
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNorsesquiterpenoids from the octocoral Paralemnalia thyrsoides (Ehrenberg 1834)†
Gia Hung Phan‡
ab,
Hao-Chun Hu‡c,
Fang-Rong Chang c,
Zhi-Hong Wende,
Jih-Jung Chenf,
Hsu-Ming Chungg,
Yu-Chi Tsai*h and
Ping-Jyun Sung*bcdij
c,
Zhi-Hong Wende,
Jih-Jung Chenf,
Hsu-Ming Chungg,
Yu-Chi Tsai*h and
Ping-Jyun Sung*bcdij
aGraduate Institute of Marine Biology, National Dong Hwa University, Pingtung 944401, Taiwan
bNational Museum of Marine Biology and Aquarium, Pingtung 944401, Taiwan. E-mail: pjsung@nmmba.gov.tw
cGraduate Institute of Natural Products, Kaohsiung Medical University, Kaohsiung 807378, Taiwan
dDepartment of Marine Biotechnology and Resources, National Sun Yat-sen University, Kaohsiung 804201, Taiwan
eInstitute of BioPharmaceutical Sciences, National Sun Yat-sen University, Kaohsiung 804201, Taiwan
fDepartment of Pharmacy, School of Pharmaceutical Sciences, National Yang Ming Chiao Tung University, Taipei 112304, Taiwan
gDepartment of Applied Chemistry, National Pingtung University, Pingtung 900391, Taiwan
hSchool of Chinese Medicine, College of Chinese Medicine, China Medical University, Taichung 404328, Taiwan. E-mail: yuchi0713@gmail.com
iChinese Medicine Research and Development Center, China Medical University Hospital, Taichung 404394, Taiwan
jPhD Program in Pharmaceutical Biotechnology, Fu Jen Catholic University, New Taipei City 242062, Taiwan
First published on 30th September 2022
Abstract
Three norsesquiterpenoids, pathyspirolactones A (1) and B (2), and napalilactone (3), featuring a γ-spirolactone moiety, were isolated from the cultured octocoral Paralemnalia thyrsoides. The structures of 1–3 were determined by analyzing spectroscopic data, DP4+ computation, specific optical rotation, and X-ray diffraction. In addition, we explored the absolute configurations of pathyspirolactone A (1) and its conformation of the cyclohexane ring to resolve the stereochemical confusion of those of norsesquiterpenoid compounds. Furthermore, we proved that pathyspirolactone B (2) was the first bromine-containing norsesquiterpenoid reported from octocorals.
1 Introduction
Octocorals of the genus Paralemnalia (family Nephtheidae) represent a rich source of natural substances with intriguing and unique structural features. Among these metabolites, sesquiterpenoids1–10 and norsesquiterpenoids11–14 are representative compounds for the natural products from Paralemnalia spp. Paralemnalia thyrsoides (Ehrenberg 1834) is one of the most common marine invertebrates natively distributed throughout tropical and subtropical regions of the Indo-Pacific Ocean. Corals were described by Shi-Zhen Li in his ancient herbal Compendium of Chinese Materia Medica, published in 1596, as “sweet, neutral and non-toxic; used to remove eye vision obstruction; clear abiding static blood; blow the powder to nose to stop nose bleeding; brighten the eye and calm the spirit; stop epileptic seizure; apply to the eye to improve floater.“15 In connection with our continuing studies of marine organisms with the aim of informing new natural products, in this research, we completed the preparation and structural identification of two nor-sesquiterpenoids, pathyspirolactones A (1) and B (2), and a known analogue, napalilactone (3) (Fig. 1), obtained from the cultured octocoral identified as P. thyrsoides.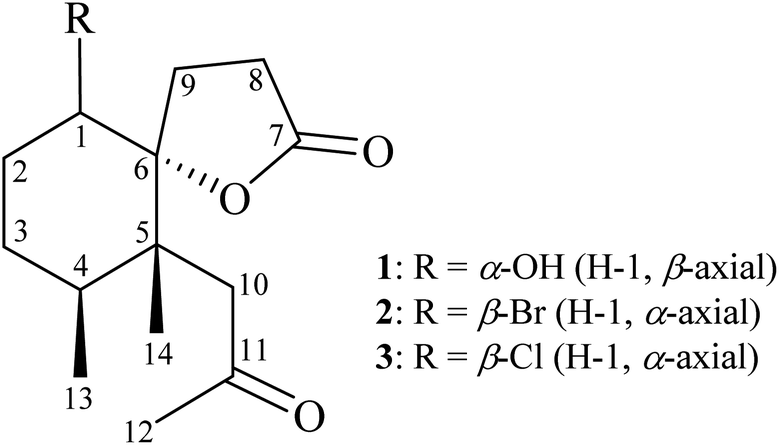 | ||
| Fig. 1 Structures of pathyspirolactones A (1) and B (2), and napalilactone (3). | ||
2 Results and discussion
Pathyspirolactone A (1) was isolated as an amorphous powder. NMR data coupled with the [M + Na]+ peak in the HRESIMS at m/z 277.14129 suggested a molecular formula C14H22O4 (calcd for C14H22O4 + Na, 277.14103) that indicated four degrees of unsaturation. IR spectrum analysis showed that 1 had absorption peaks at νmax 3422, 1762, and 1700 cm−1, suggesting that the structure of 1 included hydroxy, γ-lactone, and ketonic groups. The 13C NMR and DEPT spectra revealed that compound 1 had 14 carbons (Table 1), including three methyls, five sp3 methylenes, two sp3 methines (including one oxymethine), two sp3 quaternary carbons (including one oxygenated quaternary carbon), one ester carbonyl and one ketonic carbonyl carbon. Therefore, according to the aforementioned data, two degrees of unsaturation were accounted for, and compound 1 was identified as having two rings.| Position | 1 | 2 | 3 | epi-pathylactone | ||||
|---|---|---|---|---|---|---|---|---|
| δHa (J in Hz) | δCb, Multc | δHa (J in Hz) | δCb, Multc | δHa (J in Hz) | δCb, Multc | δHh (J in Hz) | δCh | |
| a Spectra recorded at 400 MHz in CDCl3.b Spectra recorded at 100 MHz in CDCl3.c Multiplicity deduced by DEPT and HSQC spectra and indicated by usual symbols.d Signals overlapped.e Signals overlapped.f Signals overlapped.g Signals overlapped.h Data were reported by Coelho and Diaz.16 These data were recorded at 500 MHz for 1H and 125 MHz for 13C in CDCl3.i The coupling pattern and coupling constants were assigned as “two similar coupling constants (J = 4.2, 2.9 Hz)” in the content text of the ref. 16. | ||||||||
| 1β(axial) | 3.70 dd (11.6, 4.8) | 73.4, CH | ||||||
| 1β(equiv.) | 4.27 mi | 65.0 | ||||||
| 1α(axial) | 4.59 dd (12.0, 4.4) | 58.2, CH | 4.36 dd (10.4, 4.4) | 63.8, CH | ||||
| 2α(axial) | 1.63 m | 30.4, CH2 | ||||||
| 2β(equiv.) | 1.81 m | |||||||
| 2α(equiv.)/β(axial) | 2.23 m; 2.08 ddd (14.4, 12.0, 4.0) | 29.8, CH2 | 2.12 m; 1.94 m | 28.9, CH2 | 1.78–2.0 | 27.0 | ||
| 3α(equiv.) | 1.61 m | 28.4, CH2 | ||||||
| 3β (axial) | 1.41 m | |||||||
| 3α(axial)/β(equiv.) | 1.89 m; 1.45 dddd (14.0, 4.0, 4.0, 4.0) | 28.1, CH2 | 1.83 m; 1.50 m | 27.0, CH2 | 1.44–1.79 | 26.0 | ||
| 4 | 2.17 m | 34.3, CH | 2.56 md | 33.2, CH | 2.53 m | 33.2, CH | 2.38 | 33.7 |
| 5 | 46.7, C | 46.4,f C | 46.2, C | 45.3 | ||||
| 6 | 91.8, C | 91.1, C | 91.5, C | 92.0 | ||||
| 7 | 177.9, C | 175.8, C | 175.9, C | 177.0 | ||||
| 8 | 2.74 ddd (17.2, 12.4, 8.0); 2.47 ddd (17.2, 11.2, 4.2) | 29.6, CH2 | 2.74 me; 2.53 md | 29.7, CH2 | 2.68 mg; 2.46 m | 29.4, CH2 | 2.48–2.67 | 29.0 |
| 9 | 2.37 m; 2.19 m | 26.3, CH2 | 2.48 m; 2.41 m | 26.2, CH2 | 2.39 d (10.0); 2.37 dd (10.0, 2.0) | 24.8, CH2 | 2.30 | 24.0 |
| 10 | 2.47 s | 48.9, CH2 | 2.89 d (16.0); 2.76 d (16.0e) | 46.4f, CH2 | 2.84 d (16.0); 2.70 d (16.0g) | 46.8, CH2 | 2.80–2.73 (two doublet, J = 15.1) | 47.0 |
| 11 | 208.4, C | 207.9, C | 208.0, C | 207.0 | ||||
| 12 | 2.20 s | 32.3, CH3 | 2.16 s | 33.0, CH3 | 2.16 s | 32.9, CH3 | 2.09 s | 32.2 |
| 13 | 0.99 d (7.2) | 16.6, CH3 | 1.03 d (7.6) | 15.7, CH3 | 1.01 d (7.6) | 15.7, CH3 | 0.96 d (7.7) | 15.9 |
| 14 | 0.91 s | 15.0, CH3 | 1.23 s | 18.6, CH3 | 1.18 s | 18.0, CH3 | 1.12, s | 17.2 |
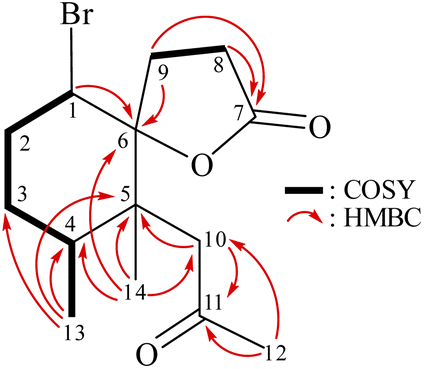
From the 1H–1H COSY spectrum (Fig. 2), the data suggested that it was possible to differentiate between the separate spin systems of H-1/H2-2/H2-3/H-4/H3-13 and H2-8/H2-9. Together with the key HMBC correlations of 1, such as H-4, H2-9, H2-10, H3-13, H3-14/C-5; H2-2, H-8, H-9, H2-10, H3-14/C-6; H2-8, H2-9/C-7; and H2-10, H3-12/C-11, these data confirmed the main carbon skeleton of 1. The HMBC correlations from H3-12/C-10, C-11; H3-13/C-3, C-4, C-5; and H3-14/C-4, C-5, C-6, C-10 indicated that CH3-12, CH3-13, and CH3-14 were placed at C-11, C-4, and C-5, respectively (Fig. 2).
 | ||
| Fig. 2 Key COSY and HMBC correlations of 1. | ||
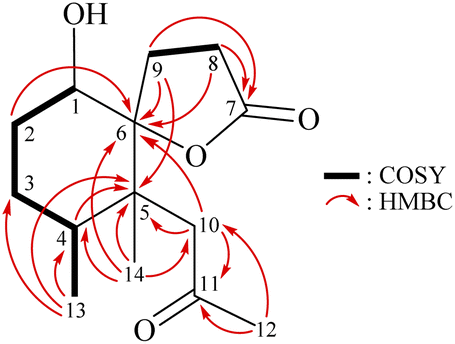
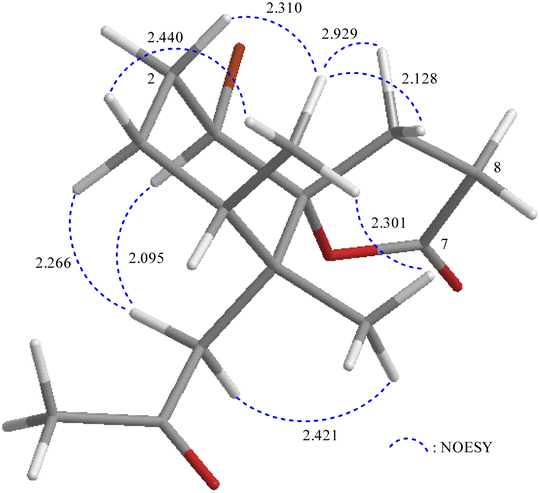
The relative stereochemistry of 1 was deduced mainly from the NOE interactions in the NOESY experiment, Chem3D Model, and vicinal 1H–1H coupling constant analysis. In accordance with convention, when analyzing the stereochemistry of 1, the tertiary C-14 methyl group at C-5 was assigned to the β-face, anchoring stereochemical analysis. In the NOESY experiment (Fig. 3), the correlations of H3-14 with H-1 and H2-9 showed that these protons were positioned on the same face of the molecule, and therefore they were assigned as β-protons. One of the methylene protons at C-3 (δH 1.41) exhibited a correlation with H3-14, leading to its assignment as H-3β, while the other was denoted as H-3α (δH 1.61). The correlation between H-3β and H3-13 reflected the β-orientation of the CH3-13 group at C-4. The configuration at the cyclohexane ring in 1 is worthy of comment. H-1 was found to exhibit correlations with H3-14, as well as coupling between H-1 and H2-2 (J = 11.6, 4.8 Hz), indicating that both H-1 and C-14 methyl at C-5 should be oriented at β-axial positions. Therefore, based on the above findings, the configurations of the stereogenic carbons of 1 were determined as (1S*, 4S*, 5R*, 6R*).
 | ||
| Fig. 3 Stereo-view of 1 (generated by computer modeling) and calculated distances (Å) between selected protons with key NOESY correlations. | ||
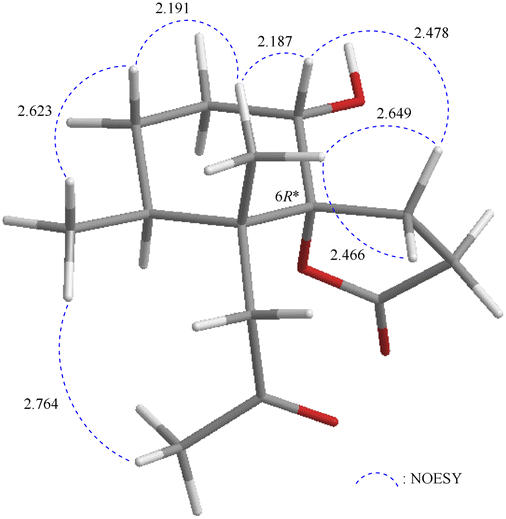
It is very interesting to note that the structure of 1 as we presented herein had been reported and named epi-pathylactone A, a synthetic product by Coelho and Diaz in 2002.16 Although these two compounds possessed the same relative configurations (1S*, 4S*, 5R*, 6S*) (Fig. 3 and 4), the different chemical shifts and coupling constants of H-1 in 1 (δH 3.70, 1H, dd, J = 11.6, 4.8 Hz) and epi-pathylactone A (δH 4.27, 1H, J = 4.2, 2.9 Hz)16 demonstrated that H-1 had significantly different dihedral angles with H-2α and H-2β in both compounds, respectively. Furthermore, the 1H-NMR spectra of compound 1 at temperatures of 0, 25, and 50 °C were also measured to discover the 1H chemical shift changes in those spectra. However, the critical proton NMR signal on C-1 (δH 3.70) in the different temperature experiments was quite similar (Fig. S27–S29†). Therefore, compound 1 was proposed as a new conformational isomer and did not undergo conformational interconversion after heat.
 | ||
| Fig. 4 Stereo-view of epi-pathylactone A (generated by computer modeling). | ||
Based on the Newman projection analysis, H-1 in 1 expressed anti with H-2α and gauch with H-2β; H-1 in epi-pathylactone A showed gauch with the methylene protons on C-2 (Fig. 5), indicating H-1 should be in a β-axial position in 1 and β-equatorial in epi-pathylactone A. In addition, the conformational variation in the cyclohexane ring led to the structural difference in those compounds, resulting in the absolute configuration of 1 being hard to discuss by the reference comparison.
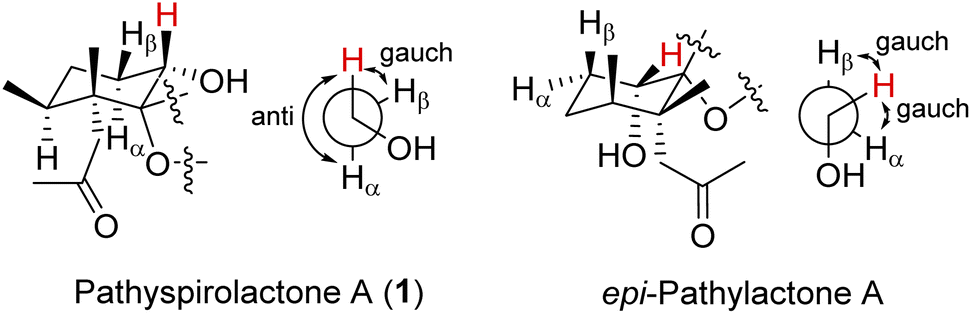 | ||
| Fig. 5 Newman projection of H-1 in 1 and epi-pathylactone A. | ||
For solving the absolute stereochemistry issue of compound 1, the DP4+ analysis was selective for double-checking the configuration of position C-1, which was the most confusing location for the type of compounds. The structures of 1-1S*, 4S*, 5R*, 6R* (diastereomer 1) and 1-1R*, 4S*, 5R*, 6R* (diastereomer 2) were computed GIAO-NMR data by Gaussian 09, and the calculated results were analysis by DP4+ (Fig. S11†). The DP4+ analysis results of 1-1S*, 4S*, 5R*, 6R* displayed the match ratio 99.94%, 99.98%, and 100.00% in sDP4+ (all data), uDP4+ (all data), and DP4+ (all data), respectively (Table S1†). Furthermore, the possible configurations of 1-1S, 4S, 5R, 6R and 1-1R, 4R, 5S, 6S were input into spartan’16 and Gaussian 09 software for calculating conformational search, structure optimization, and specific optical rotation (SOR) value. As a result, the calculated SOR value of 1-1S, 4S, 5R, 6R (66) was consistent with the experiment result of 1 (positive) (Table S2†). This is the first to clarify the absolute configuration of these norsesquiterpenoid -type compounds by in silico method and expounded on the differences in 1D-NMR data of the cyclohexane ring conformational variants.
Pathyspirolactone B (2) was isolated as an amorphous powder. ESIMS showed a pair of ion peaks at m/z 339/341 ([M + Na]+/[M + 2 + Na]+) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), with the relative intensity indicative of a bromide substituent. NMR data coupled with the [M + Na]+ peak in HRESIMS at m/z 339.05646 suggested a molecular formula C14H21BrO3 (calcd for C14H21BrO3 + Na, 339.05663), which indicated four degrees of unsaturation. IR absorptions at νmax 1777 and 1712 cm−1 suggested the presence of γ-lactone and ketonic groups. From the 1H and 13C NMR spectroscopic data, in combination with the DEPT spectrum (Table 1), two carbonyl resonances at δC 207.9 and 175.7 confirmed the presence of ketonic and γ-lactone moieties. So, from the NMR data, two degrees of unsaturation were accounted for, and 2 must be a bicyclic compound.
:1), with the relative intensity indicative of a bromide substituent. NMR data coupled with the [M + Na]+ peak in HRESIMS at m/z 339.05646 suggested a molecular formula C14H21BrO3 (calcd for C14H21BrO3 + Na, 339.05663), which indicated four degrees of unsaturation. IR absorptions at νmax 1777 and 1712 cm−1 suggested the presence of γ-lactone and ketonic groups. From the 1H and 13C NMR spectroscopic data, in combination with the DEPT spectrum (Table 1), two carbonyl resonances at δC 207.9 and 175.7 confirmed the presence of ketonic and γ-lactone moieties. So, from the NMR data, two degrees of unsaturation were accounted for, and 2 must be a bicyclic compound.
Due to electronegativity effects, the methine unit at δC 58.2 was more shielded than expected for oxygenated carbon and was correlated to the methine proton at δH 4.59 in the HSQC spectrum, proving the attachment of a bromide atom at C-1. These data, together with the 1H–1H COSY and HMBC correlations, established the molecular framework of 2 (Fig. 6). The NMR data of 2 were found to be close to those of a known halogenated norsesquiterpenoid, napalilactone (3),17 which was also isolated in this study (Fig. 1), except for replacing a chlorine atom at C-1 in 3 (δH 4.36, 1H, dd, J = 10.4, 4.4 Hz/δC 63.8, CH-1) (Fig. S25 and S26†) with a bromine atom in 2 (δH 4.59, 1H, dd, J = 12.0, 4.4 Hz 4.0 Hz/δC 58.2, CH-1). To the best of our knowledge, compound 2 is the first bromine-containing norsesquiterpenoid reported from octocorals.
 | ||
| Fig. 6 Key COSY and HMBC correlations of 2. | ||
The relative configuration of 2 was elucidated from the interactions observed in a NOESY experiment. Furthermore, it was found to be compatible with that of 2 offered by computer modeling (Fig. 7) and that obtained from vicinal proton coupling constant analysis. In the NOESY spectrum of 2, H-1 showed a correlation with one proton of CH2-10 (δH 2.76) and a large coupling constant with H-2β (J = 12.0 Hz), indicating an α-axial orientation of H-1. The methyl proton H3-14 exhibited correlations with H3-13 and H2-9 but without H2-10, revealing the β-orientations of Me-13, Me-14, and C-9 methylene at C-4, C-5, and C-7, respectively. Moreover, 2-1R*, 4S*, 5R*, 6R* (diastereomer 1) and 2-1S*, 4S*, 5R*, 6R* (diastereomer 2) were further submitted into Gaussian 09 for computed GIAO-NMR data for DP4+ analysis. The analysis results of 2-1R*, 4S*, 5R*, 6R* exhibited a 100% matched ratio with the experimental data of 2 in sDP4+ (all data), uDP4+ (all data), and DP4+ (all data) (Table S1†). Consequently, the relative configuration of 2 was elucidated to be 1R*, 4S*, 5R*, 6R*.
 | ||
| Fig. 7 Stereo-view of 2 (generated by computer modeling) and calculated distances (Å) between selected protons with key NOESY correlations. | ||
Furthermore, the SOR was used for determining the absolute configuration of 2. The calculated SOR of 2-1R, 4S, 5R, 6R and 2-1S, 4R, 5S, 6S exhibited a positive (12) and a negative (−12) value, respectively (Table S1†). The experiment SOR data of 2 (positive) was matched with the 2-1R, 4S, 5R, 6R. However, as the similar issue of 1, the absolute configuration of C-1 cast doubt on 2.
Moreover, the absolute configuration of napalilactone (3) was fully established by a single-crystal X-ray diffraction analysis with the Flack parameter x = −0.04(4).18,19 The computer-generated ORTEP diagram (Fig. 8) showed the absolute configuration of stereogenic centers of 3 were 1R, 4S, 5R, 6R. Based on the principles of biogenetics and the above DP4+ analysis, pathyspirolactone B (2) can be verified as the same absolute configuration as 3.
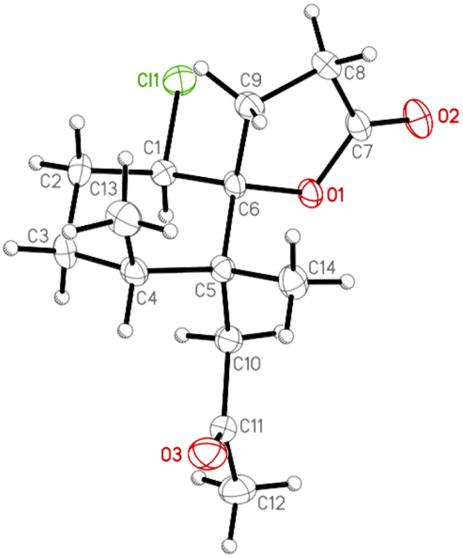 | ||
| Fig. 8 The computer-generated ORTEP diagram of 3. | ||
Based on the past reports, Paralemnalia spp. showed a promising anti-inflammatory effect and cytotoxic activity.7,8,10,14 Therefore, in in vitro anti-inflammatory activity tests, upregulation of pro-inflammatory inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) protein expression in LPS-stimulated RAW 264.7 macrophage cells were evaluated using immunoblot analysis. At a concentration of 10 μM, norsesquiterpenoids 1 and 2 were found to be inactive to reduce the level of iNOS and COX-2 in relation to control cells stimulated with LPS only. Using trypan blue staining to measure the cytotoxic effects of the compounds, it was observed that 1 and 2 did not induce cytotoxicity in RAW 264.7 macrophage cells.
3 Conclusions
In summary, we obtained three norsesquiterpenoids, including pathyspirolactones A (1) and B (2), and napalilactone (3) from octocoral P. thyrsoides. We further explored the conformation of the cyclohexane ring moiety to resolve the stereochemical confusion of those of norsesquiterpenoid compounds. Moreover, we demonstrated the absolute configurations of pathyspirolactones based on analyzing spectroscopic data, specific optical rotation, DP4+ computation, and X-ray diffraction. However, because the screening platforms were limited and lots of material were consumed in physical and spectral experiments, the other possible bioactivities for the new interesting natural substances will not be assayed at this stage.4 Experimental
4.1 General experimental procedures
Optical rotation values were measured using a JASCO P-1010 digital polarimeter. IR spectra were obtained with a Thermo Scientific Nicolet iS5 FT-IR spectrophotometer. NMR spectra were recorded on a 400 MHz Jeol ECZ NMR spectrometer using the residual CHCl3 (δH 7.26 ppm) and CDCl3 signals (δC 77.0 ppm) as internal standards for 1H and 13C NMR, respectively; coupling constants (J) are presented in Hz. ESIMS and HRESIMS were recorded using a Bruker 7 Tesla solariX FTMS system. Column chromatography was carried out with silica gel (230–400 mesh, Merck). TLC was performed on plates precoated with silica gel 60 F254 (Merck) and RP-18W/UV254 (0.15 mm-thick, Macherey-Nagel), then sprayed with 20% H2SO4 solution followed by heating to visualize the spots. NP-HPLC was performed using a system comprised of a Hitachi L-5110 pump and a Rheodyne 7725i injection port with a normal-phase column (Galaksil® EF-SiO2, 5 μm 120 Å, S/N E08210401; Galak Co., Wuxi, CN). RP-HPLC was performed using a system comprised of a Hitachi L-2130 pump, a Hitachi L-2455 photodiode array detector, and a Rheodyne 7725i injection port with a reverse-phase column (Supelco, Ascentis® C18, 581343-U, 250 mm × 10 mm, 5 μm).4.2 Animal material
Specimens of Paralemnalia thyrsoides (Ehrenberg, 1834) used for this study were collected from the culturing tank in the NMMBA in June 2021. A voucher specimen was deposited in the NMMBA (voucher no.: NMMBA-TW-SC-2021-902). Identification of the species of this organism was performed by comparison as described in previous studies.20–224.3 Extraction and isolation
The freeze-dried and sliced bodies of the coral specimen (wet/dry weight = 2031/299 g) were extracted by 95% EtOH to yield a crude extract I (31.7 g) and continued to be extracted by the mixture of MeOH/CH2Cl2 (1:1) to give an extract II (29.5 g). Then extracts I and II were partitioned with EtOAc and H2O to obtain the EtOAc-soluble layers A (8.9 g) and B (1.8 g), respectively. The EtOAc layers A and B were then combined, placed in a silica column, and eluted by hexanes/EtOAc (pure hexanes to pure EtOAc, stepwise) to yield ten fractions A–J. Fraction G (121.3 mg) was further separated by normal-phase HPLC on Galaksil® EF-SiO2 column with a mixture of n-hexane and acetone (65:35) at a rate of 5 mL min−1 to give four subfractions (G1–4). G2 (63.2 mg) was subjected to the normal-phase HPLC system with an isocratic solvent system of n-hexane and ethyl acetate (80:20, 5 mL min−1) to yield 12 fractions (G2A–L). G2L (48.6 mg) was then separated by the normal-phase HPLC system using a mixture of n-hexane and acetone (80:20, 5 mL min−1) to obtain 15 subfractions (G2L1–15). G2L10 (10.9 mg) was further purified by reverse-phase HPLC on Ascentis® C18 column with an isocratic solvent system of MeOH and H2O (55:45, 5 mL min−1) to obtain compound 1 (0.2 mg). Fraction D (710.1 mg) was subjected to the normal-phase HPLC system with a mixture of n-hexane and ethyl acetate (85:15, 5 mL min−1) to give nine subfractions (D1–9). D7 (18.2 mg) was further purified by the reverse-phase HPLC using a mixture solvent system of MeOH:H2O (50:50, 5 mL min−1) to obtain compounds 2 (0.3 mg) and 3 (11.6 mg).
4.4 Structural characterization of undescribed compounds
4.5 Single-crystal X-ray crystallography of napalilactone (3)
Suitable colorless prisms of napalilactone (3) were obtained from CDCl3. The crystal (0.599 × 0.512 × 0.081 mm3) was identified as being of the orthorhombic system, space group P212121 (#19), with a = 8.2367(2) Å, b = 9.9002(3) Å, c = 17.2237(5) Å, V = 1404.51(7) Å3, Z = 4, Dcalcd = 1.290 Mg m−3, λ (Mo Kα) = 0.71073 Å. Intensity data were obtained on a crystal diffractometer (Bruker, model: D8 Venture) up to θmax of 30.0°. All measurement data of 39483 reflections were collected, of which 4100 were independent. The structure was solved by direct methods and refined by a full-matrix least-squares on the F2 procedure.23 The refined structural model converged to a final R1 = 0.0460; wR2 = 0.1060 for 3494 observed reflections [I > 2σ(I)] and 172 variable parameters; and the absolute configuration was determined from the Flack parameter x = −0.04(4).18,19 Crystallographic data for the structure of napalilactone (3) were submitted to the Cambridge Crystallographic Data Center (CCDC) with supplementary publication number CCDC 2190441 (data can be obtained from the CCDC website at https://www.ccdc.cam.ac.uk/conts/retrieving.html).
4.6 In silico calculations
The structure was optimized the minimized energy in MM2 level and outputted as an xyz file. The file was submitted into spartan'16 software (Wavefunction Inc.; Irvine, CA, USA) at MMFF94 to generate conformational search results. The output data were imported into the Gaussian 09 software (Gaussian Inc.; Wallingford, CT, USA) and optimized using the time-dependent density functional theory (TDDFT) methodology at the B3LYP/6-31G* level in the gas phase, and at the B3LYP/6-31(d) levels in the solvent phase for SOR calculation, and the GIAO-DFT at the PCM/mpw1pw91/6-311 + g(d,p) level in the solvent phase for GIAO-NMR DP4+ analysis. All the computed SOR and NMR results were averaged by the proportion of each conformer.24,25Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Hsiao-Ching Yu and Chao-Lien Ho of the High Valued Instrument Center, National Sun Yat-sen University, for obtaining the mass (MS000600) and NMR (NMR 001100) spectra (MOST 111-2731-M-110-001). This work was mainly funded by grants from the National Museum of Marine Biology and Aquarium and the National Science and Technology Council (MOST 109-2320-B-291-001-MY3 and 111-2320-B-291-001 (1/3)), Taiwan, awarded to P.-J. S. All funding is gratefully acknowledged.Notes and references
- D. Daloze, J. C. Braekman, P. Georget and B. Tursch, Chemical studies of marine invertebrates. XXII. (1) Two novel sesquiterpenes from soft corals of the genera Lemnalia and Paralemnalia (Coelenterata, Octocorallia, Alcyonacea), Bull. Soc. Chim. Belg., 1977, 86, 47–54 CrossRef CAS.
- B. F. Bowden, J. C. Coll and S. J. Mitchell, Studies of Australian soft corals. XIX. Two new sesquiterpenes with the nardosinane skeleton from a Paralemnalia species, Aust. J. Chem., 1980, 33, 885–890 CrossRef CAS.
- J. Y. Su, Y. L. Zhong and L. M. Zeng, Two new sesquiterpenoids from the soft coral Paralemnalia thyrsoides, J. Nat. Prod., 1993, 56, 288–291 CrossRef CAS.
- H. C. Huang, C. H. Chao, J. H. Su, C. H. Hsu, S. P. Chen, Y. H. Kuo and J. H. Sheu, Neolemnane-type sesquiterpenoids from a Formosan soft coral Paralemnalia thyrsoides, Chem. Pharm. Bull., 2007, 55, 876–880 CrossRef CAS PubMed.
- A. Bishara, D. Yeffet, M. Sisso, G. Shmul, M. Schleyer, Y. Benayahu, A. Rudi and Y. Kashman, Nardosinanols A−I and lemnafricanol, sesquiterpenes from several soft corals, Lemnalia sp., Paralemnalia clavata, Lemnalia africana, and Rhytisma fulvum fulvum, J. Nat. Prod., 2008, 71, 375–380 CrossRef CAS PubMed.
- G. H. Wang, H. C. Huang, J. H. Su, Y. C. Wu, J. H. Sheu and J. -P. Paralemnolins, new sesquiterpenoids from the soft coral Paralemnalia thyrsoide, Chem. Pharm. Bull., 2010, 58, 30–33 CrossRef CAS PubMed.
- C. Y. Huang, J. H. Su, B. W. Chen, Z. H. Wen, C. H. Hsu, C. F. Dai, J. H. Sheu and P. J. Sung, Nardosinane-type sesquiterpenoids from the Formosan soft coral Paralemnalia thyrsoides, Mar. Drugs, 2011, 9, 1543–1553 CrossRef CAS PubMed.
- Y. J. Tseng, Y. S. Lee, S. K. Wang, J. H. Sheu, C. Y. Duh and A. -D. Parathyrsoidins, four new sesquiterpenoids from the soft coral Paralemnalia thyrsoides, Mar. Drugs, 2013, 11, 2501–2509 CrossRef PubMed.
- X. Han, Q. Wang, X. Luo, X. Tang, Z. Wang, D. Zhang, S. Cao, P. Li, G. Li and A. -C. Lemnalemnanes, three rare rearranged sesquiterpenoids from the soft corals Paralemnalia thyrsoides and Lemnalia sp, Org. Lett., 2022, 24, 11–15 CrossRef CAS PubMed.
- A. Alassass, M. Abubakr, W. Alarif, S. E. Ayyad and A. E. Mohammed, Anti-inflammatory, antioxidant, cytotoxic activities, and sesquiterpenoid contents of Paralemnalia thyrsoides, Pharmacogn. Mag., 2022, 18, 188–192 CrossRef CAS.
- H. C. Huang, C. H. Chao, J. H. Liao, M. Y. Chiang, C. F. Dai, Y. C. Wu and J. H. Sheu, A novel chlorinated norsesquiterpenoid and two related new metabolites from the soft coral Paralemnalia thyrsoides, Tetrahedron Lett., 2005, 46, 7711–7714 CrossRef CAS.
- R. R. Izac, P. Schneider, M. Swain and W. Fenical, New nor-sesquiterpenoids of apparent nardosinane origin from the pacific soft-coral Paralemnalia thyrsoides, Tetrahedron Lett., 1982, 23, 817–820 CrossRef CAS.
- J. Y. Su, Y. L. Zhong, J. Q. Wu and L. M. Zeng, A new nor-sesquiterpenoid from the soft coral Paralemnalia thyrsoids, Chin. Chem. Lett., 1991, 2, 785–786 CAS.
- Z. J. Zhang, W. F. Chen, B. R. Peng, Z. H. Wen and P. J. Sung, (+)-Pathylactone A, a new natural norsesquiterpenoid from the octocoral Paralemnalia thyrsoides, Nat. Prod. Commun., 2018, 13, 5–7 Search PubMed.
- Y. C. Chang, C. C. Chiang, Y. S. Chang, J. J. Chen, W. H. Wang, L. S. Fang, H. M. Chung, T. L. Hwang and P. J. Sung, Novel caryophyllane-related sesquiterpenoids with anti-inflammatory activity from Rumphella antipathes (Linnaeus, 1758), Mar. Drugs, 2020, 18, 554 CrossRef CAS PubMed.
- F. Coelho and G. Diaz, Studies on the synthesis of (±)-pathylactone A, a norsesquiterpene lactone isolated from marine sources, Tetrahedron, 2002, 58, 1647–1656 CrossRef CAS.
- J. R. Carney, A. T. Pham, W. Y. Yoshida and P. J. Scheuer, Napalilactone, a new halogenated norsesquiterpenoid from the soft coral Lemnalia africana, Tetrahedron Lett., 1992, 33, 7115–7118 CrossRef CAS.
- H. D. Flack, On enantiomorph-polarity estimation, Acta Crystallogr., 1983, A39, 876–881 CrossRef CAS.
- H. D. Flack and G. Bernardinelli, Absolute structure and absolute configuration, Acta Crystallogr., 1999, A55, 908–915 CrossRef CAS.
- C. F. Dai and C. H. Chin, Octocorallia Fauna of Kenting National Park. Kenting National Park Headquarters: Kenting, Pingtung, Taiwan, 2019, pp. 344–345 Search PubMed.
- C. F. Dai, Ocean Center, National Taiwan University, Taipei, Taiwan, 2021, pp. 436–437.Octocorallia Fauna of Taiwan Search PubMed.
- C. F. Dai, Corals of Taiwan: Octocorallia, Owl Publishing House Co. Ltd., Taipei, Taiwan, 2022, 2, p. 267 Search PubMed.
- G. M. Sheldrick, SHELXT - Integrated space-group and crystal-structure determination, Acta Crystallogr., 2015, A71, 3–8 CrossRef PubMed.
- N. Grimblat, M. M. Zanardi and A. M. Sarotti, Beyond DP4: an improved probability for the stereochemical assignment of isomeric compounds using quantum chemical calculations of NMR shifts, J. Org. Chem., 2015, 80, 12526–12534 CrossRef CAS.
- H. C. Hu, Y. H. Tsai, Y. C. Chuang, K. H. Lai, Y. M. Hsu, T. L. Hwang, C. C. Lin, F. Fülöp, Y. C. Wu, S. Y. Yu, Y. T. Kuo and F. R. Chang, Estrogenic and anti-neutrophilic inflammatory phenanthrenes from Juncus effusus L, Nat. Prod. Res., 2022, 36, 3043–3053 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: HRESI-MS, 1D and 2D-NMR spectra of 1 and 2; ESI-MS and 1D-NMR spectra of 3; experimental and calculated SOR values of 1 and 2; X-ray crystallo-graphic data of 3; DP4+ analysis of 1 and 2. CCDC 2190441. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2ra05015c |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |