
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel immunochemotherapy based on immunogenicity-activated and immunosuppression-reversed biomimetic nanoparticles†
Huaqin Zuo‡
 *a,
Junxian Tao‡b,
Manli Wangc,
Xiaoyan Xiea and
Mei Sun*a
*a,
Junxian Tao‡b,
Manli Wangc,
Xiaoyan Xiea and
Mei Sun*a
aDepartment of Hematology, Northern Jiangsu People's Hospital Affiliated to Yangzhou University, Yangzhou, Jiangsu 225001, P. R. China. E-mail: sybilzuo@163.com; smzgwj@126.com
bDepartment of Endocrinology, Northern Jiangsu People's Hospital Affiliated to Yangzhou University, Yangzhou, Jiangsu 225001, P. R. China
cGraduate School of Dalian Medical University, Dalian, Liaoning 116044, P. R. China
First published on 3rd October 2022
Abstract
Studies show that infiltrated myeloid-derived suppressor cells (MDSCs) are vital in the immunosuppressive tumor microenvironment and account for lymphoma refractoriness and recurrence. Here, we developed a biomimetic nanoplatform (PM–PLGA–DOX/GEM) in which platelet membranes (PM) wrap PLGA nanoparticles co-loaded with doxorubicin (DOX) and gemcitabine (GEM). PM–PLGA–DOX/GEM would accumulate in tumor tissues because of the enhanced permeation and retention (EPR) effect and the tumor cell-induced platelet aggregation (TCIPA) effect. GEM could eliminate the MDSCs in tumor tissues, thereby reversing the immunosuppressive tumor microenvironment. Furthermore, DOX could invoke the immunogenic cell death (ICD) of lymphoma cells. Consequently, numerous T cells were recruited and activated to improve the therapeutic effects. This study will offer a potential platform for clinical treatment of lymphoma and other solid tumors.
Introduction
Lymphoma, originating from lymphoid hematopoietic tissues, is one of the top ten malignant tumors with high mortality. Its incidence is increasing year by year and appears to be a trend amongst the younger population.1 Until now, chemotherapy has remained the mainstay for lymphoma treatment. Nevertheless, conventional chemotherapy has limited efficacy due to cumulative dose effects and non-specific biodistribution of agents.2 In recent years, the treatment of lymphoma has entered a new era with the rapid development of tumor immunotherapy.3 However, clinical results of immunotherapy are far from satisfactory compared with its remarkable achievements in preclinical research.4 Studies have shown that the immunosuppressive tumor microenvironment accounts for the unsatisfactory outcome of immunotherapy in the clinic.5,6Myeloid derived suppressor cells (MDSCs), a heterogeneous and immature population of myeloid cells with suppressive properties, are considered as the essential cells to maintain the immunosuppressive tumor microenvironment.7 MDSCs can suppress both innate and adaptive immune responses, and lymphoma prognosis is negatively correlated with the infiltrated number of MDSCs in tumor tissues.8,9 MDSCs can strongly suppress the activation and cytotoxicity of T cells and NK cells, which directly evoke the anti-tumor immune responses.10,11 Furthermore, MDSCs also can stimulate recruitment of regulatory T cells, another population of immunosuppressive cells, in tumor sites.12 The wide suppressive activity of MDSCs in immunosuppressive tumor microenvironment leads to tumor progression and refractoriness to a great extent, which renders MDSCs as a potential target for the effective tumor immunotherapy. Recently, some therapeutic methods are raised based on MDSCs, such as gemcitabine that directly damages MDSC, tyrosine kinase inhibitor that induces MDSC differentiation, and phosphodiesterase type 5 inhibitor that reduces the activity of MDSCs, etc.12,13 However, these methods have side effects on normal tissues because of the poor selectivity. Moreover, as MDSCs are a double-edged sword, excessive clearance of systemic MDSCs takes a risk of evoking autoimmune diseases.14 Therefore, it is important to develop new strategies for selectively depleting MDSCs in tumor tissues rather than systemic MDSCs, which is still challenging unfortunately.15
Recently, nanoparticles coated with cell membranes are widely developed as biomimetic drug delivery systems. In particular, platelet membranes-enveloped nanoparticles have drawn much attention by virtue of unique features of platelets.16–18 As a natural population of blood cells, platelets are produced numerously every day with a circulating lifespan of 7–10 days.19 Additionally, platelets can adhere to cancer cells by tumor cell-induced platelet aggregation (TCIPA),20 which may be attributed to protein molecule binding such as P-selectin on platelet membranes (PM) and podocalyxin-like protein 1 (PCLP1) on tumor cells.21 These proteins are retained in PM that prepared from natural platelets.22 PM-enveloped nanoparticles are expected to actively target tumor sites through the TCIPA effect. Consequently, platelet membranes endow the coated nanoparticles with essential properties such as good biocompatibility, excellent biodegradability, immune escape, prolonged circulation time and tumor accumulation.16,22
In this study, we establish a biomimetic nanoplatform (PM–PLGA–DOX/GEM) that PM cloak PLGA nanoparticles co-loaded with gemcitabine (GEM) and doxorubicin (DOX). For the PM–PLGA–DOX/GEM, on one hand, platelet membranes can protect normal cells from the encapsulated drugs, thereby notably reducing the side effects. On the other hand, platelet membranes can prevent nanoparticles from immune surveillance and physical clearance. PM–PLGA–DOX/GEM can accumulate in the tumor tissues because of the enhanced permeation and retention (EPR) effect and TCIPA effect,23,24 which will greatly increase the drug concentration in tumor tissues. GEM can selectively eliminate the MDSCs in tumor tissues, hence relieving tumor immunosuppression. Furthermore, DOX can induce immunogenic cell death (ICD) of lymphoma cells.25 ICD has been considered to dramatically strengthen the tumor cell immunogenicity via arising damage-associated molecular patterns (DAMPs) and incite T cell activation finally.26,27 Taken together, the therapeutic effects will be improved remarkably because of the enormously enhanced anti-tumor immunity (Fig. 1). This study is supposed to offer a novel strategy for lymphoma treatment based on immunogenic activation and immunosuppressive reversion.
