-ylmethyl) substituted (pyrazolo/benzo)triazinone derivatives were designed and evaluated in silico against CM as potential inhibitors of this enzyme. The docking of target molecules was performed at the interface site of MtbCM (PDB: 2FP2), which is a homodimer, using the open-source tool AutoDock Vina. All the best ranked molecules participated in a strong H-bonding with the ILE67 of B chain at the backbone position in addition to several hydrophobic/van der Waals interactions with the hydrophobic residues. Based on encouraging docking results, the one-pot synthesis of newly designed benzofuran derivatives were carried out using the tandem Pd/Cu-catalyzed Sonogashira cross-coupling followed by intramolecular cyclization of 2-iodophenols with appropriate terminal alkynes. The reaction was carried out in the presence of (PPh3)2PdCl2 and CuI in Et3N at 60 °C for 2–3 h when a range of novel 3-(benzofuran-2-ylmethyl) substituted (pyrazolo/benzo)triazinone derivatives were obtained in high (>80%) yields. Three molecules i.e. 3h, 3i and 3m that participated in good interaction with CM in silico showed encouraging (64–65%) inhibition of this enzyme when tested at 30 μM in vitro. An SAR within this class of molecules suggested that the inhibitory activity was influenced by the aryl/heteroaryl ring fused with the triazinone moiety and in general the benzotriazinone series emerged as better active molecules than that from pyrazolotriazinone series. The lower activity observed for compound 3a and 3d was reflected by the lack of key H-bond in addition to the lower number of hydrophobic interactions noted for these compounds in silico. Considering the molecular docking in silico, CM inhibition in vitro and computational ADME prediction the benzofuran derivatives 3i and 3m were selected as potential hit molecules for further pharmacological studies. Overall, the current efforts not only disclose the Pd/Cu-catalyzed convenient access to novel 3-(benzofuran-2-ylmethyl) substituted (pyrazolo/benzo)triazinone derivatives but also highlight the utility of the related framework for the identification of new inhibitors of CM.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SS thanks ICMR, New Delhi, India for a Senior Research Fellowship (TB/41/2020-ECD-I). Authors thank Prof. Gautham G. Shenoy of MAHE, Manipal, Karnataka, India, the Management of DRILS, Hyderabad, India and Manipal University, Manipal, India for encouragement and support.Notes and references
- WHO, Global tuberculosis report 2020, World Health Organization, Geneva, Switzerland, 2020, ISBN 978-92-4-001313-1 Search PubMed.
- For a review, see: M. Khanapur, M. Alvala, M. Prabhakar, K. S. Kumar, R. K. Edwin, P. S. Saranya, R. K. Patel, G. Bulusu, P. Misra and M. Pal, Bioorg. Med. Chem., 2017, 25, 1725–1736, DOI:10.1016/j.bmc.2017.02.001.
- E. Haslam, Shikimic Acid: Metabolism and Metabolites, Wiley, New York, 1993 Search PubMed.
- End to killer TB? Scientists identify an enzyme that may hold clue, https://researchmatters.in/article/end-killer-tb-scientists-identify-enzymemay-hold-clue Search PubMed.
- G. S. Reddy and M. Pal, Curr. Med. Chem., 2021, 28, 4531–4568, DOI:10.2174/0929867327666200918144709.
- K. S. Kumar, R. Adepu, S. Sandra, D. Rambabu, G. R. Krishna, C. M. Reddy, P. Misra and M. Pal, Bioorg. Med. Chem. Lett., 2012, 22, 1146–1150 CrossRef.
- A. Nakhi, B. Prasad, U. Reddy, R. M. Rao, S. Sandra, R. Kapavarapu, D. Rambabu, G. R. Krishna, C. M. Reddy, K. Ravada, P. Misra, J. Iqbal and M. Pal, Med. Chem. Commun., 2011, 2, 1006–1010 RSC.
