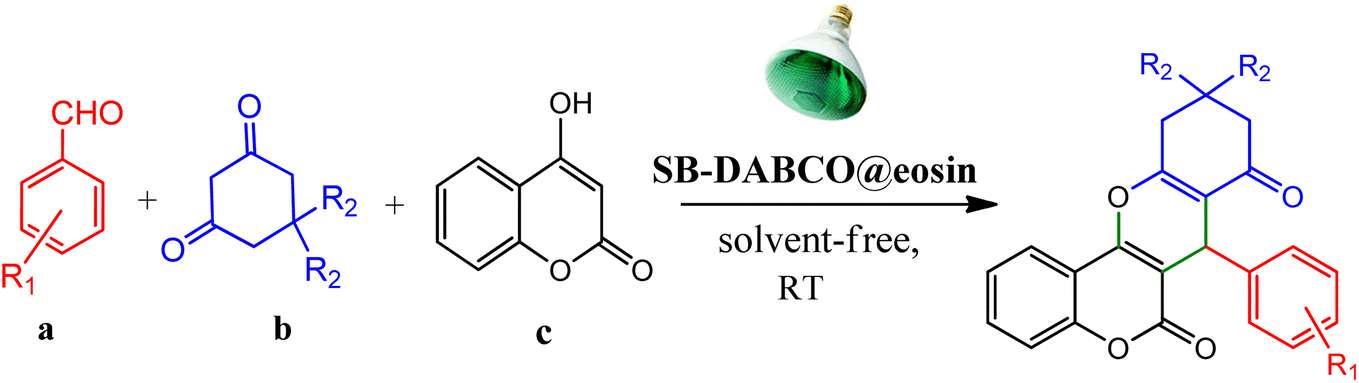DOI:
10.1039/D2RA05122B
(Paper)
RSC Adv., 2022,
12, 28886-28901
Magnetic nanoparticle-supported eosin Y salt [SB-DABCO@eosin] as an efficient heterogeneous photocatalyst for the multi-component synthesis of chromeno[4,3-b]chromene in the presence of visible light
Received
16th August 2022
, Accepted 25th September 2022
First published on 11th October 2022
Abstract
Heterogeneous photocatalysts present a favourable procedure to realize green and eco-friendly organic reactions. We have demonstrated an SB-DABCO@eosin catalyst in a green one-pot multi-component protocol for the production of various chromeno[4,3-b]chromenes via condensation of aromatic aldehydes and dimedone under the photo-redox catalyst bearing eosin Y using visible light. The synthesized nanocatalyst was characterized using various physicochemical techniques such as FT-IR, XRD, EDX, UV-vis, SEM, TGA and DRS. The significant advantages of the present methodology include excellent yield, cost-effectiveness, easy work-up, 100% atom economy, broad substrate scope, easy separation and efficient recycling. Furthermore, the evidence showed that the investigated condensation reaction proceeds via a radical mechanism, which proved the need for reactive species such as OH˙ and ˙O2− in the photocatalytic process. In addition to the improved handling and process control, the yield of products and the rate of reactions have increased considerably in the present strategy. Reproducibility studies also guarantee good reusability and stability of the nanocatalyst for at least five runs.
1. Introduction
The development of multi-component reactions (MCRs) has attracted attention in an environment friendly pathway. Multi-component reactions merge at least three reactants in a single reaction vessel to produce a product containing most atoms of the starting materials in a convergent way. MCRs as an efficient tool in drug discovery fields and synthetic organic chemistry have gained preponderance over customary multistep reactions.1–6 MCRs possess many apparent advantages over multistep processes, such as atom economy, high efficiency, reduced waste generation, quick and simple implementation, time-saving and energy economy. Simple separation and small amounts of used solvents or chromatographic materials are other advantages of MCRs.7–9
In the past few years, magnetic nanoparticles (MNPs) appeared as a new kind of semi-heterogeneous support for catalysts, and the intrinsic magnetic properties of the support allow functionally convenient separation using an external magnet. Magnetic nanoparticles are important catalysts for the synthesis of various compounds because they have a considerable advantages such as non toxicity, enhanced water solubility, elevated stability and reduced particle aggregation and reusability compared to other catalysts.10–16 Among different types of nanoparticles, iron oxide nanoparticles have been widely explored by researchers because they do not retain any magnetization when the magnetic field is removed. In fact, iron oxide nanoparticles (Fe3O4) have been widely applied due to their easy functionalization with polymers and other materials.17,18
The chromenes that are crucial oxygenated heterocyclic compounds mostly come up as an important structural component in biologically active natural compounds such as anthocyanins, alkaloids, tocopherols and flavonoids. In addition, chromene compounds have played a remarkable role in the synthesis of favorable compounds in the fields of pharmacology and drug chemistry.19–21 These compounds exhibit diverse biological activities and act as anti-cancer,22 anti-tumor,23 anti-HIV,24 anti-Alzheimer,25 anti-bacterial,26 anti-leishmanial,27 antitubercular,28 antihypertensive,29 antioxidative,30 and anti-inflammatory31 agents. Chromanes have been shown to have the ability to activate potassium channels and impede phosphodiesterase IV and dihydrofolate reductases and have been employed as colored optical lasers, pigments, fluorescence markers and potential biodegradable agrochemicals.32–35 4H-Chromenes can develop the apoptotic procedure in the cancerous cells by binding to Bcl-2 anti-apoptotic proteins and therefore show anti-cancer activity.36–39
However, in recent years, owing to the increasing attention on the organic synthesis under green chemistry, chemists redesign the synthesis methods by focusing on the visible light. Visible light has the potential to serve as an inexpensive, ubiquitous, sustainable, plentiful, unlimited, universally available source of energy as well as a nonpolluting agent for chemical reactions.40–43 However, most organic molecules cannot absorb visible wavelengths and this is one of the major obstacles in the development of photochemical processes which prompts the growth of efficient visible light photocatalysts in organic synthesis. Among the metal-free organic dyes, EY has been used as a profitable and environmentally friendly alternative in the structure of many photoreduction catalysts.44 In the mechanism for the photodegradation of organic dyes, upon irradiation with incident photons, electrons are excited to the conduction band (CB) of the photocatalyst, while holes are formed in the valence band (VB).45–47 Since many organic photocatalysts (and among that eosin Y) change their photophysical properties under the influence of acid–base equilibria, determining the actual nature of the dye used in the reaction conditions is of fundamental importance. It is able to transfer the activation energy to the reactants in chemical reactions.48 Due to its ability to absorb green light, various applications have emerged for EY as an organic photosensitizer in visible light-promoted organic synthesis.49,50 In continuation, we introduce a simple, more efficient, visible light promoted procedure for the synthesis of different substituted chromeno[4,3-b]chromene derivatives via combination of aryl aldehydes, 4-hydroxy coumarin and 1,3-cyclohexanedione in the presence of SB-DABCO@eosin as a green heterogeneous nanocatalyst under solventless conditions (Scheme 1). The present strategy promotes the cyclization of chromeno[4,3-b] chromenes via the formation of C–C and C–O bonds to obtain the products with excellent yield during a simple one-pot operation under mild reaction conditions. The extended procedure is an affordable alternative to devices involving metal base catalysts. Its atom economy, mild conditions, high convergence, near stoichiometric use of reactants without any additives and concomitant step economy in combination with their general compatibility rank it among sustainable synthetic methods.
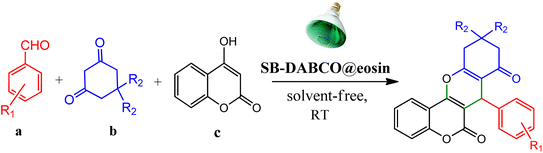 |
| | Scheme 1 Multi-component reaction of aldehydes (a), cyclic 1,3-dicarbonyl (b) and 4-hydroxycoumarin (c) promoted by SB-DABCO@eosin. | |
2. Experimental section
2.1. Materials and methods
All materials and solvents were purchased from Merck, Sigma-Aldrich, and Fluka and were employed as received without further purification. Fourier transform infrared (FT-IR) was performed in the range 400 to 4000 cm−1 (PerkinElmer) using the KBr disc technique. Morphologies of the synthesized nanomaterials were analyzed using a Quantum 2000 field emission scanning electron microscope (FESEM). 1H- and 13C-NMR spectra were recorded on a 300 MHz Bruker AVANCE III spectrometer using TMS as the internal reference. A Photonix UV-visible array spectrophotometer was used to record UV-visible spectra. Elemental analyses were carried out using a Thermo Finnigan (Flash-1112EA) microanalyzer. Energy dispersive X-ray spectroscopy (EDS, Bruker QUANTAX 200) was run with an electron probe microanalyser (JEOL JXA-8230) equipped with an energy dispersive spectrometer. Melting points were determined in open capillary tubes on Stuart BI Branstead Electrothermal IA9200 apparatus. X-ray diffraction (XRD) patterns were acquired at a scanning rate of 3° min−1 using a Philips PW1730 diffractometer at 40 keV and 30 mA for monochromatized Cu Kα radiation in the 2θ domain from 10° to 80°. Thermogravimetric analyses were performed with a TGA 92 Setaram with a heating rate of 10 °C per minute.
2.2. General method for the synthesis of the Fe3O4 nanoparticles
Fe3O4 magnetic nanoparticles (MNPs) were synthesized according to a previous literature report with some modification. A mixture of 2.25 g FeCl3·6H2O and 0.9 g FeCl2·4H2O was dissolved in deionized water (40 ml) in a 200 ml flask. The solution was stirred vigorously (800 rpm) at RT for 10 min. Then, 7 ml of solution of 25% (w/w) ammonia was added into the mixture in a drop-wise manner over a 3 min period by means of a dropping funnel. The reaction system turned black quickly. The mixture of magnetite nanoparticles was stirred for 4 h at RT. The supernatant was decanted and magnetic Fe3O4 nanoparticles were collected by means of an external magnet and rinsed exhaustively with distilled water (20 ml) and ethanol (20 ml) until the pH of the solution reached 7. The washing operation was continued until the pH reached 7. Finally, the black MNPs were washed with 20 ml of acetone and dried in an oven at 50 °C.51
2.3. Synthesis of the Fe3O4·SiO2 nanoparticles (SMNPs)
0.25 g of Fe3O4 was added to a 100 ml balloon containing 40 ml of deionized water and 10 ml of 96% ethanol, and the contents of the balloon were sonicated for 30 min or more to disperse the nanoparticles well. We continued the irradiation process until we observed the dispersion of the nanoparticles well. Then 7 ml of ammonia was added dropwise to the contents of the 100 ml balloon in one min. This operation was performed under ultrasound radiation. In this case, it was exposed to ultrasound again for another 10 min. Then tetraethyl orthosilicate (TEOS, 4 ml) was added to the solution drop by drop under ultrasonic irradiation for 2 min. After adding all the TEOS to the 100 ml balloon, it was irradiated for another 5 min. So for all the work steps, the 100 ml balloon was inside the ultrasonic bath. After this, we took the balloon out of the ultrasonic bath and rotated it with a magnetic stirrer for 24 h at room temperature. Then, we washed and dried the sample as described for Fe3O4.52
2.4. Synthesis of Fe3O4@SiO2@Pr–Cl (SMNPs@Pr–Cl)
0.8 g SMNPs was added to a 50 ml balloon containing 30 ml of dried toluene and the contents of the balloon were sonicated for 30 min or more to disperse the fine nanoparticles. We continued the irradiation process until we observed the dispersion of the nanoparticles well. Then 4 ml of 3-chloropropyl trimethoxysilane was added to the 50 ml balloon by drop syringe over 2 min and allowed to disperse for another 10 min. The contents of the balloon were then removed from the ultrasonic water bath and rotated at 110 °C using a magnetic stirrer for 48 h under reflux conditions. The nanoparticles were then removed using a magnet and first washed with 10 ml of ordinary toluene, and then washed with deionized water and then ethanol, as in the above method. Finally, they were dried in an oven at a temperature of 50 °C for 4 to 5 h.53
2.5. Preparation of silica bonded 1,4-diazabicyclo[2.2.2]octane (SB-DABCO)
30 ml dry acetone was poured into a round-bottomed flask equipped with a refluxing condenser and 0.56 g DABCO (5 mmol) and 1 g SMNPs@Pr–Cl were added and the mixture was refluxed. After 36 h the reaction mixture was cooled at room temperature and dried under vacuum on a glass filter, and then washed with acetone, ethanol, and methanol in turn. The product obtained from this step was dried for 5 h under vacuum at 50 °C to give SB-DABCO as a white powder.54
2.6. Surface modification of SB-DABCO nanoparticles with eosin
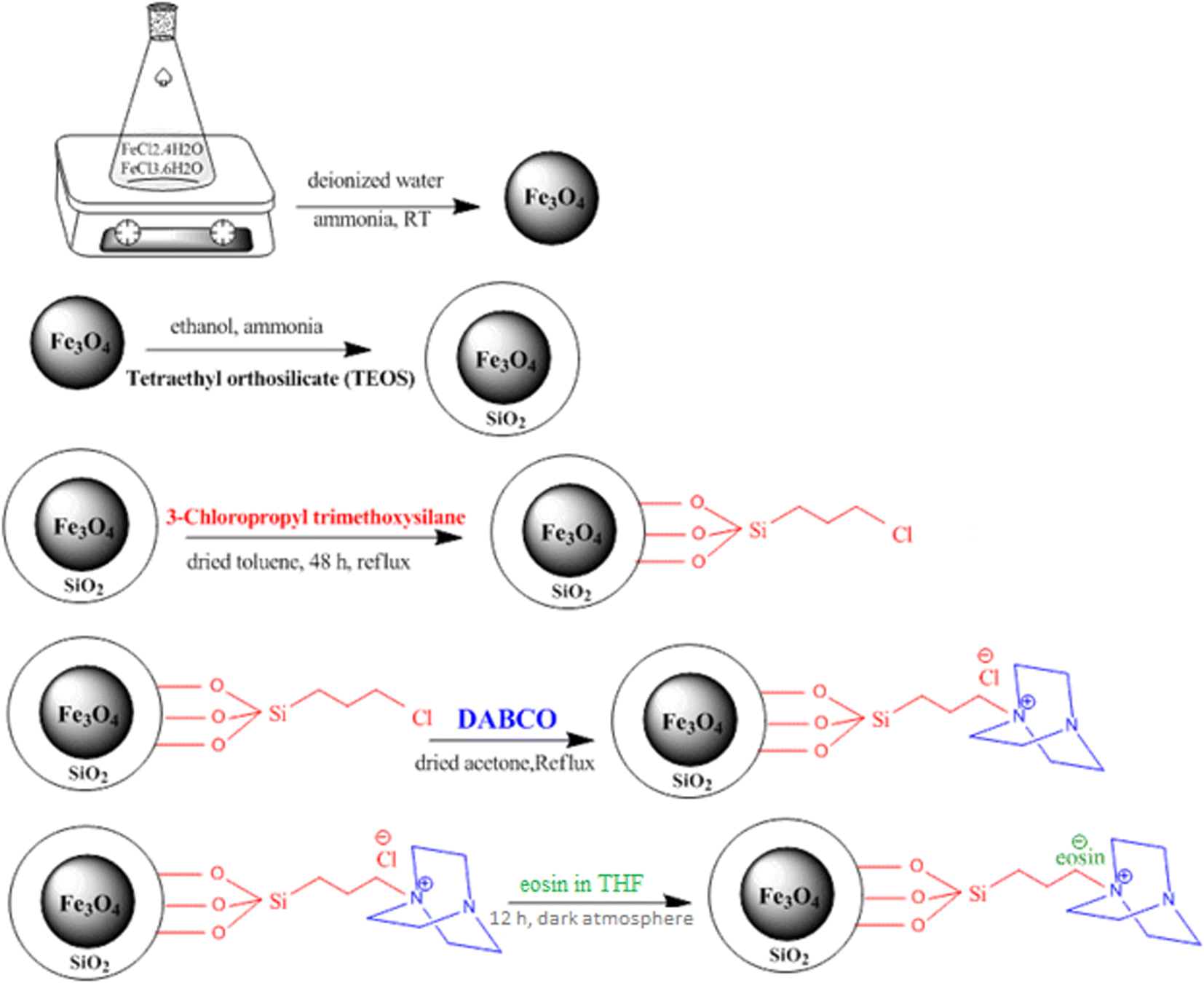
The SB-DABCO@eosin nanohybrid material was obtained from the well-known wet impregnation procedure. Hence, 0.02 ml eosin was dissolved in THF. Then, nano SB-DABCO (0.5 g) was slowly added to the obtained solution for 12 h under a dark atmosphere. The resulting powder was dried at 80 °C under air for 24 h. All steps of the SB-DABCO@eosin synthesis are summarized in Scheme 2.
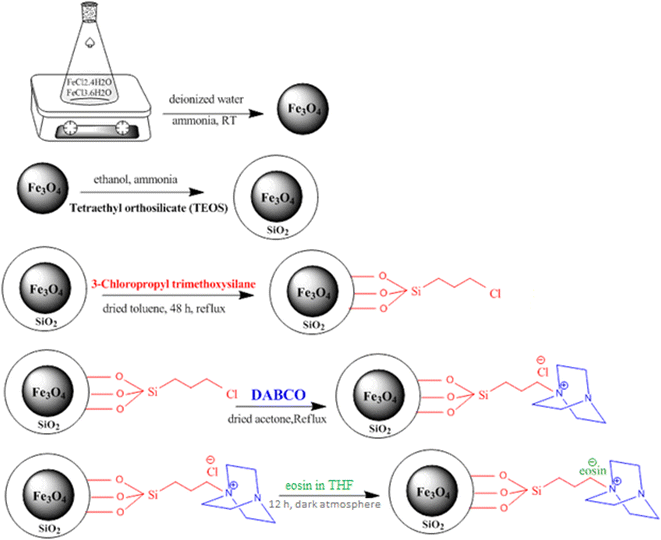 |
| | Scheme 2 Schematic illustration of the synthesis of SB-DABCO@eosin NPs. | |
2.7. General procedure for synthesis of substituted chromeno[4,3-b]chromene
1 mg SB-DABCO@eosin nanoparticles, 100 mg benzaldehyde (1 mmol), 162 mg 4-hydroxycoumarin (1 mmol) and 140 mg dimedone (1 mmol) were added to a 25 ml round bottom flask. The mixture was irradiated with a green light-emitting diode (λmax = 535 nm) under open air conditions at room temperature. After completion of the reaction (controlled by TLC, n-hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate, 3:1), 3 ml H2O was added to the mixture and the desired chromene product was dissolved in EtOAc. After extracting the aqueous fraction with ethyl acetate (3 × 3 ml), the obtained organic layer was dried over sodium sulfate and concentrated in vacuo. The raw accomplished product was purified through column chromatography (100–200 mesh silica gel; ethyl acetate/hexane) to supply the pure chromene. The product was confirmed by 1H and 13C NMR spectra.
:ethyl acetate, 3:1), 3 ml H2O was added to the mixture and the desired chromene product was dissolved in EtOAc. After extracting the aqueous fraction with ethyl acetate (3 × 3 ml), the obtained organic layer was dried over sodium sulfate and concentrated in vacuo. The raw accomplished product was purified through column chromatography (100–200 mesh silica gel; ethyl acetate/hexane) to supply the pure chromene. The product was confirmed by 1H and 13C NMR spectra.
2.8. Spectral NMR data selected of the prepared chromenes
2.8.1 Table 5, entry 1. 1H NMR (300 MHz, DMSO-d6); δ: 7.79 (1H, d, J = 7.8 Hz), 7.50 (1H, d, J = 7.2 Hz), 7.02–7.32 (7H, m), 4.65 (1H, s), 2.63 (2H, s), 2.21 (1H, d, J = 15.9 Hz), 2.09 (1H, d, J = 16.2 Hz), 1.04 (3H, s), 0.93 (3H, s) ppm; 13C NMR (75 MHz, DMSO-d6); δ: 196.2, 162.1, 159.5, 153.5, 152.2, 142.4, 133.1, 129.0, 128.0, 126.8, 124.2, 122.3, 116.1, 114.2, 113.4, 105.8, 51.5, 33.0, 31.9, 28.6, 26.5 ppm.
2.8.2 Table 5, entry 2. 1H NMR (300 MHz, DMSO-d6) δ (ppm): 8.05 (2H, d, J = 8.7 Hz), 7.87 (1H, s), 7.86 (1H, d, J = 7.8 Hz), 7.52 (1H, d, J = 7.2 Hz), 7.44 (1H, d, J = 8.7 Hz), 7.35 (1H, t, J = 7.5 Hz), 7.29 (1H, d, J = 8.1 Hz), 4.80 (1H, s), 2.68 (2H, s), 2.25 (1H, d, J = 16.2 Hz), 2.14 (1H, d, J = 16.2 Hz), 1.09 (3H, s), 0.97 (3H, s); 13C NMR (75 MHz, MSO-d6) δ (ppm): 195.5, 163.3, 160.7, 154.8, 152.7, 150.5, 147.0, 133.4, 129.8, 124.5, 123.8, 123.3, 117.2, 13.9, 113.6, 105.3, 50.7, 33.4, 32.6, 28.5, 27.8.
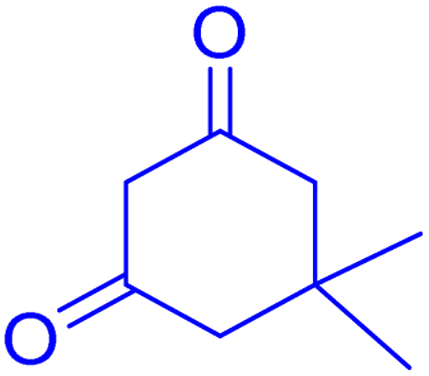
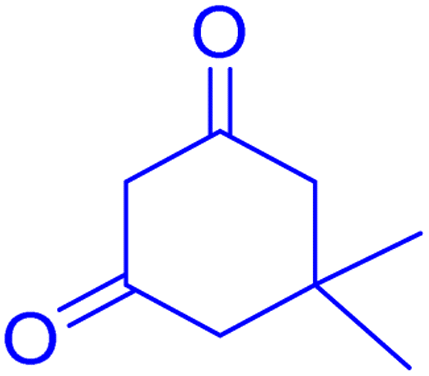
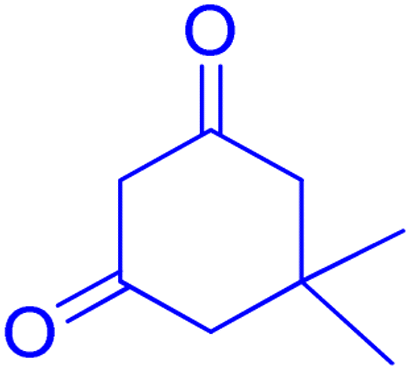
2.8.3 Table 5, entry 4. 1H NMR (300 MHz, DMSO-d6) δ (ppm): 7.85–7.89 (1H, m), 7.50–7.56 (1H, m), 7.29–7.40 (2H, m), 7.15 (2H, d, J = 9.0 Hz), 7.09 (1H, d, J = 9.0 Hz), 6.66–6.73 (1H, m), 4.70 (1H, s), 3.69 (3H, s), 2.72 (2H, s), 2.21 (1H, d, J = 16.2 Hz), 2.09 (1H, d, J = 16.2 Hz), 1.12 (3H, s), 0.99 (3H, s); 13C NMR (75 MHz, DMSO-d6) δ (ppm): 196.3, 161.9, 160.8, 158.8, 154.0, 152.9, 136.0, 133.0, 129.9, 129.5, 124.7, 123.0, 116.9, 115.7, 113.7, 113.4, 106.5, 55.5, 50.9, 32.7, 32.5, 29.5, 27.6.
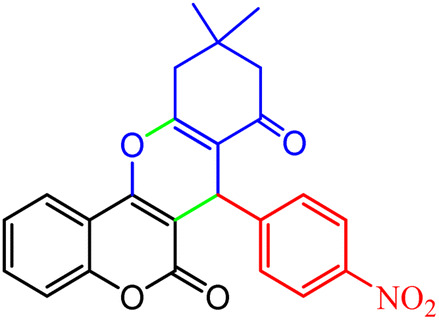
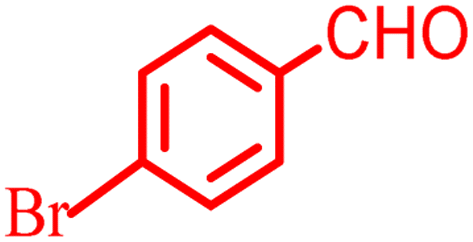
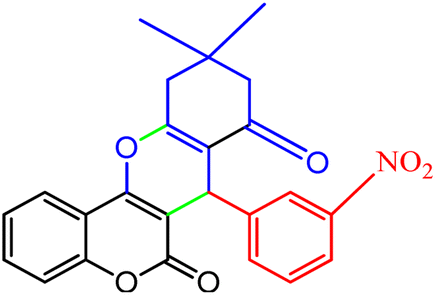
2.8.4 Table 5, entry 5. 1H NMR (300 MHz, DMSO-d6) δ (ppm): 8.09 (1H, s), 7.89 (1H, d, J = 8.1 Hz), 7.83 (1H, d, J = 7.8 Hz), 7.65 (1H, d, J = 6.6 Hz), 7.54 (1H, t, J = 7.5 Hz), 7.25–7.39 (2H, m), 7.17 (1H, d, J = 8.1 Hz), 4.88 (1H, s), 2.70 (1H, d, J = 18 Hz), 2.53 (1H, d, J = 18 Hz), 2.15 (1H, d, J = 16.5 Hz), 2.12 (1H, d, J = 16.5 Hz), 1.05 (3H, s), 0.99 (3H, s); 13C NMR (75 MHz, DMSO-d6) δ (ppm): 196.2, 163.3, 159.6, 154.0, 152.2, 147.9, 145.0, 135.4, 133.0, 129.7, 124.9, 123.5, 123.3, 121.9, 117.0, 113.4, 105.0, 50.4, 39.3, 33.6, 32.3, 28.9, 27.3.
2.8.5 Table 5, entry 6. 1H NMR (300 MHz, DMSO-d6) δ (ppm): 7.97–8.03 (2H, m), 7.88–7.90 (2H, m), 7.57–7.61 (1H, m), 7.29–7.51 (3H, m), 5.10 (1H, s), 2.83–2.99 (2H, m), 2.43–2.49 (2H, m), 2.09–2.23 (2H, m); 13C NMR (75 MHz, DMSO-d6) δ (ppm): 196.3, 164.9, 153.0, 145.0, 136.3, 132.6, 129.1, 124.2, 123.0, 122.9, 122.4, 117.3, 115.5, 113.6, 105.5, 36.9, 33.8, 27.5, 20.4.
2.8.6 Table 5, entry 7. 1H NMR (300 MHz, DMSO-d6) δ (ppm): 8.08 (1H, d, J = 7.8 Hz), 7.79 (1H, t, J = 7.5 Hz), 7.49–7.59 (2H, m), 7.37 (2H, d, J = 8.4 Hz), 6.95 (2H, d, J = 7.8 Hz), 3.89 (3H, s), 4.94 (1H, s), 2.95–3.13 (2H, m), 2.58 (2H, t, J = 6.3 Hz), 2.19–2.33 (2H, m); 13C NMR (75 MHz, DMSO-d6) δ (ppm): 195.9, 163.9, 160.8, 159.9, 153.9, 153.0, 135.9, 133.2, 130.2, 125.2, 123.4, 121.2, 116.9, 116.5, 114.0, 113.5, 112.0, 106.9, 55.6, 37.2, 32.9, 27.0, 20.5.
3. Results and discussion
In this paper, we have tried to synthesize substituted chromeno[4,3-b]chromenes using SB-DABCO@eosin NPs. Some procedures have been reported for the production of chromeno[4,3-b]chromene derivatives via combination of arylaldehydes with 4-hydroxycoumarine and cyclic-1,3-diketone compounds in the presence of p-TSA, Fe[DS]3, [DMDBS]·2HSO4, and I2/HOAc.54–57 However, most of these techniques bear at least one of the following disadvantages: difficult and boring work-up procedures, long reaction time, high operating temperature, non-recyclable or expensive metal catalysts, moderate yields and poor selectivity, commercially unavailable compounds, corrosive and harmful organic solvents, and limited substrate scope.
Therefore, it seems that the main work is to replace inefficient methods with more plausible methods based on recyclable, stable, and improved catalysts. Environmental and economic concerns have turned researchers' attention to green chemical processes in organic synthesis. In recent years, photochemical techniques, especially using visible light, have become an efficient tool for chemical reactions. The superiority of using a visible light photo redox catalyst is its ability to activate atmospheric oxygen, a natural green oxidizing agent in visible light, controlled by organic transformations involving single electron transfer (SET). Among the organic dyes of the fluorescein family, the organic dye eosin Y has found a better place in photocatalyzed organic reactions due to its better performance capacity.48
3.1. Characterization and physicochemical properties of [SB-DABCO@eosin] nanoparticles
The synthesized supported [SB-DABCO@eosin] nanocatalyst was characterized by means of XRD, UV-vis, DRS, FT-IR, SEM, TGA and EDX. The surface interaction between eosin and [SB-DABCO@eosin] can be investigated by using FT-IR and UV-vis analyses. The material was analyzed by different analytical and spectroscopic techniques. The XRD patterns of the materials reported the formation of nano sized particles. The morphology and particle size of the [SB-DABCO@eosin] NPs were further confirmed by scanning electron microscopic (SEM) study. Energy dispersive X-ray analysis through SEM obviously showed the high purity of the sample.
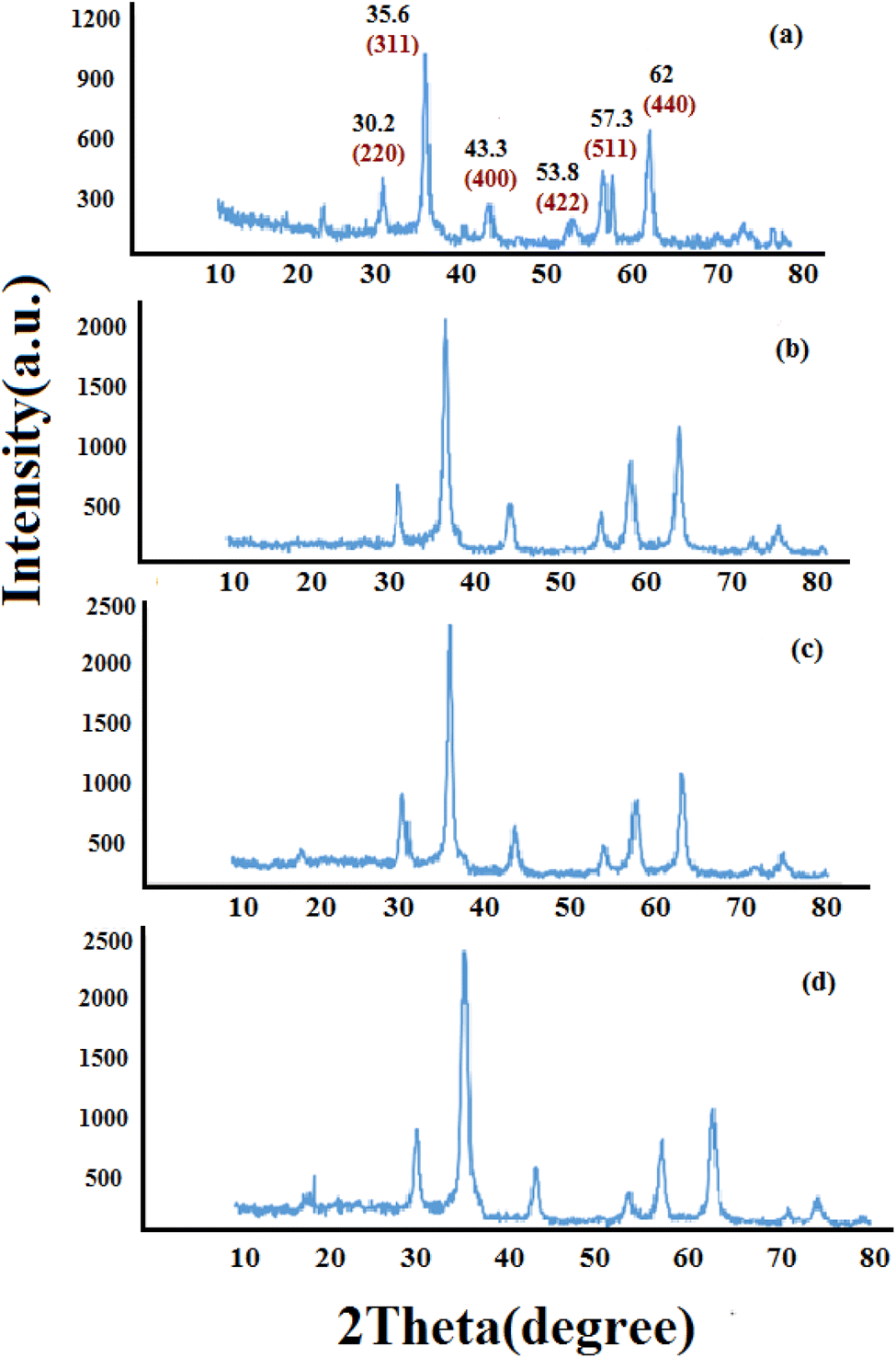
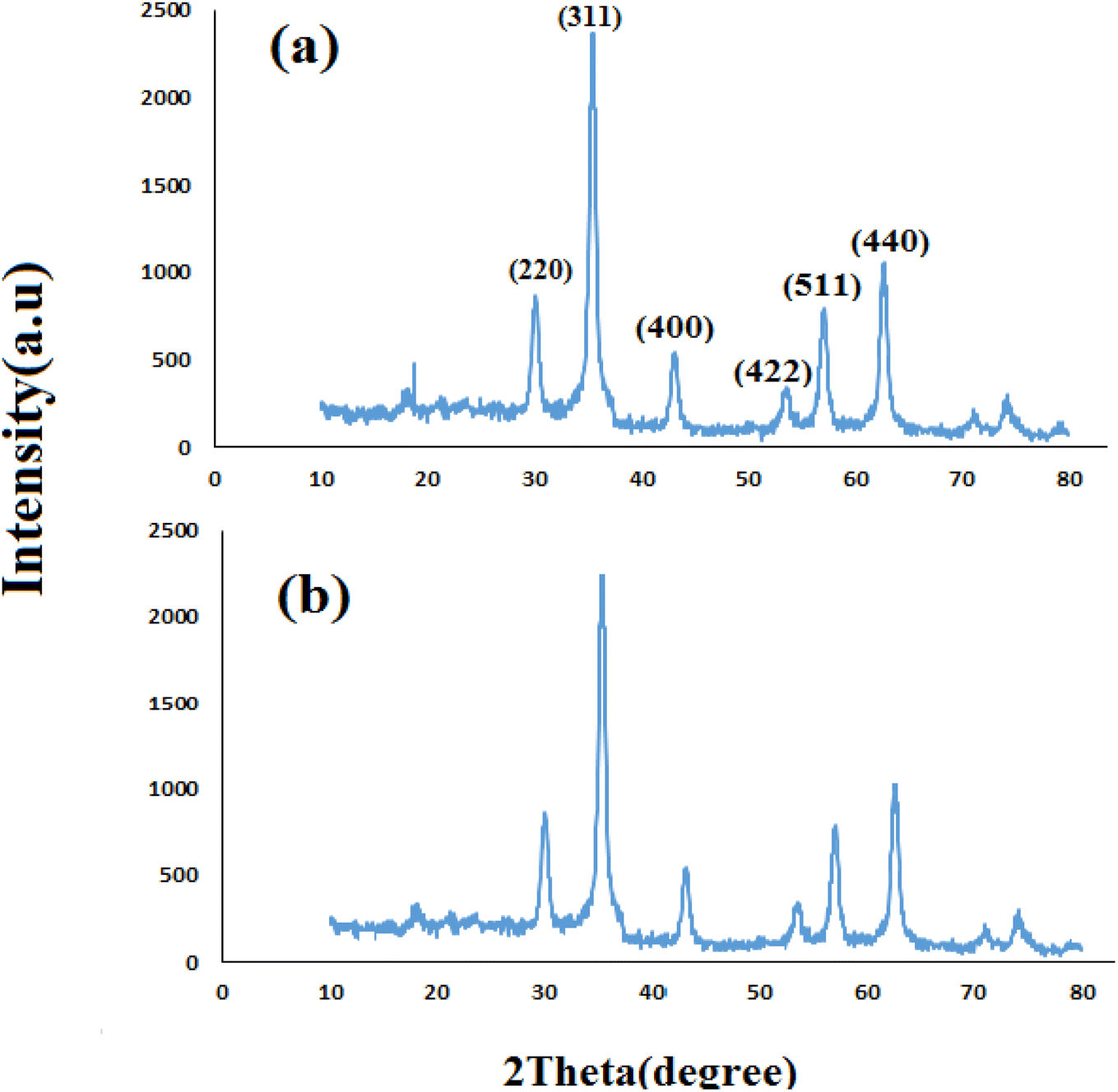
3.1.1 XRD analyses. Fig. 1 shows the XRD pattern of synthesized nanomaterials Fe3O4 (MNPs), SMNPs, SMNPs@DABCO and SMNPs@DABCO@eosin. The positions and relative intensities of all diffraction peaks in the XRD pattern of MNPs matched with the reference pattern of the cubic inverse spinel structure of magnetite (JCPDS card number 19-0629). Six sharp Bragg diffraction peaks at the 2θ of 30.2°, 35.6°, 43.3°, 53.8°, 57.3°, and 62° are the corresponding peaks of (220), (311), (400), (422), (511), and (440) miller indices, respectively (Fig. 1a). Repetition of the peaks mentioned above, without any additional peaks, in the XRD patterns of the products of different stages of the catalyst synthesis indicates the preservation of the magnetite structure in the nanoparticles even after the surface modification.58 However, there was no detectable diffraction, indicating the presence of silica, which could be attributed to its high dispersity over the nanocomposites. The presence of DABCO does not have a significant effect on the cubic spinel crystal structure of Fe3O4 nanoparticles and as a result in their XRD pattern, again, it shows no phase change after surface modification of magnetite nanoparticles. Moreover, the size of crystalline magnetite nanoparticles can be calculated by using the XRD data in the Debye–Scherrer formula (D = kλ/βcosθ, λ = 0.15406 nm); here, the peak of the highest intensity (311, 2θ = 35.6) was chosen to define the diameter of the nanoparticles. Thus, the size of SMNPs@DABCO@eosin particles was attained to be ∼80 nm, in accord with the FESEM analysis.
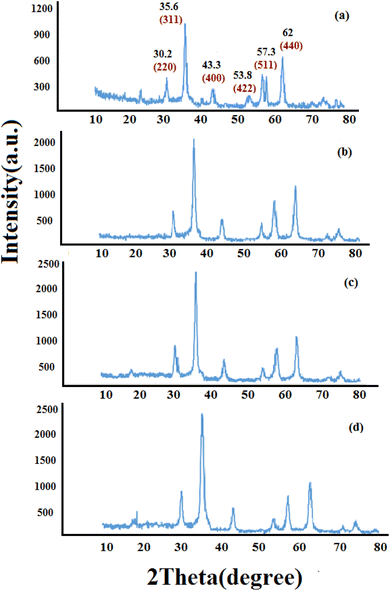 |
| | Fig. 1 Experimental XRD patterns for the synthesized Fe3O4 (a), SMNPs (b), and SMNPs@DABCO (c) compared with the simulated SMNPs@DABCO@eosin (d). | |
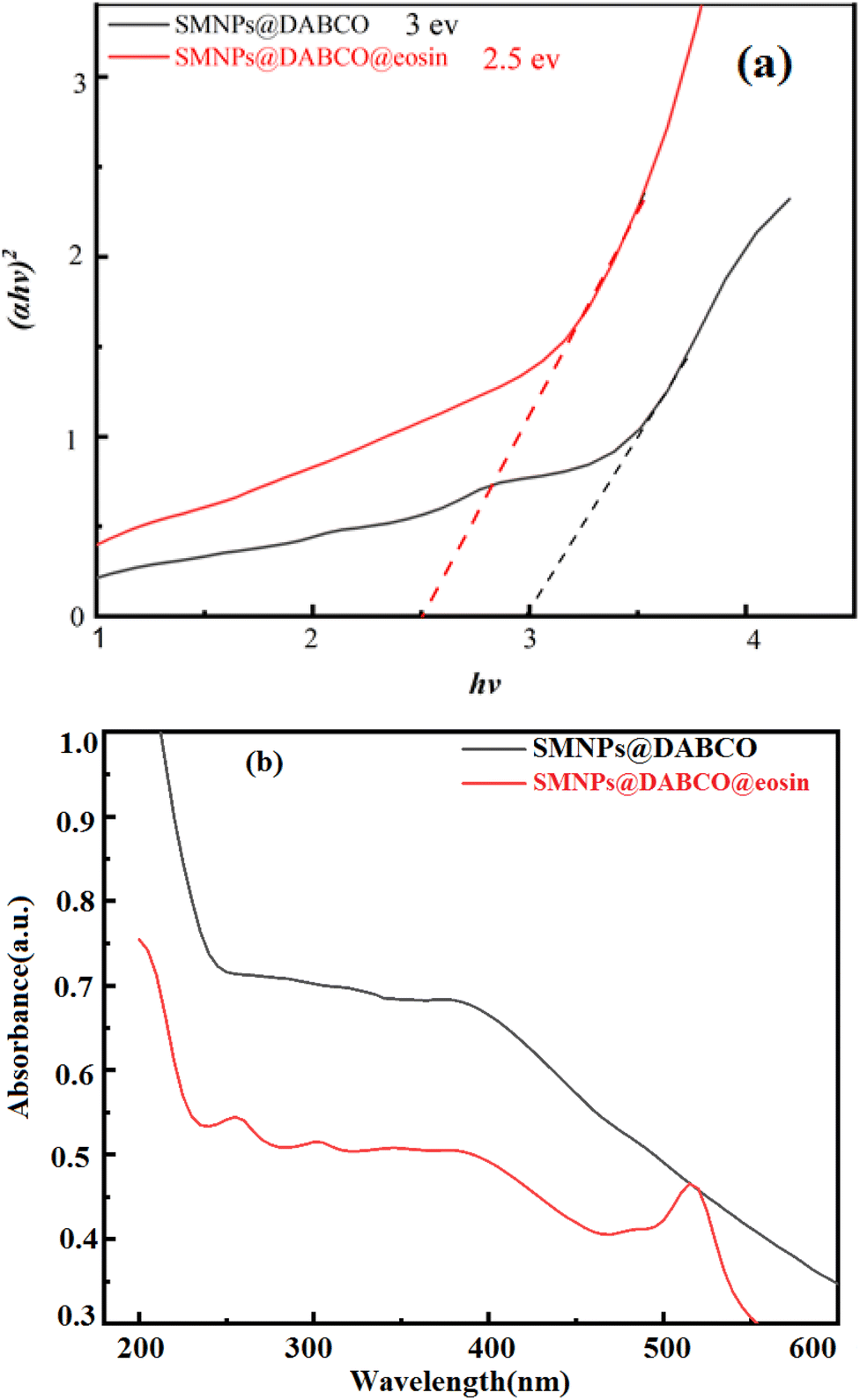
3.1.2 UV-vis and DRS analysis. The UV-vis absorption spectra of SMNPs@DABCO and SMNPs@DABCO@eosin were studied with the purpose of investigating the mechanism involved in the efficient visible light absorption. Diffuse reflectance spectra were recorded to estimate the observed band-gap of the catalyst. Estimation of the optical band gaps of samples to photon energy was done using the extrapolation of the linear slope of the Tauc plot (Fig. 2). The calculated band-gap energy of SMNPs@DABCO and SMNPs@DABCO@eosin was found to be 3 and 2.5 eV, respectively. To obtain the optical energy band gap (Eg), the extrapolation plot of hν versus (αhν)2 was used (Fig. 2b). In this relation, the analysis is carried out based on the equation αhν = A(hν − Eg)n, where A is a constant and α, hν, and Eg refer to the absorption coefficient, light frequency and band-gap, respectively.59 We think that eosin reduces the optical band gap and, therefore, reduction of absorbance. In this case, photons cannot excite electrons from the valence band (VB) to the conduction band (CB). In addition, functionalization of catalyst nanoparticles with eosin affects the electronic features of surface electrons.60–62
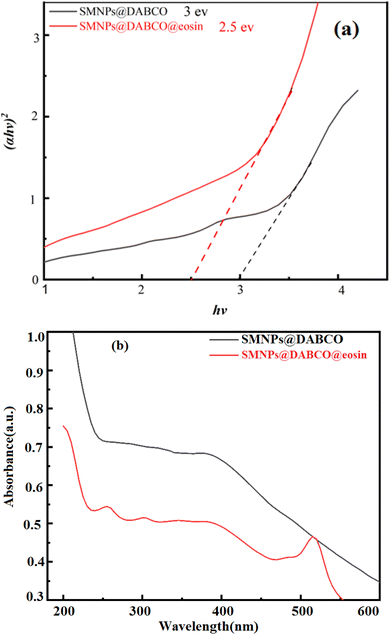 |
| | Fig. 2 (a) DRS of SMNPs@DABCO and SMNPs@DABCO@eosin and (b) UV-vis absorption spectrum. | |
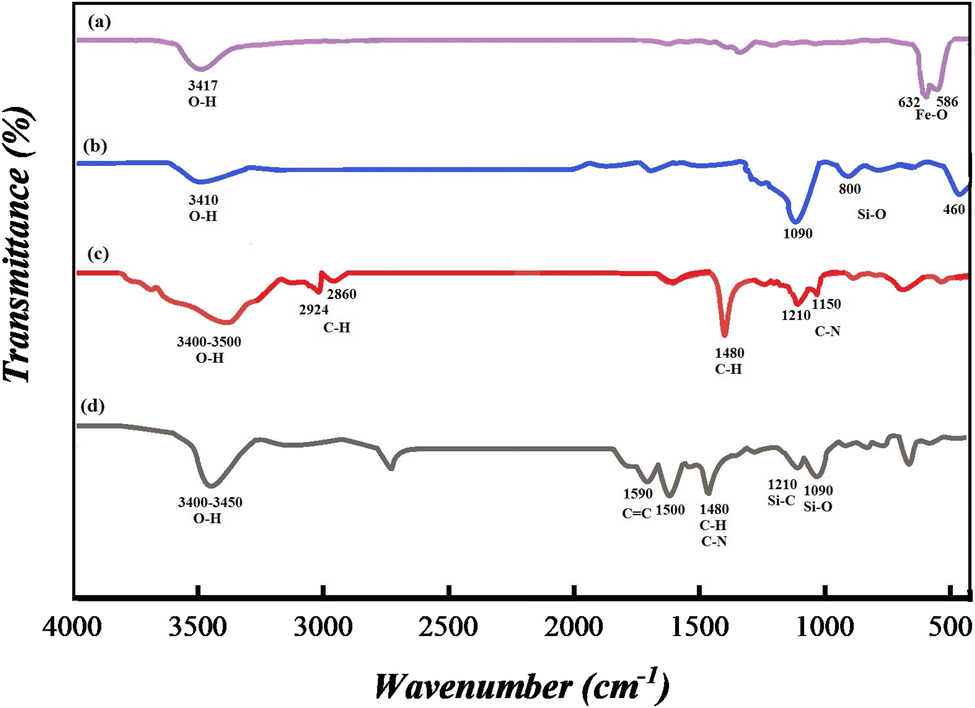
3.1.3 FT-IR spectroscopy. FT-IR spectroscopy was applied to recognize the successful synthesis of SMNPs@DABCO@eosin. Hence all fragments including (a) Fe3O4, (b) SMNPs, (c) SMNPs@DABCO and (d) SMNPs@DABCO@eosin were studied. Intense peaks near 586 and 632 cm−1 correspond to Fe–O stretching vibration (Fig. 3a). In the spectra displayed in Fig. 3b, the advent of characteristic peaks at 1090, 800 and 460 cm−1 is related to Si–O–Si stretching, Si–O bending, and Si–O–Si bending vibrations respectively. The anchoring of DABCO on the SMNP fragment is confirmed by the appearance of symmetric and asymmetric CH2 of the alkyl chains at 2860 and 2924 cm−1, CH2 sharp bending peak at 1480 cm−1 and C–N stretching peaks at 1150 and 1210 cm−1 (Fig. 3c).58,63,64 In Fig. 3d, the presence of absorption bands at 1590 and 1500 cm−1, which are related to the carboxyl functional group and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C of aromatic phenyl rings, respectively, confirms the loading of EY on the SMNP@DABCO surface.65
C of aromatic phenyl rings, respectively, confirms the loading of EY on the SMNP@DABCO surface.65
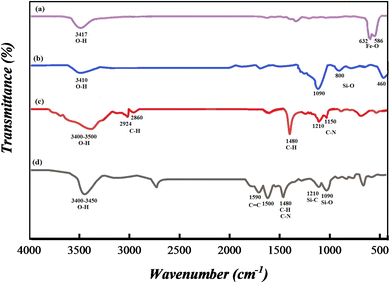 |
| | Fig. 3 FT-IR spectra of (a) Fe3O4, (b) SMNPs, (c) SMNPs@DABCO and (d) SMNPs@DABCO@eosin. | |
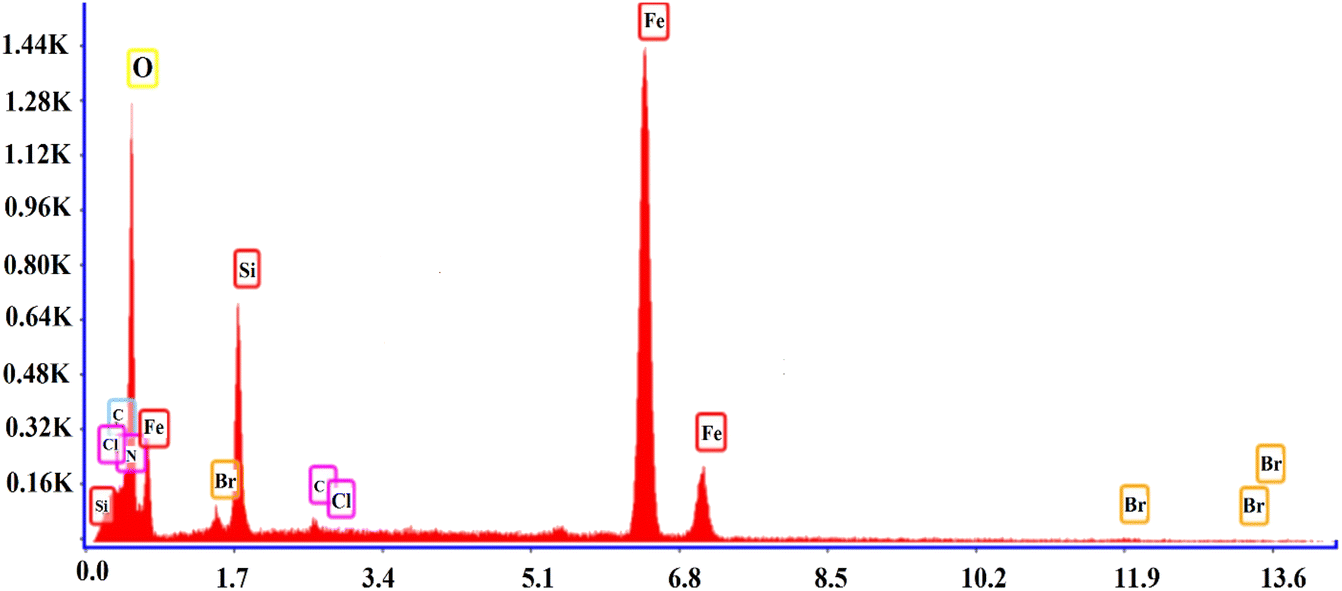
3.1.4 FESEM and EDS of SMNPs@DABCO@eosin. FESEM is an efficient method to characterize the morphological features of the synthesized structure. The surface morphologies of SMNPs, SMNPs@Pr–Cl, SB-DABCO and SMNPs@DABCO@eosin are provided in Fig. 4a–d. By consideration of the SEM image, it can be derived that the synthesized nanocatalyst has almost a nanosphere framework and the diameter of these nanospheres was less than 100 nm. EDS analysis confirmed the symbiosis of Fe, Si and O in the nanocomposite. Energy-dispersive X-ray spectroscopy is often utilized to present beneficial data about the elemental composition of the compounds. According to Fig. 5, it is detailed that SMNPs@DABCO@eosin possess all predicted elemental cases including C, N, O, Si, Cl, Fe, and Br in the data from the EDS coupled SEM analysis.
 |
| | Fig. 4 The SEM images of SMNPs (a), SMNPs@Pr–Cl (b), SB-DABCO (c) and SMNPs@DABCO@eosin (d). | |
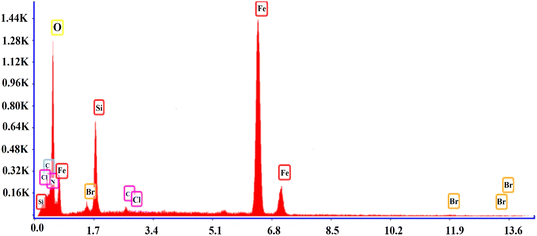 |
| | Fig. 5 The EDS analysis of SMNPs@DABCO@eosin. | |
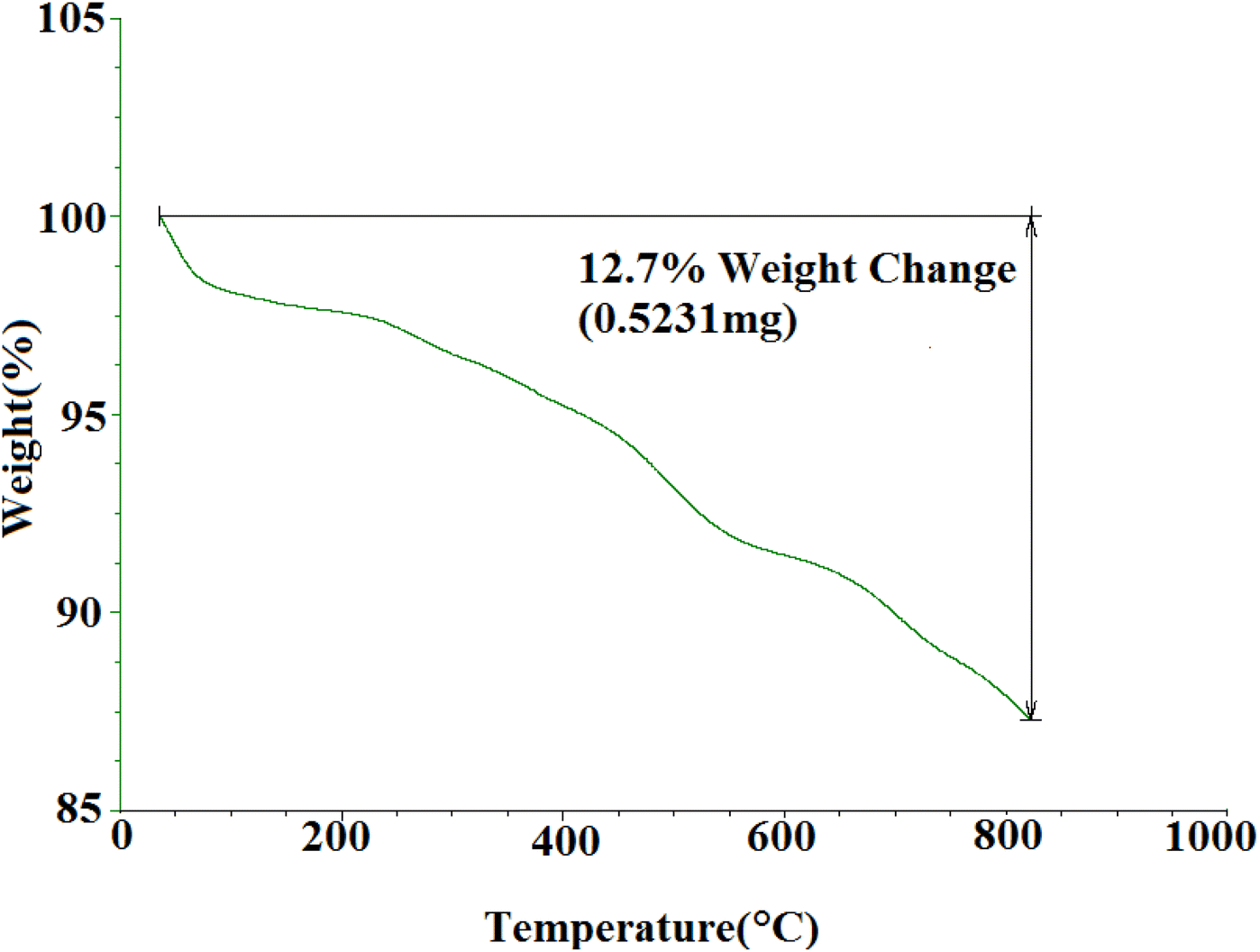
3.1.5 TGA curve of SMNPs@DABCO@eosin. TGA was used to investigate the thermal stability of synthesized SMNPs@DABCO@eosin (Fig. 6). As the temperature rises, several steps of decomposition are apparent from Fig. 6. The initial weight loss observed below 200 °C is most probably caused by the water desorption from the nanoparticle surface.66 The EY segment was destructed at about 200–380 °C.65 Thermal decomposition of the 3-chloropropyl silane groups was also done between 270 and 480 °C.67 Research has determined that MNPs-DABCO has been degraded at 250–500 °C and the DABCO fragment is separated from MNPs.67 Also the latest weight loss, above 600 °C, has indicated the presence of remaining silica and Fe3O4 nanoparticles.
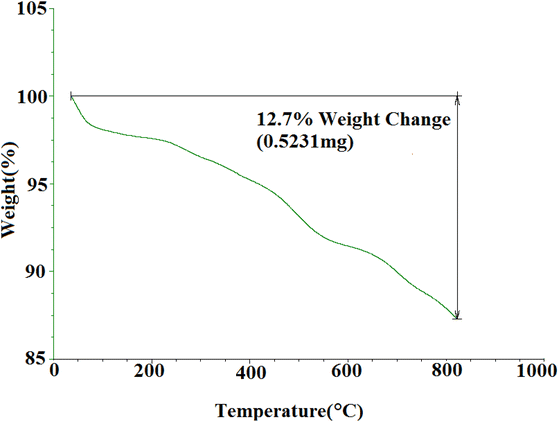 |
| | Fig. 6 TGA profile for SMNPs@DABCO@eosin. | |
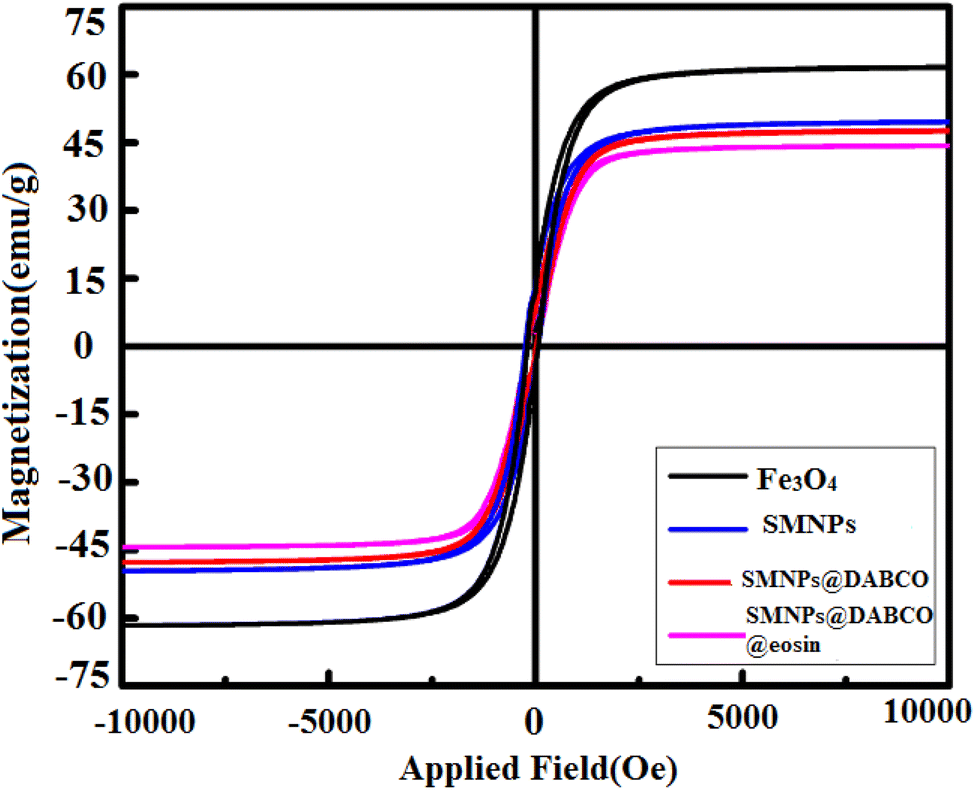
3.1.6 VSM analysis. VSM analysis was performed to display the magnetic features of SMNPs@DABCO@eosin that should be compared to those obtained for the primary core at room temperature. In Fig. 7, the hysteresis loops at room temperature are drawn to estimate the magnetic behavior of these synthesized nanocompounds. As forecast, anchoring the consecutive layers on the Fe3O4 nanoparticles drops the magnetic saturation values of Fe3O4 nanoparticles (from 60 emu g−1 to 41 emu g−1). In addition, due to the magnetic core–shell nanocomposite, the as-synthesized photocatalyst can be simply gathered from aqueous solution by an external magnetic field for a little while and then can be redispersed by lightly shaking it. The outcomes are consistent with the rising number of layers onto the primary magnetic core surface. In total, the obtained evidence proves the successful immobilization on modified Fe3O4 magnetic nanoparticles.
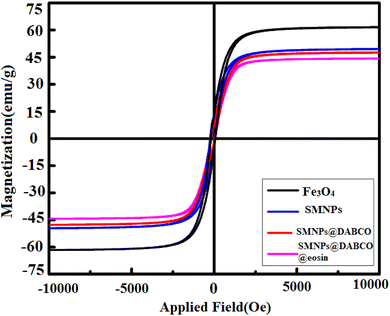 |
| | Fig. 7 Room-temperature magnetization curves of Fe3O4, SMNP, SMNPs@DABCO, and SMNPs@DABCO@eosin. | |
3.2. Catalytic tests
To investigate the photocatalytic activity of SMNPs@DABCO@eosin, a model reaction was designed using 1 mmol benzaldehyde, 1 mmol dimedone, and 1 mmol 4-hydroxycoumarin in the presence of the photocatalyst under irradiation with LED light. The reaction mixture was magnetically stirred in an open air test tube. To derive the best reaction conditions, various parameters of condensation reaction, such as irradiation source, amount of photocatalyst, reaction time, type of solvents and temperature, were optimized.
3.2.1 Studying the effect of the light source. One of the main factors affecting the efficiency of photocatalytic reactions is the irradiation source; hence, the model reaction was subjected to different light irradiations (Table 1). The results of this survey showed that if the reaction be done under the dark and some color light LED conditions, different yields would be obtained under the same conditions and the best efficiency was obtained for green LED light irradiation (λ = 535 nm).
Table 1 Optimization of the irradiation source in condensation reactiona
| Entry |
Light source |
Light specification |
SMNPs@DABCO@eosin |
Time (h) |
Yield (%) |
| Reaction conditions: benzaldehyde (1 mmol), 4-hydroxycoumarin (1 mmol), dimedone (1 mmol), SMNPs@DABCO@eosin (1 mg), solvent free, room temperature, open atmosphere. |
| 1 |
Dark |
— |
0.001 |
2 |
15 |
| 2 |
White LED |
20 × 1 W |
0.001 |
2 |
19 |
| 3 |
Blue |
2.5 W, λ = 431 nm |
0.001 |
2 |
47 |
| 4 |
Red |
2.5 W, λ = 695 nm |
0.001 |
2 |
44 |
| 5 |
Green |
2.5 W, λ = 535 nm |
0.001 |
2 |
62 |
| 6 |
Violet |
2.5 W, λ = 455 nm |
0.001 |
2 |
40 |
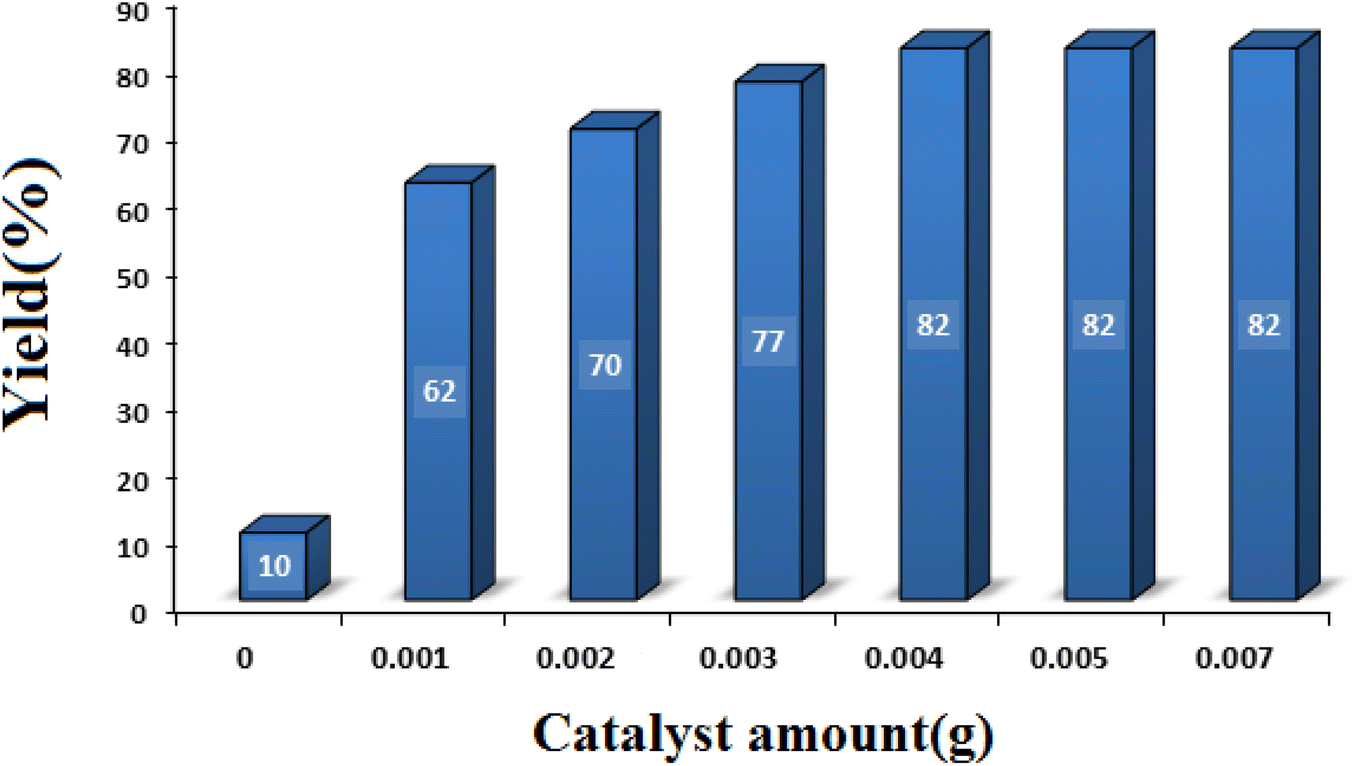
3.2.2 Studying the effect of catalyst amount. With the aim of achieving the optimal amount of catalyst, the model reaction was performed in the presence of different amounts of SMNPs@DABCO@eosin (0–0.005 g). As seen in Fig. 8, 0.004 g of SMNPs@DABCO@eosin gave the best efficiency (82%) under the chosen reaction conditions. Nonetheless, extra enhancement of the catalyst amount modified neither the reaction time nor the efficiency.
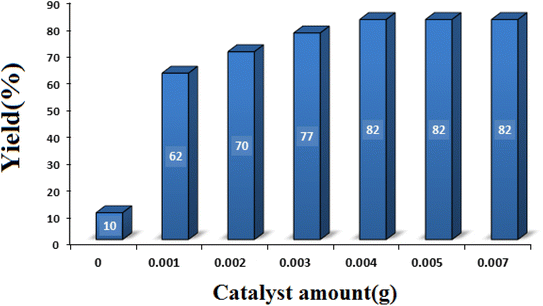 |
| | Fig. 8 Optimization of the catalyst amount in condensation reaction [reaction conditions: benzaldehyde (1 mmol), 4-hydroxycoumarin (1 mmol), dimedone (1 mmol), solvent free, room temperature, open atmosphere, green LEDs (2.5 W, λ = 535 nm), time: 2 h]. | |
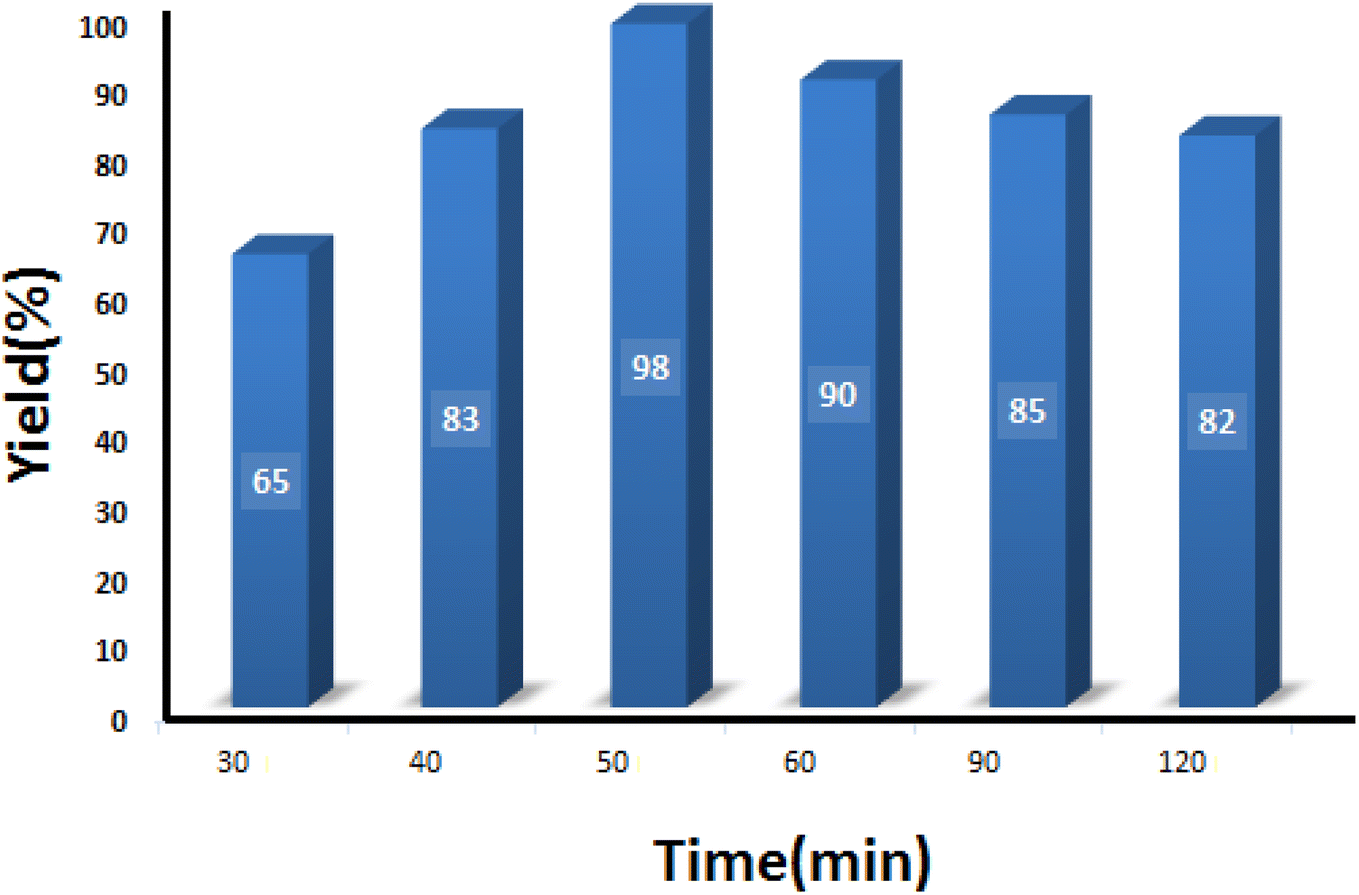
3.2.3 Studying the effect of reaction time. The model reaction was conducted at different reaction times to obtain the optimum time recommended to get the maximum product. As can be seen in Fig. 9, 50 min was sufficient to attain the maximum output of 98%. It is worth noting that increasing the reaction time caused a slight decrease in the yield percentage, which can be related to the poisoning of the catalyst surface.
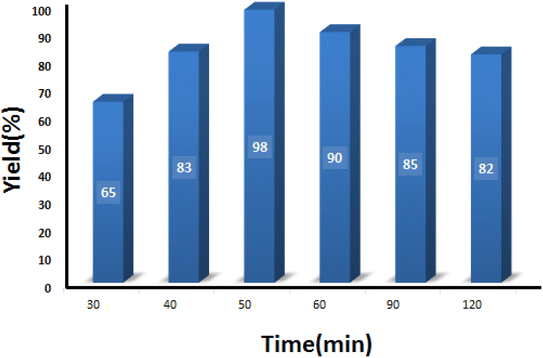 |
| | Fig. 9 Optimization of the reaction time in condensation reaction [benzaldehyde (1 mmol), 4-hydroxycoumarin (1 mmol), dimedone (1 mmol), SMNPs@DABCO@eosin (0.004 g), solvent free, room temperature, open atmosphere, green LEDs (2.5 W, λ = 535 nm)]. | |
3.2.4 Studying the effect of solvents. To optimize the reaction solvent, the condensation reaction of benzaldehyde, 4-hydroxycoumarin and dimedone was done in some chosen solvents such as H2O, toluene, ethanol and acetonitrile under optimum conditions. The results showed that the reaction has the best yield under solvent-free conditions (98% after 50 min, Table 2, entry 1).
Table 2 Optimization of solvent in condensation reactiona
| Entry |
SMNPs@DABCO@eosin (g) |
Solvent |
Time (min) |
Yieldb (%) |
| Reaction conditions: benzaldehyde (1 mmol), 4-hydroxycoumarin (1 mmol), dimedone (1 mmol), SMNPs@DABCO@eosin (0.004 g), room temperature, open atmosphere, green LEDs (2.5 W, λ = 535 nm). Isolated yield of product. |
| 1 |
0.004 |
— |
50 |
98 |
| 2 |
0.004 |
H2O |
50 |
86 |
| 3 |
0.004 |
Toluene |
50 |
62 |
| 4 |
0.004 |
Ethanol |
50 |
84 |
| 5 |
0.004 |
CH3CN |
50 |
70 |
3.2.5 Studying the effect of temperature. To elevate the product and achieve the best reaction conditions, finally we focused on the effect of temperature on the reaction condensation of benzaldehyde with 4-hydroxycoumarin and dimedone (Table 3). As can be seen an increase in temperature didn't increase the yield and the best outcome was gained at 25 °C after 50 min; therefore, 25 °C (room temperature) was kept as the reaction temperature for all runs.
Table 3 Optimization of temperature in condensation reactiona
| Entry |
SMNPs@DABCO@eosin (g) |
Temperature (°C) |
Time (min) |
Yieldb (%) |
| Reaction conditions: benzaldehyde (1 mmol), 4-hydroxycoumarin (1 mmol), dimedone (1 mmol), SMNPs@DABCO@eosin (0.004 g), solvent free, open atmosphere, green LEDs (2.5 W, λ = 535 nm). Isolated yield of product. |
| 1 |
0.004 |
25 |
50 |
98 |
| 2 |
0.004 |
55 |
50 |
88 |
| 3 |
0.004 |
82 (reflux) |
50 |
90 |
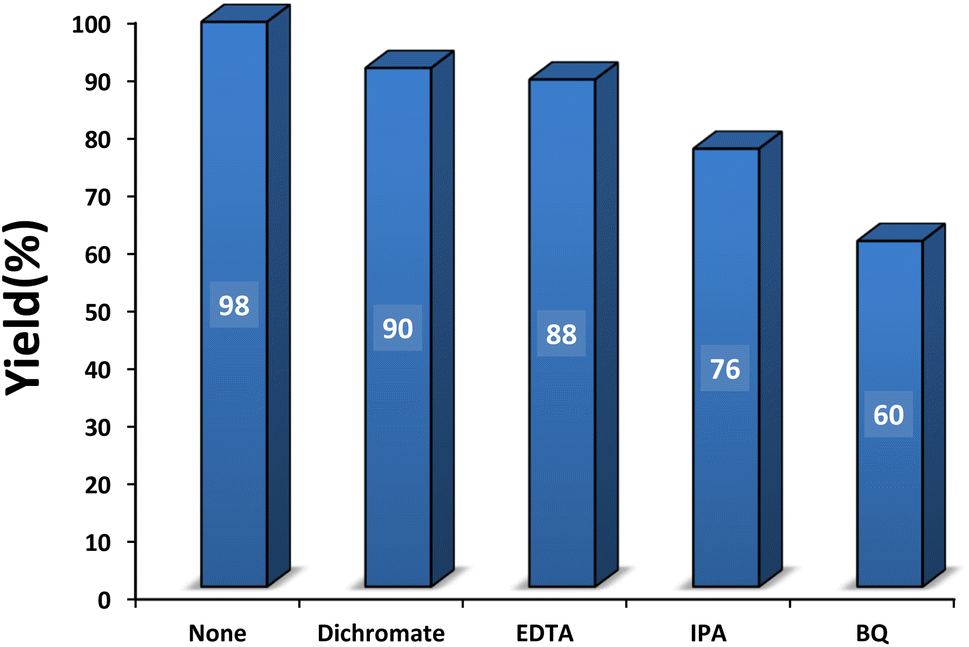
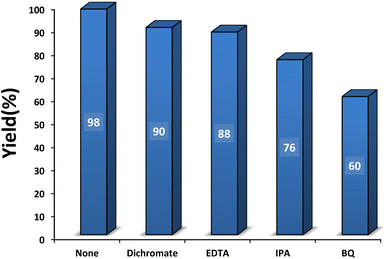
3.2.6 Role of hole and electron scavengers. Hole and electron scavengers were involved in the photocatalytic condensation of benzaldehyde with 4-hydroxycoumarin and dimedone in the presence of SMNPs@DABCO@eosin as a heterogeneous nanophotocatalyst (Fig. 10). Broadly, superoxide anion-radical, hydroxyl radical and holes are effective radical species in a photocatalysis reaction. Consequently, several common scavengers were investigated in the present study. In these experiments dichromate and ethylenediaminetetraacetic acid (EDTA) were employed as an electron scavenger68 and h+ scavenger,69 respectively. In this case, yield% was decreased 8% and 10% respectively, which confirmed probable contribution of e− and h+ in the photocatalysis reaction. In the presence of isopropyl alcohol (IPA), as a hydroxyl free radical scavenger,70 yield% was reduced from 98 to 76%. In addition, a great decrease of yield% (38%) in the presence of p-benzoquinone (BQ) as an ˙O2− scavenger71 proved that ˙O2− would strongly affect the photocatalytic process. Overall, it can be concluded from the data that OH˙ and ˙O2− would be the main species in the photocatalytic procedure.72,73
 |
| | Fig. 10 Effect of some familiar scavengers on condensation reaction [benzaldehyde (1 mmol), 4-hydroxycoumarin (1 mmol), dimedone (1 mmol), SMNPs@DABCO@eosin (0.004 g), solvent free, room temperature, open atmosphere, green LEDs (2.5 W, λ = 535 nm), time: 50 min]. | |
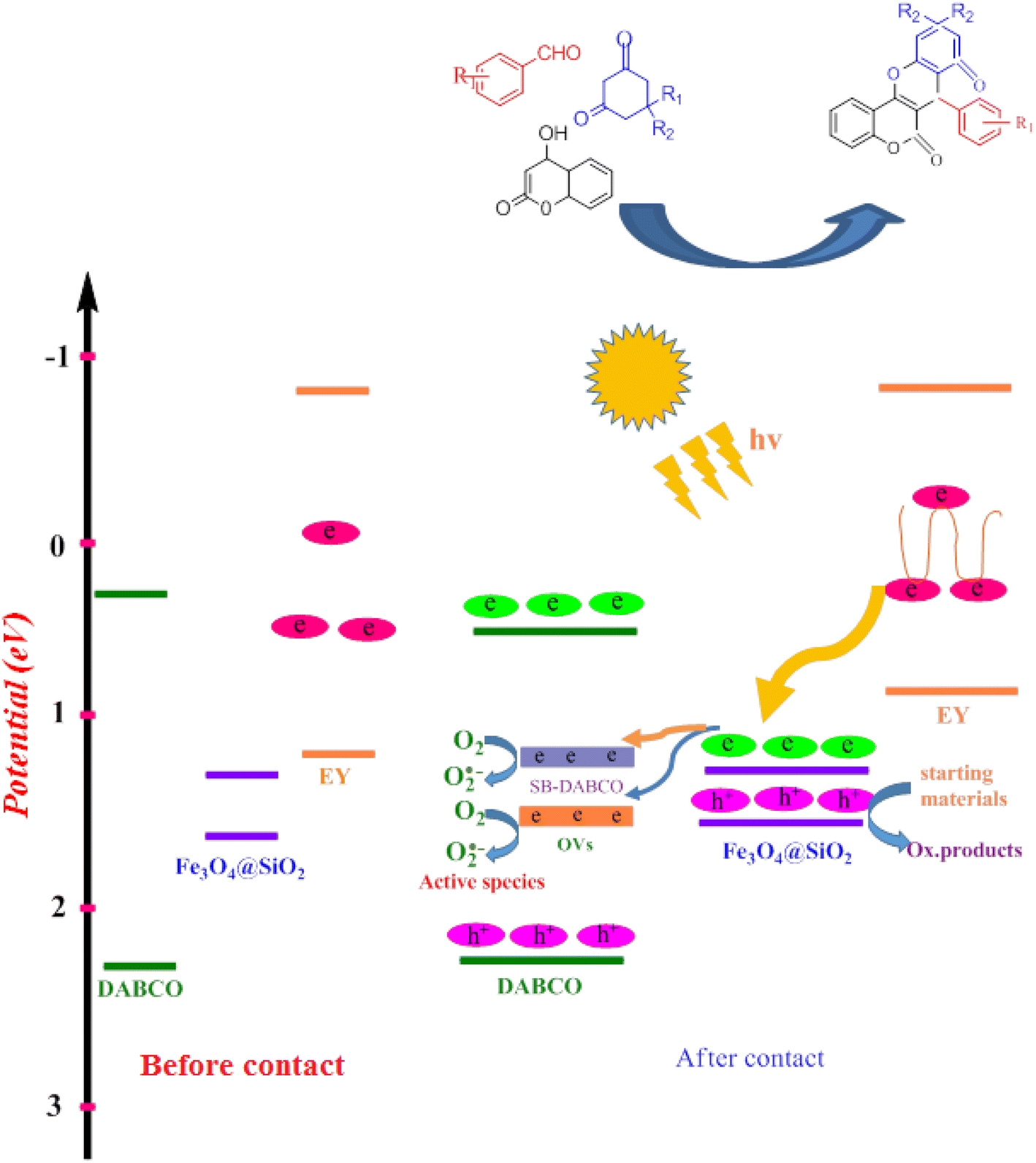
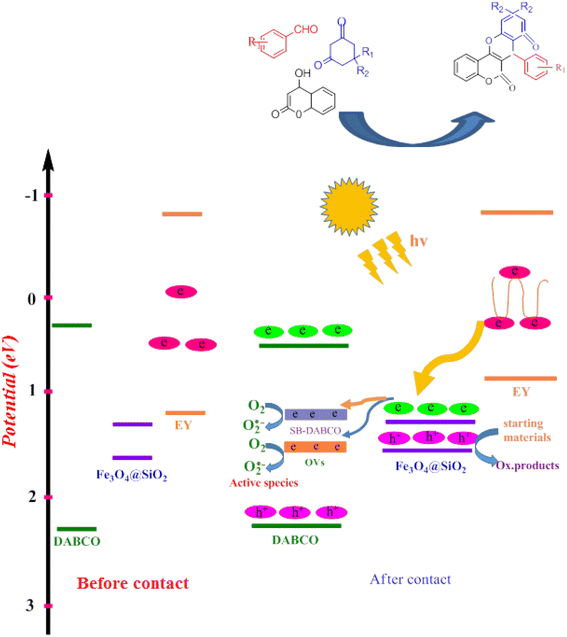
In a common photocatalytic system, choosing an appropriate electron carrier is necessary to facilitate effective transfer of charge carriers and enhance the stability of the photocatalyst. Fe3O4·SiO2@DABCO (SMNPs@DABCO) and eosin fragments absorb sufficient energy to produce electron–hole pairs upon exposure to the green LEDs. The results of this study show that light absorption remarkably increased with the as-prepared hierarchical SMNPs@DABCO@eosin nanocomposite. The photoinduced electrons in SMNPs@DABCO CB tend to shift toward the interface of the heterojunction and endeavor to combine with the holes of eosin (EY) VB. Thus, SMNPs contribute to the process and bridges between the DABCO and EY, enhancing the combination rate. Therefore, the recombination rate of charge carriers drastically suppresses within SMNPs@DABCO and EY, leading to accumulation of electrons in EY CB and holes in the VB of SMNPs@DABCO. Ultimately, the accumulated electrons with strong reducibility would reduce O2 to ˙O2− (Scheme 3).74
 |
| | Scheme 3 Band position of DABCO, EY, SMNPs and the produced heterojunctions. | |
3.2.7 Comparison of the present protocol with some reported similar methods. The gained results for the catalytic activity of SMNPs@DABCO@eosin were compared with a number of similar reports on the preparation of chromeno[4,3-b]chromenes in terms of catalyst amount, reaction time, temperature, solvent and yield%, as shown in Table 4. Due to the use of a very small amount of catalyst, relatively low time, low temperature, no use of solvent and high efficiency, compared to the investigated methods, the present method is a special case that provides a successful route for the one-step/one-pot synthesis of the desired products under very mild conditions.
Table 4 Comparison of the catalytic activity of SMNPs@DABCO@eosin with some reported catalysts in the synthesis of chromeno[4,3-b]chromenea
| Catalyst |
Time (min) |
Temp (°C) |
Solvent |
Yield (%) |
Ref. |
| Benzaldehyde was used in all cases. |
| SMNPs@DABCO@eosin (4 mg) |
50 |
r.t. |
— |
98 |
This work |
| Eosin Y (2 mol%) |
210 |
r.t. |
CH3CN |
77 |
19 |
| Fe3O4@SiO2@(CH2)3OMoO3H (2 mg) |
45 |
80 |
— |
90 |
75 |
| t-ZrO2 np (10 mol%, 12.3 mg) |
30 |
80 |
H2O (5 ml) |
91 |
76 |
| CuFe2O4@SO3H (0.05 g) |
150 |
70 |
EtOH |
90 |
77 |
| Fe(DS)3 (10 mol%) |
120 |
70 |
H2O |
87 |
57 |
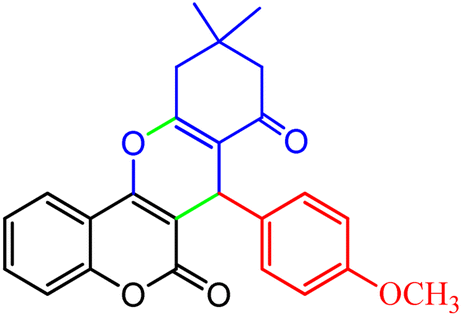
3.2.8 Synthesis of different chromeno[4,3-b]chromenes catalyzed by SMNPs@DABCO@eosin. To define the extent, limitations and versatility of the present strategy, various aromatic aldehydes were reacted with diketone and 4-hydroxycoumarin for production of different chromeno[4,3-b]chromenes under solvent free conditions and green LEDs (2.5 W, λ = 535 nm). As mentioned in Table 5, aromatic aldehydes bearing different electron releasing/withdrawing substituents were able to create the desired product in good to excellent yield in short reaction time with superior selectivity. The products were purified by recrystallization from ethanol and the second step from ethyl acetate.62 The products were characterized by spectroscopic and spectrometric methods.
Table 5 Preparation of chromeno[4,3-b]chromenea
| Entry |
Aldehyde |
Dimedone |
Product |
Yield (%) |
M.p |
| Found, reported78–80 |
| Reaction conditions: benzaldehyde (1 mmol), 4-hydroxycoumarin (1 mmol), dimedone (1 mmol), SMNPs@DABCO@eosin (0.004 g), solvent free, room temperature, open atmosphere, green LEDs (2.5 W, λ = 535 nm), time: 50 min. |
| 1 |
 |
 |
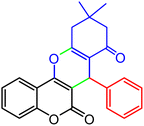 |
90 |
223–224, 220–222 |
| 2 |
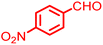 |
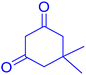 |
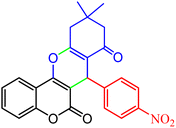 |
96 |
209–211, 208–210 |
| 3 |
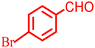 |
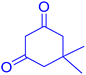 |
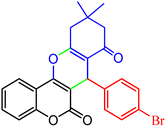 |
76 |
227–229, 228–230 |
| 4 |
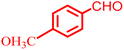 |
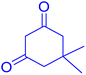 |
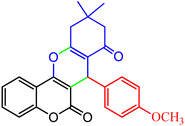 |
75 |
190–191, 187–189 |
| 5 |
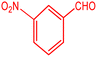 |
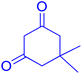 |
 |
79 |
233–234, 230–232 |
| 6 |
 |
 |
 |
77 |
220–222, 218–220 |
| 7 |
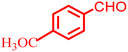 |
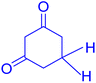 |
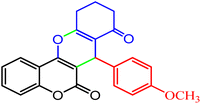 |
78 |
196–198, 196–197 |
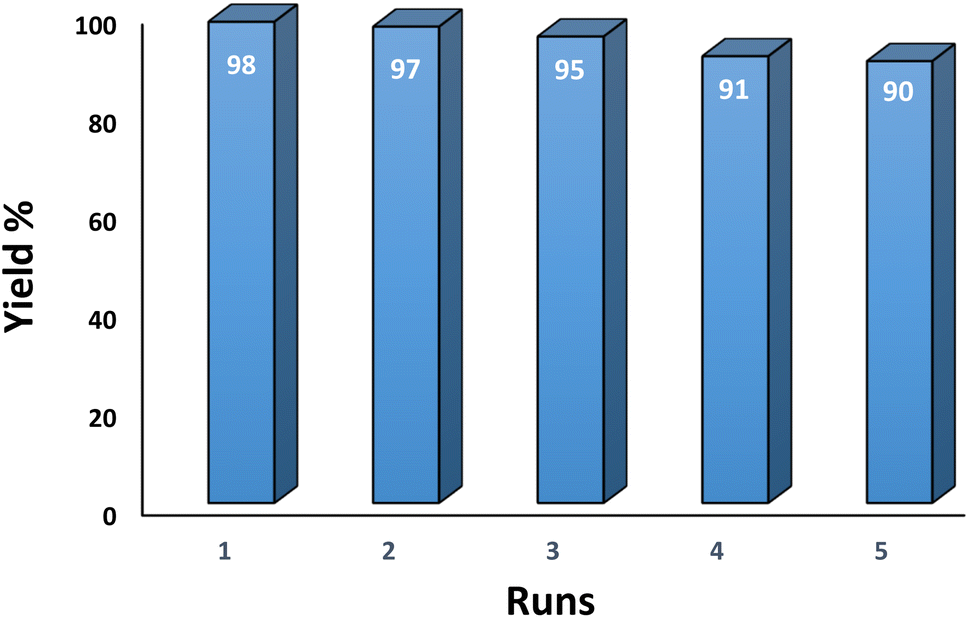
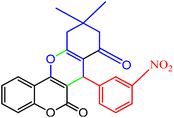
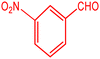
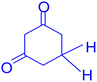
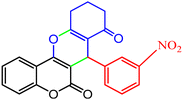
3.2.9 Studying the stability and reusability of nano SMNPs@DABCO@eosin. Reusability is one of the key parameters for evaluating the performance of catalysts. A catalyst is of great importance in the industry when it can be easily recovered and reused. Therefore to study the stability and reusability of nanoparticles of SMNPs@DABCO@eosin, after completion of the first run, toluene was appended and the catalyst was detached from the reaction mixture via a simple filtration. The filtered catalyst was washed with a copious amount of toluene, then dried at room temperature and reused in the same reaction for five consecutive runs (Fig. 11). It was found that SMNPs@DABCO@eosin could be reused five times without a significant decrease in extraction efficiency. The results proved acceptable reusability of the SMNPs@DABCO@eosin photocatalyst. XRD of the final reused nanocatalyst was compared with that of the fresh one. Interestingly, the nature and chemical composition of the reused nanomaterial after the fifth run was closely comparable with that of the fresh one. This study proved the stability of the nanophotocatalyst after at least five runs (Fig. 12).
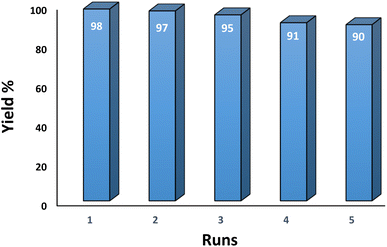 |
| | Fig. 11 Recyclability of the SMNPs@DABCO@eosin nanophotocatalyst. | |
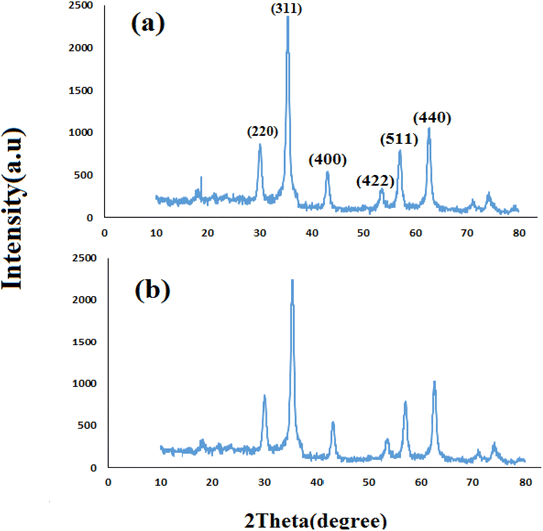 |
| | Fig. 12 X-ray diffractions (XRDs) of the fresh (a) and reused (b) SMNP@DABCO@eosin nanophotocatalyst. | |
3.3. Suggesting a plausible reaction pathway
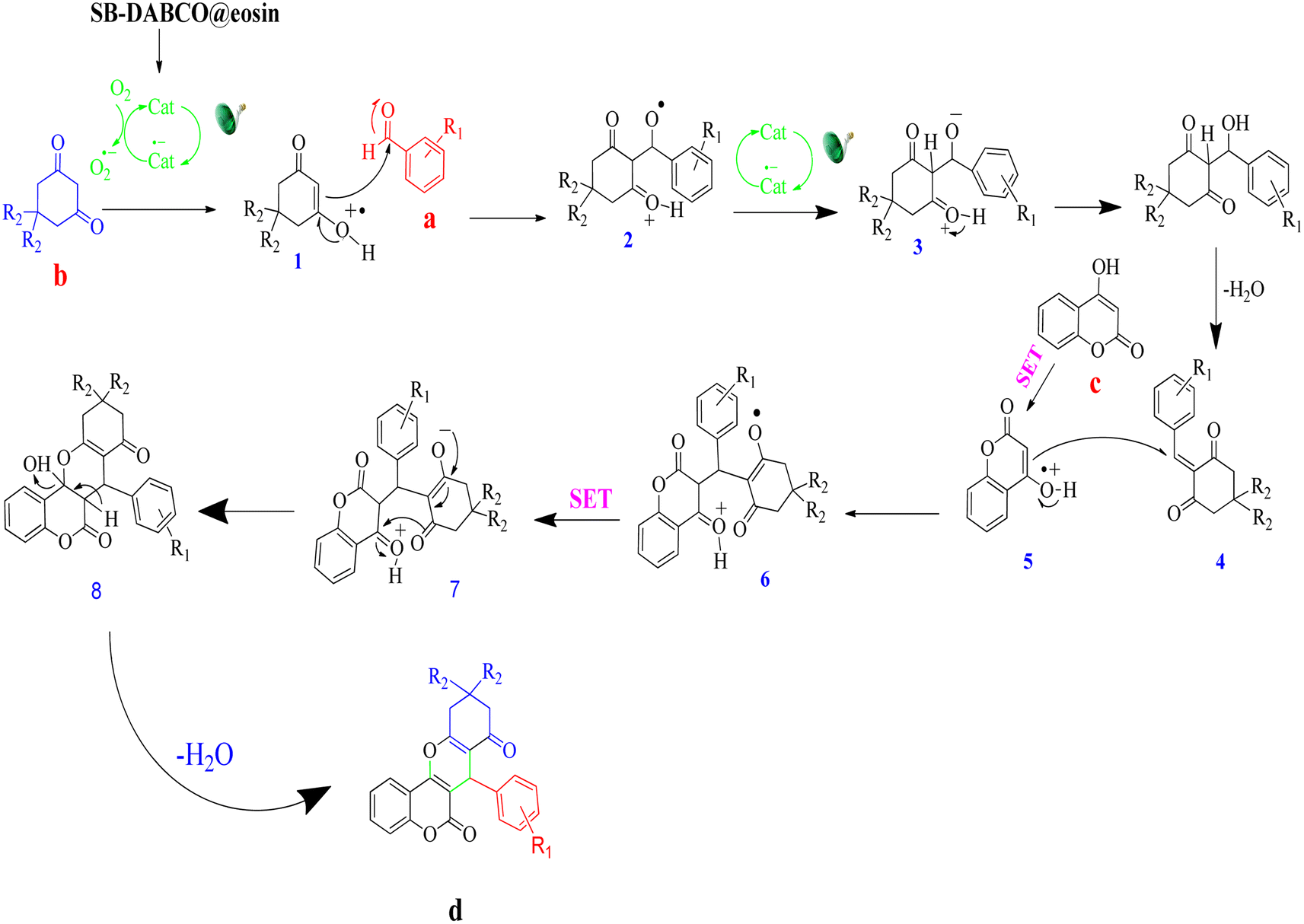
A feasible mechanism for the generation of chromeno[4,3-b]chromene by SMNPs@DABCO@eosin is given in Fig. 13. It is proposed that the photoredox catalyst eosin Y is converted to excited state eosin Y* upon visible light irradiation, and then this eosin Y* undergoes reductive quenching by 1,3-dicarbonyl compound (b) to create the radical cation (1) and converts to the radical anion of eosin Y. Now this radical cation (1) is reacted with aldehyde (a) to form (2), and simultaneously eosin Y undergoes single electron transfer (SET) to produce compound (2) to (3). Dehydration of the compound (3) produces the β-dicarbonyl-enone (4) which can act as a Michael acceptor. Again 4-hydroxycoumarin (c) converts into a radical cation (5) by the photo-redox catalyst eosin Y via the single electron transfer (SET), and now this radical cation (5) undergoes Michael type addition reaction with β-dicarbonyl-enone (4) and furnishes a compound (6). At the same time, eosin Y undergoes single electron transfer (SET) to convert (6) into (7). Finally (7) on a simple dehydration reaction leads to the targeted molecule (d).81
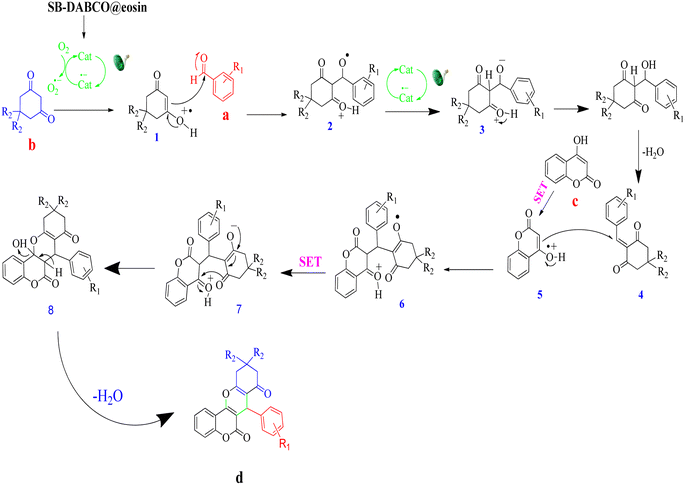 |
| | Fig. 13 A probable reaction pathway for the condensation reaction. | |
4. Conclusions
In conclusion, we have successfully introduced an impressive route for the production of chromeno[4,3-b] chromene in the presence of a metal free catalyst under green LED irradiation. Eosin Y as a low cost and readily available photoredox catalyst has surely proven to be a superior green replacing reagent. We are very hopeful that this methodology will play a fundamental role in medicinal chemistry, natural product chemistry and organic synthesis by serving as a robust and economical alternative way to synthesize target molecules.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The work was supported by Hakim Sabzevari University.
References
- D. Insuasty, J. Castillo, D. Becerra, H. Rojas and R. Abonia, Molecules, 2020, 25(3), 505 CrossRef PubMed.
- C. G. Neochoritis, T. Zarganes-Tzitzikas, K. Katsampoxaki-Hodgetts and A. Dömling, Kinderleicht, J. Chem. Educ., 2020, 97(10), 3739–3745 CrossRef CAS PubMed.
- S. Zhi, X. Ma and W. Zhang, Org. Biomol. Chem., 2019, 17(33), 7632–7650 RSC.
- C. S. Graebin, F. V. Ribeiro, K. R. Rogério and A. E. Kümmerle, A Review, Curr. Org. Synth., 2019, 16(6), 855–899 CrossRef CAS PubMed.
- R. C. Cioc, E. Ruijter and R. V. Orru, Green Chem., 2014, 16, 2958–2975 RSC.
- M. B. Gawande, V. D. Bonifácio, R. Luque, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 5522–5551 RSC.
- A. R. Salih and Z. A. K. Al-Messri, Eurasian Chem. Commun., 2021, 3, 533–541 CAS.
- M. Nikpassand and L. Z. Fekri, Chem. Methodol., 2020, 4, 437–446 CrossRef CAS.
- E. Haddazadeh and M. K. Mohammadi, Chem. Methodol., 2020, 4, 324–332 Search PubMed.
- S. Shylesh, V. Schnemann and W. R. Thiel, Angew. Chem., Int. Ed., 2010, 49, 3428–3459 CrossRef CAS PubMed.
- R. Hudson, Y. Feng, R. S. Varma and A. Moores, Green Chem., 2014, 16, 4493–4505 RSC.
- M. B. Gawande, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 3371–3393 RSC.
- S. Sajjadifar, I. Amini, S. Habibzadeh, G. Mansouri and E. Ebadi, Chem. Methodol., 2020, 4, 623–634 CAS.
- A. H. Karimi, A. Hekmat-Ara, A. Zare, M. Barzegar, R. Khanivar and M. Sadeghi-Takallo, Eurasian Chem. Commun., 2021, 3, 360–368 CAS.
- S. A. M. Ziabari, M. Babamoradi, Z. Hajizadeh and A. Maleki, Eurasian Chem. Commun., 2020, 2, 456–464 CrossRef CAS.
- H. Aghahosseini and A. Ramazani, Eurasian Chem. Commun., 2020, 2, 410–419 CrossRef CAS.
- E. M. Materón, C. M. Miyazaki, O. Carr, N. Joshi, P. H. S. Picciani, C. J. Dalmaschio, F. Davis and F. M. Shimizu, Applied Surface Science Advances, 2021, 6, 100163 CrossRef.
- H. Su, M. Yang, L. Zhao, Sh. Yao, Q. Geng, L. Wang and G. Li, Eur. Polym. J., 2022, 177, 111476 CrossRef CAS.
- A. K. Sharma, J. Tiwari, D. Jaiswal, S. Singh, J. Singh and J. Singh, Curr. Organocatal., 2019, 6, 222–230 CrossRef CAS.
- R. Tayebee, A. Pejhan, H. Ramshini, B. Maleki, N. Erfaninia, Z. Tabatabaie and E. Esmaeili, Appl. Organomet. Chem., 2018, 32(1), 3924 CrossRef.
- F. Javadi and R. Tayebee, Iran. J. Catal., 2017, 7(4), 283–292 CAS.
- S. F. Razavi, M. Khoobi, H. Nadri, A. Sakhteman, A. Moradi, S. Emami, A. Foroumadi and A. Shafiee, Eur. J. Med. Chem., 2013, 64, 252–259 CrossRef CAS PubMed.
- M. Suzuki, K. Nakagawa-Goto, S. Nakamura, H. Tokuda, S. L. Morris-Natschke, M. Kozuka, H. Nishino and K. H. Lee, Pharm. Biol., 2006, 44, 178–182 CrossRef CAS.
- Z. Q. Xu, M. G. Hollingshead, S. Borgel, C. Elder, A. Khilevich and M. T. Flavin, Bioorg. Med. Chem. Lett., 1999, 9, 133–138 CrossRef CAS.
- C. Bruhlmann, F. Ooms, P. Carrupt, B. Testa, M. Catto, F. Leonetti, C. Altomare and A. Cartti, J. Med. Chem., 2001, 44, 3195–3198 CrossRef CAS PubMed.
- S. Emami, A. Foroumadi, M. A. Faramarzi and N. Samadi, Arch. Pharm., 2008, 341, 42–48 CrossRef CAS PubMed.
- L. Firoozpour, N. Edraki, M. Nakhjiri, S. Emami, M. Safavi, S. K. Ardestani, M. Khoshneviszadeh, A. Shafiee and A. Foroumadi, Arch. Pharmacal Res., 2012, 35(12), 2117–2125 CrossRef CAS PubMed.
- R. K. Nimesh, D. H. Dhaval, T. M. Prashant and K. P. Saurabh, Med. Chem. Res., 2011, 20, 854–864 CrossRef.
- A. Amr, M. Abdalla, S. Essaouy, M. Areef, M. Elgamal, T. Nassear and A. Haschich, Russ. J. Gen. Chem., 2017, 87, 1826–1833 CrossRef CAS.
- S. W. Ng, L. H. Chung, C. F. Yeung, H. S. Lo, H. L. Shek, T. S. Kang, C. H. Leung, D. L. Ma and C. Y. Wong, Chem.–Eur. J., 2018, 24, 1779–1783 CrossRef CAS PubMed.
- K. Nitin, K. Sushil, G. Himanshu and P. K. Sharma, WRJB, 2012, 1, pp. 1–5 Search PubMed.
- R. O. S. Kitamura, P. Romoff, M. C. M. Young, M. J. Kato and J. H. G. Lago, Phytochemistry, 2006, 67, 2398–2402 CrossRef PubMed.
- N. Thomas and S. M. Zachariah, Asian J. Pharm. Clin. Res., 2013, 6, 11–15 CAS.
- M. K. Hussain, M. F. Khan, S. Khatoon, A. G. Al-Sehemi and M. Saquib, Chromenes: Phytomolecules with Immense Therapeutic Potential, Springer, Singapore, 2020, pp. 185–204 Search PubMed.
- V. Raj and J. Lee, Front. Chem., 2020, 8, 623 CrossRef CAS PubMed.
- M. Mahdavi, J. Davoodi, M. R. Zali and A. Foroumadi, Biomed. Pharmacother., 2011, 65, 175–182 CrossRef CAS PubMed.
- J. L. Wang, D. Liu, Z.-J. Zhang, S. Shan, X. Han, S. M. Srinivasula, C. M. Croce, E. S. Alnemri and Z. Huang, Proc. Natl. Acad. Sci., India, 2000, 97, 7124–7129 CAS.
- S. A. Patil, J. Wang, X. S. Li, J. Chen, T. S. Jones, A. Hosni-Ahmed, R. Patil, W. L. Seibel, W. Li and D. D. Miller, Bioorg. Med. Chem. Lett., 2012, 22, 4458–4461 CrossRef CAS PubMed.
- A. M. Shestopalov, Y. M. Litvinov, L. A. Rodinovskaya, O. R. Malyshev, M. N. Semenova and V. V. Semenov, ACS Comb. Sci., 2012, 14, 484–490 CrossRef CAS PubMed.
- S. K. Sahoo, Renew. Sust. Energ. Rev., 2016, 59, 927–939 CrossRef.
- B. Li, R. Tayebee, E. Esmaeili, M. S. Namaghi and B. Maleki, RSC Adv., 2020, 10(67), 40725–40738 RSC.
- M. Hricovíni, J. Asher and M. Hricovíni, RSC Adv., 2020, 10(10), 5540–5550 RSC.
- S. Aguado, L. Casarrubios, C. R. de Arellano and M. A. Sierra, RSC Adv., 2020, 10(50), 29855–29867 RSC.
- Y. Zhang, C. Ye, S. Li, A. Ding, G. Gu and H. Guo, RSC Adv., 2017, 7, 13240–13243 RSC.
- Y. Chiu, T. Chang, Ch. Chen, M. Sone and Y. Hsu, Catalysts, 2019, 9, 430 CrossRef CAS.
- M. Fang, C. Tsao and Y. Hsu, J. Phys. D: Appl. Phys., 2020, 53, 143001 CrossRef CAS.
- Y. Chen, Y. Wang, H. Moon, K. Yong and Y. Hsu, RSC Adv., 2021, 11, 12288–12305 RSC.
- M. Majek, F. Filace and A. J. von Wangelin, Beilstein J. Org. Chem., 2014, 10, 981–989 CrossRef PubMed.
- V. Srivastava and P. P. Singh, RSC Adv., 2017, 7, 31377–31392 RSC.
- S. Yadav, M. Srivastava, P. Rai, B. P. Tripathi, A. Mishra, J. Singh and J. Singh, New J. Chem., 2016, 40, 9694–9701 RSC.
- M. C. Mascolo, Y. Pei and T. A. Ring, Materials, 2013, 6, 5549–5567 CrossRef CAS PubMed.
- Q. Zhang, H. Su, J. Luo and Y. Wei, Catal. Sci. Technol., 2013, 3, 235–243 RSC.
- A. A. Elmekawy, J. B. Sweeney and D. R. Brown, Catal. Sci. Technol., 2015, 5, 690–696 RSC.
- A. Hasaninejad, M. Shekouhy, N. Golzar, A. Zare and M. M. Doroodmand, Appl. Catal., A, 2011, 402, 11–22 CrossRef CAS.
- X. J. Sun, J. F. Zhou and S. J. Zhi, Synth. Commun., 2012, 42, 1987–1994 CrossRef CAS.
- H. Anaraki-Ardakani, R. Ghanavatian and M. Akbari, World Appl. Sci. J., 2013, 22, 802–808 CAS.
- K. Pradhan, S. Paul and A. R. Das, Tetrahedron Lett., 2013, 54, 3105–3110 CrossRef CAS.
- M. A. Nasseri, B. Zakerinasab and M. M. Samieadel, RSC Adv., 2014, 4, 41753–41762 RSC.
- X. Zhong, W. T. Wu, H. N. Jie, W. Y. Tang, D. Y. Chen, T. Ruan and H. P. Bai, RSC Adv., 2020, 10, 38024 RSC.
- E. Koushki, RSC Adv., 2021, 11, 23390–23399 RSC.
- L. Liu, E. Koushki and R. Tayebee, J. Mol. Liq., 2021, 330, 115542 CrossRef CAS , ISSN 0167-7322..
- H. A. Doost, A. Ghasedi and E. Koushki, J. Mol. Liq., 2021, 323, 115074 CrossRef CAS , ISSN 0167-7322..
- S. Luo, X. Zheng, H. Xu, X. Mi, L. Zhang and J.-P. Cheng, Adv. Synth. Catal., 2007, 349, 2431–2434 CrossRef CAS.
- C. Dálaigh, S. A. Corr, Y. Gunko and S. J. Connon, Angew. Chem., Int. Ed., 2007, 46, 4329–4332 CrossRef PubMed.
- Z. Jalili, R. Tayebee and F. M. Zonoz, RSC Adv., 2021, 11(29), 18026–18039 RSC.
- R. Gupta, M. Yadav, R. Gaur, G. Arora, P. Rana, P. Yadav, A. Adholeya and K. Sharma, ACS Omega, 2019, 4, 21529–21539 CrossRef CAS PubMed.
- A. Rostami, Y. Navasi, D. Moradi and A. Ghorbani-Choghamarani, Catal. Commun., 2014, 43, 16–20 CrossRef CAS.
- S. Naraginti, Y. Li, Y. Wu, C. Zhang and A. R. Upreti, RSC Adv., 2016, 6, 87246 RSC.
- X. Zheng, J. Yuan, J. Shen, J. Liang, J. Che, B. Tang, G. He and H. Chen, J. Mater. Sci.: Mater. Electron., 2019, 30, 5986–5994 CrossRef CAS.
- R. K. Sharma, B. Arora, S. Sharma, S. Dutta, A. Sharma, S. Yadav and K. Solanki, Mater. Chem. Front., 2020, 4, 605–620 RSC.
- Z. Yin, M. Han, Z. Hu, L. Feng, Y. Liu, Z. Du and L. Zhang, Chem. Eng. J., 2020, 390, 124532 CrossRef CAS.
- Y. Lin, C. P. Yang, Q. Y. Niu and S. L. Luo, Adv. Funct. Mater., 2022, 32, 2108814 CrossRef CAS.
- Y. Lin, X. Wu, Y. Han, Ch. Yang, Y. Ma, Ch. Du, Q. Teng, H. Liu and Y. Zhong, Appl. Catal., B, 2019, 258, 117969 CrossRef CAS.
- C. Liu, S. Abbaspour, M. Rouki, R. Tayebee, M. Jarrahi and E. E. Shahri, Appl. Organomet. Chem., 2022, 36(11), e6878 CAS.
- F. Khosravian, B. Karami and M. Farahi, New J. Chem., 2017, 41, 11584–11590 RSC.
- A. Saha, S. Payra and S. Banerjee, RSC Adv., 2015, 5, 101664–101671 RSC.
- S. Vajar and M. Mokhtary, Polycyclic Aromat. Compd., 2019, 39, 111–123 CrossRef CAS.
- H. Emtiazi and M. A. Amrollahi, S. Afr. J. Chem., 2014, 67, 175–179 Search PubMed.
- Y. K. Al-Majedy, A. A. Al-Amiery, A. A. H. Kadhum and A. B. Mohamad, Biomed Res. Int., 2016, 2016, 5891703 Search PubMed.
- R. Motamedi, S. Baghbani and F. F. Bamoharram, Synth. Commun., 2012, 42(11), 1604–1612 CrossRef CAS.
- B. Maleki, M. Chahkandi, R. Tayebee, S. Kahrobaei, H. Alinezhad and S. Hemmati, Appl. Organomet. Chem., 2019, 33, 5118 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Behrooz Maleki
*a,
Behrooz Maleki