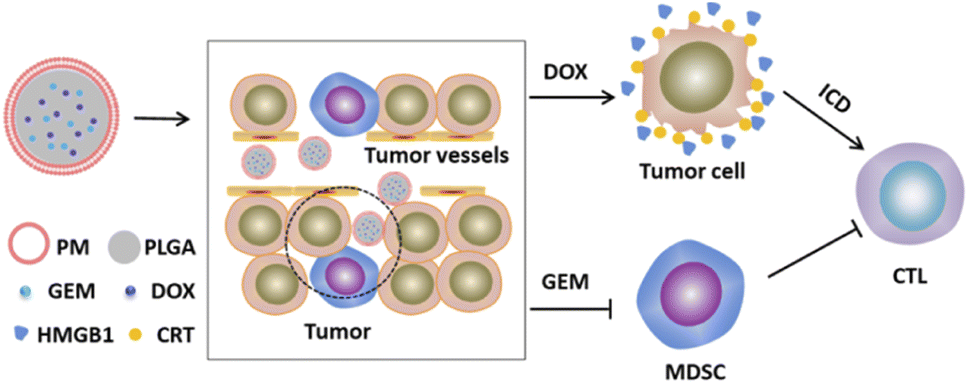 | ||
| Fig. 1 Schematic illustration of PM–PLGA–DOX/GEM structure and therapeutic strategy. PM–PLGA–DOX/GEM nanoparticles are synthesized by platelet membranes (PM) wrapping PLGA nanoparticles co-carried with doxorubicin (DOX) and gemcitabine (GEM). They can accumulate in tumor tissues through the EPR effect and TCIPA effect. Herein, DOX can induce immunogenic cell death (ICD) of tumor cells, and GEM can eliminate the filtrated MDSCs in tumors, thus relieving tumor immunosuppression. The anti-tumor immune responses are enhanced by the recruited and activated cytotoxic T lymphocytes (CTLs) based on immunogenic activation and immunosuppressive reversion. | ||
Materials and methods
Materials and reagents
DOX and GEM were obtained from MedChemExpress (NJ, USA). PLGA was bought from Sigma-Aldrich (MO, USA). Cell Counting Kit-8 (CCK-8) was purchased from Dojindo (Kyushu, Japan). The Annexin V-FITC Apoptosis Detection Kit was bought from BD Biosciences (NJ, USA). Monoclonal antibodies to CD8, Gr-1, CD11b and calreticulin (CRT) were bought from BioLegend (CA, USA). ELISA kits for transforming growth factor-β (TGF-β), interleukin-10 (IL-10) and high mobility group protein B1 (HMGB1) were bought from CUSABIO (Wuhan, China).Platelet membrane preparation
Platelets were obtained through centrifugation of whole blood. Briefly, the whole blood of C57BL/6J mice was drawn with acid-citrate anticoagulation and centrifuged at 200g for 10 min at room temperature. The supernatant was collected to obtain platelet-rich plasma (PRP). Then platelets were collected after PRP was suffered to centrifugation with 2000g for 20 min and PBS washing. Platelet membranes were prepared by a multigelation process.22 Platelet suspensions added with protease inhibitors were frozen at −80 °C and then thawed at 25 °C. This freeze–thaw process was repeated three times to acquire platelet membranes.Synthesis of PM–PLGA–DOX/GEM nanoparticles
PLGA–DOX/GEM nanoparticles were synthesized through a double emulsion and solvent evaporation method. DOX and GEM were added to 1 M NaOH solution with the concentrations of 20 mg ml−1. PLGA particles were dissolved in dichloromethane (50 mg ml−1). These two kinds of solution were mixed at the volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 to prepare the first emulsion by 2 min sonication. Next, 1% poloxamer aqueous solution was added for another 2 min sonication to obtain the water/oil/water emulsion. The solvent was evaporated through gently stirring for 3 h in a fume hood. PLGA–DOX/GEM nanoparticles were collected by centrifugation at 80000g for 5 min, washed by water and filtrated by 0.2 μm filter membrane. The prepared platelet membranes as described above were mixed with PLGA–DOX/GEM nanoparticles in deionized water. The mixture was stirred overnight after sonication for 3 min. A “right-side-out” assembly was obtained because the outer membranes of platelets are negatively charged and PLGA particles are negatively charged.22 The final PM–PLGA–DOX/GEM nanoparticles were synthesized after centrifugation.
:2 to prepare the first emulsion by 2 min sonication. Next, 1% poloxamer aqueous solution was added for another 2 min sonication to obtain the water/oil/water emulsion. The solvent was evaporated through gently stirring for 3 h in a fume hood. PLGA–DOX/GEM nanoparticles were collected by centrifugation at 80000g for 5 min, washed by water and filtrated by 0.2 μm filter membrane. The prepared platelet membranes as described above were mixed with PLGA–DOX/GEM nanoparticles in deionized water. The mixture was stirred overnight after sonication for 3 min. A “right-side-out” assembly was obtained because the outer membranes of platelets are negatively charged and PLGA particles are negatively charged.22 The final PM–PLGA–DOX/GEM nanoparticles were synthesized after centrifugation.
Characterization
The morphological characteristics of PM–PLGA–DOX/GEM nanoparticles were observed using a transmission electron microscope (TEM-2100, JEOL, Japan). The diameter distribution was detected by dynamic light scattering (BI-9000AT, Brookhaven, USA). Drug loading (DL) and encapsulation efficiency (EE) were analyzed via high performance liquid chromatography (HPLC). Drug release behavior was determined by dynamic dialysis as described previously.28Cell culture
As a lymphoma cell line induced in a C57BL mouse, EL4 cells were obtained from National Collection of Authenticated Cell Cultures (Shanghai, China). They were cultured in DMEM medium supplemented with 10% fetal bovine serum (FBS) at 37 °C and passaged every three days.Induction of MDSCs in vitro
Sterile bone marrow cells were collected from femurs and tibias of C57BL/6J mice. After erythrocyte lysis, the resulting cells were plated into 6 well plates at a density of 1.5 × 106 cells per well and induced through 40 ng ml−1 IL-6 and 40 ng ml−1 GM-CSF in the medium for 4 days. MDSCs (CD11b+Gr-1+) were obtained via flow cytometry sorting and cultured in the RPMI 1640 medium added with 10% FBS, 10 mM HEPES, 2 mM L-glutamine, and 20 μM 2β-mercaptoethanol.CCK-8 assay
To determine the cytotoxicity of PM–PLGA–DOX/GEM on EL4 cells and MDSCs, CCK-8 assay was carried out. Cells were seeded into 96-well plates at 1 × 104 cells per well and treated with PBS (as the control group), DOX, PLGA–DOX, DOX + GEM, PLGA–DOX/GEM, and PM–PLGA–DOX/GEM, respectively. The absolute concentrations for DOX and GEM were 0.25 μg ml−1 and 0.20 μg ml−1, respectively. After 24 hour treatment, 10 μl CCK-8 solutions were added to each well and incubated for another 3 h. A microplate reader was utilized to determine the optical density (OD) at 450 nm. Cell viability (%) was calculated as the formula: ODtreatment/ODcontrol × 100%.Apoptosis assay
Cell apoptosis was detected by the Annexin V-FITC Apoptosis Detection Kit. EL4 cells and MDSCs were seeded into in 6-well plates with a density of 2 × 105 cells per well and treated with PBS, DOX, PLGA–DOX, DOX + GEM, PLGA–DOX/GEM, and PM–PLGA–DOX/GEM, respectively. After 24 hour incubation, cells were collected by centrifugation and washing. Cold binding buffer was used to resuspend the cells. Afterwards, each tube was added with 5 μl Annexin V-FITC for 15 minute incubation in the dark. Apoptosis was analyzed quantitatively using flow cytometry.ICD biomarker determination in vitro
The EL4 cells treated as above were fixed by 4% paraformaldehyde and then incubated with the anti-CRT primary antibodies for 1 h at 37 °C. Thereafter, the cells were added with FITC-conjugated secondary antibodies for additional one hour incubation away from light. Cells were counterstained with DAPI to present nuclei and further observed with a confocal microscope to estimate the CRT expression levels. Additionally, HMGB1 released from EL4 cells was analyzed by HMGB1 ELISA kit.MDSC suppressive assay
The treated MDSCs as above were co-cultured with autologous T cells at different ratios of 1:1, 1:5, and 1:10 for 4 days. Culture medium was supplemented with anti-CD3/CD28 antibodies as T-cell stimulators. The MDSC suppressive effect on T-cell proliferation was measured using 3H-thymidine incorporation assay that cells were pulsed with 0.5 μCi per well 3H-thymidine. 3H-Thymidine incorporation was measured using a multi-purpose scintillation counter.
Establishment of lymphoma-bearing mouse model
8 week old female C57BL/6J mice were purchased from the Comparative Medicine Center of Yangzhou University (Yangzhou, China). All animal experiments were approved by the Animal Ethics Committee of Yangzhou University and performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Yangzhou University. EL4 cells (100 μl) at the density of 2 × 107 cells per ml were administrated subcutaneously to establish lymphoma-bearing mouse model. Tumor sizes were measured every two days and tumor volumes were figured using the formula: 1/2 × length × width2. When the volume reached 80–150 mm3, the model was considered to be established successfully.In vivo distribution of PM–PLGA–DOX/GEM
After being injected intravenously with PLGA–DOX/GEM or PM–PLGA–DOX/GEM nanoparticles (DOX 5 mg kg−1), the lymphoma-bearing mice were sacrificed at different time points (2, 4, 8, 12 and 24 h). Their major organs and tumors were separated, weighed, lysed and homogenized. Acidified isopropanol was used to extract DOX in the tissue homogenate. The extracted supernatant was analyzed using HPLC after centrifugation.Therapeutic efficacy in vivo
Lymphoma-bearing mice were randomly allocated into 6 groups, namely control group (PBS), DOX group, PLGA–DOX group, DOX + GEM group, PLGA–DOX/GEM group, and PM–PLGA–DOX/GEM group. Each group had 5 mice and each mouse was injected intravenously every 2 days via tail vein. For a single dose, the equivalent dosage of DOX was 1 mg kg−1 and GEM was 2 mg kg−1. Relative tumor volume (RTV) was calculated as V/V0, where V0 signifies the initial volume before treatment. Tumor tissues were isolated after the mice were sacrificed on day 12 post treatment. The tumors isolated from mice were sectioned after formalin fixing paraffin embedding. Staining with hematoxylin and eosin (H&E), Ki67 and TUNEL was performed to evaluate the therapeutic effects.Anti-tumor immunity in vivo
Tumor tissue sections were suffered to immunofluorescence assay to access the anti-tumor immunity in tumors. The paraffin section were deparaffinized and retrieved. After BSA blocking, they were incubated with primary antibodies (CRT, CD8, CD11b and Gr-1) overnight in a humidified box at 4 °C. PE-conjugated goat anti-rabbit antibodies were used as secondary antibodies for CRT, CD8 and CD11b. The secondary antibody for Gr-1 was labelled with FITC. With nuclei stained with DAPI, immunofluorescence was observed using confocal microscopy and typical images were taken.Safety assessment
To estimate the in vivo safety of PM–PLGA–DOX/GEM, healthy C57BL/6J mice were administrated intravenously with PBS, DOX, PLGA–DOX, DOX + GEM, PLGA–DOX/GEM, and PM–PLGA–DOX/GEM, respectively. On day 12 after treatments, blood serum was obtained for biochemical test. Creatine kinase-MB (CK-MB), alanine transaminase (ALT) and creatinine (CRE), which respectively reflects myocardial injury, hepatic function, and renal function, were determined. Major organs, including heart, lung, liver, spleen and kidney, were isolated from the sacrificed mice and made into section. After H&E staining, an optical microscope was used to capture histopathological changes.Statistical analysis
One-way ANOVA followed by SNK test was utilized to analyze data by the SPSS software (version 13.0). A P-value < 0.05 was considered statistically significant (*P < 0.05, **P < 0.01, ***P < 0.001).Results and discussion
Characterization of PM–PLGA–DOX/GEM nanoparticles
The morphology of PLGA–DOX/GEM and PM–PLGA–DOX/GEM nanoparticles was detected by TEM. As displayed in Fig. 2A, both PLGA–DOX/GEM and PM–PLGA–DOX/GEM nanoparticles showed a homogeneously dispersed spherical morphology with diameter of about 150 nm and 180 nm, respectively. For drug delivery, the diameters are desired because particles with sizes less than 10 nm could be quickly removed by renal clearance while particles more than 200 nm could not passively target tumors through the EPR effect.29 Furthermore, platelets could bind to tumor cells (Fig. S1†) while platelet membranes that we prepared retained these specific proteins (Fig. S2†), suggesting that platelet membranes would facilitate accumulation of the cloaked nanoparticles in tumors. As shown in Fig. 2B, platelet membrane-cloaked PLGA nanoparticles indicated favorable stability in serum and PBS without aggregation and precipitation. The drug loading (DL) and encapsulation efficiency (EE) of PM–PLGA–DOX/GEM nanoparticles were detected by HPLC (Fig. 2C). For DOX, the DL was 3.81 ± 0.27% and EE was 79.16 ± 0.35%. For GEM, the DL and EE were 3.67 ± 0.32% and 75.43 ± 0.48%, respectively. Furthermore, the in vitro release profiles of PM–PLGA–DOX/GEM nanoparticles were determined (Fig. 2D). Nearly 70% of both DOX and GEM were released within 12 h in PBS at pH = 5.5, while the cumulative release of DOX and GEM were only approximately 30% in 24 h when PM–PLGA–DOX/GEM nanoparticles dispersed in PBS with pH = 7.4. These results demonstrate that PM–PLGA–DOX/GEM nanoparticles have a pH-triggered release pattern, resulting in high concentration of drugs in tumor tissues on account of tumor acidic microenvironment.30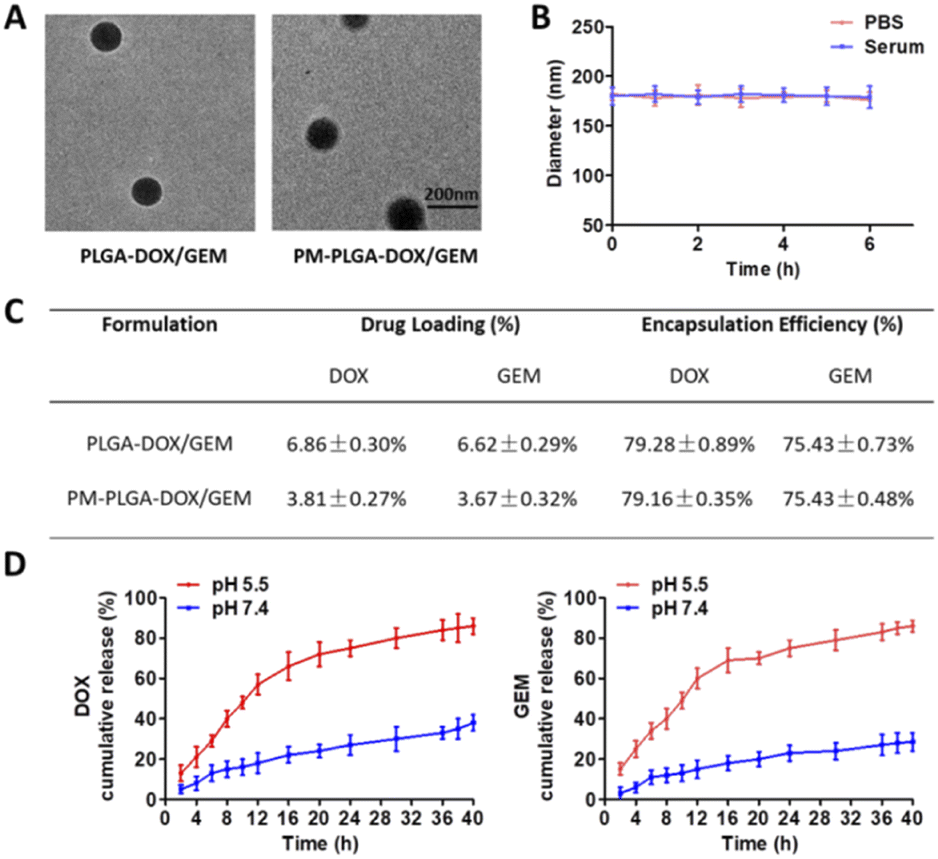 | ||
| Fig. 2 Characterization of PM–PLGA–DOX/GEM nanoparticles. (A) TEM images of PLGA–DOX/GEM and PM–PLGA–DOX/GEM nanoparticles. (B) Stability of PM–PLGA–DOX/GEM in PBS and 20% serum at 37 °C. (C) Drug loading (DL) and encapsulation efficiency (EE) of PLGA–DOX/GEM and PM–PLGA–DOX/GEM nanoparticles. (D) Drug release profiles of PM–PLGA–DOX/GEM nanoparticles in PBS at pH = 7.4 and pH = 5.5. | ||
In vitro anti-tumor efficacy
The cytotoxicity of PM–PLGA–DOX/GEM nanoparticles on EL4 cells was assessed using CCK-8 assay. As shown in Fig. 3A, the cell viabilities of EL4 cells treated with DOX, DOX + GEM, PLGA–DOX, PLGA–DOX/GEM, and PM–PLGA–DOX/GEM were obviously inhibited compared with the control group and there was a higher inhibition rate in PM–PLGA–DOX/GEM group than any other groups (P < 0.05). Moreover, flow cytometric analysis of EL4 cell apoptosis was conducted. EL4 cells were cultured with the indicted treatments for 24 hour and then stained with Annexin V-FITC (Fig. 3B). The apoptosis rates were significantly increased in treatment groups (P < 0.05). Particularly, the PM–PLGA–DOX/GEM group had a higher rate compared with PLGA–DOX/GEM (P < 0.05) while there was no significant difference between the DOX + GEM and PLGA–DOX/GEM groups.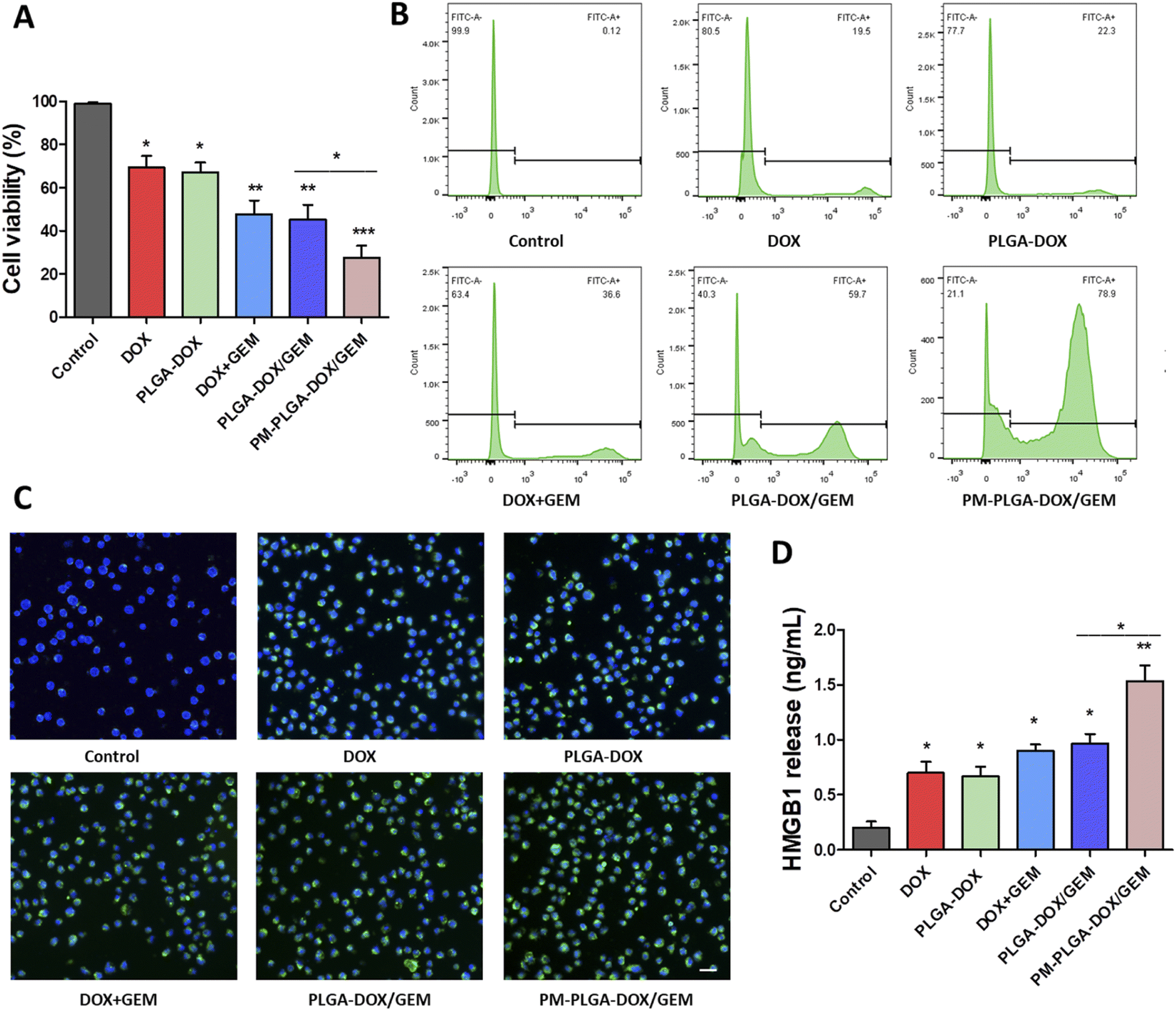 | ||
| Fig. 3 Cytotoxicity of PM–PLGA–DOX/GEM on EL4 cells. (A) Cell viability of EL4 cells subjected to PBS (control), DOX, PLGA–DOX, DOX + GEM, PLGA–DOX/GEM, and PM–PLGA–DOX/GEM, respectively. (B) Flow cytometric analysis of EL4 cell apoptosis. EL4 cells were cultured with the indicted treatments for 24 h and then stained with Annexin V-FITC. (C) CRT immunofluorescence on EL4 cells with different treatments. Scale bars: 20 μm. (D) HMGB1 levels released from EL4 cells with different treatments were detected by ELISA in the supernatant. All experiments were repeated three times. *P < 0.05, **P < 0.01, ***P < 0.001. | ||
This phenomenon can be explained by the EPR effect of nanoparticles failed in vitro.31 The consistent results of cell viability inhibition and apoptosis reveal that platelet membrane-coated nanoparticles could enhance drug toxicity through relatively targeted drug delivery to tumor cells, thus improving the anti-tumor effects.
Recently, immunogenic cell death (ICD) has emerged emphatically and been recognized to remarkably aggrandize the tumor cell immunogenicity through arising damage-associated molecular patterns (DAMPs) and ultimately activating effector T cells. Therefore, ICD plays an essential role in anti-tumor immunity.32 It has been reported that some chemotherapeutic agents including DOX can evoke ICD.33 To investigate whether ICD was similarly elicited by our DOX formulation, we next investigate CRT and HMGB1 that reflect the degree of ICD using immunofluorescence and ELISA, respectively. As Fig. 3C and S3† illustrate, the fluorescence signals of CRT on EL4 cell surface in treatment groups were obviously stronger than the control group, and the PM–PLGA–DOX/GEM group appeared the best outcome among all groups (P < 0.01). The HMGB1 levels released from EL4 cells were in accordance with the CRT fluorescence with the highest concentration in PM–PLGA–DOX/GEM group (Fig. 3D, P < 0.01). Given these results, PM–PLGA–DOX/GEM nanoparticles could elicit favorable ICD to further activate effector T cells.
Cytotoxic activity of PM–PLGA–DOX/GEM nanoparticles on MDSCs in vitro
GEM, as an antimetabolite chemotherapeutic agent, was reported to be particularly promising in eliminating MDSCs compared with other chemotherapeutics,34,35 and we further assessed the efficacy of PM–PLGA–DOX/GEM nanoparticles to MDSCs. Bone marrow cells of mice were induced by IL-6 and GM-CSF and then sorted by flow cytometry to obtain MDSCs, as previously described.36 As shown in Fig. 4A, DOX + GEM, PLGA–DOX/GEM and PM–PLGA–DOX/GEM nanoparticles dramatically reduced the cell viability of MDSCs (P < 0.01), while free DOX and PLGA–DOX nanoparticles slightly impaired MDSCs compared with the control. Consistently, a considerable MDSC apoptosis was detected in DOX + GEM, PLGA–DOX/GEM and PM–PLGA–DOX/GEM groups (Fig. 4B, P < 0.01). These results suggest that GEM in the nanoparticles retained the ability of eliminating MDSCs.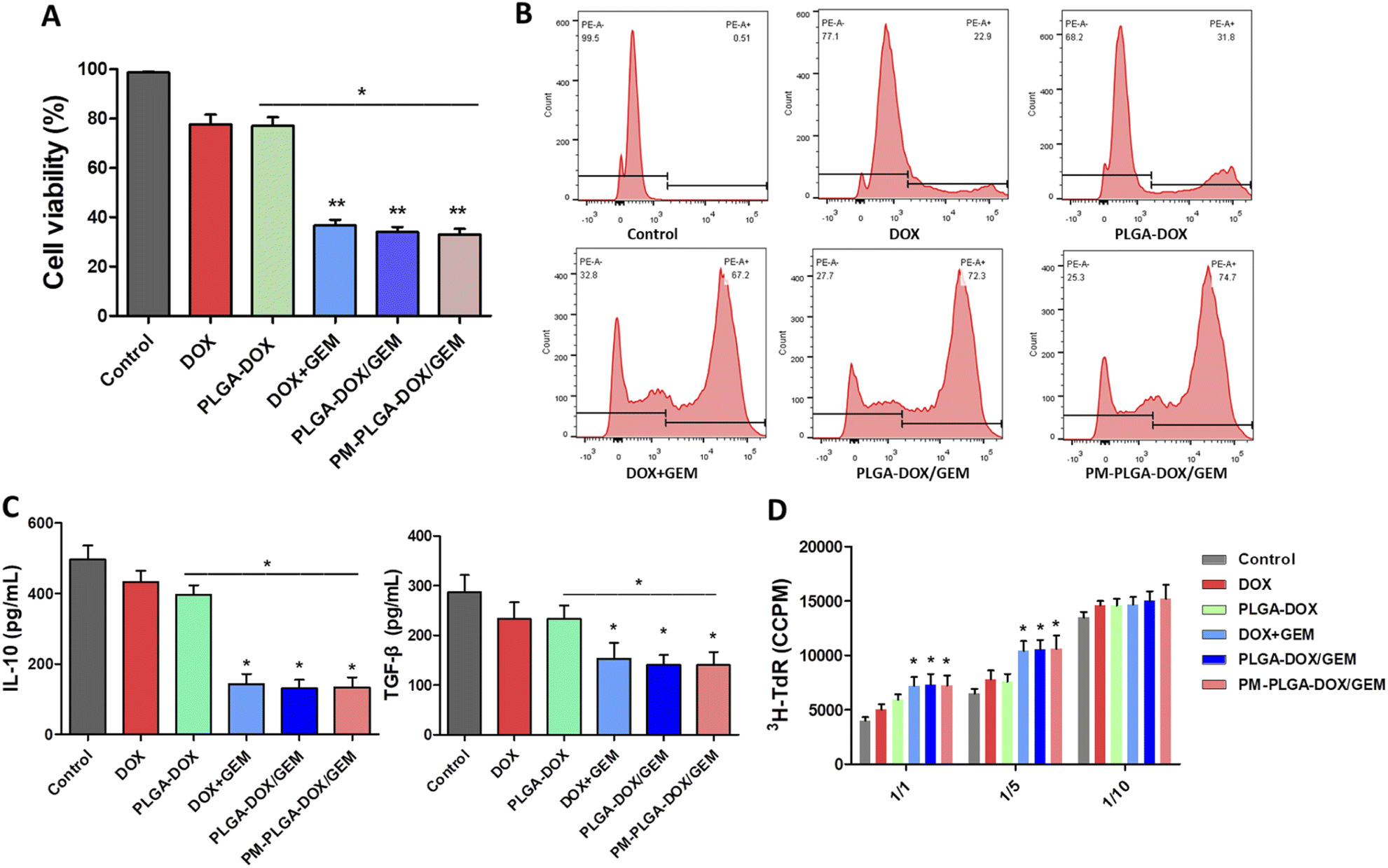 | ||
| Fig. 4 Cytotoxic effects of PM–PLGA–DOX/GEM on MDSCs in vitro. (A) Cell viability of MDSCs respectively treated with PBS (control), DOX, PLGA–DOX, DOX + GEM, PLGA–DOX/GEM, and PM–PLGA–DOX/GEM for 24 h. (B) MDSC apoptosis after 24 hour treatments was investigated by flow cytometry with Annexin V-FITC staining. (C) IL-10 and TGF-β secreted by MDSCs in the medium were detected by ELISA. (D) Inhibitory effect of MDSCs on T cells was investigated using MDSC suppressive assay. All experiments were repeated three times. *P < 0.05, **P < 0.01. | ||
MDSCs have a wide and cross-talk immunosuppressive activity as a critical component in immunosuppressive tumor microenvironment. Through secreting some cytokines, like TGF-β and IL-10, MDSCs can induce the expansion of regulatory T cells and compromise the function of T cells and natural killer cells. MDSCs also can directly suppress T cell proliferation, hence inhibiting the anti-tumor immune responses prominently.36,37 In order to evaluate the impact of PM–PLGA–DOX/GEM nanoparticles on MDSC immunosuppressive efficacy, we further determined the IL-10 and TGF-β secretion and performed T cell suppression assays. After MDSCs were cultured for 24 h with different treatments, the supernatant was detected using ELISA. As Fig. 4C exhibits, the secretion of both IL-10 and TGF-β was decreased by DOX + GEM, PLGA–DOX/GEM and PM–PLGA–DOX/GEM nanoparticles (P < 0.05). MDSCs induced from bone marrow after different treatments were co-cultured with autologous T cells stimulated by anti-CD3/CD28 Abs at ratios of 1:1, 1:5, and 1:10, respectively. The T cell proliferation was determined by incorporated 3H-thymidine. As shown in Fig. 4D, the 3H-thymidine incorporation was increased notably in the DOX + GEM, PLGA–DOX/GEM and PM–PLGA–DOX/GEM groups at ratios of 1:1 and 1:5 (P < 0.05), implying that inhibitory effect of MDSCs on T cell proliferation was impaired by GEM in different formulations.
In vivo anti-tumor efficacy
PM–PLGA–DOX/GEM nanoparticles are expected to achieve enrichment in the tumor sites by taking advantage of the EPR effect and TCIPA effect, thus improving the drug concentration in tumor tissues. Therefore, in vivo distribution profiles of PM–PLGA–DOX/GEM nanoparticles were detected (Fig. 5A and B). In the PM–PLGA–DOX/GEM group, the concentrations of DOX in tumor tissues were about 2 folds as the PLGA–DOX/GEM group, which reveals that platelet membrane can greatly improve the concentration of tumor tissues by active targeting.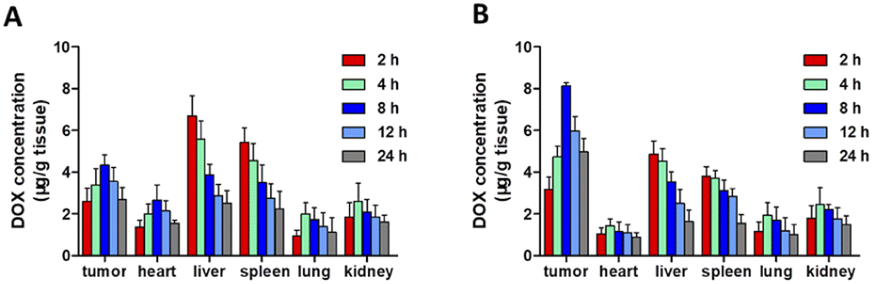 | ||
| Fig. 5 Distribution of PM–PLGA–DOX/GEM nanoparticles in vivo. Lymphoma-bearing mice were treated with PLGA–DOX/GEM nanoparticles (A) and PM–PLGA–DOX/GEM nanoparticles (B) and sacrificed at different time points (n = 5). | ||
Next, the in vivo anti-tumor effects of PM–PLGA–DOX/GEM nanoparticles were further investigated. EL4-lymphoma-bearing mice were established in this experiment. As shown in Fig. 6A and B, tumors in the control group displayed a marked growth. In contrast, the tumor sizes in all treatment groups decreased compared with the control group. The average tumor volumes that were treated with DOX, PLGA–DOX, PLGA–DOX/GEM, and PM–PLGA–DOX/GEM successively decreased, along with the smallest one in the PM–PLGA–DOX/GEM group (P < 0.01). To further evaluate the therapeutic effects, tumor tissues in different groups were sectioned and suffered to different staining. In Fig. 6C for H&E staining, tumor tissues presented extensive necrosis and apoptosis in the PM–PLGA–DOX/GEM group. From the Ki67 staining images (Fig. 6D), we could found the tumor tissues had the least Ki67 expression, a proliferation marker, after PM–PLGA–DOX/GEM treatment. Meanwhile, the massive apoptosis in tumor tissues treated with PM–PLGA–DOX/GEM nanoparticles were observed (P < 0.01), as the TUNEL staining exhibited (Fig. 6E and S4†). These results consistently revealed the superior therapeutic efficacy of PM–PLGA–DOX/GEM.
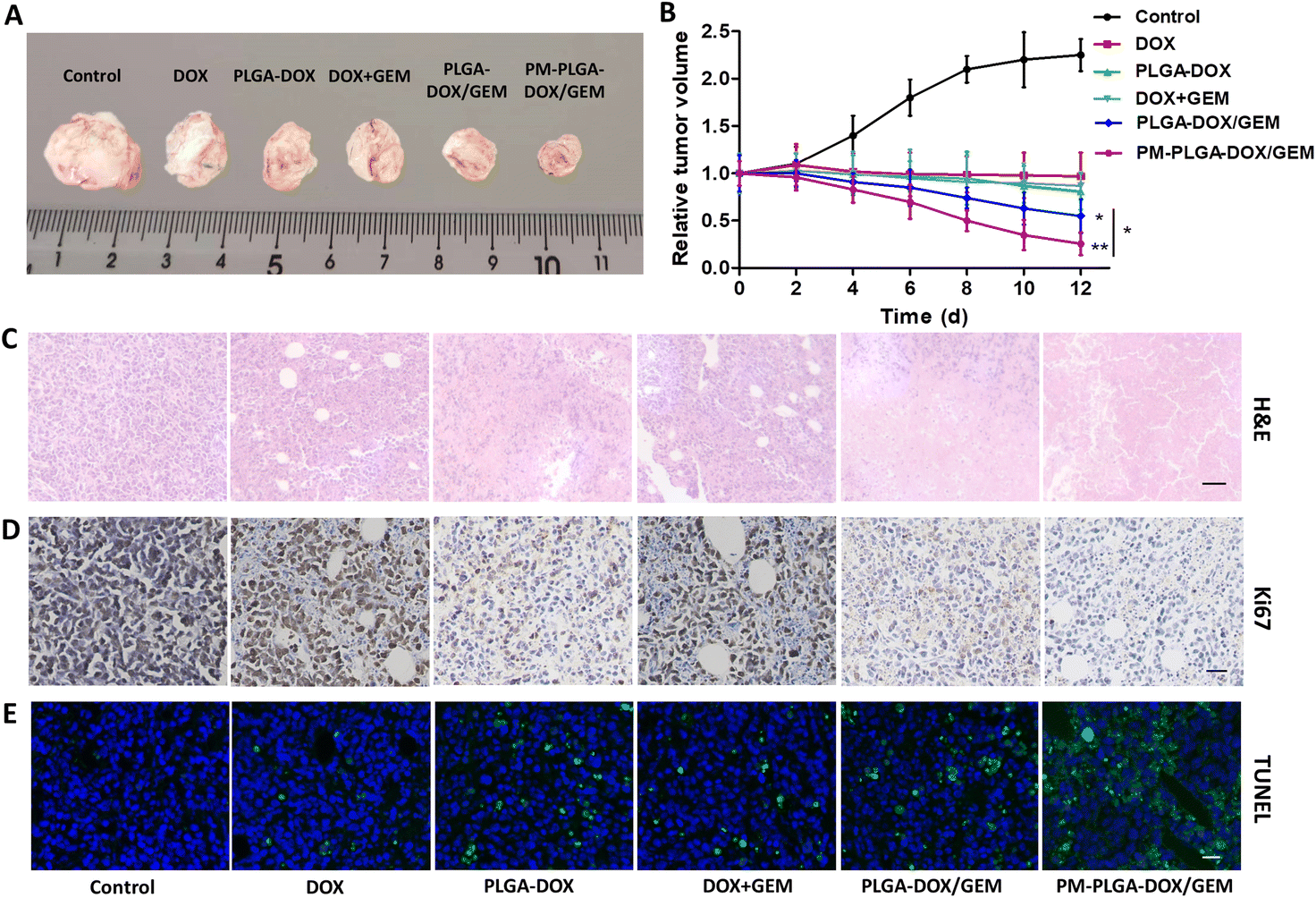 | ||
| Fig. 6 Therapeutic effects of PM–PLGA–DOX/GEM nanoparticles in vivo. (A) Representative subcutaneous tumors isolated from lymphoma-bearing mice with different treatments at day 12. (B) Relative tumor volume–time curves after treatments in different groups. (C) Histopathological changes of tumors from lymphoma-bearing mice treated with different formulations for 12 days. Scale bar: 50 μm. (D) Immunohistochemical staining images for Ki67 in tumors from different groups. Scale bar: 20 μm. (E) Fluorescence images for TUNEL staining in tumors isolated from mice that received different treatments. DAPI was utilized to counterstain nuclei. Scale bar: 20 μm, *P < 0.05, **P < 0.01 (n = 5). | ||
In vivo immune microenvironment
Since the favorable therapeutic efficacy of PM–PLGA–DOX/GEM has been verified, the tumor immune microenvironment was further investigated. Firstly, the expression of CRT exposed on the cell surfaces was determined in tumor tissues, which reflected the levels of ICD that could make the dead cells ‘visible’ to antigen presenting cells and then stimulate T cell activation. In Fig. 7A and S5A,† we can find that green fluorescence in treatment groups were stronger than that in control group with the strongest fluorescence in the PM–PLGA–DOX/GEM group (P < 0.01). This result demonstrates that PM–PLGA–DOX/GEM remarkably increased the CRT exposure and improved the ICD level of tumor cells.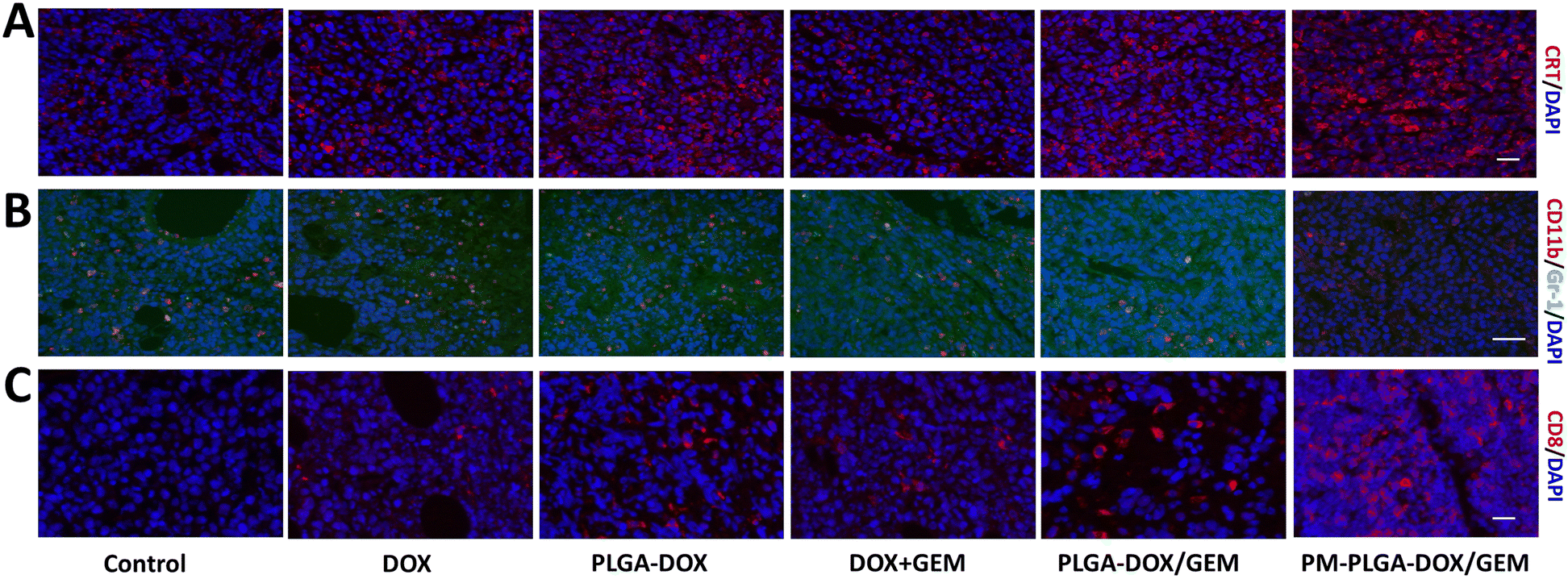 | ||
| Fig. 7 Effects of PM–PLGA–DOX/GEM nanoparticles on anti-tumor immunity in vivo. (A) CRT immunofluorescence of EL4 tumor sections from different groups. CRT was labelled with PE and DAPI was used to counterstain nuclei. Scale bar: 20 μm. (B) Immunofluorescence for MDSCs (CD11b+Gr-1+) infiltrated in tumor tissues that subjected to indicated treatments. CD11b was marked by PE and Gr-1 was labelled by FITC. Scale bar: 50 μm. (C) Detection of CD8+ T cells in tumor tissues with PE-labelled CD8 and DAPI staining. Scale bars: 20 μm (n = 5). | ||
Besides the predominant ICD level in vivo, the introduction of GEM was hypothesized to reduce the infiltrated MDSCs, which are recognized as crucial immunosuppressive cells. After different treatments, MDSCs infiltrated in tumor tissues were detected by immunofluorescence and flow cytometry with PE-labelled CD11b and FITC-labelled Gr-1. As shown in Fig. 7B, S5B and S6,† the number of MDSCs (CD11b+Gr-1+) in tumor tissues was decreased in the DOX + GEM, PLGA–DOX/GEM and PM–PLGA–DOX/GEM groups, especially in the PM–PLGA–DOX/GEM group. MDSCs can suppress the anti-tumor immune responses by secreting a lot of cytokines, such as TGF-β and IL-10. Therefore, we detected their secretion from MDSCs to further reveal the immunosuppressive function (Fig. S7†). As we can see, PM–PLGA–DOX/GEM treatment significantly reduced both TGF-β and IL-10 secretion from MDSCs than other groups (P < 0.05), thus reversing MDSC immunosuppression. To evaluate the anti-tumor immunity, we further assessed the infiltrated cytotoxic T lymphocytes (CTLs, CD8+) in tumors in different groups. From Fig. 7C, S5C and S8,† we can obviously find many CTLs in tumor tissues in the PLGA–DOX/GEM and PM–PLGA–DOX/GEM groups. Moreover, the PM–PLGA–DOX/GEM group had more CTLs than PLGA–DOX/GEM group (P < 0.05), which demonstrates the superiority of platelet membranes in drug delivery. Taken together, PM–PLGA–DOX/GEM nanoparticles could enhance anti-tumor immunity through immunogenic activation and immunosuppressive reversion, thus improving the anti-tumor effects.
In vivo safety assessment
Healthy C57BL/6J mice were utilized in this experiment. After different treatments, the general conditions of mice, including eating, behaviour, and physical features, had no apparent changes.Additionally, there was no significant difference in CK-MB, ALT and CRE levels among all groups (Fig. 8A). The toxicity to major organs was also evaluated by H&E staining. As Fig. 8B shows, no obvious pathological changes happened in any organs from PM–PLGA–DOX/GEM treated mice. As a result, PM–PLGA–DOX/GEM nanoparticles are promising for tumor clinical treatment with good biocompatibility.
 | ||
| Fig. 8 In vivo toxicity of various formulations on normal cells and tissues. (A) CK-MB, ALT, and BUN concentrations in serum at day 12 post-injection. (B) H&E-staining images of heart, lung, liver, spleen and kidney. Scale bar: 200 μm (n = 5). | ||
Conclusions
In summary, we successfully synthesized PM–PLGA–DOX/GEM biomimetic nanoparticles with high encapsulation efficiency and good biocompatibility. PM–PLGA–DOX/GEM can make drugs accumulate in tumor tissues via EPR and TCIPA effects to improve the drug concentration. Hereinto, DOX can induce immunogenic cell death of lymphoma cells, while GEM can eliminate the MDSCs in tumor tissues. Therefore, PM–PLGA–DOX/GEM nanoparticles could enhance anti-tumor immune responses through immunogenic activation and immunosuppressive reversion, thus improving the anti-tumor effects. This study provides a potential strategy for tumor clinical treatment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of Jiangsu Province (BK20190908).References
- K. D. Miller, M. Fidler-Benaoudia, T. H. Keegan, H. S. Hipp, A. Jemal and R. L. Siegel, Ca-Cancer J. Clin., 2020, 70, 443–459 CrossRef PubMed.
- V. Bachanova, M.-A. Perales and J. S. Abramson, Blood Rev., 2020, 40, 100640 CrossRef CAS.
- X. Wang, B. C. Waschke, R. A. Woolaver, S. M. Y. Chen, Z. Chen and J. H. Wang, Protein Cell, 2020, 11, 472–482 CrossRef CAS PubMed.
- M. J. V. Elsas, T. V. Hall and S. H. V. D. Burg, Cancers, 2020, 12, 935 CrossRef.
- N. Mcgranahan and C. Swanton, Cell, 2017, 170, 825–827 CrossRef CAS PubMed.
- F. Wei, D. Wang, J. Wei, N. Tang, L. Tang, F. Xiong, C. Guo, M. Zhou, X. Li, G. Li, W. Xiong, S. Zhang and Z. Zeng, Cell. Mol. Life Sci., 2021, 78, 173–193 CrossRef CAS PubMed.
- R. J. Tesi, Trends Pharmacol. Sci., 2019, 40, 4–7 CrossRef CAS PubMed.
- A. Betsch, O. Rutgeerts, S. Fevery, B. Sprangers, G. Verhoef, D. Dierickx and M. Beckers, Blood Rev., 2018, 32, 490–498 CrossRef CAS PubMed.
- R. Sun, Z. Zheng, L. Wang, S. Cheng, Q. Shi, B. Qu, D. Fu, C. Leboeuf, Y. Zhao, J. Ye, A. Janin and W.-L. Zhao, Mol. Oncol., 2021, 15, 246–261 CrossRef CAS PubMed.
- L. Bird, Nat. Rev. Immunol., 2020, 20, 352–353 CrossRef CAS PubMed.
- A. Sacchi, N. Tumino, A. Sabatini, E. Cimini, R. Casetti, V. Bordoni, G. Grassi and C. Agrati, Front. Immunol., 2018, 9, 1271 CrossRef PubMed.
- S. Huo, L. Liu, Q. Li and J. Wang, Discov. Med., 2020, 30, 119–128 Search PubMed.
- A. L. Chang, J. Miska, D. A. Wainwright, M. Dey, C. V. Rivetta, D. Yu, D. Kanojia, K. C. Pituch, J. Qiao and P. Pytel, Cancer Res., 2016, 76, 5671–5682 CrossRef CAS.
- J. Zhang, A. Hodges, S. H. Chen and P. Y. Pan, Cell. Immunol., 2021, 362, 104300 CrossRef CAS PubMed.
- Y. Liu, G. Wei, W. A. Cheng, Z. Dong, H. Sun, V. Y. Lee, S.-C. Cha, D. L. Smith, L. W. Kwak and H. Qin, Cancer Immunol. Immunother., 2018, 67, 1181–1195 CrossRef CAS PubMed.
- S. