DOI:
10.1039/D2RA04408K
(Paper)
RSC Adv., 2022,
12, 23839-23844
Enzymatic synthesis of vitexin glycosides and their activity†
Received
17th July 2022
, Accepted 15th August 2022
First published on 23rd August 2022
Abstract
Vitexin is an active component of many traditional chinese medicines, and is found in various plants. The low solubility of vitexin limits its pharmaceutical usage. In this study, solvent-stable β-fructosidase was used to glycosylate vitexin in organic solvents. The β-fructosidase showed high activity and stability in 30–80% (v/v) ethyl acetate with 90–99% yields of vitexin glycosides. Highly efficient synthesis of β-D-fructofuranosyl-(2→6)-vitexin (1.04 g L−1) and β-D-difructofuranosyl-(2→6)-vitexin (0.45 g L−1) was attained in 50% (v/v) ethyl acetate solvent system from 1.5 g L−1 vitexin. Two novel vitexin glycosides showed higher anti-tumor activities compared to that of vitexin by employing a human breast cancer cytotoxicity assay.
1. Introduction
Vitexin (apigenin-8-C-glucoside) is a bioactive ingredient found in many traditional chinese medicines. As a C-glycosylated flavone, vitexin was isolated from various plants, such as hawthorn, mung bean, pearl millet, Passiflora, pigeon pea, bamboo, wheat leaves, mimosa, chaste tree, etc.1–5 It was reported to have anti-tumor,6–8 anti-oxidant,9 anti-viral,10 and hepatoprotective activities.11,12 Thus, vitexin is a potential phytochemical constituent for pharmaceutical usage. However, the poor aqueous solubility of vitexin is a serious issue which limits its pharmaceutical application. Glycosylation has been used to increase the solubility and bioactivity of various natural products.13,14 However, the chemical method requires glycosyl activation and complicated reaction steps to control regio-selectivity. In contrast, enzymatic glycosylation has attracted extensive interest because of its exquisite selectivity and mild reaction conditions.15,16 There are two types of enzymes used for glycosylation: glycosyltransferases and glycosidases.17,18 Glycosyltransferases catalyze the formation of glycosidic bonds needed to supply an activated sugar donor, such as uridine-diphosphate glucose (UDP-G), which constrained its practical application. Glycosylation with glycosidases is under thermodynamic control using a non-activated sugar donor, which made these enzymes attractive in the low-cost synthesis of glycosides.18 Enzymatic synthesis of vitexin glucosides has been reported by Chang;19 two glycosidases and four glycosyltransferases were selected to glycosylate vitexin. The glycosyltransferase (BtGT_16345) from Bacillus thuringiensis showed glycosylation activity, catalyzing the conversion of vitexin-4′-O-β-glucoside (17.5% yield) and vitexin-5-O-β-glucoside (18.6% yield).19 However, the glycosidases (amylosucrase and maltogenic amylase) did not catalyze the glycosylation of vitexin.19 The reported enzymatic glycosylation of vitexin is not efficient with lower yields mainly due to the poor solubility issue of the substrate. The above results indicate that efficient glycosylation of vitexin is still more challenging for glycosyltransferases and glycosidases.
Interestingly, enzymatic glycosylation in organic solvents provides some industrially attractive advantages, such as reversing the thermodynamic equilibrium of glycosidases hydrolysis reactions, improving the aqueous solubility of hydrophobic substrates, eliminating microbial contamination, and lowing industrial cost process.20,21 By using this approach, solvent-stable glycosidase (β-fructosidase from Arthrobacter nicotianae) showed high activity and stability in hydrophilic organic solvents with more than 90% yield of puerarin glycosides.20 In this study, we demonstrate that the solvent-stable β-fructosidase can be efficiently used for the glycosylation of vitexin, and evaluate the anti-tumor activities of vitexin glycosides.
2. Materials and methods
2.1 Materials and regents
Vitexin was purchased from Zelang (Jiangsu, China). Methanol used for HPLC analysis was of chromatographic grade from Sinopharm (Shanghai, China). n-butanol, ethyl acetate, hexane, ethanol, methanol, DMSO were of analytical grade and purchased from Beijing Chemical Factory (Beijing, China). Water used for deionized by an osmosis Milli-Q system (Millipore, USA). All other chemicals were of analytical grade and purchased from Sunshine (Nanjing, China). The β-fructosidase was purified from Arthrobacter nicotianae XM6 (CCTCC M2010164).20
2.2 Enzymatic glycosylation of vitexin
The glycosylation reaction (1.0 L) was conducted in 1/15 M Na2HPO4/KH2PO4 buffer (pH 6.47) containing 0.25 U mL−1 β-fructosidase, 10% (w/v) sucrose and 1.5 g L−1 vitexin and 10–80% (v/v) organic solvent. The reaction mixture was conducted at 35 °C with shaking at 180 rpm for 24 h, then the reaction was stopped at 100 °C for 5 min.
