
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBenzodioxole grafted spirooxindole pyrrolidinyl derivatives: synthesis, characterization, molecular docking and anti-diabetic activity†
Narayanasamy Nivethaa,
Reshma Mary Martizb,
Shashank M. Patilb,
Ramith Ramub,
Swamy Sreenivasac and
Sivan Velmathi *a
*a
aOrganic and Polymer Synthesis Laboratory, Department of Chemistry, National Institute of Technology, Tiruchirappalli, 620 015, Tamil Nadu, India. E-mail: velmathis@nitt.edu
bDepartment of Biotechnology and Bioinformatics, School of Life Sciences, JSS Academy of Higher Education and Research, Mysuru, 570 015, Karnataka, India
cDepartment of Chemistry, University College of Science, Tumkur University, Tumkur, 572 103, Karnataka, India
First published on 25th August 2022
Abstract
A highly stereoselective, three-component method has been developed to synthesize pyrrolidine and pyrrolizidine containing spirooxindole derivatives. The interaction between the dipolarophile α,β-unsaturated carbonyl compounds and the dipole azomethine ylide formed in situ by the reaction of 1,2-dicarbonyl compounds and secondary amino acids is referred to as the 1,3-dipolar cycloaddition reaction. The reaction conditions were optimized to achieve excellent stereo- and regioselectivity. Shorter reaction time, simple work-up and excellent yields are the salient features of the present approach. Various spectroscopic methods and single crystal X-ray diffraction examinations of one example of compound 6i validated the stereochemistry of the expected products. The anti-diabetic activity of the newly synthesized spirooxindole derivatives was tested against the α-glucosidase and α-amylase enzymes. Compound 6i was found to exhibit potent inhibition activity against α-glucosidase and α-amylase enzymes which is further evidenced by molecular docking studies.
Introduction
Synthetic organic chemistry has been pursuing the efficient transformation of basic starting materials into highly functionalized complex products that combine environmental and economic benefits. In this context, multicomponent reactions (MCRs) have emerged as a particularly effective synthetic tool for achieving this aim. The field of 1,3-dipolar cycloaddition reactions has recently exploded in popularity for the multicomponent synthesis of spirocyclic motifs. Spiro compounds are ubiquitously essential core units of many natural or synthesised substrates, and they include a wide range of organic molecules of chemical, industrial, and medicinal importance.1,2Spirooxindoles are a unique family of heterocyclic scaffolds since they have been used in drug development as structural and adjacent units at the perimeter of substrates. Because of their unusual three-dimensional structural characteristics, spiro compounds are predicted to bind adequately with protein active sites more easily than flat aromatic ring systems. The inclusion of the oxindole core and additional heterocyclic moieties (typically fused at the oxindole's C-3 position) makes these motifs attractive drug development targets.3 Among them, the spiro-pyrrolidinyl oxindoles were utilized as promising synthetic intermediates4 and exhibited notable biological activities, including anti-diabetic,5 antifungal6 anti-inflammatory, anti-tumour,7 anti-tubercular, antimalarial,8 acetylcholinesterase (AChE), and antiviral9 inhibition properties. For instance, Spirotryprostatin A and B are inhibitors of microtubule assembly and also display antibiotic as well as anti-cancer properties.10 Further, pteropodine works as a positive modulator of 5-HT2 and muscarinic M2 receptors (Fig. 1).11 Spirooxindole pyrrolidinyl frameworks consisting of Horsifiline and Coerulescine are employed as an analgesic and local anesthetic, respectively.
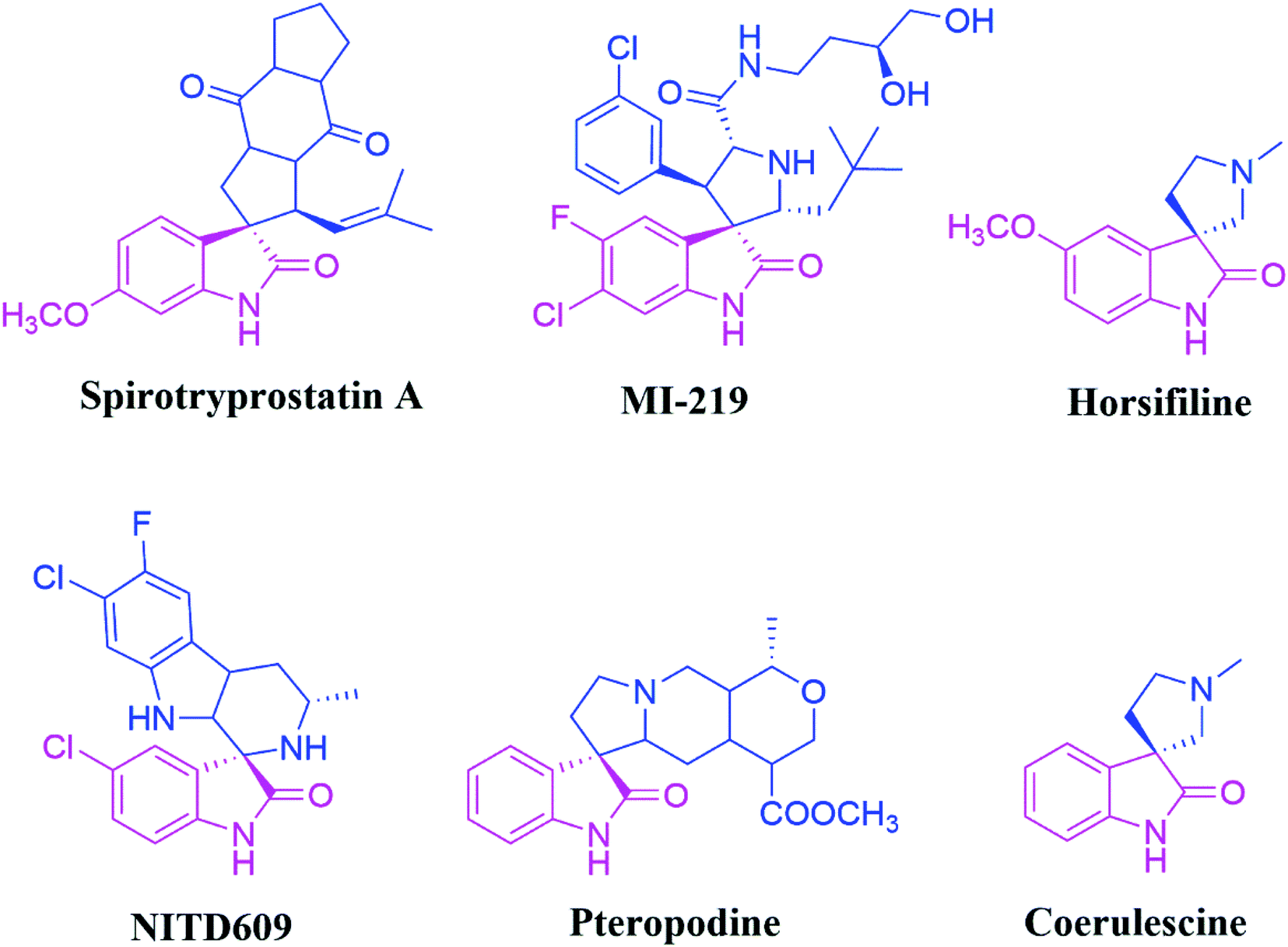 | ||
| Fig. 1 Spirooxindole core consisting of some bioactive compounds. | ||
Through C–C and C–X bond forms, 1,3-dipolar cycloadditions12 play a crucial role in the building of complex heterocyclic motifs. The multicomponent 1,3-dipolar cycloaddition of azomethine ylides generated in situ from the decarboxylative condensation of 1,2-dicarbonyl compounds and α-amino acids to exocyclic olefinic dipolarophiles have attracted a great deal of attention.13 Through two C–C and one C–N bond-forming processes, as well as the development of four new conterminous stereocentres in a single transformation, this sort of one-pot three-component technique allows the construction of architecturally intriguing spirocyclic hybrids.14
MCRs (multicomponent reactions) are environmentally friendly reactions in which three or more reactants combine to produce complex products with high atom economy by combining all of the starting ingredients. In organic synthesis and drug discovery programs, MCRs are cost-effective, time-saving, and provide the required product in high yield under simple and moderate reaction conditions.15,16 These synthetic procedures provide for faster access to a more robust platform for building structurally complicated molecule libraries.17
Diabetes is one of the worldwide prevalent epidemic diseases. Globally, around 463 million people were living with diabetes as of 2019, whereas it is estimated that by 2045 the count will be raised to 700 million, approximately.18 Increment of blood glucose levels, a common cause of uncontrolled diabetes, may, on the extension of time lead to serious consequences, like liver damage, strokes, peripheral nephropathy, coronary heart disease, nephropathy and retinopathy.19 Diverse digestive enzymes incorporated in starch hydrolysis are present in the small intestine and oral cavity.20,21 Among them, α-glucosidase and α-amylase are essential enzymes in the digestion of glycogen and starch22 and play significant roles in controlling the postprandial glucose concentration.23 Thus, inhibition of either or both α-glucosidase and α-amylase is an efficacious way to diminish postprandial glycaemia. The inhibitory activity of these enzymes can impede the transport of carbs into the circulation, reducing the postprandial rise in blood glucose following a mixed carbohydrate meal, and hence can be an important approach in the management of type 2 diabetes.24
Anti-hyperglycemic medicines like acarbose, voglibose, and miglitol are used to treat type 2 diabetes because they block α-glucosidase or α-amylase and are effective at controlling postprandial blood glucose levels. However, the negative side effects and expensive cost have limited their use.25 Because diabetes affects about 5% of the global population, its management without side effects remains a challenge for the medical community. As a result, research into agents for this purpose has grown in importance, and researchers are competing to find new effective and safe therapeutic agents for diabetes treatment.26–31
In continuation of our interest towards the synthesis of potent bioactive spirocyclic hybrids by developing biocompatible methodologies,32 we report herein the regio- and stereoselective three-component synthesis of a novel class of spirooxindole pyrrolidine and pyrrolizidine moieties. The synthesis was accomplished by 1,3-dipolar cycloaddition of benzodioxole chalcones to azomethine ylides generated in situ from isatin and sarcosine/L-proline. Further, the synthesized compounds were subjected to anti-diabetic activity against α-glucosidase and α-amylase enzymes.
Results and discussion
Chemistry
In the presence of appropriate reaction conditions, a one-pot, three-component, 1,3-dipolar cycloaddition process combining the dipolarophiles as benzodioxole chalcones and the dipole as azomethine ylide was used to synthesise desirable spirooxindole derivatives.The benzodioxole chalcones (1a–l) were prepared according to the literature procedure33 with slight modification. Herein, the reaction involves Claisen–Schmidt condensation of 3,4-(methylenedioxy)acetophenone and different substituted benzaldehydes in alkaline medium (10 percent KOH) at room temperature, and were further purified by recrystallization from ethanol in good yield.
We investigated the reaction conditions for the synthesis of spirooxindole pyrrolidine and pyrrolizidine derivatives through the 1,3-dipolar cycloaddition reaction using in situ generated azomethine ylide, which results in the formation of three new bonds, one spirocyclic quaternary carbon, and four stereocenters in a single step using these benzodioxole chalcones as dipolarophiles.
An equimolar quantity of dipolarophile (1a), isatin (2) and sarcosine (3) were taken as a model substrate for examining the synthesis of benzodioxole substituted spirooxindole pyrrolidine (5a) as shown in Table 1.
|
||||
|---|---|---|---|---|
| Entry | Solvent | Temperature (oC) | Time (h) | Yieldb (%) |
| a All reactions were carried out with 1a (1 mmol), 2 (1 mmol) and 3 (1 mmol).b Isolated yields.c RT = room temperature.d NR = no reaction. The entry in bold indicates optimal condition. | ||||
| 1 | DMF | 120 | 8.5 | 50 |
| 2 | Acetonitrile | 85 | 5 | 65 |
| 3 | DCM | 40 | 6 | 55 |
| 4 | 1,4-Dioxane | 90 | 4 | 70 |
| 5 | Methanol: 1,4-Dioxane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
80 | 6.5 | 74 |
| 6 | Toluene | 100 | 10 | 45 |
| 7 | Methanol | 67 | 1.5 | 80 |
| 8 | Methanol | RTc | 12 | NRd |
| 9 | Ethanol | 80 | 1.5 | 76 |
| 10 | Water | 90 | 10 | NRd |
| 11 | Methanol: Water (1:1) |
75 | 5 | 60 |
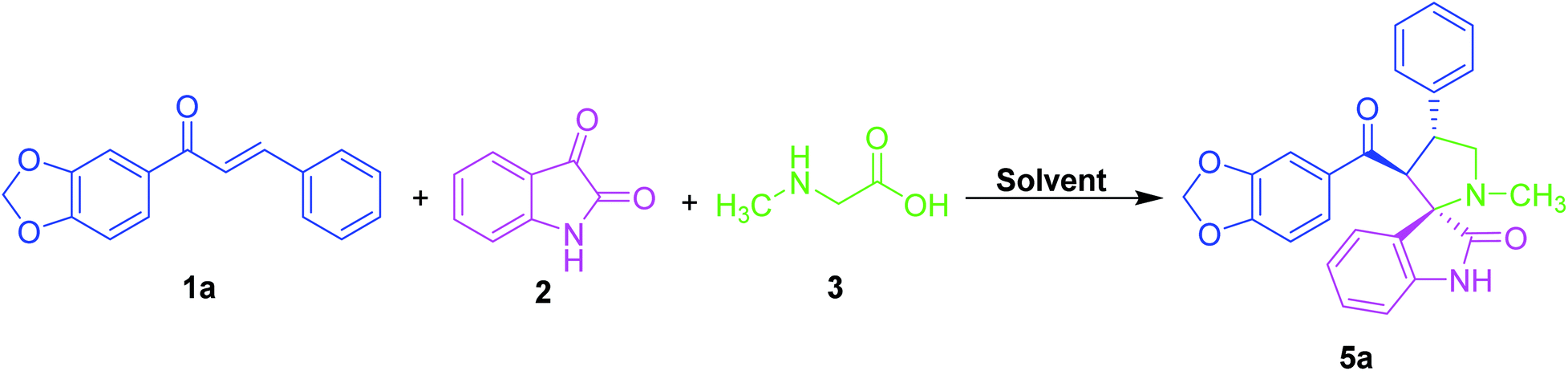
Polar aprotic solvents including dimethylformamide (DMF), acetonitrile, dichloromethane (DCM) and 1,4-dioxane and non-polar aprotic solvent like toluene produced desired product in lesser yield, due to the reason that the starting materials were sparingly soluble in these solvents even at the reflux temperature and prolonged time (Table 1, entry 1–6). We further investigated with various polar protic solvents such as methanol, ethanol, water and methanol:water systems to monitor the progress of the reaction (Table 1, entry 7–11). The reaction proceeded well in the case of methanol as the solvent to furnish the desired product in good yield and shorter time (Table 1, entry 7). Besides, by increasing or decreasing the mole ratio of amino acids and 1,2-dicarbonyl compound as well as the reaction temperature, there is no significant enhancement in the efficiency and yield of the reaction.
Consequently, unstabilized azomethine ylides were formed in situ by the decarboxylative condensation of isatin (2) and sarcosine (3), which rapidly reacted with the dipolarophiles (1a–l) under reflux in methanol by 1,3-dipolar cycloaddition reaction to afford novel spirooxindole pyrrolidine derivatives (5a–l) in excellent yields (Table 2).
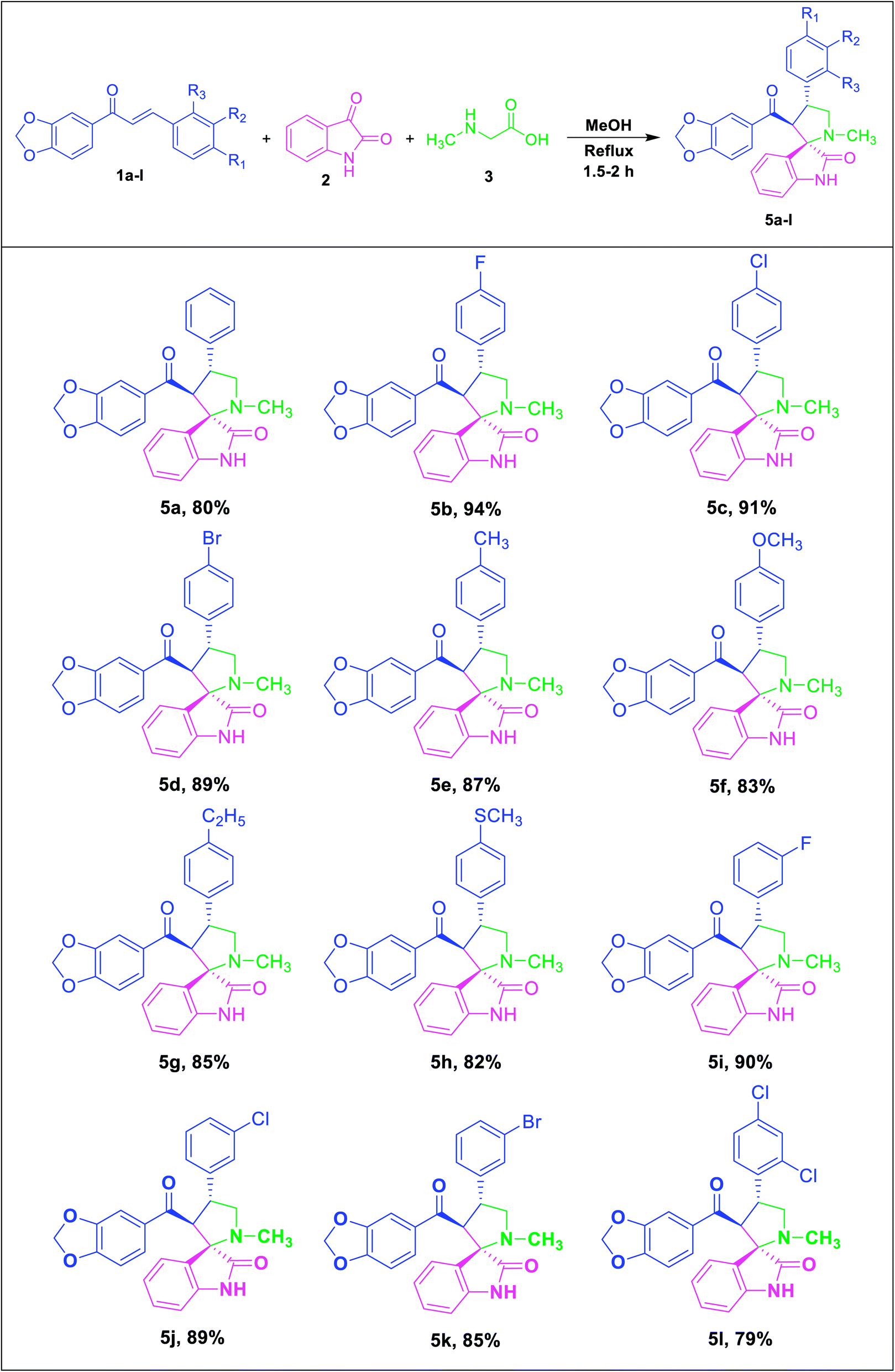 |
This cycloaddition reaction is stereo- and regioselective, with the β-carbon of the α,β-unsaturated compounds of the dipolarophile adding to electron-rich carbon of the dipole. The successful synthesis of the spirooxindole pyrrolidine (5a–l) and pyrrolizidine (6a–l) derivatives were established by spectroscopic and crystallographic studies. Scrutiny of all spectral data confirmed the formation of the spirooxindole derivatives obtained from the benzodioxole chalcones as dipolarophiles.
The molecular structure of the newly synthesized spirooxindole pyrrolidine derivatives 5a–l was elucidated by FT-IR, proton NMR, carbon NMR and High-resolution Mass Spectrometry (HR-MS). The FT-IR spectrum of the compound 5c showed oxindole –NH stretching at 3348 cm−1, benzodioxole –OCH2O- exhibited characteristic stretching vibration at 2916 cm−1 and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching at 1720 and 1659 cm−1 for oxindole and benzodioxole carbonyls, respectively.
O stretching at 1720 and 1659 cm−1 for oxindole and benzodioxole carbonyls, respectively.
In the 1H NMR spectrum, compound 5c exhibited a singlet at δ 2.06 for pyrrolidine N–CH3 protons. The doublet for the methylene protons (H-5) appeared at δ 3.39 and the methine proton (H-4) of the pyrrolidine ring resonated as a multiplet at δ 4.33–4.35 (Fig. 2). The methylenedioxy protons of benzodioxole appeared at δ 6.02 as a doublet. A broad singlet at δ 10.59 was due to the oxindole –NH proton and the rest of the aromatic protons resonated between the region of δ 6.55–7.43. The 13C NMR spectrum of 5c showed the presence of 26 carbons including the carbon peaks at δ 195.13 and 179.40 for benzodioxole and oxindole ring carbonyls (Fig. 2). The methylenedioxy carbon appeared at δ 102.47, spiro carbon (C-2) at δ 73.19 and pyrrolidine N–CH3 carbon at d 34.92, respectively. The peaks found at δ 44.03 and 60.02 corresponds to pyrrolidine ring methine (C-4) and methylene (C-5) carbons respectively. Additionally, the formation of compound 5c was evidenced by the HR-MS analysis, exhibiting a characteristic [M + H]+ peak at m/z 461.1269, which exactly matches with the calculated mass of the compound. Based on the above spectral data information confirmed the structure as 3′-(benzo[d][1,3]dioxole-5-carbonyl)-4′-(4-chlorophenyl)-1′-methylspiro[indoline-3,2′-pyrrolidin]-2-one (5c).
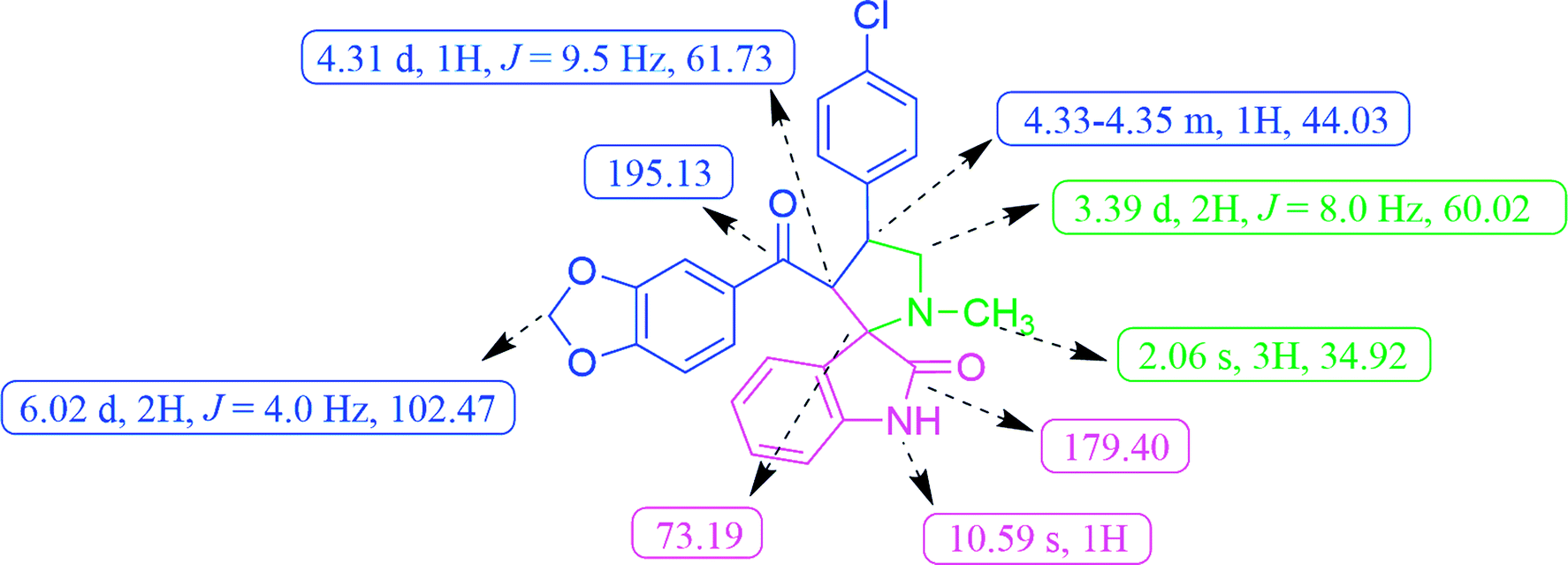 | ||
| Fig. 2 Selected 1H and 13C NMR chemical shifts of spirooxindole pyrrolidine 5c. | ||
To broaden the scope and reproducibility of the above outcomes, the cycloaddition reaction was further widened. Herein, we have proposed the reaction between dipolarophile derivatives of benzodioxole chalcones (1a–l) with azomethine ylide generated in situ from isatin (2) and L-proline (4) to obtain the corresponding spirooxindole pyrrolizidine derivatives 6a–l in good yields with high selectivity (Table 3).
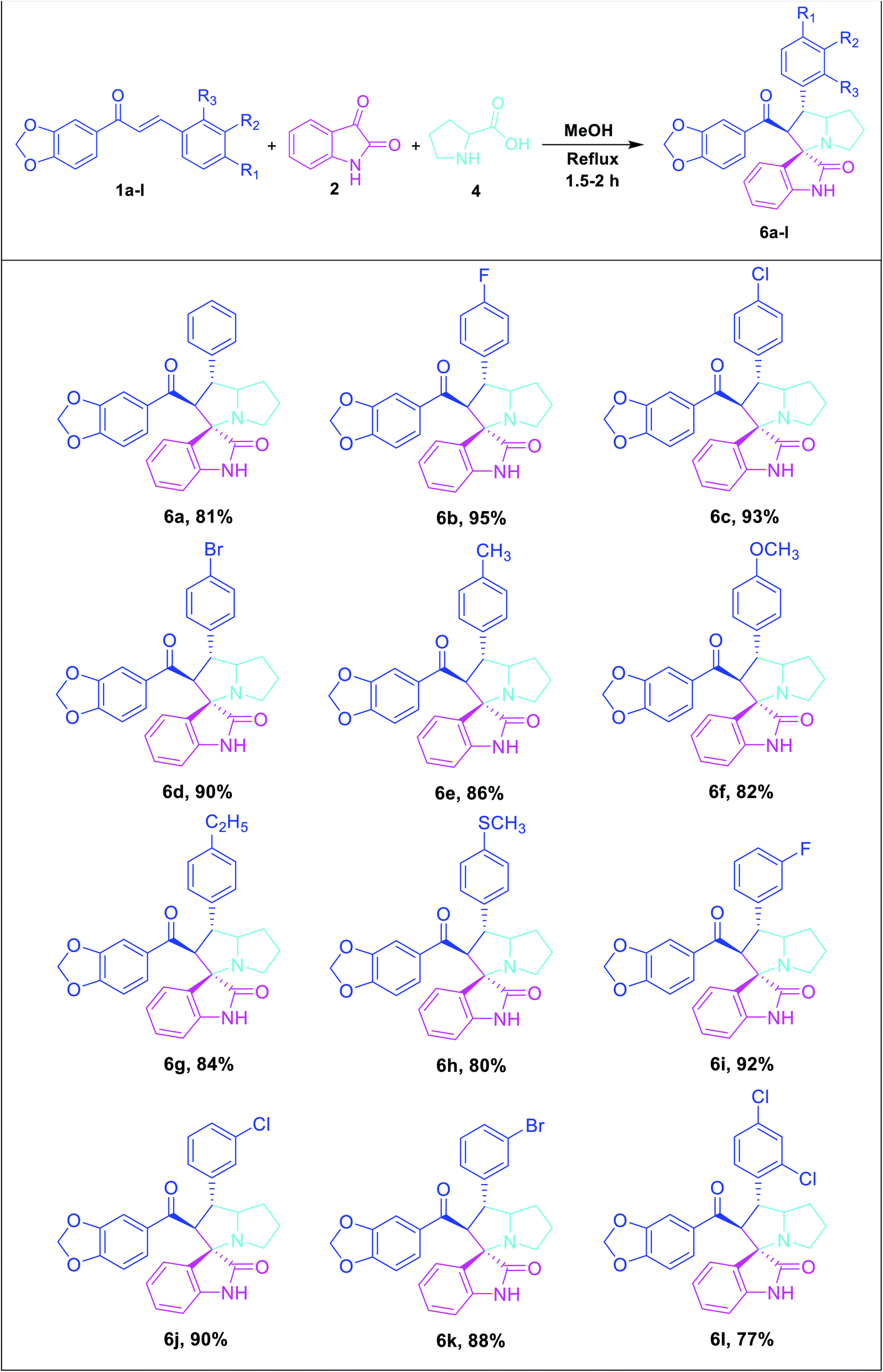 |
The FT-IR spectrum of the compound 6c displayed –NH group stretching at 3120 cm−1 and O–CH2–O stretching at 2968 cm−1. The benzodioxole and oxindole ring carbonyls appeared at 1721 and 1652 cm−1, respectively. The 1H NMR spectrum exhibited two multiplets at δ 1.70–1.78 and 1.84–1.89 were due to the pyrrolidine ring H-6 and H-7 methylene protons. A multiplet at δ 2.33–2.36 and a doublet of doublet at δ 2.58 were assigned to the pyrrolizidine ring H-8 methylene protons. The methine proton (H-4) was resonated as a doublet at δ 3.86, indicating the regioselectivity of the product. A multiplet that appeared at d 3.89–3.91 was due to the H-5 proton of the pyrrolizidine ring. The methylenedioxy protons (O–CH2–O) was resonated as a doublet at d 6.04. A broad singlet at d 10.30 was observed due to the oxindole –NH proton and the rest of the aromatic protons resonated between the region of δ 6.59–7.48. The 13C NMR spectrum showed the presence of 28 carbons, including the benzodioxole and oxindole ring carbonyl carbons at δ 194.76 and 179.86, methylenedioxy carbon at δ 102.45, and spiro carbon (C-2) at d 73.00, respectively. The pyrrolizidine ring of two methines (C-4 & C-5) and three methylene carbons (C-6, C-7 & C-8) were observed at δ 48.00, 71.69, 29.81, 27.11 and 52.21, respectively (Fig. 3). Moreover, the structure of 6c was further confirmed by HR-MS analysis, displaying a characteristic [M + H]+ peak at m/z 487.1431.
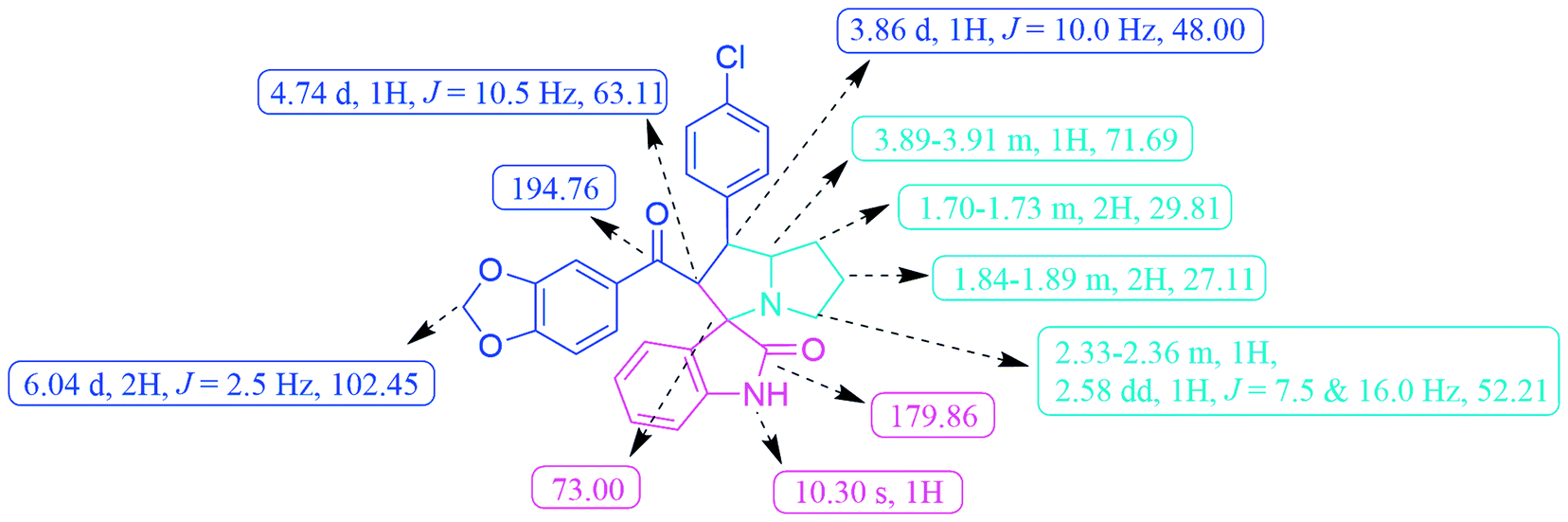 | ||
| Fig. 3 Selected 1H and 13C NMR chemical shifts of spirooxindole pyrrolizidine 6c. | ||
Similarly, the formation of all other spirooxindole pyrrolizidine derivatives was confirmed by FT-IR, 1H and 13C NMR and HR-MS spectral data. Finally, the regio- and stereochemical outcomes of the achieved product 6i was determined unambiguously by single crystal X-ray diffraction studies (Fig. 4).
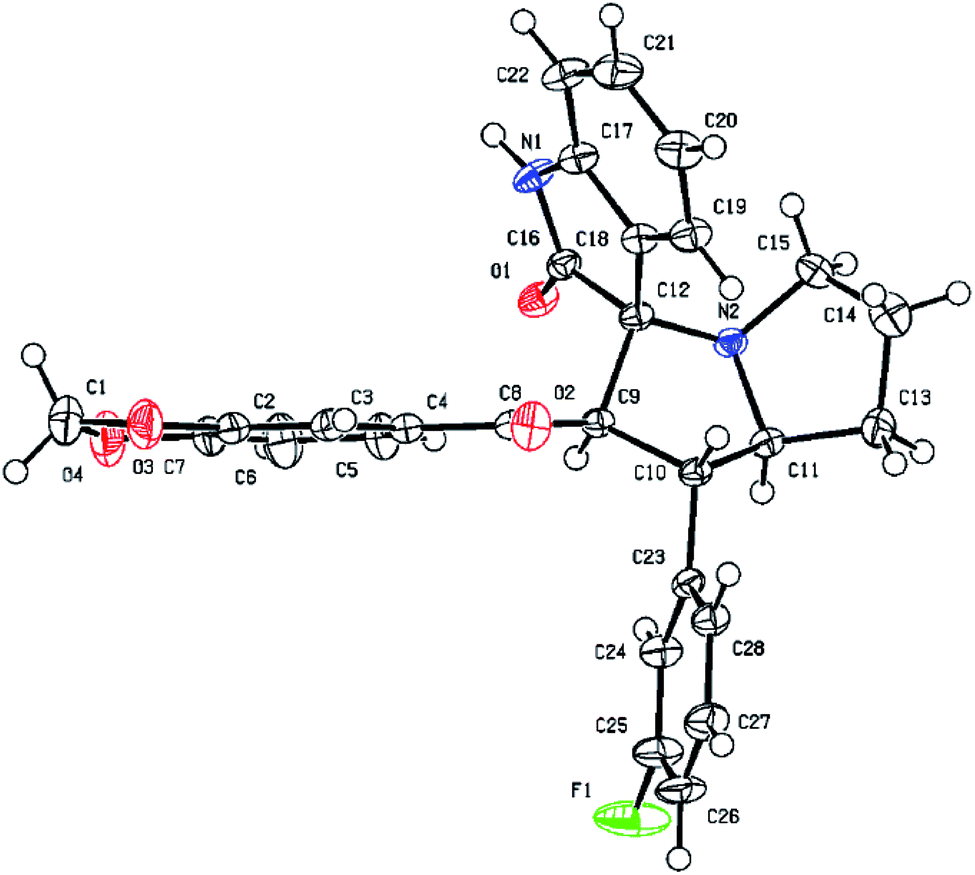 | ||
| Fig. 4 ORTEP diagram of compound 6i. | ||
It is expected that the nucleophilic attack of sarcosine (3) into the active carbonyl of isatin (2) involves the conversion of the carbonyl group to alcohol. The resulting OH will further attack the carboxylic group in sarcosine to obtain lactone functionality (I). Thus, the reactive azomethine ylide (II) was generated by in situ decarboxylative condensation reaction. Subsequently, the azomethine ylide undergoes a 1,3-dipolar cycloaddition reaction with dipolarophile (1a) to afford the spirooxindole pyrrolidine derivative (5a) in a diastereoselective and regioselective manner (Scheme 1). However, it is accepted that the cycloaddition of azomethine ylide proceeds preferentially via an S-shaped ylide than a W-shaped ylide because of the higher resonance energy and stability of the S-shaped ylide.34
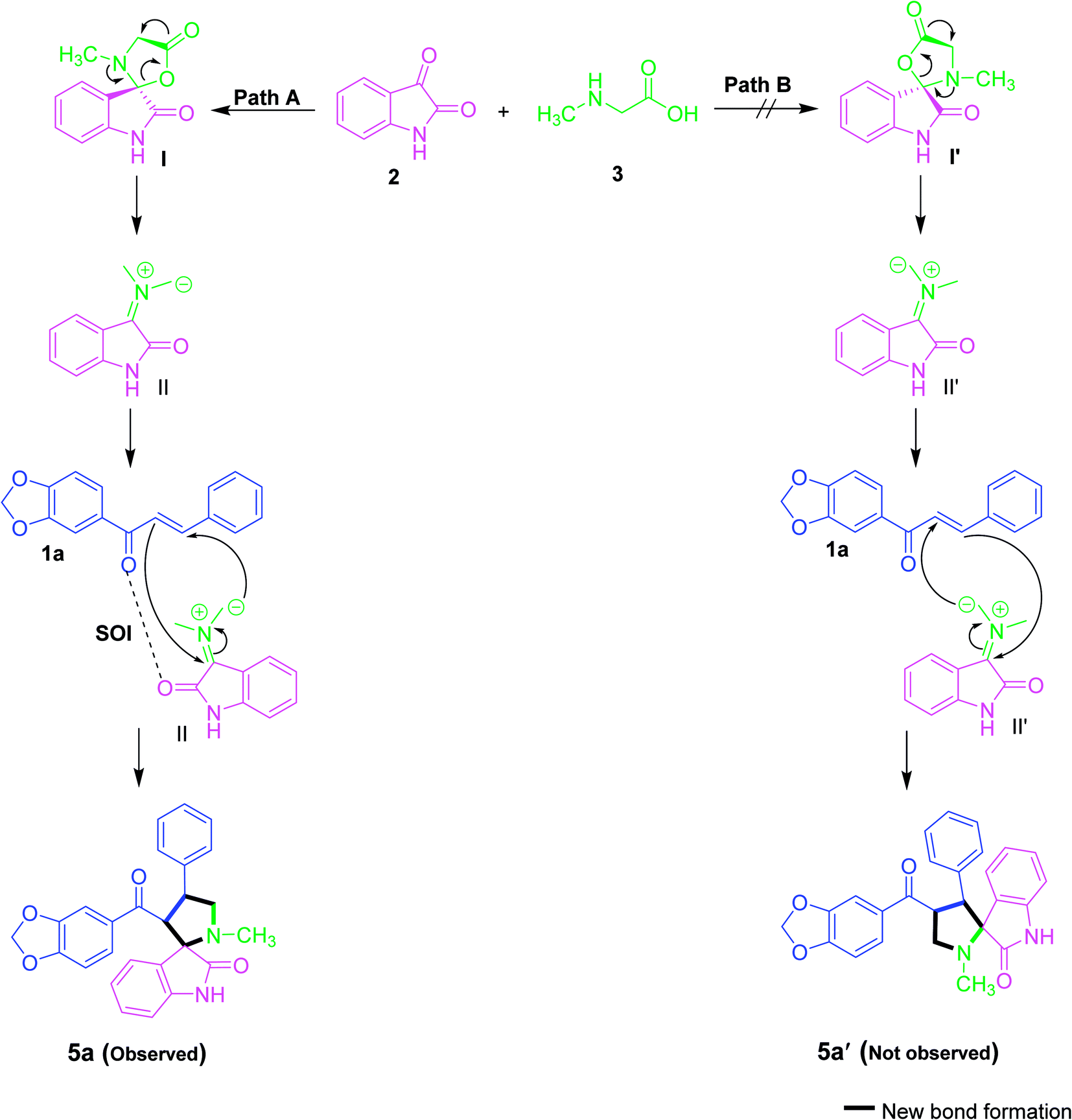 | ||
| Scheme 1 Proposed reaction mechanism for the synthesis of 5. | ||
The formation of regioisomer 5 could be described given the secondary orbital interaction (SOI) between the orbital of the carbonyl group of dipolarophiles with respect to azomethine ylide. The reaction between the dipolarophile and azomethine ylide is more favourable for regioisomer 5 via path A due to the secondary orbital interaction35,36 while the regioisomer 5′ is not possible via path B (Scheme 1). Consequently, the orientation of the C2 stereocentre is always pre-established, hence confirming the stereochemistry of the spirooxindole pyrrolidine 5. Overall, the reaction is demonstrated to be highly regio- and stereoselective, leading to the construction of only one stereoisomer.
Single crystal X-ray diffraction studies
Single crystal X-ray diffraction analysis confirmed the structure of compound 6i and molecular structure is shown in Fig. 4.Collected crystal data and the refinement parameters are tabulated (See ESI†). Compound 6i crystallized in a triclinic crystal system and space group P-1. Two molecular units are present in a unit cell and selected bond lengths and bond angles are shown in the ESI.† The bond lengths of C(10)–C(11); 1.535(2) Å, C(12)–C(9); 1.556 Å and N(2)–C(12); 1.472 Å were confirmed the formation of new spirooxindole pyrrolizidine derivative. Two spiro-moieties are combined with C12 asymmetric centre. From the crystal packing of 6i molecules have a strong intermolecular hydrogen bond between C16–N1H1⋯O1.
In vitro biological studies
| Test Compounds | Enzymes IC50a, b (μg mL−1) | |
|---|---|---|
| α-amylase | α-glucosidase | |
| a All the values are expressed as mean ± SE. Means in the same column with diverse superscripts are significantly different (p ≤ 0.05) as per separated by Duncan multiple range test.b The IC50 value is defined as the inhibitor concentration to inhibit 50% of enzyme activity under assay conditions.c Standard: Acarbose (positive control). | ||
| 5b | 70.50 ± 0.50 f | 46.15 ± 1.30 g |
| 5c | 46.00 ± 0.44 c | 17.66 ± 1.90 b |
| 5d | 35.78 ± 2.06 b | 12.05 ± 0.13 b |
| 5e | 36.80 ± 1.04 b | 14.75 ± 0.02 c |
| 5g | 45.50 ± 2.31 c | 24.05 ± 1.86 e |
| 5j | 62.04 ± 0.81 e | 30.00 ± 1.77 f |
| 5k | 50.75 ± 0.07 d | 25.03 ± 0.63 e |
| 5l | 44.80 ± 0.32 c | 18.20 ± 0.86 d |
| 6a | 63.00 ± 0.36 e | 26.00 ± 1.02 e |
| 6c | 44.25 ± 0.05 c | 18.03 ± 1.00 d |
| 6g | 35.00 ± 0.35 b | 14.00 ± 0.50 c |
| 6i | 32.40 ± 0.86 a | 10.00 ± 0.16 a |
| 6j | 50.50 ± 1.00 d | 15.00 ± 0.00 c |
| 6k | 62.05 ± 1.11 e | 29.55 ± 0.08 f |
| Standard c | 30.05 ± 0.05 a | 10.15 ± 0.06 a |
Similar tests were carried out to see if series 5 (5b, 5c, 5d, 5e, 5g, 5j, 5k, 5l) and series 6 (6a, 6c, 6g, 6i, 6j, 6k) inhibited α-amylase, another important carbohydrate hydrolyzing enzyme. In comparison to the other compounds tested, 6i (IC50:32.40 g mL−1) had the strongest inhibitory action against α-amylase, whereas 5b (IC50:70.50 g mL−1) had the lowest inhibitory impact, as indicated in Table 4. All of the substances examined had IC50 values that were significantly higher (p < 0.05) than the medicinal medication acarbose (IC50:30.05 g mL−1).
The inhibitory effects of various doses of test compounds and aminoguanidine (25, 50, and 100 g mL−1) on AGE after 3 weeks of incubation are shown in Fig. 5. Albumin glycation was reduced in a dose-dependent manner after incubation with series 5 (5b, 5c, 5d, 5e, 5g, 5j, 5k, 5l) and series 6 (6a, 6c, 6g, 6i, 6j, 6k). By the end of the trial, it was obvious that 6i had greater inhibitory activity against AGE than the other test compounds tested at varying doses. The fluorescence experiments on AGE, as shown in Fig. 5A and B concluded that compounds inhibit in the range of 50–75% at a concentration of 100 g mL−1. On a 21 day incubation period, 6i showed stronger inhibition than a well-known inhibitor, aminoguanidine, at various doses.
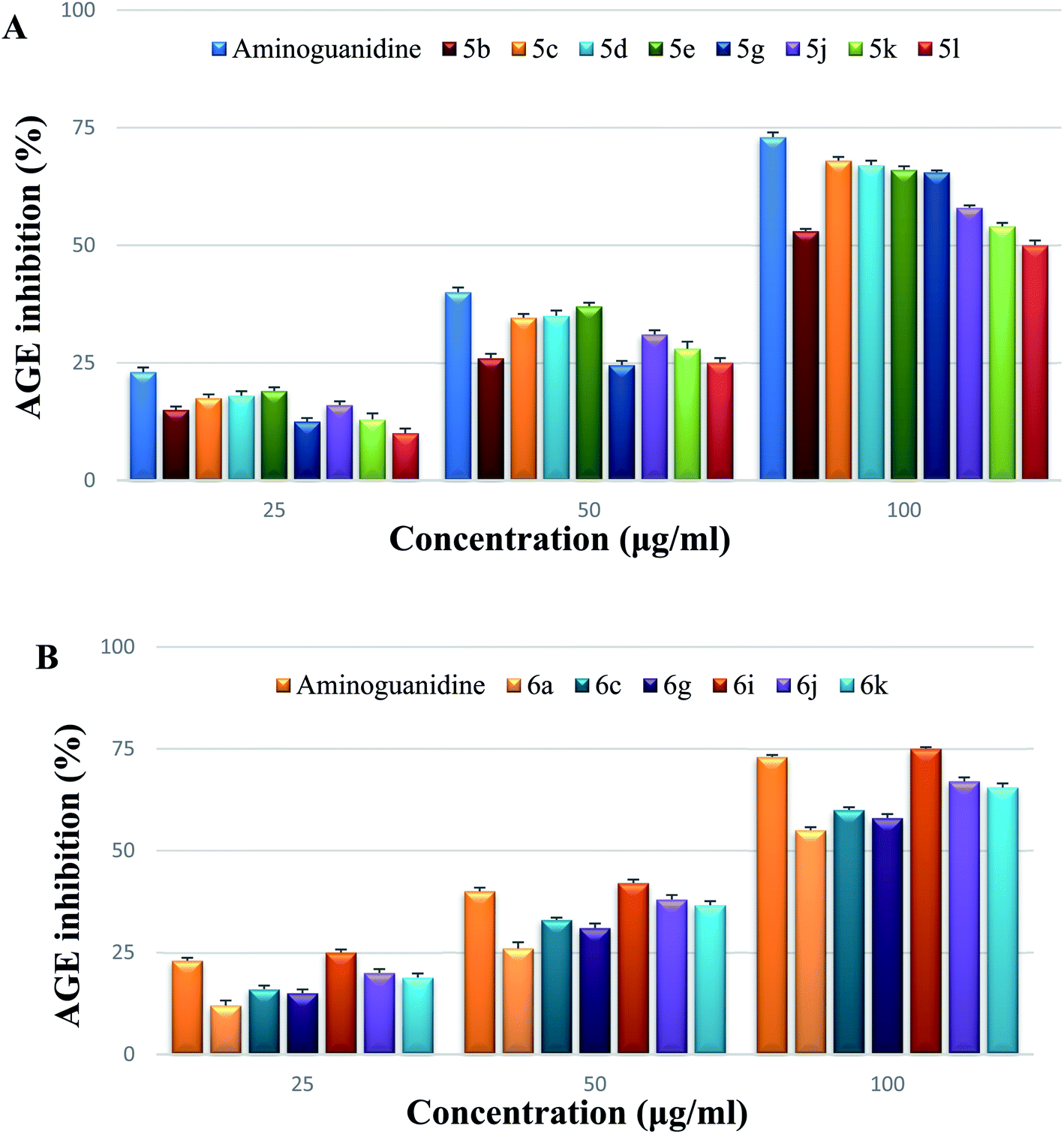 | ||
| Fig. 5 (A) Inhibitory effects of series 5 spirooxindole pyrrolidine derivatives on AGE formation at diverse concentrations. (B) Inhibitory effects of series 6 spirooxindole pyrrolizidine derivatives on AGE formation at diverse concentrations. | ||
In silico biological studies
| Sl. No. | Name of the compound | Binding affinity (kcal mol−1) | Total no. of non-bonded interactions | Total no. of hydrogen bonds | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| α-glucosidase | α-amylase | Human serum albumin | α-glucosidase | α-amylase | Human serum albumin | α-glucosidase | α-amylase | Human serum albumin | ||
| 1 | 5b | −10.2 | −8.4 | −9.1 | 12 | 8 | 9 | 4 | 3 | 2 |
| 2 | 5c | −9.5 | −8.1 | −8.8 | 16 | 9 | 5 | 3 | 2 | 1 |
| 3 | 5d | −11.1 | −8.6 | −9.2 | 14 | 8 | 10 | 3 | 3 | 2 |
| 4 | 5e | −10.2 | −8.4 | −9.7 | 21 | 10 | 12 | 9 | 4 | 4 |
| 5 | 5g | −9.6 | −8.1 | −8.9 | 11 | 9 | 12 | 1 | 3 | — |
| 6 | 5j | −9.5 | −7.9 | −8.8 | 9 | 11 | 12 | 1 | 2 | 2 |
| 7 | 5k | −9.7 | −8.0 | −9.1 | 15 | 7 | 14 | 4 | 2 | — |
| 8 | 5l | −9.9 | −8.5 | −8.6 | 16 | 14 | 12 | 4 | 7 | 3 |
| 9 | 6a | −10.0 | −8.3 | −9.5 | 15 | 9 | 16 | 2 | 2 | 1 |
| 10 | 6c | −10.1 | −8.2 | −9.3 | 15 | 11 | 16 | 4 | 1 | 1 |
| 11 | 6g | −10.1 | −8.9 | −10.0 | 15 | 13 | 19 | 5 | 4 | 8 |
| 12 | 6i | −10.1 | −8.5 | −9.9 | 13 | 11 | 13 | 3 | 2 | 1 |
| 13 | 6j | −10.5 | −8.2 | −9.7 | 17 | 12 | 14 | 3 | 3 | 3 |
| 14 | 6k | −10.0 | −8.2 | −8.8 | 15 | 8 | 6 | 2 | 2 | 1 |
| 15 | Acarbose | −8.6 | −6.0 | — | 10 | 6 | — | 9 | 6 | — |
| 16 | Aminoguanidine | — | — | −4.1 | — | — | 6 | — | — | 4 |
with α–glucosidase along with their respective distance
| Sl. No. | Name of the compound | Binding affinity (kcal mol−1) | Hydrogen bonds | Electrostatic bonds | Hydrophobic bonds | |||
|---|---|---|---|---|---|---|---|---|
| Pi-sigma | Pi- Pi bond | Alkyl | Pi-alkyl | |||||
| 1 | 6i | −10.1 | HIS A: 279 (2.86), HIS A: 279 (2.86), ARG A: 312 (2.93), ARG A: 312 (2.45), PHE A: 157 (3.47) | HIS A: 279 (4.60), GLU a:304 (4.10) | — | PHE A: 157 (4.73), UNL1 (5.37) | ARG A: 312 (4.31) | LEU A: 218 (5.42), ALA A: 278 (3.07), LEU A: 218 (4.06), ALA A: 278 (3.04), ARG A: 312 (4.81) |
| 2 | Acarbose | −8.6 | ASN A: 241 (2.31), HIS A: 279 (2.77), ARG A: 439 (2.48), PRO A: 309 (2.33), HIS A: 279 (2.82), PRO A: 309 (2.33), HIS A: 239 (2.25), HIS A: 279 (2.98), ARG A: 439 (2.44) | — | HIS A: 279 (3.58) | — | — | — |
| Sl. No. | Name of the compound | Binding affinity (kcal mol−1) | Hydrogen bonds | Electrostatic bonds | Halogen | Hydrophobic bonds | |||
|---|---|---|---|---|---|---|---|---|---|
| Pi-sigma | Pi–Pi bond | Alkyl | Pi-alkyl | ||||||
| 1 | 6i | −8.9 | LYS A: 457 (2.91), LYS A: 457 (2.82), SER A: 494 (2.71), SER A: 494 (2.32) | LYS A: 457 (3.73), LYS A: 457 (3.43) | GLU A: 493 (3.14) | — | TRP A: 396 (3.92), TRP A: 396 (5.21) | — | TRP A: 396 (4.91), TRP A: 396 (4.32), LYS A: 457 (4.54), LYS A: 457 (5.03) |
| 2 | Acarbose | −6.0 | ARG A: 343 (1.88), ARG A: 343 (6.15), ARG A: 392 (2.14), CYS A: 378 (2.46), GLU A: 390 (2.96), ASP A: 456 (2.56) | — | — | — | — | — | — |
| Sl. No. | Name of the compound | Binding affinity (kcal mol−1) | Hydrogen bonds | Electrostatic bonds | Hydrophobic bonds | ||||
|---|---|---|---|---|---|---|---|---|---|
| Amide-Pi Stacked | Pi-sigma | Pi–Pi bond | Alkyl | Pi-alkyl | |||||
| 1 | 6i | −10.0 | LYS A: 195 (2.34), ARG A: 218 (2.43), ARG A: 218 (3.09), ARG A: 218 (2.46) | — | PRO A: 447 (4.11) | — | HIS A: 440 (4.51), UNL1 (5.14) | LYS A: 195 (4.43), VAL A: 455 (4.51) | TYR A: 452 (5.00), LYS A: 436 (5.01), CYS A: 448 (5.42), PRO A: 447 (5.16), CYS A: 448 (4.04), LYS A: 444 (4.80), PRO A: 447 (5.34), CYS A: 448 (3.99), LYS A: 195 (5.19) |
| 2 | Aminoguanidine | −4.1 | ASP A: 108 (1.97), LYS A: 106 (2.48), LEU A: 103 (2.66), LYS A: 106 (1.91) | GLU A: 465 (2.39), ASP A: 108 (2.89) | — | — | — | — | — |
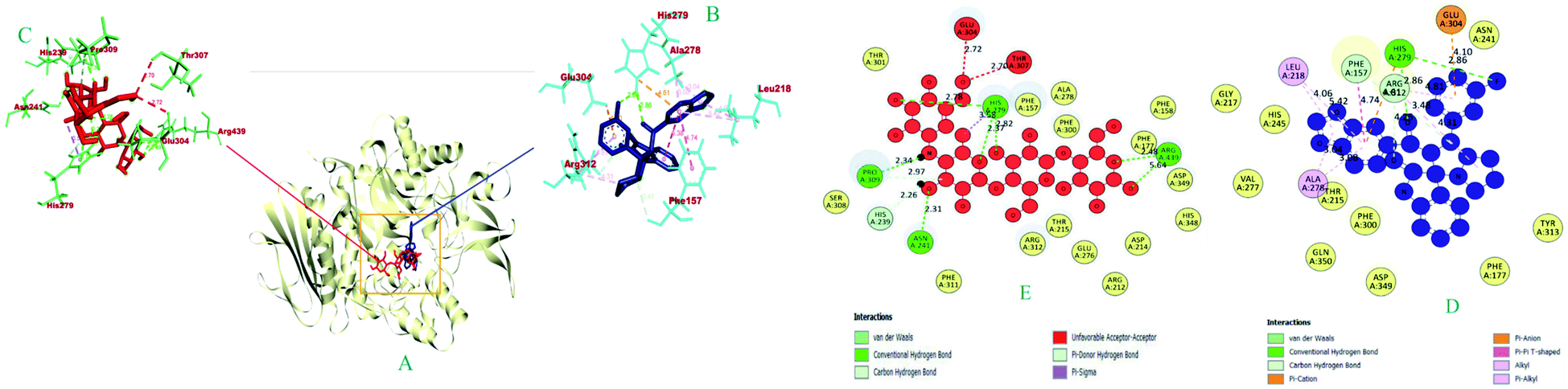 | ||
| Fig. 6 The 3D and 2D interaction view of compound 6i with the binding sites of α-glucosidase. | ||
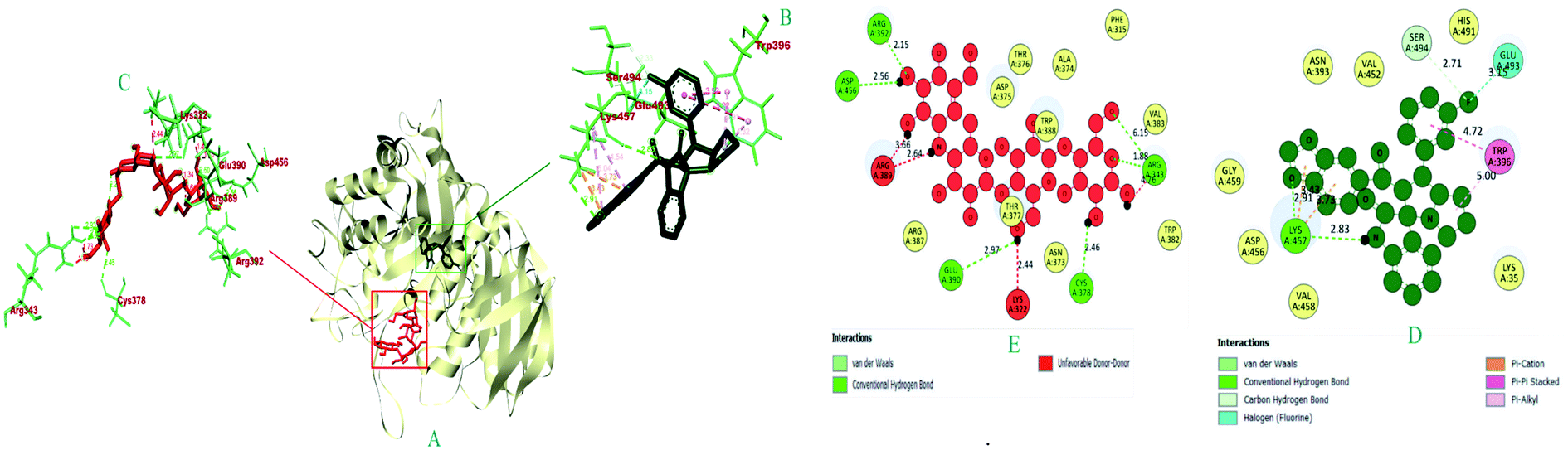 | ||
| Fig. 7 The 3D and 2D interaction view of compound 6i with the binding sites of α-amylase. | ||
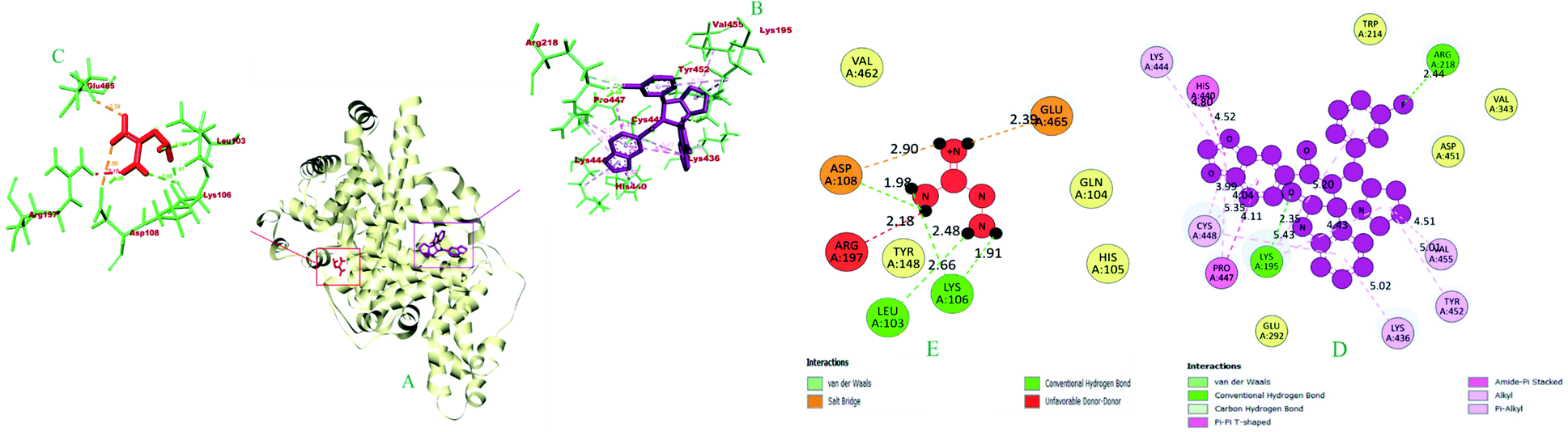 | ||
| Fig. 8 The 3D and 2D interaction view of compound 6i with the binding site of Human serum albumin. | ||
Molecular dynamics simulation
Molecular dynamics simulation is performed to evaluate the conformational stabilities of protein-ligand complexes. For all the protein-ligand complexes, the RMSD, RMSF, Rg, and SASA graphs were plotted. The graphical representation of molecular dynamics simulation for α-glucosidase, α-amylase, and human serum albumin has been given in Figures 9–11, respectively. The RMSD plot of the protein-ligand complex displays the stability of the ligand and protein over the course of a 100 ns simulation. All the RMSD trajectories found in this simulation were found to be stable after 20 ns, free from initial fluctuations. In the case of α-glucosidase, RMSD of both protein-6i and protein-acarbose complexes were predicted to be within the range of 0.30–0.40 nm. However, protein backbone atoms were also found with the same pattern as both complexes. In the case of α-amylase, the protein-6i complex was found to be in the range of 0.4–0.5 nm. Yet both protein backbone atoms and protein-acarbose complex were in the range of 0.3–0.4 nm. Further, RMSD of protein-6i and protein backbone atoms in case of human serum albumin were found to be in the range of 0.5–0.6 nm. Yet, the protein-aminoguanidine complex predicted showed the fluctuation in the RMSD of 0.75 nm at 62 ns.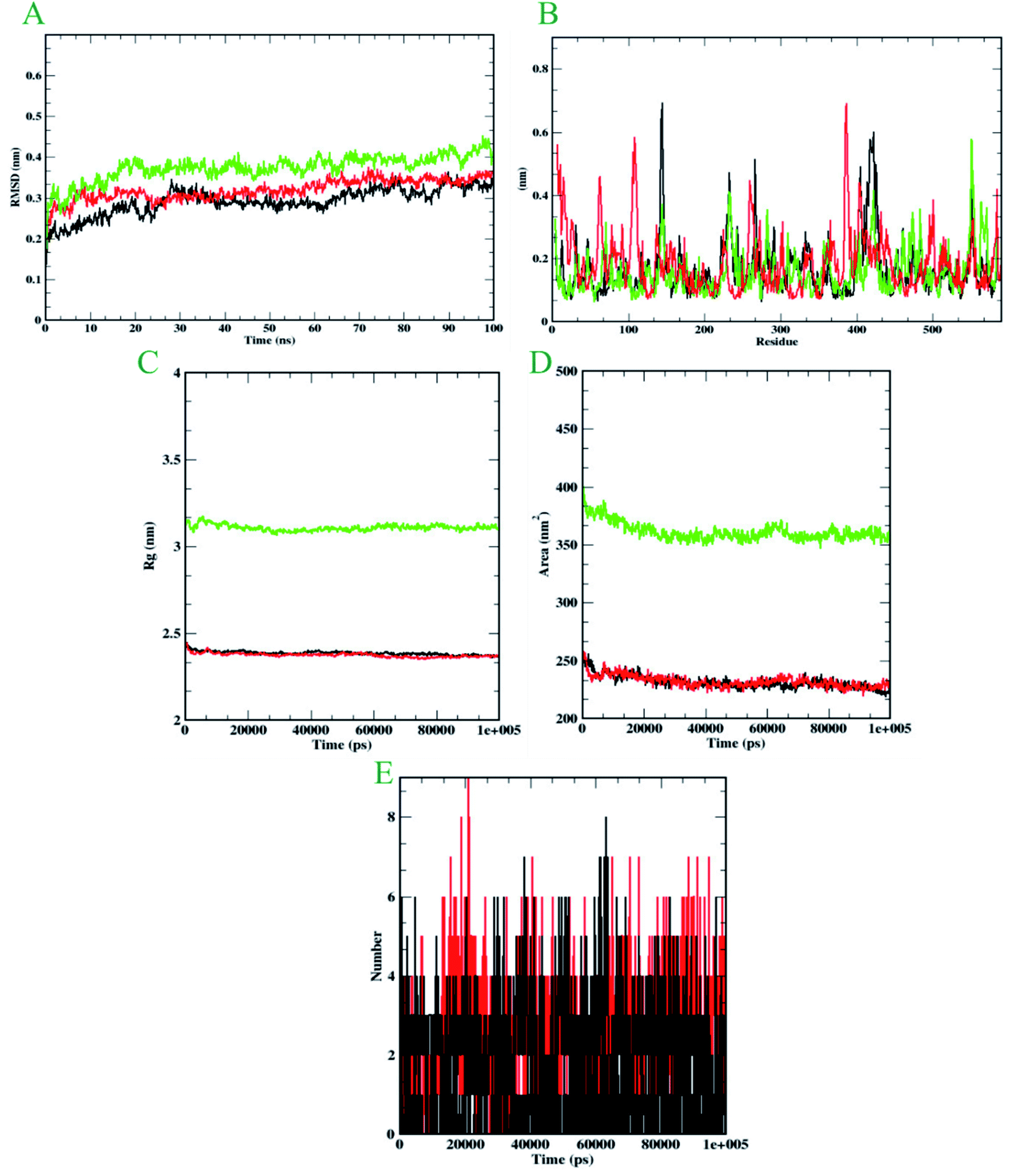 | ||
| Fig. 9 Analysis of RMSD, RMSF, Rg, SASA, and number of hydrogen bonds of 6i (black) and acarbose (red) with α–glucosidase (green) at 100 ns. (A) Time evolution of backbone RMSD of the complex structure. (B) RMSF of protein and ligand. (C) The radius of gyration (Rg) (D) SASA (E) Hydrogen bonds occurring over the time of simulation between protein and ligand. | ||
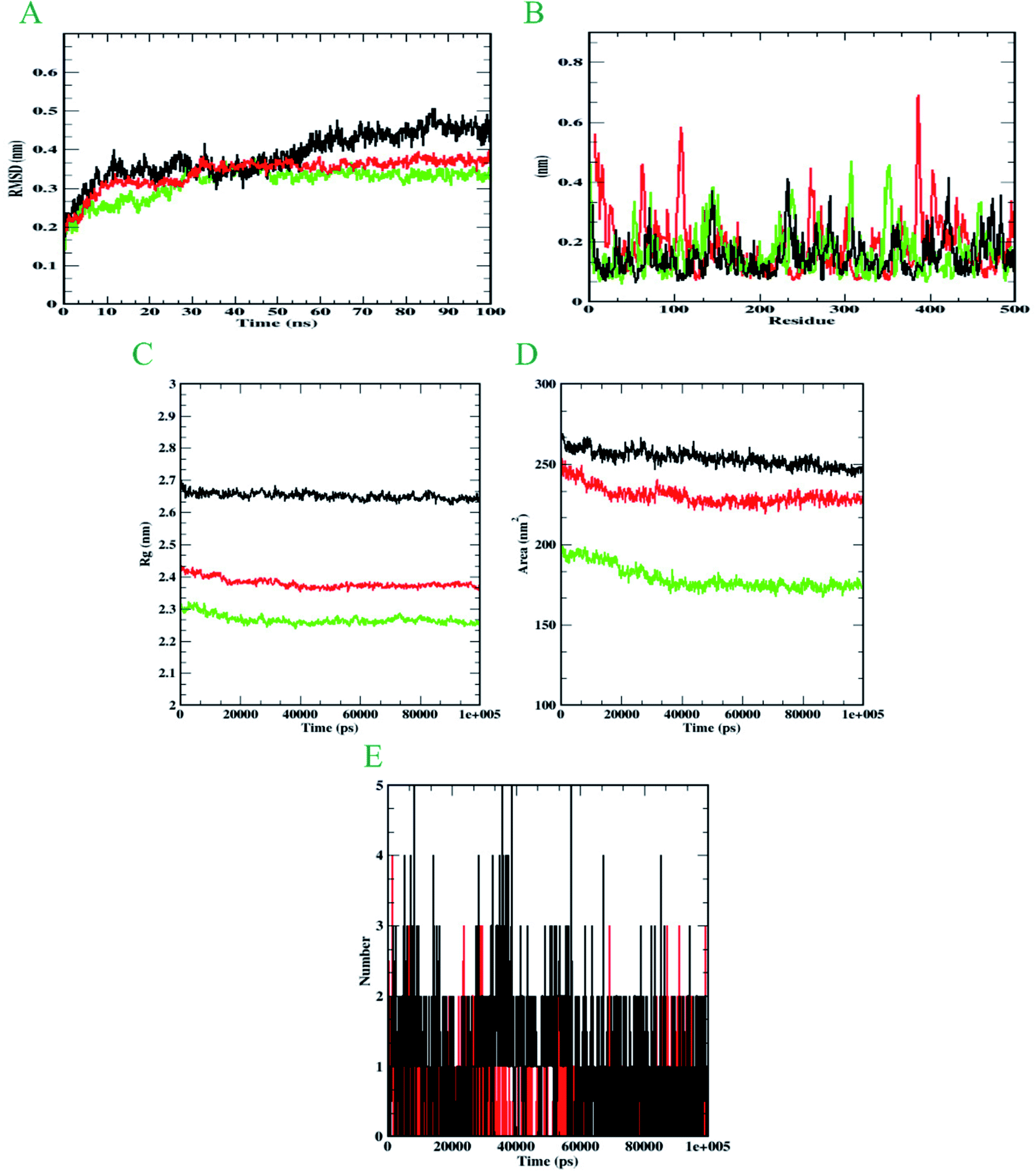 | ||
| Fig. 10 Analysis of RMSD, RMSF, Rg, SASA, and number of hydrogen bonds of 6i (black) and acarbose (red) with α-amylase (green) at 100 ns. (A) Time evolution of backbone RMSD of the complex structure. (B) RMSF of protein and ligand. (C) The radius of gyration (Rg) (D) SASA (E) Hydrogen bonds occurring over the time of simulation between protein and ligand. | ||
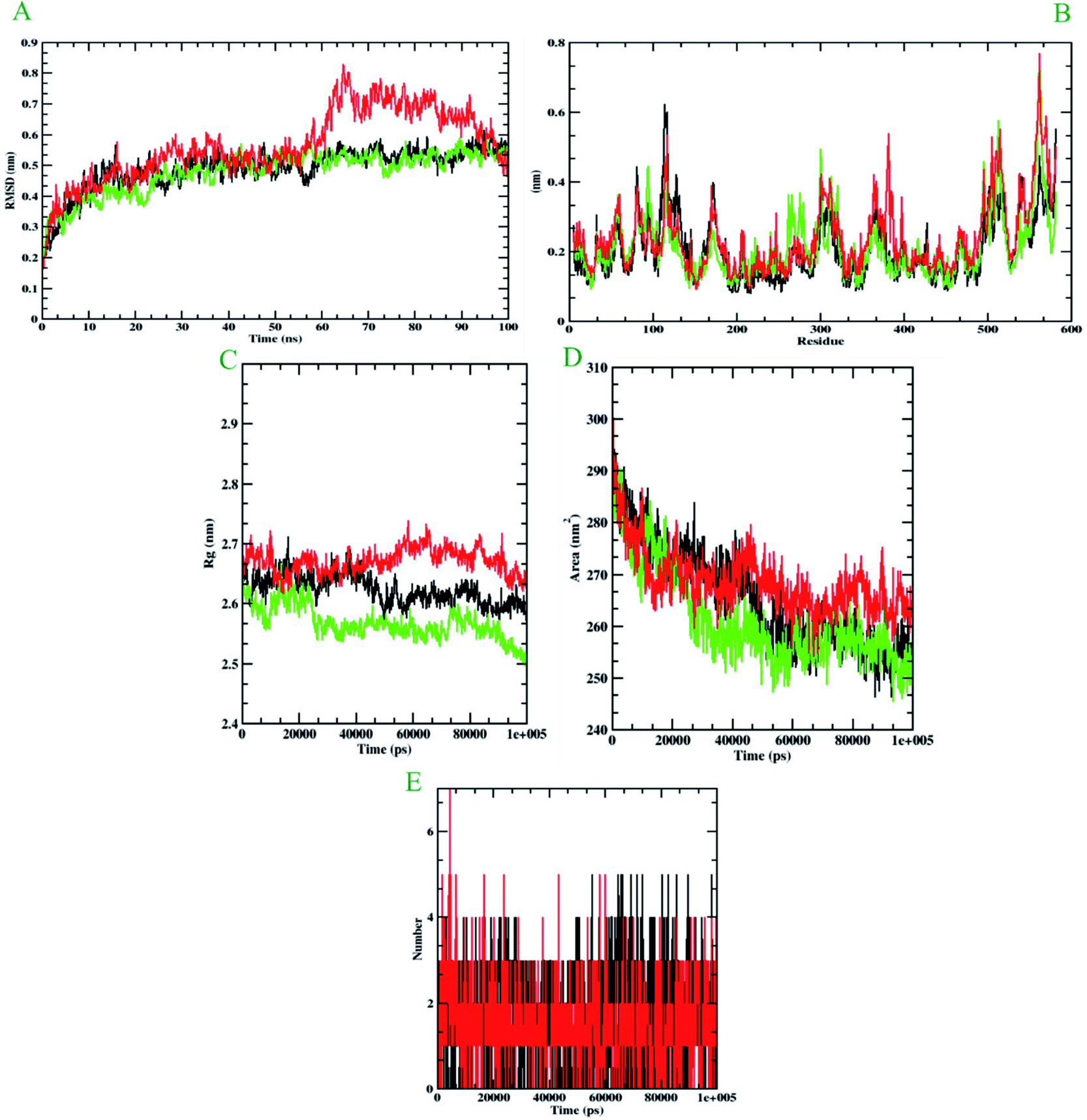 | ||
| Fig. 11 Analysis of RMSD, RMSF, Rg, SASA, and the number of hydrogen bonds of 6i (black) and Aminoguanidine (red) with Human serum albumin (green) at 100 ns. (A) Time evolution of backbone RMSD of the complex structure. (B) RMSF of protein and ligand. (C) The radius of gyration (Rg) (D) SASA (E) hydrogen bonds occurring over the time of simulation between protein and ligand. | ||
The average deviation of a particle (e.g., a protein residue) from a reference location over time is calculated using RMSF. As a result, RMSF focuses on the regions of the protein structure that deviate the most/least from the mean. In the case of all the three protein backbone atoms, fluctuations were observed only during N- and C-terminals as well as loop regions. However, protein-6i complexes were found with minimal fluctuations in all the 3 simulations run. Whereas, protein-acarbose and protein-aminoguanidine complexes were found with more fluctuations.
The radius of gyration (Rg) of proteins and protein-ligand complexes was plotted because it reflects the structural compactness of the molecules by considering the variable masses determined to root mean square distances with respect to the central axis of rotation. In this study, Rg of α-glucosidase protein backbone atoms was found to be ranging between 3.0–3.5 nm. Whereas, Rg of both protein-6i and the protein–acarbose complex was found to be in the same pattern, between 2.0-2.5 nm. In the case of α-amylase, Rg of protein backbone was found to be ranging between 2.2-2.3 nm, protein-6i between 2.6–2.7, and protein-acarbose between 2.3–2.4 nm. All three trajectories were predicted with different Rg values. However, in the case of human serum albumin, protein-6i complex the average Rg was found to be between 2.6–2.66 and protein-aminoguanidine had the same pattern of Rg with 2.68–2.7 nm. Whereas, protein backbone atoms were predicted with 2.6 nm of Rg value.
Furthermore, the area around the hydrophobic core created between protein-ligand complexes was shown in SASA plots for all of the protein-ligand complexes. In the case of α-glucosidase, the protein backbone atoms were found with the SASA value of ∼350 nm2 and both ligand complexes were predicted with 200–250 nm2. In the case of α-amylase, all the three SASA directories had different values. Protein backbone atoms were predicted with 155–200 nm2, protein-6i complex with ∼250 nm2, and protein-acarbose with 200–250 nm2 SASA value. Conversely, all three trajectories had the same pattern of SASA value in case of human serum albumin. All the trajectories were predicted to have the SASA value between 250–290 nm2.
Furthermore, it is essential to calculate the number of H-bonds formed during the simulation, as few bonds were simultaneously broken and rebuilt. During the simulation with α-glucosidase, acarbose had formed 9, while 6i formed 8 hydrogen bonds. In the case of α-amylase, 6i edged over acarbose to form 5 bonds, whereas, acarbose had 4 hydrogen bonds. Whereas, in the case of human serum albumin, where aminoguanidine had formed 7 hydrogen bonds, however, 6i formed 5 of them. Therefore, by considering all the MD trajectories, it can be deduced that compound 6i had stability and extensive interaction in the case of all three proteins.
Binding free energy calculations
Evaluation of binding free energies is essential, as the concept dwells with the stability and extent of binding interaction of ligands with the protein. In all the protein-ligand complexes formed, acarbose, aminoguanidine and 6i used van der Waals's energy to form the complexes. This was followed by binding energy. Polar solvation energy was found to be not involved with the protein-ligand complexes, as most of the values were positive. Moreover, the binding free energies of the protein-6i complex were comparatively more negative (stable complex) than both the controls. In addition, the binding free energies shown by the protein-control drug complexes were deviating from the real values. This shows that the binding of 6i with all the 3 target proteins is stable and stronger, in comparison with the control drugs. The details of binding free energy calculations are given in Table 9.| 6i- α–glucosidase complex | Acarbose- α-glucosidase complex | 6i- α-amylase complex | Acarbose- α-glucosidase complex | 6i- Human serum albumin complex | Aminoguanidine- Human serum albumin complex | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Values (kJ mol−1) | Standard deviation (kJ mol−1) | Values (kJ mol−1) | Standard deviation (kJ mol−1) | Values (kJ mol−1) | Standard deviation (kJ mol−1) | Values (kJ mol−1) | Standard deviation (kJ mol−1) | Values (kJ mol−1) | Standard deviation (kJ mol−1) | Values (kJ mol−1) | Standard deviation (kJ mol−1) | |
| van der Waal energy | −316.391 | ±15.473 | −218.605 | ±145.706 | −169.669 | ±101.479 | −150.112 | ±115.233 | −231.156 | ±12.251 | −37.584 | ±17.357 |
| Electrostatic energy | −21.871 | ±5.801 | −4.761 | ±6.221 | −6.992 | ±11.374 | −10.911 | ±6.801 | −23.992 | ±10.977 | −19.990 | ±12.655 |
| Polar solvation energy | 107.897 | ±13.989 | 103.307 | ±55.952 | 79.945 | ±50.793 | 60.951 | ±25.681 | 125.581 | ±30.228 | 38.908 | ±29.180 |
| SASA energy | −21.576 | ±0.997 | −17.835 | ±13.498 | −12.899 | ±7.329 | −15.929 | ±6.997 | −19.988 | ±1.220 | −5.720 | ±1.880 |
| Binding energy | −251.941 | ±22.094 | −137.894 | ±122.951 | −109.615 | ±73.901 | −112.119 | ±46.114 | −149.555 | ±26.000 | −24.385 | +/20.763 |
Conclusion
In summary, we have developed a highly efficient synthesis of spirooxindole pyrrolidine (5a–l) and pyrrolizidine (6a–l) by 1,3-dipolar cycloaddition reaction of benzodioxole chalcones (1a–l) with azomethine ylide generated by the reaction of isatin (2) and sarcosine (3)/L-proline (4) in presence of methanol to afford desired products in high yield. The stereochemistry of the products was unambiguously confirmed by FT-IR, NMR and HR-MS techniques. Additionally, the formation of a single regio- and stereoselective of the desired products were confirmed by single crystal X-ray diffraction studies of compound 6i (CCDC: 2143927). Among the synthesized monospirooxindole compounds, compound 6i showed stronger inhibition against α-glucosidase and α-amylase enzymes. This can be attributed to the presence of the fluoro-group in the C-3 position, which results in the enhanced activity. Among the synthesized spiro heterocyclic motifs, compounds 6 exhibited stronger activity against α-glucosidase and α-amylase enzymes. This is probably due to the presence of oxindole and pyrrolizidine rings, which enhanced the diabetic activity. Further, comparing the IC50 values of standard drugs under similar conditions shown that, the IC50 values for the spirooxindole derivatives are significantly lower than their conventional counterparts, demonstrating excellent diabetic efficacy of the spirooxindole substrates.Conflicts of interest
There are no conflicts to declare.Notes and references
- B. Ganem, Acc. Chem. Res., 2009, 42, 463–472 CrossRef CAS PubMed.
- J. D. Sunderhaus and S. F. Martin, Chem.–Eur. J., 2009, 15, 1300–1308 CrossRef CAS PubMed.
- B. Yu, D. Q. Yu and H. M. Liu, Eur. J. Med. Chem., 2015, 97, 673–698 CrossRef CAS PubMed.
- C. Marti and E. M. Careira, Eur. J. Med. Chem., 2003, 12, 2209–2219 Search PubMed.
- (a) C. Teja, S. N. Babu, A. Noor, J. A. Daniel, S. A. Devi, F. Rahman and N. Khan, RSC Adv., 2020, 10, 12262–12271 RSC; (b) A. Toumi, S. Boudriga, K. Hamden, M. Sobeh, M. Cheurfa, M. Askri, M. Knorr, C. Strohmann and L. Brieger, Bioorg. Chem., 2021, 106, 104507 CrossRef CAS PubMed.
- A. Thangamani, Eur. J. Med. Chem., 2010, 45, 6120–6126 CrossRef CAS PubMed.
- (a) K. Ding, Y. Lu, Z. Nikolovska-Coleska, G. P. Wang, S. Qiu, S. Shangary, W. Gao, D. G. Qin, J. Stuckey, K. Krajewski, P. P. Roller and S. M. Wang, J. Am. Chem. Soc., 2005, 127, 10130–10131 CrossRef CAS PubMed; (b) K. Ding, Y. P. Lu, Z. Nikolovska-Coleska, G. P. Wang, S. Qiu, S. Shangary, W. Gao, D. G. Qin, J. Stuckey, K. Krajewski, P. P. Roller and S. M. Wang, J. Med. Chem., 2006, 49, 3432–3435 CrossRef CAS PubMed.
- B. K. S. Yeung, B. Zou, M. Rottmann, S. B. Lakshminarayana, S. H. Ang, S. Y. Leong, J. Tan, J. Wong, S. Keller-Maerki, C. Fischi, A. Goh, E. K. Schmitt, P. Krastel, E. Francotte, K. Kuhen, D. Plouffe, K. Henson, T. Wagner, E. A. Winzeler, F. Petersen, B. Reto, V. Dartois, T. T. Diagana and T. H. Keller, J. Med. Chem., 2010, 53, 5155–5164 CrossRef CAS PubMed.
- K. Lundahl, J. Schut, J. L. M. A. Schlatmann, G. B. Paerels and A. Peters, J. Med. Chem., 1972, 15, 129–132 CrossRef CAS PubMed.
- (a) M. M. Khafagy, A. H. F. A. El Wahas, F. A. Eid and A. M. El Agrody, Farmaco, 2002, 57, 715–722 CrossRef CAS PubMed; (b) P. R. Sebahar and R. M. Williams, J. Am. Chem. Soc., 2000, 122, 5666–5667 CrossRef CAS; (c) A. Bertamino, C. Aquino, M. Sala, N. D. Simone, C. A. Mattia, L. Erra, S. Musella, P. Iannelli, A. Carotenuto, P. Grieco, E. Novellino, P. Campiglia and I. Gomez-Monterrey, Bioorg. Med. Chem., 2010, 18, 4328–4337 CrossRef CAS PubMed.
- (a) T. H. Kang, K. Matsumoto, Y. Murakami, H. Takayama, M. Kitajima, N. Aimi and H. Watanabe, Eur. J. Pharmacol., 2002, 444, 39–45 CrossRef CAS PubMed; (b) C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748–8758 CrossRef CAS PubMed.
- J. Rodriguez and D. Bonne, Stereoselective Multiple Bond-Forming Transformations in Organic Synthesis, John Wiley & Sons, Inc., New Jersey, 1st edn, 2015 Search PubMed.
- (a) A. R. Suresh Babu, R. Raghunathan, G. Gayatri and G. N. Sastry, J. Heterocycl. Chem., 2006, 43, 1467–1472 CrossRef; (b) A. A. Watson, G. W. J. Fleet, N. Asano, R. J. Molyneux and R. J. Nash, Phytochemistry, 2001, 56, 265–295 CrossRef CAS PubMed; (c) D. O'Hagan, Nat. Prod. Rep., 1997, 14, 637–651 RSC; (d) S. Horri, H. Fukase, T. Matsuo, Y. Kameda, N. Asano and K. Matsui, J. Med. Chem., 1986, 29, 1038–1046 CrossRef PubMed; (e) M. A. Spearman, J. C. Jamieson and J. A. Wright, Exp. Cell Res., 1987, 168, 116–126 CrossRef CAS PubMed; (f) A. Karpas, G. W. J. Fleet, R. A. Dwek, S. Petursson, S. K. Mamgoong, N. G. Ramsden, G. S. Jacob and T. W. Rademacher, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 9229–9233 CrossRef CAS PubMed; (g) J. R. Liddell, Nat. Prod. Rep., 1998, 15, 363–370 RSC; (h) J. P. Michael, Nat. Prod. Rep., 1997, 14, 619–636 RSC.
- (a) T. Hashimoto and K. Maruoka, Chem. Rev., 2015, 115, 5366–5412 CrossRef CAS PubMed; (b) Y. Gu, Green Chem., 2012, 14, 2091–2128 RSC; (c) R. C. Cioc, E. Ruijter and R. V. A. Orru, Green Chem., 2014, 16, 2958–2975 RSC; (d) A. Nagaraju, B. J. Ramulu, G. Shukla, A. Srivastava, G. K. Verma, K. Raghuvanshi and M. S. Singh, Green Chem., 2015, 17, 950–958 RSC; (e) M. Li, A. Taheri, M. Liu, S. Sun and Y. Gu, Adv. Synth. Catal., 2014, 356, 537–556 CrossRef CAS.
- (a) A. Padwa, Chem. Soc. Rev., 2009, 38, 3072–3081 RSC; (b) A. Domling, Chem. Rev., 2006, 106, 17–89 CrossRef PubMed; (c) D. M. D'Souza and T. J. J. Muller, Chem. Soc. Rev., 2007, 36, 1095–1108 RSC; (d) D. Tejedor and F. G. Tellado, Chem. Soc. Rev., 2007, 36, 484–491 RSC; (e) V. Polshettiwar and R. S. Varma, Chem. Soc. Rev., 2008, 37, 1546–1557 RSC.
- C. Hulme and V. Gore, Curr. Med. Chem., 2003, 10, 51–80 CrossRef CAS PubMed.
- (a) C. J. O'Connor, H. S. G. Beckmann and D. R. Spring, Chem. Soc. Rev., 2012, 41, 4444–4456 RSC; (b) E. Ruijter, R. Scheffelaar and R. V. A. Orru, Angew. Chem., Int. Ed., 2011, 50, 6234–6246 CrossRef CAS PubMed.
- J. Elflein, Pharma and Medtech, 2021 Search PubMed.
- M. Marcovecchio, A. Mohn and F. Chiarelli, J. Endocrinol. Invest., 2005, 28, 853–863 CrossRef CAS PubMed.
- H. Rasouli, S. M. Hosseini-Ghazvini, H. Adibi and R. Khodarahmi, Food Funct., 2017, 8, 1942–1954 RSC.
- J. R. N. Taylor, M. N. Emmambux and J. Kruger, Starch-Starke, 2015, 67, 79–89 CrossRef CAS.
- P. M. Sales, P. M. Souza, L. A. Simenoni, P. O. Magalhaes and D. Sliveria, J. Pharm. Pharm. Sci., 2012, 15, 141–183 Search PubMed.
- Y. Tan, S. K. C. Chang and Y. Zhang, Food Chem., 2017, 214, 259–268 CrossRef CAS PubMed.
- W. Puls, U. Keup, H. Krause, P. G. Thomas and F. Hoffmeister, Naturwissenschaften, 1977, 64, 536–537 CrossRef CAS PubMed.
- K. V. Dileep, K. Nithiyanandan and C. Remya, J. Biomol. Struct. Dyn., 2018, 36, 3354–3361 CrossRef CAS PubMed.
- O. J. Afonne, O. E. Orisakwe, E. Obi, C. Orish and D. D. Akumka, Indian J. Pharmacol., 2000, 32, 239–241 Search PubMed.
- P. Daisy, R. Jasmine, S. Ignacimuthu and E. Murugan, J. Phytomed., 2009, 16, 252–257 CrossRef CAS PubMed.
- A. Shirwaikar, K. Rajendran and I. S. R. Punitha, J. Ethnopharmacol., 2005, 97, 369–374 CrossRef PubMed.
- O. Akerele, World Health Forum, 1993, 14, 390–395 CAS.
- R. Geethalakshmi, D. V. L. Sarada, P. Marimuthu and K. Ramasamy, Int. J. Biotechnol. Biochem., 2010, 6, 369–376 Search PubMed.
- S. Dewanjee, A. K. Das, R. Sahu and M. Gangopadhyay, Food Chem. Toxicol., 2009, 47, 2679–2685 CrossRef CAS PubMed.
- N. Nivetha and A. Thangamani, J. Mol. Struct., 2021, 1242, 130716 CrossRef CAS.
- L. D. Chiaradia, P. G. A. Martins, M. N. S. Cordeiro, R. V. C. Guido, G. Ecco, A. D. Andricopulo, R. A. Yunes, J. Vernal, R. J. Numes and H. Terenzi, J. Med. Chem., 2012, 55, 390–402 CrossRef CAS PubMed.
- C. E. P. Galvis and V. V. Kouznetsov, Org. Biomol. Chem., 2013, 11, 7372–7386 RSC.
- (a) A. Dandia, A. K. Jain and D. S. Bhati, Tetrahedron Lett., 2011, 52, 5333–5337 CrossRef CAS; (b) H. Liu, G. Dou and D. Shi, J. Comb. Chem., 2010, 12, 633–637 CrossRef CAS PubMed.
- R. Satheeshkumar, L. Edatt, V. B. S. Kumar and K. J. R. Prasad, ChemistrySelect, 2017, 2, 2626–2633 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2143927. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2ra04452h |
| This journal is © The Royal Society of Chemistry 2022 |