- G. S. Reddy, A. V. Snehalatha, R. K. Edwin, K. A. Hossain, V. B. Giliyaru, R. C. Hariharapura, G. G. Shenoy, P. Misra and M. Pal, Bioorg. Chem., 2019, 91, 103155, DOI:10.1016/j.bioorg.2019.103155.
- For a review, see: Z. Xu, S. Zhao, Z. Lv, L. Feng, Y. Wang, F. Zhang, L. Bai and J. Deng, Eur. J. Med. Chem., 2019, 162, 266–276, DOI:10.1016/j.ejmech.2018.11.025.
- V. M. Rao, A. S. Rao, S. S. Rani and M. Junaid, Int. J. Res. Pharm. Chem., 2018, 8, 112–122 Search PubMed , http://www.ijrpc.com/files/13-01-18/14.pdf.
- S. D. Gorman, D. S. Winston, D. Sahu and D. D. Boehr, Biochemistry, 2020, 59, 2528–2540 CrossRef CAS PubMed.
- P. Prakash, B. Aruna, A. A. Sardesai and S. E. Hasnain, J. Biol. Chem., 2005, 280, 19641–19648 CrossRef CAS.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- P. Csizmadia, The Third International Electronic Conference On Synthetic Organic Chemistry, 1999, September 1–30, p. 367–369 Search PubMed.
- R. Huey and G. M. Morris, Using AutoDock 4 with AutoDockTools: A Tutorial, The Scripps Research Institute, La Jolla, CA, USA, 2003 Search PubMed.
- W. L. DeLano, CCP4 Newsletter On Protein Crystallography, 2002, vol. 40, pp. 82–92 Search PubMed.
- For a recent review, see: L. Chiummiento, R. D'Orsi, M. Funicello and P. Lupattelli, Molecules, 2020, 25, 2327, DOI:10.3390/molecules25102327.
- G. J. S. Doad, J. A. Barltrop, C. M. Pretty and T. C. Owen, Tetrahedron Lett., 1989, 30, 1597–1598 CrossRef CAS.
- K. Okuro, M. Furuune, M. Enna, M. Miura and M. Nomura, J. Org. Chem., 1993, 58, 4716–4721 CrossRef CAS.
- N. G. Kundu, M. Pal, J. S. Mahanty and M. De, J. Chem. Soc., Perkin Trans., 1997, 1, 2815–2820 RSC.
- J. H. Li, J. L. Li, D. P. Wang, S. F. Pi, Y. X. Xie, M. B. Zhang and X. C. Hu, J. Org. Chem., 2007, 72, 2053–2057 CrossRef CAS PubMed.
- C. G. Bates, P. Saejueng, J. M. Murphy and D. Venkataraman, Org. Lett., 2002, 4, 4727–4729 CrossRef CAS PubMed.
- E. A. Jaseer, D. J. C. Prasad and G. Sekar, Tetrahedron, 2010, 66, 2077–2082 CrossRef CAS.
- W. Zeng, W. Wu, H. Jiang, L. Huang, Y. Sun, Z. Chen and X. Li, Chem. Commun., 2013, 49, 6611–6613 RSC.
- F. Chahdoura, S. Mallet-Ladeira and M. Gomez, Org. Chem. Front., 2015, 2, 312–318 RSC.
- A. Ohtaka, T. Teratani, R. Fujii, K. Ikeshita, T. Kawashima, K. Tatsumi, O. Shimomura and R. Nomura, J. Org. Chem., 2011, 76, 4052–4060 CrossRef CAS PubMed.
- S. Priyadarshini, P. A. Joseph, P. Srinivas, H. Maheswaran, M. L. Kantam and S. Bhargava, Tetrahedron Lett., 2011, 52, 1615–1618 CrossRef CAS.
- P. Saejueng, C. G. Bates and D. Venkataraman, Synthesis, 2005, 10, 1706–1712 Search PubMed.
- R. Wang, S. Mo, Y. Lu and Z. Shen, Adv. Synth. Catal., 2011, 353, 713–718 CrossRef CAS.
- J. Gil-Moltó and C. Nájera, Eur. J. Org. Chem., 2005, 19, 4073–4081 CrossRef.
- R. Cano, M. Yus and D. J. Ramon, Tetrahedron, 2012, 68, 1393–1400 CrossRef CAS.
- E. K. Yum, O. K. Yang, J. E. Kim and H. J. Park, Bull. Korean Chem. Soc., 2013, 34, 2645–2649 CrossRef CAS.
- A. Bruneau, K. P. J. Gustafson, N. Yuan, C. W. Tai, I. Persson, X. Zou and J. E. Bäckvall, Chem.–Eur. J., 23, 12886–12891 CrossRef CAS PubMed.
- M. Bhadra, H. S. Sasmal, A. Basu, S. P. Midya, S. Kandambeth, P. Pachfule, E. Balaraman and R. Banerjee, ACS Appl. Mater. Interfaces, 2017, 9, 13785–13792 CrossRef CAS PubMed.
- P. K. Mandali and D. K. Chand, Synthesis, 2015, 47, 1661–1668 CrossRef CAS.
- (a) L. D. Caspers and B. J. Nachtshei, Chem.–Asian J., 2018, 13, 1231–1247 CrossRef CAS; (b) B. Panda, Asian J. Org. Chem, 2020, 9, 492–507 CrossRef CAS; (c) R. Chinchilla and C. Nájera, Chem. Soc. Rev., 2011, 40, 5084–5121 RSC; (d) B. Panda and T. K. Sarkar, Synthesis, 2013, 45, 817–829 CrossRef CAS.
- (a) N. G. Kundu, M. Pal, J. S. Mahanty and S. K. Dasgupta, J. Chem. Soc., Chem. Commun., 1992, 41–42 RSC; (b) M. Pal, V. Subramanian and K. R. Yeleswarapu, Tetrahedron Lett., 2003, 44, 8221–8225 CrossRef CAS.
- B. V. D. Rao, S. V. Vardhini, D. Kolli, M. V. B. Rao and M. Pal, Anti-Cancer Agents Med. Chem., 2020, 20, 580–588, DOI:10.2174/1871520620666200128120356 , http://www.eurekaselect.com/178730/article.
- R. M. Rao, B. J. Luther, C. S. Rani, N. Suresh, R. Kapavarapu, K. V. L. Parsa, M. V. B. Rao and M. Pal, Bioorg. Med. Chem. Lett., 2014, 24, 1166–1171 CrossRef CAS PubMed.
- A. Nakhi, M. S. Rahman, G. P. K. Seerapu, R. K. Banote, K. L. Kumar, P. Kulkarni, D. Haldar and M. Pal, Org. Biomol. Chem., 2013, 11, 4930–4934 RSC.
- D. S. Laxmi, S. V. Vardhini, V. R. Guttikonda, M. V. B. Rao and M. Pal, Anti-Cancer Agents Med. Chem., 2020, 20, 932–940, DOI:10.2174/1871520620666200311102304.
- S. K. Kim, S. K. Reddy, B. C. Nelson, G. B. Vasquez, A. Davis, A. J. Howard, S. Patterson, G. L. Gilliland, J. E. Ladner and P. T. Reddy, J. Bacteriol., 2006, 188, 8638–8648 CrossRef CAS.
- S. Sasso, C. Ramakrishnan, M. Gamper, D. Hilvert and P. Kast, FEBS J., 2005, 272, 375–389 CrossRef CAS.
- H. Agrawal, A. Kumar, N. C. Bal, M. I. Siddiqi and A. Arora, Bioorg. Med. Chem. Lett., 2007, 17, 3053–3058 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 1–13 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectral data (file type: word). Copies of spectra of all target compounds synthesized. See https://doi.org/10.1039/d2ra05255e |
| This journal is © The Royal Society of Chemistry 2022 |