2.3 Kinetic analysis of β-fructosidase
The kinetic parameters were determined in the reaction mixture containing vitexin as substrate and 50 mM sucrose in Na2HPO4/KH2PO4 buffer (1/15 M, pH 6.47). Substrate concentration was varied in the range 20–200 μM. The purified β-fructosidase (0.25 U mL−1) was then added, and the reaction was incubated at 35 °C for 10 min, and 30 min for organic solvent reaction systems. Then, the reaction was stopped by adding 900 μL of methanol to 100 μL of reaction solution. The reaction products were analyzed by HPLC. One unit (U) of activity was equivalent to 1 μM of vitexin converted by the amount of enzyme per minute at described conditions. Each analysis was carried out in triplicate.
2.4 Analysis, isolation and identification of products
In general, products were analyzed by reversed-phase liquid chromatography on Agilent 1260 HPLC, using a XBridge C18 column (5 μm, 4.6 × 250 mm). The HPLC analysis method was performed using the following gradient: 0.0–13.0 min, 10–100% B; 13.0–20.0 min, 100% B; 20.0–20.1 min, 100–10% B; 20.1–23.0 min, equilibration with 10% B. The mobile phase consisted of solvents A (water) and B (methanol). The column was operated with a constant flow rate of 0.8 mL min−1 at 30 °C and a sample injection volume of 20 μL. The liquid chromatography hyphened with the high-resolution electrospray ionization mass spectrometry (HR-ESI-MS) was run on an Agilent 6546 Accurate-Mass Q-TOF LC/MS instrument with a Poroshell 120 EC-C18 column (2.7 μm, 4.5 × 50 mm). The LC-HR/MS analysis of products was performed using the “A + B” mobile phase as specified below: 0.0–13.0 min, 10–100% B. The column was operated with a constant flow rate of 0.3 mL min−1 at 30 °C. Glycosylation products were purified by semipreparative HPLC on an Agilent Technologies 1260 Infinity LC System (Agilent, California, USA) by an Agilent Eclipse XDB-C18 column (5 μm, 9.4 × 250 mm) at a flow rate of 2.0 mL min−1. For structure elucidation, NMR spectra were recorded on a Bruker AV-600 NMR spectrometer (Bruker, Fällanden, Switzerland). Samples were dissolved in DMSO-d6 at room temperature with tetramethylsilane (Me4Si) as the chemical shift reference.
2.5 Determination of aqueous solubility
The aqueous solubility of vitexin and vitexin glucosides was determined as follows. Each compound was vortexed in double-deionized H2O for 30 min at 25 °C. The mixture was centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 20 min at 25 °C. The supernatant was filtered through a 0.2 μm nylon membrane, mixed with an equal volume of methanol, and analyzed by HPLC. The concentration of these compounds was determined on the basis of the peak areas, using calibration curves prepared by HPLC analyses of authentic samples.
000 × g for 20 min at 25 °C. The supernatant was filtered through a 0.2 μm nylon membrane, mixed with an equal volume of methanol, and analyzed by HPLC. The concentration of these compounds was determined on the basis of the peak areas, using calibration curves prepared by HPLC analyses of authentic samples.
2.6 Cell lines and culture conditions
Human breast cancer cells MCF-7 and MDA- MB-231 were purchased from the Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, China). Two cells were maintained in 90% DMEM (HyClone, USA) supplemented with 2 mM glutamine, 100 U mL−1 penicillin, 100 μg mL−1 streptomycin and 10% heat-inactivated fetal bovine serum at 37 °C in a humidified incubator with 5% CO2.
2.7 Growth inhibition assay
The cytotoxicity effects of β-D-fructofuranosyl-(2→6)-vitexin, β-D-difructofuranosyl-(2→6)-vitexin and vitexin were determined by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide; solarbio, China) assay.22 Approximately 1 × 104 cells suspended in 100 μL of growth medium were added to each well of a 96 well plates and were incubated. The cells were treated with vitexin, β-D-fructofuranosyl-(2→6)-vitexin and β-D-difructofuranosyl-(2→6)-vitexin at final concentrations of 10, 20, 40, 80, 160, 250 μM for 48 h. Cells treated with dimethyl sulfoxide (DMSO) alone (0.01%) were used as control. Each concentration was tested with 6 replicate wells. After 48 h, the medium containing the treatments was removed, and 100 μL of MTT solution (0.5 mg mL−1) was added to each well and incubated at 37 °C for 3 h. The formazan crystals were dissolved in 100 μL of DMSO, and the absorbance was measured at 490 nm with ELISA plate reader. The experiment was performed in triplicate.
3. Results and discussion
3.1 The structural identification
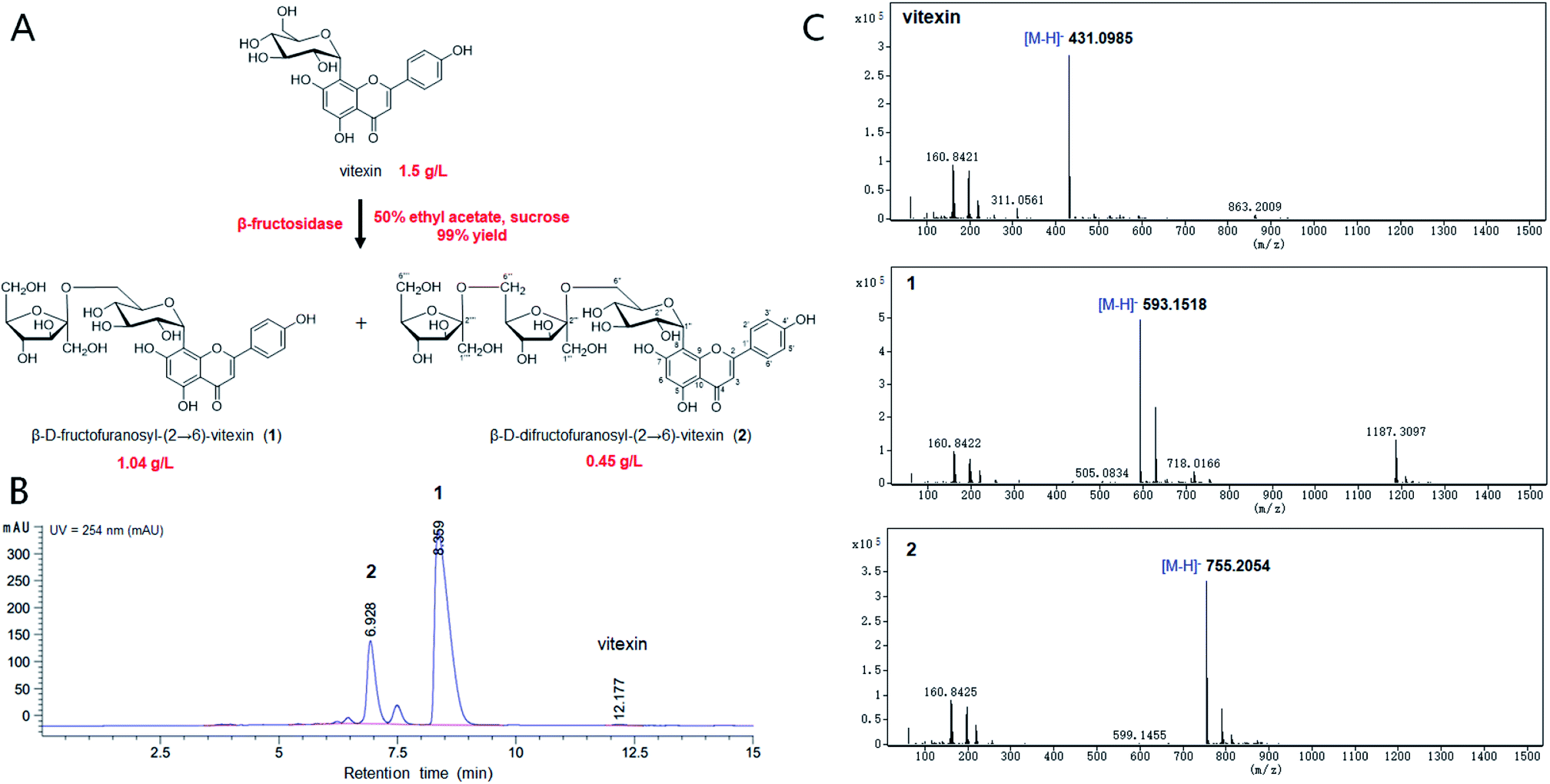
The chemical structure of the peak fractions was further identified by LC-HR/MS, 1H NMR, 13C NMR, and 2D NMR spectra. The HR-ESI mass spectrum of compound 1 showed an ion peak [M–H]− at m/z 593.1518 (calculated for C27H30O15, 594.1585) (Fig. 1). Compared with vitexin, the compound 1 has an additional formula C6H10O5, suggesting that 1 may be a vitexin monosaccharide derivative. The 1H NMR spectra in low field showed six ethylenic signals [δ 6.77(1H, s), 6.27 (1H, s), 7.95 (2H, d, 8.8 Hz), 6.91 (2H, d, 8.8 Hz)] (Table 1). These were the characteristic vitexin signals. The high field peak δ 4.70 (d, 9.9 Hz) was the core vitexin glucose terminal hydrogen signal. The δ 73.9 ppm methylene may be β-sugar terminal carbon, which was connected with C–C bond. The δ 104.8 ppm quaternary carbon must be the other sugar terminal carbon. Data from HSQC spectra can classify all carbon signals except quaternary carbon. The connections of each self-spinning segment can be inferred by HMBC spectra. From the 13C NMR, 1H NMR and 2D NMR spectra, we speculated that the monosaccharide was β-D-fructofuranose. A comparison analysis between the NMR data of compound 1 and vitexin revealed that the NMR data of two compounds were almost identical except for the signals due to a fructose moiety. The other diagnostic difference between 1 and vitexin was the carbon signal at C-6'' (δC 62.8) of the glucose moiety, which was about 1.1 ppm downfield from those of vitexin (δC 61.7) due to the known effects of O-glycosylation. Furthermore, the carbon signal of C-5'' (δC 75.8) was about 6.5 ppm upfield from those of vitexin (δC 82.3). The HMBC spectrum (Fig. S7†) showed long-range correlations between C-2′′′ (δC 104.8) and H-6′′ of glucose moiety and H-1′′′ of fructose moiety, thus proving the correlation between C-2′′′ and H-6′′ of compound 1. Therefore, the chemical structure of 1 was elucidated as β-D-fructofuranosyl-(2→6)-vitexin.
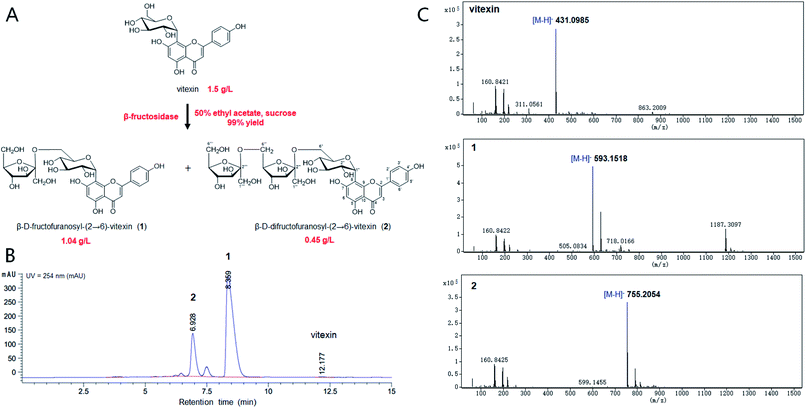 |
| | Fig. 1 Bioconversion of vitexin glycosides by β-fructosidase. (A) The chemical structures of vitexin and vitexin glycosides. (B) HPLC profiling of biotransformation for vitexin glycosides. (C) The LC-HR/MS analysis of the reaction products. | |
Table 1 NMR (600 MHz) spectral data of vitexin and glucosides in DMSO-d6
| Position |
Vitexin |
β-D-Fructofuranosyl-(2→6)-vitexin |
β-D-Difructofuranosyl-(2→6)-vitexin |
| δC |
δH |
δC |
δH |
δC |
δH |
| 2 |
164.4 |
|
164.3 |
|
164.4 |
|
| 3 |
102.9 |
6.79 (s) |
103.0 |
6.77 (s) |
102.9 |
6.77 (s) |
| 4 |
182.5 |
|
182.6 |
|
182.6 |
|
| 5 |
160.8 |
|
161.0 |
|
161.0 |
|
| 6 |
98.6 |
6.28 (s) |
98.7 |
6.27 (s) |
98.7 |
6.28 (s) |
| 7 |
163.1 |
|
163.3 |
|
163.2 |
|
| 8 |
105.1 |
|
104.8 |
|
104.6 |
|
| 9 |
156.4 |
|
156.5 |
|
156.5 |
|
| 10 |
104.5 |
|
104.5 |
|
104.5 |
|
| 1′ |
122.1 |
|
122.1 |
|
122.1 |
|
| 2′ |
129.4 |
8.03 (d, J = 8.7 Hz) |
129.1 |
7.95 (d, J = 8.8 Hz) |
129.2 |
7.95 (d, J = 8.5 Hz) |
| 3′ |
116.3 |
6.90 (d, J = 8.7 Hz) |
116.3 |
6.91 (d, J = 8.8 Hz) |
116.3 |
6.91 (d, J = 8.0 Hz) |
| 4′ |
161.6 |
|
161.6 |
|
161.6 |
|
| 5′ |
116.3 |
6.90 (d, J = 8.7 Hz) |
116.3 |
6.91 (d, J = 8.8 Hz) |
116.3 |
6.91 (d, J = 8.0 Hz) |
| 6′ |
129.1 |
8.03 (d, J = 8.7 Hz) |
129.1 |
7.95 (d, J = 8.8 Hz) |
129.2 |
7.95 (d, J = 8.5 Hz) |
| 1′′ |
73.8 |
4.69 (d, J = 9.9 Hz) |
73.9 |
4.70 (d, J = 9.9 Hz) |
73.9 |
4.71 (d, J = 9.9 Hz) |
| 2′′ |
71.3 |
3.84 (t) |
71.7 |
|
71.4 |
|
| 3′′ |
79.1 |
3.26 (m) |
79.0 |
|
78.9 |
|
| 4′′ |
71.0 |
3.39 (m) |
71.2 |
|
71.2 |
|
| 5′′ |
82.3 |
3.77 (m) |
75.8 |
|
75.6 |
|
| 6′′ |
61.7 |
3.53 (m) |
62.8 |
|
63.0 |
|
| 1′′′ |
|
|
63.0 |
|
63.1 |
|
| 2′′′ |
|
|
104.8 |
|
104.7 |
|
| 3′′′ |
|
|
80.4 |
|
80.7 |
|
| 4′′′ |
|
|
77.4 |
|
77.0 |
|
| 5′′′ |
|
|
82.5 |
|
76.2 |
|
| 6′′′ |
|
|
62.0 |
|
62.9 |
|
| 1′′’ |
|
|
|
|
61.4 |
|
| 2′′’ |
|
|
|
|
104.6 |
|
| 3′′’ |
|
|
|
|
80.3 |
|
| 4′′’ |
|
|
|
|
76.9 |
|
| 5′′’ |
|
|
|
|
82.5 |
|
| 6′′’ |
|
|
|
|
61.9 |
|
| 13-OH |
|
13.18 (s) |
|
13.17 (s) |
|
13.17 (s) |
The HR-ESI mass spectrum of compound 2 showed an ion peak [M–H]− at m/z 755.2054 (calculated for C33H40O20, 756.2113) (Fig. 1). Compared with vitexin, the compound 1 has an additional formula C12H20O10, suggesting that 1 may be a vitexin disaccharide derivative. The 1H NMR spectra in low field showed six ethylenic signals [δ 6.77(1H, s), 6.28 (1H, s), 7.95 (2H, d, 8.5 Hz), 6.91 (2H, d, 8.0 Hz)] (Table 1). These were the characteristic vitexin signals. The high field peak δ 4.71 (d, 9.9 Hz) was the core vitexin glucose terminal hydrogen signal. The δ 73.9 ppm methylene should be β-sugar terminal carbon, which was connected with C–C bond. The δ 104.7 ppm and δ 104.6 ppm quaternary carbon should be the fructose terminal carbon. The difference between 2 and vitexin was the carbon signal at C-6'' (δC 63.0) of the glucose moiety, which was about 1.3 ppm downfield from those of vitexin (δC 61.7) due to the known effects of O-glycosylation. And other diagnostic difference between 2 and 1 were carbon signal at C-6′′′ (δC 62.9) and C-5′′′ (δC 76.2) of the fructose, which were about 0.9 ppm downfield and 6.3 ppm upfield from those of compound 1 (δC 62.0 and δC 82.5), respectively. The HMBC spectrum also showed long-range correlations between C-2′′′' (δC 104.6) and H-6′′′ of fructose moiety and H-1′′′' of fructose moiety (Fig. S14†), thus proving the correlation between C-2′′′' and H-6′′′ of compound 2. Therefore, the chemical structure of 2 was elucidated as β-D-difructofuranosyl-(2→6)-vitexin. The spectroscopic data for 1H and 13C-NMR (600 MHz, DMSO-d6) were summarized in Table 1.
3.2 Efficient glycosylation in hydrophobic organic solvents
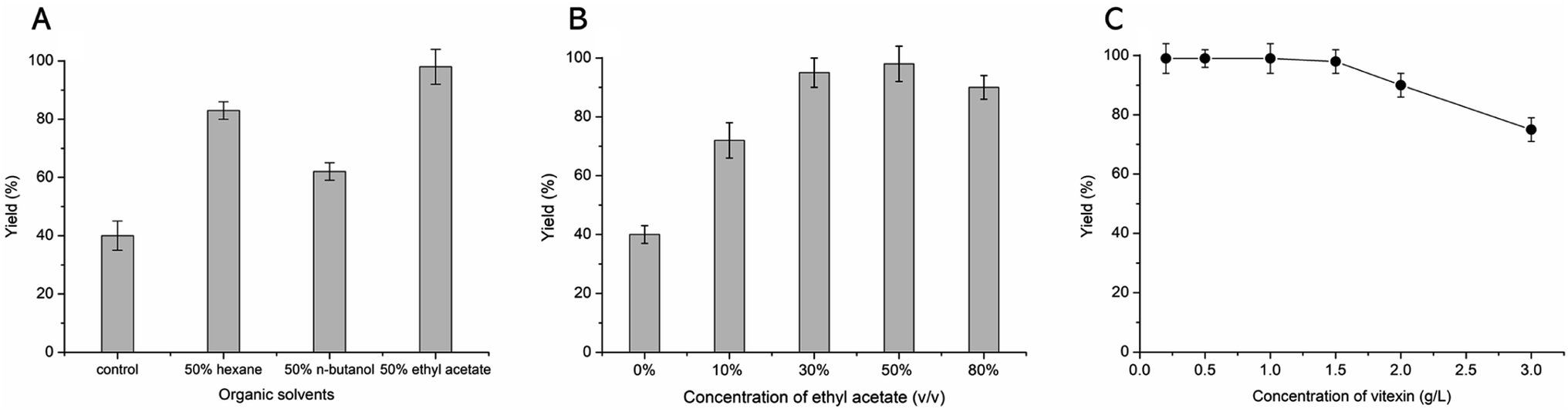
The β-fructosidase from Arthrobacter nicotianae has been shown excellent synthesis of puerarin glycosides in hydrophilic organic solvent.20 However, the solubility of vitexin is not obvious increased in hydrophilic organic solvent. In order to improve the solubility of vitexin, the enzymatic synthesis was investigated in hydrophobic organic solvents. The low yield (40%) of glycosylation was observed in aqueous solution because of the poor solubility of vitexin (Fig. 2A). Supplement with several hydrophobic organic solvents (50%, v/v) led to a significant improvement in the glycosylation of vitexin. The higher yield (99%) of vitexin glycosides was obtained in 50% ethyl acetate solvent system compared with hexane and n-butanol solvent systems (Fig. 2A). The effect of ethyl acetate proportion on the vitexin glycosylation was studied with the increase of the concentration from 0% to 80%. The glycosylation reaction system with addition of 10%, 30% and 50% of ethyl acetate led to 1.8, 2.3 and 2.4 times yield than in aqueous solution (Fig. 2B). Then the inhibition effect on glycosylation was appeared when the concentration of ethyl acetate up to 80%. Furthermore, the optimization of vitexin concentration was conducted shown in Fig. 2C. The efficient glycosylation was achieved in 1.5 g L−1 substrate concentration with 99% yield of vitexin glycosides. The β-D-fructofuranosyl-(2→6)-vitexin (1.04 g L−1) and β-D-difructofuranosyl-(2→6)-vitexin (0.45 g L−1) was attained in 50% (v/v) ethyl acetate solvent after 48 h of bioconversion.
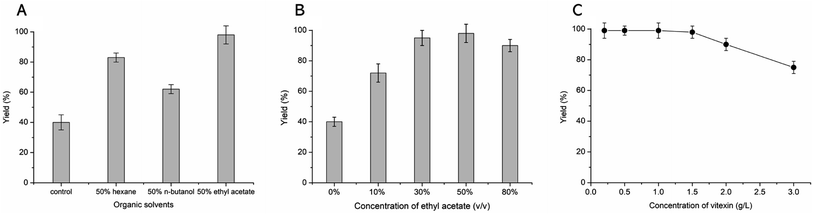 |
| | Fig. 2 Optimized bioconversion conditions for the glycosylation of vitexin. The effect of organic solvents (A), concentration of ethyl acetate (B) and concentration of substrate (C) on the vitexin glycosides yield. | |
3.3 Kinetic analysis of β-fructosidase in organic solvent systems
The kinetic parameters were determined using the purified β-fructosidase in various systems are listed in Table 2. Compared to the buffer media, Vmax of the β-fructosidase to vitexin slight slowed from 5.6 μM s−1 to 4.9, 3.8 and 5.5 μM s−1 in 50% (v/v) of hexane, n-butanol and ethyl acetate solvent, respectively. This observation suggests that β-fructosidase possesses great solvent tolerance, and especially shows excellent activity in 50% (v/v) of ethyl acetate. The Km values in organic solvents (Table 2) suggest that β-fructosidase showed almost equal affinity for vitexin in water and 50% (v/v) of ethyl acetate solvent. The catalytic turnover constant (Kcat, 64.3 s−1 and 62.8 s−1) and the catalytic efficiency (Kcat/Km, 555.3 s−1 mM−1 and 549.4 s−1 mM−1) were found to be nearly the same in water and 50% (v/v) of ethyl acetate. The excellent values of Kcat and Kcat/Km in 50% (v/v) of ethyl acetate suggest that the β-fructosidase may be beneficial for improving the yield of vitexin glycosides in the solvent system.
Table 2 Kinetic parameters of β-fructosidase in different media
| Solvent |
Vmaxa [μM s−1] |
Km [μM] |
Kcatb [s−1] |
Kcat/Km [s−1 mM−1] |
| Sucrose fixed at 50 mM, vitexin varied from 20 μM to 200 μM. Kcat values were calculated using a molecular mass of 55 KDa for the β-fructosidase. |
| Water |
5.6 |
115.8 |
64.3 |
555.3 |
| 50% hexane |
4.9 |
112.7 |
58.1 |
515.5 |
| 50% n-butanol |
3.8 |
82.6 |
41.4 |
501.2 |
| 50% ethyl acetate |
5.5 |
114.3 |
62.8 |
549.4 |
3.4 Solubility of vitexin glucosides
The aqueous solubility of vitexin glucosides were obviously improved through glycosylation (Table 3). The solubility of β-D-fructofuranosyl-(2→6)-vitexin and β-D-difructofuranosyl-(2→6)-vitexin was over 100 times and 300 times than vitexin, respectively. The increase aqueous solubility of vitexin glucosides could be better improved its clinical application.
Table 3 Aqueous solubility of vitexin and vitexin glycosides
| Compound |
Aqueous solubilitya (mg L−1) |
| Deionized H2O at pH 7.0. |
| Vitexin |
28.2 ± 2.5 |
| β-D-Fructofuranosyl-(2→6)-vitexin |
2961.0 ± 98.5 |
| β-D-Difructofuranosyl-(2→6)-vitexin |
8911.2 ± 637.4 |
3.5 Cytotoxicity assay
MTT assay was performed to compare the cell cytotoxicity induced between vitexin and its glycosides. As shown in Table 4, vitexin was showed effective cytotoxicity on MCF-7 cells (IC50 98.5 μg mL−1) and MDA-MB-231 cells (IC50 65.8 μg mL−1) after 48 h of incubation. The cytotoxicity of vitexin glycosides were better than that of vitexin, β-D-fructofuranosyl-(2→6)-vitexin presented IC50 values of 81.9 and 52.4 μg mL−1 for MCF-7 and MDA-MB-231 cell lines, and β-D-difructofuranosyl-(2→6)-vitexin presented IC50 values of 81.3 and 52.1 μg mL−1, respectively. However, there no obvious difference of antitumor activity between β-D-fructofuranosyl-(2→6)-vitexin and β-D-difructofuranosyl-(2→6)-vitexin. This suggests that glycosylation of vitexin can enhances the antitumor activity.
Table 4 Cytotoxicity of vitexin and vitexin glycosides
| Compound |
IC50 (Mean ± SD, μg mL−1) |
| MCF-7 |
MDA-MB-231 |
| Vitexin |
98.5 ± 6.2 |
65.8 ± 3.4 |
| β-D-Fructofuranosyl-(2→6)-vitexin |
81.9 ± 5.6 |
52.4 ± 2.5 |
| β-D-Difructofuranosyl-(2→6)-vitexin |
81.3 ± 6.7 |
52.1 ± 3.8 |
4. Conclusions
The aim of this study was to investigate the applicability of hydrophobic organic solvents for enzymatic glycosylation of vitexin. The yield of vitexin glycosides was significantly improved by using the 50% (v/v) ethyl acetate solvent system compared to the aqueous solution. Moreover, the β-fructosidase showed high stability and activity in 50% (v/v) ethyl acetate solvent system with 99% yield of glycoside products. Two vitexin glycosides exhibited excellent aqueous solubility and better anti-tumor activity.
Author contributions
X.M. Wu conceived and supervised the project. T.T. Xu designed and performed experiments, and wrote the manuscript. All authors contributed to the data analysis and manuscript preparation.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The work was financed by National Natural Science Foundation of China (82174089 and 81991524).
References
- K. Masike, A. de Villiers, D. de Beer, E. Joubert and M. A. Stander, J. Food Compos. Anal., 2022, 106, 104308 CrossRef CAS.
- T. T. Phan, L. T. M. Do, T. M. Lien, T. V. Phung, T. T. H. Nguyen, V. N. Huynh, D. T. T. Ngo, K. P. P. Nguyen and T. T. A. Nguyen, Nat. Prod. Res., 2022, 36, 171–176 CrossRef CAS PubMed.
- M. He, J. W. Min, W. L. Kong, X. H. He, J. X. Li and B. W. Peng, Fitoterapia, 2016, 115, 74–85 CrossRef CAS PubMed.
- Y. J. Fu, Y. G. Zu, W. Liu, L. Zhang, M. H. Tong, T. Efferth, Y. Kong, C. L. Hou and L. Y. Chen, J. Sep. Sci., 2008, 31, 268–275 CrossRef CAS PubMed.
- E. Gaitan, R. H. Lindsay, R. D. Reichert, S. H. Ingbar, R. C. Cooksey, J. Legan, E. F. Meydrech, J. Hill and K. Kubota, J. Clin. Endocrinol. Metab., 1989, 68, 707–714 CrossRef CAS PubMed.
- K. K. Khoja, R. M. J. Howes, R. Hider, P. A. Sharp, I. W. Farrell and G. O. Latunde-Dada, Nutrients, 2022, 14, 784 CrossRef CAS PubMed.
- J. Huang, Y. Zhou, X. Zhong, F. Su and L. Xu, Oxid. Med. Cell. Longevity, 2022, 3129155 CAS.
- P. Q. D. Nguyen, H. T. Nguyen, L. T. K. Nguyen, H. Q. Vo, A. T. Le, T. T. Do and D. V. Ho, Trop. J. Nat. Prod. Res., 2020, 4, 703–707 CrossRef CAS.
- N. Yan, H. Zhang, Z. Zhang, H. Zhang, L. Zhou, T. Chen, S. Feng, C. Ding and M. Yuan, Arabian J. Chem., 2022, 15, 103707 CrossRef CAS.
- J. Tang, B. Dong, M. Liu, S. Liu, Xi. Niu, C. Gaughan, A. Asthana, H. Zhou, Z. Xu, G. Zhang, R. H. Silverman and H. Huang, J. Med. Chem., 2022, 65, 1445–1457 CrossRef CAS PubMed.
- K. K. Noor, M. U. Ijaz, N. Ehsan, A. Tahir, D. K. Yeni, S. M. N. K. Zihad, S. J. Uddin, A. Ashraf and J. Simal-Gandara, Biomed. Pharmacother., 2022, 150, 112934 CrossRef CAS PubMed.
- A. Farooq, A. Iqbal, N. F. Rana, M. Fatima, T. Maryam, F. Batool, Z. Rehman, F. Menaa, S. Azhar, A. Nawaz, F. Amin, Z. M. Mohammedsaleh and S. S. Alrdahe, Int. J. Mol. Sci., 2022, 23, 3131 CrossRef CAS PubMed.
- G. Huang, M. Lv, J. Hu, K. Huang and H. Xu, Mini-Rev. Med. Chem., 2016, 16, 1013–1016 CrossRef CAS PubMed.
- J. Zhao, J. Yang and Y. Xie, Int. J. Pharm., 2019, 570, 118642 CrossRef CAS PubMed.
- D. Piedrabuena, A. Rumbero, E. Pires, A. Leal-Duaso, C. Civera, M. Fernandez-Lobato and M. Hernaiz, RSC Adv., 2021, 11, 24312–24319 RSC.
- P. Nekvasilová, M. Hovorková, Z. Mészáros, L. Petrásková, H. Pelantová, V. Křen, K. Slámová and P. Bojarová, Int. J. Mol. Sci., 2022, 23, 4106 CrossRef PubMed.
- L. Mestrom, M. Przypis, D. Kowalczykiewicz, A. Pollender, A. Kumpf, S. R. Marsden, I. Bento, A. B. Jarzebski, K. Szymanska, A. Chrusciel, D. Tischler, R. Schoevaart, U. Hanefeld and P. L. Hagedoorn, Int. J. Mol. Sci., 2019, 20, 5263 CrossRef CAS PubMed.
- C. Moulis, D. Guieysse, S. Morel, E. Séverac and M. Remaud-Siméon, Curr. Opin. Chem. Biol., 2021, 61, 96–106 CrossRef CAS PubMed.
- J. Y. Wu, T. Y. Wang, H. Y. Ding, Y. R. Zhang and T. T. Chang, Molecules, 2021, 26, 6274 CrossRef CAS PubMed.
- X. M. Wu, J. L. Chu, B. Wu, S. Zhang and B. F. He, Bioresour. Technol., 2013, 129, 659–662 CrossRef CAS PubMed.
- T. T. Xu, C. Wang, S. J. Jiang, T. T. Yang and X. M. Wu, RSC Adv., 2022, 12, 18232–18237 RSC.
- L. Zhang, X. La, J. Tian, H. Li, A. Li, Y. Liu, C. Wu and Z. Li, J. Funct. Foods, 2021, 85, 104620 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b
*b