
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fe(III)-catalysed selective C–N bond cleavage of N-phenylamides by an electrochemical method†
Yiwen Xua,
Yang Longa,
Runyou Yea,
Qiang Lia,
Fang Ke b and
Xiangge Zhou*a
b and
Xiangge Zhou*a
aCollege of Chemistry, Sichuan University, Wangjiang Road 29, Chengdu 610064, China. E-mail: zhouxiangge@scu.edu.cn
bSchool of Pharmacy, Fujian Provincial Key Laboratory of Natural Medicine Pharmacology, Fujian Medical University, Fuzhou 350004, China. E-mail: kefang@mail.fjmu.edu.cn
First published on 25th August 2022
Abstract
An Fe(III)-catalysed transformation of secondary N-phenyl substituted amides to primary amides by an electrochemical method is developed. Regioselective aryl C–H oxygenation occurs during the reaction, promoting selective C(phenyl)-N bond cleavage to form primary amides in yields of up to 92%.
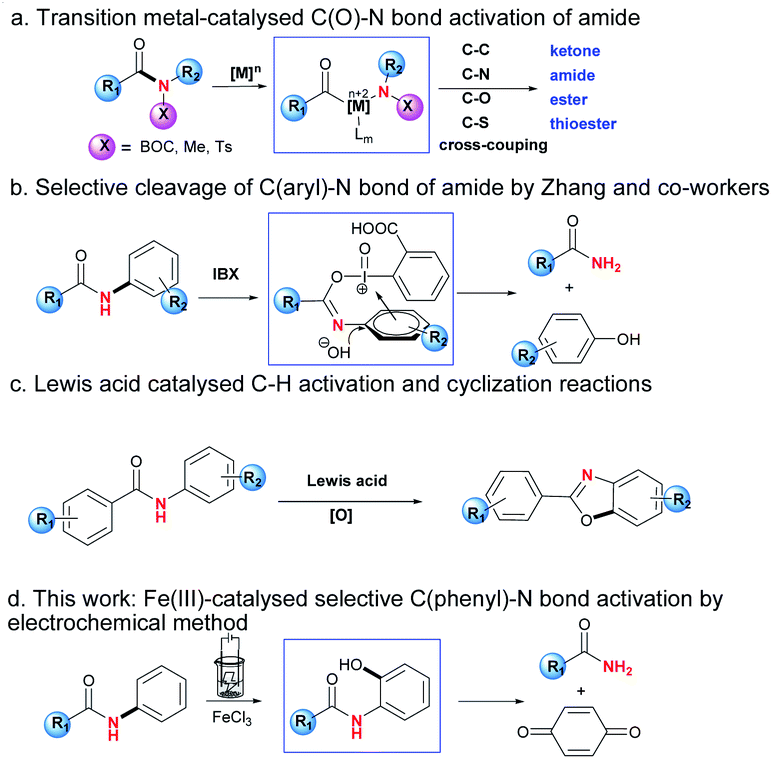
Amido groups are important structural units in organic compounds, which can be widely found in organic molecules, bioactive drugs, polymer functional materials etc.1 The research on modification or derivation of amides has flourished recently. For example, Szostak and co-workers reported C(O)–N bond activation of amides by nickel, palladium and rhodium catalysts, which could be followed by coupling reactions to form a series of carbonyl compounds including ketones, amides, esters and thioesters (Scheme 1a).2 Meanwhile, in the case of N-phenyl substituted amide substrates, which contain two C–N bonds in the same molecule, the selective C(phenyl)–N bond cleavage is scarcely reported compared with that of the C(O)–N bond.3 One of the possible reasons was reported to be the greater conjugation ability of nN → π*aryl than that of nN → π*C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/i_char_e001.gif) O, which might cause the bond energies of the C(phenyl)–N and C(O)–N bonds to be 104 and 95 kcal mol−1 respectively.4 The selective C(phenyl)–N bond cleavage is highly challenging and of interest to academic as well as industrial chemists. Zhang and co-workers unveiled an example of IBX-promoted selective cleavage of the C(phenyl)–N bond, and the usage of a stoichiometric oxidant during the reaction would limit its practical applications, which makes the selective cleavage of the C(phenyl)–N bond challenging (Scheme 1b).5
O, which might cause the bond energies of the C(phenyl)–N and C(O)–N bonds to be 104 and 95 kcal mol−1 respectively.4 The selective C(phenyl)–N bond cleavage is highly challenging and of interest to academic as well as industrial chemists. Zhang and co-workers unveiled an example of IBX-promoted selective cleavage of the C(phenyl)–N bond, and the usage of a stoichiometric oxidant during the reaction would limit its practical applications, which makes the selective cleavage of the C(phenyl)–N bond challenging (Scheme 1b).5
 | ||
| Scheme 1 Strategies for activation of amido C–N bonds. | ||
On the other hand, with the rapid revival of electrochemistry, which is regarded as an environmentally benign and powerful tool for a sustainable alternative to conventional redox approaches, impressive progress has been recently achieved for redox art through an electrochemistry.6 Especially, metal electrocatalytic reaction could serve as an attractive synthetic route to realize reactions that cannot be performed under conventional metal catalysis.7 In these reactions, metal catalyst could be coordinated with substrate to generate stable intermediate under constant current, affording highly value-added products.
Furthermore, Nagasawa and co-workers recently developed a Lewis acid/O2 catalytic system, which realised the synthesis of heterocycles by C–H functionalisation/C–O bond formation protocol (Scheme 1c).8 In these reactions, ground-state-stabilisation of amide was destroyed by strong affinitie of Lewis acid toward oxygen atom of amido group, lowering the resonance effects and thus electronic delocalisation.9
In continuation of our work on transition metal-catalysed inert bond activation and electrochemical work,10 herein is reported the selective C(phenyl)–N bond cleavage by the redox direct-oxidation of metal oxygenation catalysts at the electrode surface, which transformed secondary N-phenylamides to be primary amides (Scheme 1d). This protocol features the following items: cheap FeCl3 as catalyst; regioselective C–H oxygenation assisted chemoselective C(phenyl)–N bond cleavage of secondary phenylamide; avoidance of addition of oxidant; mild reaction conditions including room temperature and without inert atmosphere.
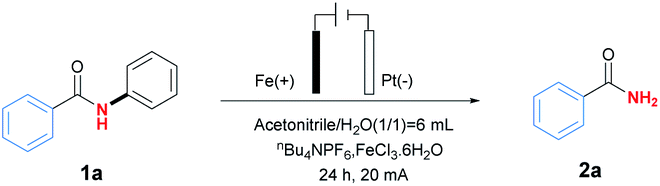
At the beginning of our exploration, N–phenylbenzamide (1a) was chosen as template substrate to identify optimal reaction conditions (for details, see Table S1 in ESI†). As shown in Table 1, the desired primary amide product 2a was obtained in 82% yield after 24 h under 20 mA with Fe plate as anode and Pt net as cathode, together with nBu4NPF6 (0.05 M) as electrolyte in mixture solvent of MeCN (3 mL) and H2O (3 mL) at room temperature in the presence of FeCl3·6H2O (0.2 equiv.) as catalyst (Table 1, entry 1). Control experiments showed that electricity and supporting electrolyte was essential for the reaction, and nBu4NPF6 was found to be more fitful for the reaction than other phase transfer reagents including nBu4NBr and nBu4NClO4 (Table 1, entries 2–4). After screening of different transition metal salts, FeCl3·6H2O was proved to be proper choice compared with ZnCl2, Cu(OTf)2, Fe(OAc)2 and Fe(acac)3 (Table 1, entries 5–8). Meanwhile, solvent seemed to be an important factor to affect the results, and the mixture solvent of MeCN and H2O in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was the best one. Attempts to replace H2O by other protic solvents resulted in inferior yields (Table 1, entries 9–14). Subsequently, the influence of reaction time was tested, and longer reaction time than 24 h seemed to be unfavourable for the reaction, which might be caused by the hydrolysis of product 2a to be benzoic acid monitored by GC/Mass (Table 1, entries 15 and 16). The electrode material sacrificial anode also had an important impact on the reaction (Table 1 entry 17 and 18). At last, it was reported the addition of DABCO (1,4-diaza-bicyclo[2.2.2]octane) as sacrificial reductant would prevent the hydrolysis of product,11 however, it seemed to be unnecessary in this reaction, which would result in inferior yield 30% (Table 1, entry 19).
:1 was the best one. Attempts to replace H2O by other protic solvents resulted in inferior yields (Table 1, entries 9–14). Subsequently, the influence of reaction time was tested, and longer reaction time than 24 h seemed to be unfavourable for the reaction, which might be caused by the hydrolysis of product 2a to be benzoic acid monitored by GC/Mass (Table 1, entries 15 and 16). The electrode material sacrificial anode also had an important impact on the reaction (Table 1 entry 17 and 18). At last, it was reported the addition of DABCO (1,4-diaza-bicyclo[2.2.2]octane) as sacrificial reductant would prevent the hydrolysis of product,11 however, it seemed to be unnecessary in this reaction, which would result in inferior yield 30% (Table 1, entry 19).
|
||
|---|---|---|
| Entry | Changes from standard conditions | Yield (%)b |
| a Standard conditions: 1a (0.6 mmol), H2O (3 mL), MeCN (3 mL), nBu4NPF6 (0.05 M), FeCl3·6H2O (0.2 equiv.), Pt net and Fe plate electrodes (1.0 × 1.0 cm2), 20 mA, undivided cell, room temperature, air, 24 h.b Yields were determined using GC analysis with 1H-benzo[d]imidazole as internal standard.c Isolated yield. | ||
| 1 | None | 89/82c |
| 2 | Without electricity | N.D |
| 3 | nBu4NBr instead of nBu4NPF6 | 10 |
| 4 | nBu4NClO4 instead of nBu4NPF6 | 18 |
| 5 | ZnCl2 instead of FeCl3·6H2O | 58 |
| 6 | Cu(OTf)2 instead of FeCl3·6H2O | 40 |
| 7 | Fe(OAc)2 instead of FeCl3·6H2O | 44 |
| 8 | Fe(acac)3 instead of FeCl3·6H2O | 63 |
| 9 | Without MeCN | Trace |
| 10 | Without H2O | N.D. |
| 11 | Ethylene carbonate instead of MeCN | 18 |
| 12 | DMF instead of MeCN | 11 |
| 13 | Methanol instead of H2O | 32 |
| 14 | Acetic acid instead of H2O | Trace |
| 15 | 14 h instead of 24 h | 24 |
| 16 | 36 h instead of 24 h | 62 |
| 17 | Pt(+) instead of Fe(+) | 60 |
| 18 | Ni(+) instead of Fe(+) | 87 |
| 19 | DABCO as additives | 30 |
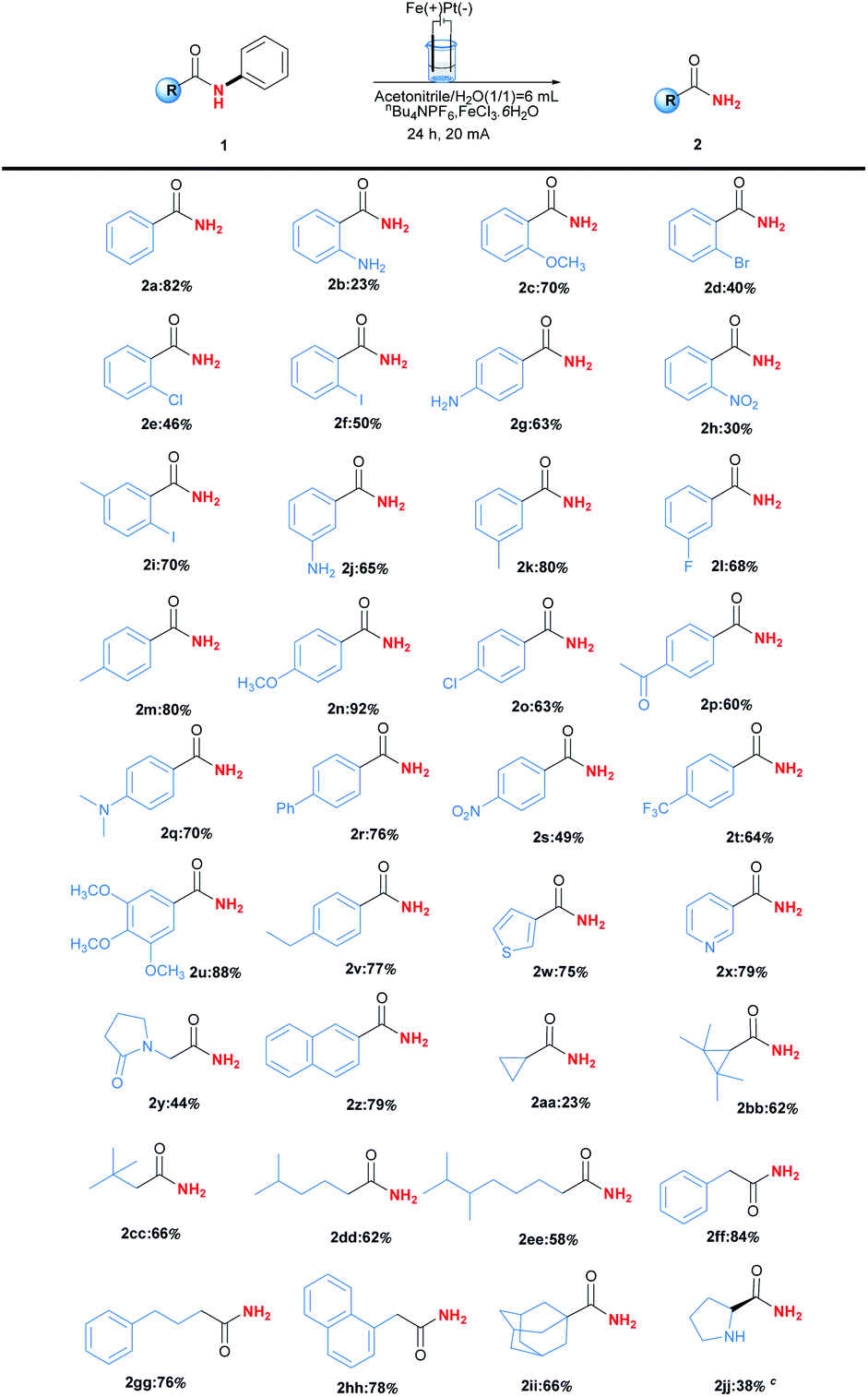
With the optimised reaction conditions in hand, the substrate scope was then explored as shown in Table 2. In general, this transformation was amenable to substrates containing different substituents, and a series of N-phenylbenzamides efficiently underwent C(aryl)–N bond cleavage dephenylation reactions to afford primary amides 2. Steric hindrance played an important role in the reaction. For example, the ortho and para-chloro substituted substrates gave the corresponding products 2e and 2o in yields 46% and 63% respectively. Other halogen substituents including Br and I could also be applied in this reaction and delivered the desired products 2d, 2f and 2i in moderate yields ranging from 40%–70%, providing the potentiality for late-stage functionalisation when combing with traditional cross-coupling strategies. Meanwhile, electron-rich substituents seemed to be beneficial to the reaction, and the best yield 92% was obtained in the case of product 2n containing para-methoxyl substituent. Furthermore, substrates bearing heterocyclic or naphthalene moiety could also be tolerated in this transformation to give the corresponding products, including niacinamide 2w in yield of 79%. More importantly, the aliphatic amides were also applied in this reaction, affording the desired products 2aa-2hh in yields ranging from 23% to 84%. The stable rigid structure of adamantane could undergo this dephenylation reaction to give 66% yields (2ii). Further studies revealed that chirality could be retained under the reaction conditions (2jj).
| a Standard conditions: 1a (0.6 mmol), H2O (3 mL), MeCN (3 mL), nBu4NPF6 (0.05 M), FeCl3·6H2O (0.2 equiv.), Pt net and Fe plate electrodes (1.0 × 1.0 cm2), 20 mA, undivided cell, room temperature, air, 24 h.b Yields were determined using GC analysis with 1H-benzo[d]imidazole as internal standard.c Isolated yield. |
|---|
 |
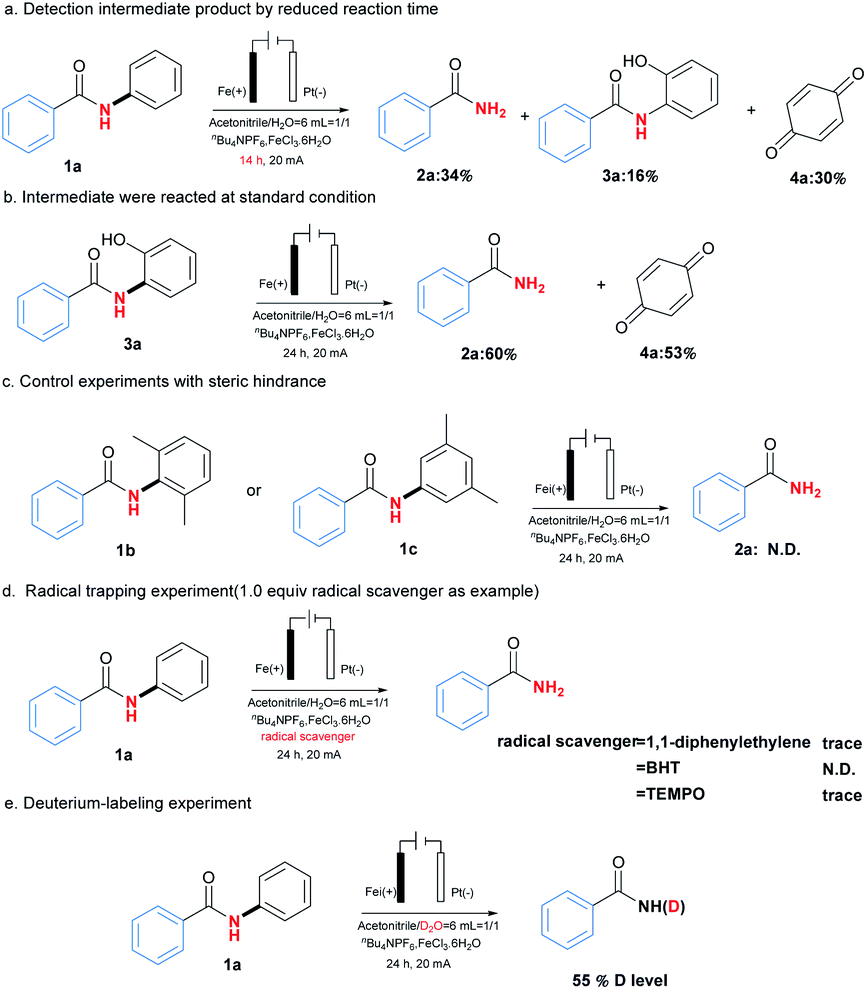
To clarify the possible reaction pathway, a series of experiments were then carried out as shown in Scheme 2. At first, N-(2-hydroxyphenyl)benzamide (3a) and p-benzoquinone (4a) were detected during the reaction, and decrement of reaction time from 24 hours to 14 hours resulted in the formation of 2a, 3a and 4a in yields 34%, 16% and 30%, which indicated 3a would be intermediate during reaction (Scheme 2a). Further experiment by using 3a as substrate also afforded the corresponding product 2a smoothly (Scheme 2b). Meanwhile, when ortho or meta position of N-phenyl group was occupied by substituents, no product could be formed, which suggested the necessity of vacancy of ortho and meta position for C–H oxygenation process (Scheme 2c).
 | ||
| Scheme 2 Control experiments for reaction pathway studies. | ||
Next, the addition of radical scavengers such as 1,1-diphenylethylene, butylated hydroxytoluene (BHT) or 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) would completely inhibit the reaction, suggesting the reaction might involve a radical pathway (Scheme 2d).12 At last, when D2O was used instead of H2O as solvent, 55% of deuterium incorporation was observed, which demonstrated the reaction would involve a hydrolysis process (Scheme 2e).
Meanwhile, the cyclic voltammetric (CV) experiments were carried out as shown in Fig. 1. Curves a, b and c exhibited redox peaks for blank, Fe(II) and Fe(III), which indicated that Fe(I) would not be involved in this reaction. Meanwhile, curve b showed Fe(II)ipa at −0.49 mA, Fe(II)ipc at 0.15 mA and Fe(III)ipa at −0.10 mA, while curve c showed Fe(II)ipa at −0.30 mA, Fe(II)ipc at 0.07 mA and Fe(III)ipa at −0.30 mA, which indicated Fe(II) would participate in the reaction to form product from intermediate 3a.
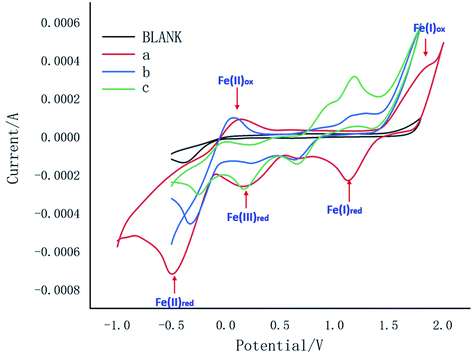 | ||
| Fig. 1 Cyclic voltammograms experiment (Blank: 0.1 M nBu4NPF6, MeCN/H2O = 1/1; a: Blank + FeCl3·6H2O 0.02 mmol; b: Blank + 1a 0.1 mmol + FeCl3·6H2O 0.02 mmo; c: Blank + 3a 0.1 mmol + FeCl3·6H2O 0.02 mmol). | ||
Based on the above experimental results and literature survey,13 a plausible reaction pathway for Fe(III)-catalysed selective C–N bond activation of N-phenyl amides promoted by regioselective C–H oxygenation was proposed (Fig. 2). At first, coordination of Fe(III) with 1a14 would activate amide carbonyl to lose electron efficiently at anode, forming free radical A.15 Meanwhile, solvent H2O would be oxidised to be radical ˙OH in the reaction. Next, the radical coupling reaction between A and ˙OH would form intermediate 3a.16 Then, 3a would be reacted with iron salt to form complex B,9(d) which was further transformed to be intermediates C and D after a single electron transfer (SET) process and a radical reaction with ˙OH.17 At last, reduction of D resulted in the generation of intermediate E at cathode, which was finally converted to be product 2a by a hydrogen reduction process.18
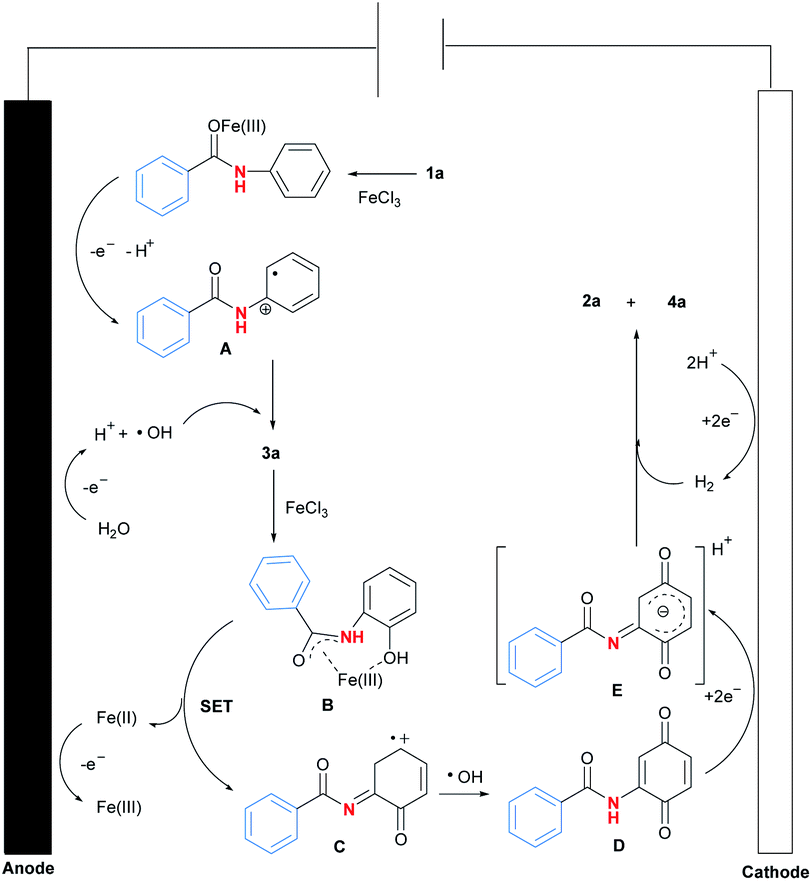 | ||
| Fig. 2 Possible reaction pathway. | ||
The applications of this transformation were then carried out. As shown in Scheme 3a, telmisartan derivative could be easily transformed to be free amide 2kk in 42% yield, which could broaden N-phenylamides' further applications. Moreover, this methodology could be applied for the synthesis of trimethobenzamide 5,19 a powerful antihistamine, starting from 3,4,5-trimethoxybenzamide (2u) (Scheme 3b). These results indicated this transformation expanded the type of synthetic methodologies with phenyl groups as removable protecting groups under mild reaction conditions. At last, the gram-scale synthesis of 1a (6 mmol) with 60 mA under the optimal reaction conditions was performed, furnishing the desired product 2a in 72% yield (Scheme 4).
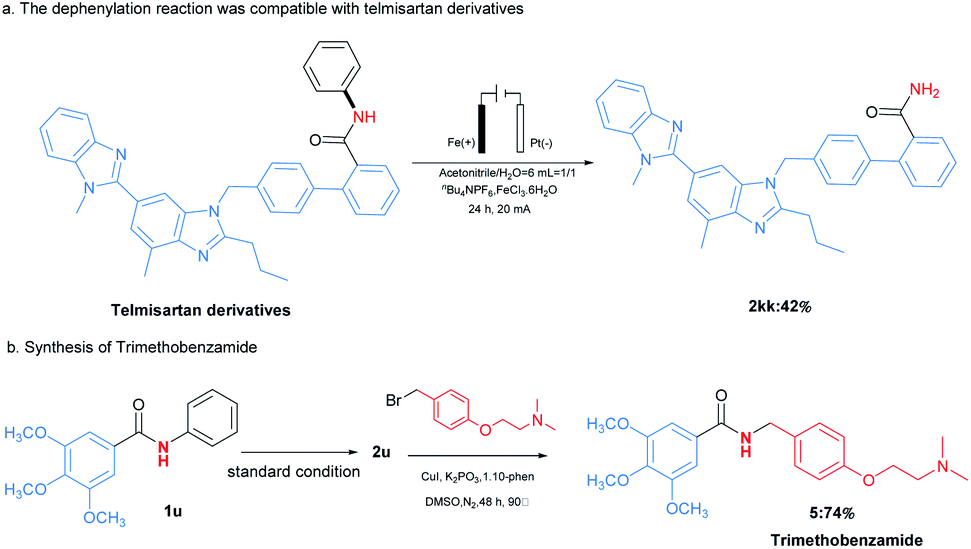 | ||
| Scheme 3 Applications in the synthesis of selected compounds. | ||
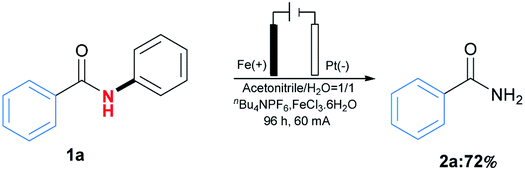 | ||
| Scheme 4 Gram-scale experiment. | ||
In summary, we have developed an iron-catalysed, electrochemical oxidation mediated strategy for selective C–N activation dephenylation of secondary N-phenylamide. This transformation exhibits an ample scope, high yield, and broad functional group tolerance. A more distinct feature of this work pertains to its applicability to selective dephenylation of N-phenylamides without pre-activated amides. The development of new reactions based on this strategy is currently in progress.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Natural Science Foundation of China (Grant no. 22071159) for financial support. We also acknowledge Comprehensive Training Platform of Specialised Laboratory, College of Chemistry, Sichuan University, for HRMS analysis.Notes and references
- (a) Y. Qin, G. Li, T. Qi and H. Huang, Mater. Chem. Front., 2020, 4, 1554 RSC; (b) S. Mahesh, K.-C. Tang and M. Raj, Molecules, 2018, 23, 2615 CrossRef PubMed; (c) P. Dydio, D. Lichosyt and J. Jurczak, Chem. Soc. Rev., 2011, 40, 2971 RSC; (d) Z.-T. Li, J.-L. Hou and C. Li, Acc. Chem. Res., 2008, 41, 1343 CrossRef CAS PubMed.
- (a) G. Li and M. Szostak, Chem. Rec., 2020, 20, 649 CrossRef CAS PubMed; (b) G. Li and M. Szostak, Nat. Commun., 2018, 9, 4165 CrossRef PubMed; (c) P. Gao and M. Szostak, Org. Lett., 2020, 22, 6010 CrossRef CAS PubMed; (d) M. M. Rahman and M. Szostak, Org. Lett., 2021, 23, 4818 CrossRef CAS PubMed; (e) G. Meng and M. Szostak, Org. Lett., 2016, 18, 796 CrossRef CAS PubMed; (f) P. Lei, G. Meng, S. Shi, Y. Ling, J. An, R. Szostak and M. Szostak, Chem. Sci., 2017, 8, 6525 RSC; (g) T. Zhou, G. Li, S. P. Nolan and M. Szostak, Org. Lett., 2019, 21, 3304 CrossRef CAS PubMed.
- (a) M. Tobisu, K. Nakamura and N. Chatani, J. Am. Chem. Soc., 2014, 136, 5587 CrossRef CAS PubMed; (b) M. Wang, X. Zhang, Y.-X. Zhuang, Y.-H. He and T.-P. Loh, J. Am. Chem. Soc., 2015, 137, 1341 CrossRef CAS PubMed; (c) Y. Lei, A. D. Wrobleski, J. E. Golden, D. R. Powell and J. Aube, J. Am. Chem. Soc., 2005, 127, 4552 CrossRef CAS PubMed.
- (a) R. T. Sanderson, Polar Covalence; Academic Press: New York, 1983 Search PubMed; (b) R. T. Sanderson, Chemical Bonds and Bond Energy; Academic Press: New York, 1976 Search PubMed.
- (a) Z. Zhang, D. Zheng, Y. Wan, G. Zhang, J. Bi, Q. Liu, T. Liu and L. Shi, J. Org. Chem., 2018, 83, 1369 CrossRef CAS PubMed; (b) Z. Zhang, X. Li, M. Song, Y. Wang, D. Zheng, G. Zhang and G. Chen, J. Org. Chem., 2019, 84, 12792 CrossRef CAS PubMed.
- (a) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230 CrossRef CAS PubMed; (b) C. Kingston, M. D. Palkowitz, Y. Takahira, J. C. Vantourout, B. K. Peters, Y. Kawamata and P. S. Baran, Acc. Chem. Res., 2020, 53, 72 CrossRef CAS PubMed.
- (a) P. Gandeepan, L. H. Finger, T. H. Meyer and L. Ackermann, Chem. Soc. Rev., 2020, 49, 4254 RSC; (b) L. Ackermann, Acc. Chem. Res., 2020, 53, 84 CrossRef CAS PubMed; (c) A. Jutand, Chem. Rev., 2008, 108, 2300 CrossRef CAS PubMed.
- (a) S. Ueda and H. Nagasawa, Angew. Chem., Int. Ed., 2008, 47, 6411 CrossRef CAS PubMed; (b) S. K. Alla, P. Sadhu and T. Punniyamurthy, J. Org. Chem., 2014, 79, 7502 CrossRef CAS PubMed; (c) S. Ueda and H. Nagasawa, J. Org. Chem., 2009, 74, 4272 CrossRef CAS PubMed; (d) K. Sahoo and N. Panda, Adv. Synth. Catal., 2022, 364, 1023 CrossRef CAS; (e) T. Gieshoff, A. Kehl, D. Schollmeyer, K. D. Moeller and S. R. Waldvogel, Chem. Commun., 2017, 53, 2974 RSC.
- (a) H. T. Clarke, J. R. Johnson and R. Robinson, Eds., The Chemistry of Penicillin; Princeton University Press: Princeton, 1949 CrossRef; (b) K. Tani and B. M. Stoltz, Nature, 2006, 441, 731 CrossRef CAS PubMed; (c) A. J. Kirby, I. V. Komarov, P. D. Wothers and N. Feeder, Angew. Chem., Int. Ed., 1998, 37, 785 CrossRef CAS.
- (a) Y. Long, Z. Su, Y. Zheng, S. He, J. Zhong, H. Xiang and X. Zhou, ACS Catal., 2020, 10, 3398 CrossRef CAS; (b) F. Ke, Y. Xu, S. Zhu, X. Lin, C. Lin, S. Zhou and H. Su, Green Chem., 2019, 21, 4329 RSC; (c) X. Yao, X. Weng, K. Wang, H. Xiang and X. Zhou, Green Chem., 2018, 20, 2472 RSC.
- X. Zhang, C. Yang, H. Gao, L. Wang, L. Guo and W. Xia, Org. Lett., 2021, 23, 3472 CrossRef CAS PubMed.
- R. D. Little, J. Org. Chem., 2020, 85, 13375 CrossRef CAS PubMed.
- (a) A. Volkov, F. Tinnis, T. Slagbrand, P. Tnllo and H. Adolfsson, Chem. Soc. Rev., 2016, 45, 6685 RSC; (b) C.-A. Wang, M. M. Rahman, E. Bisz, B. Dziuk, R. Szostak and M. Szostak, ACS Catal., 2022, 12, 2426 CrossRef CAS.
- X. Huang, Q. Zhang, J. Lin, K. Harms and E. Meggers, Nat. Catal., 2019, 2, 34 CrossRef CAS.
- V. Pace, W. Holzer and B. Olofsson, Adv. Synth. Catal., 2014, 356, 3697 CrossRef CAS.
- Y.-H. Sun, T.-Y. Sun, Y.-D. Wu, X. Zhang and Y. Rao, Chem. Sci., 2016, 7, 2229 RSC.
- K. C. Nicolaou, K. Sugita, P. S. Baran and Y.-L. Zhong, J. Am. Chem. Soc., 2002, 124, 2221 CrossRef CAS PubMed.
- (a) G. W. Kabalka and S. T. Summers, J. Org. Chem., 1981, 46, 1217 CrossRef CAS; (b) J. Justicia, J. L. Oller-López, A. G. Campan, J. E. Oltra, J. M. Cuerva, E. Bunuel and D. J. Cardenas, J. Am. Chem. Soc., 2005, 127, 14911 CrossRef CAS PubMed; (c) L. Zhou, Z. Liu, Y. Liu, Y. Zhang and J. Wang, Tetrahedron, 2013, 69, 6083 CrossRef CAS.
- C. Tang and N. Jiao, Angew. Chem., Int. Ed., 2014, 53, 6528 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra04709h |
| This journal is © The Royal Society of Chemistry 2022 |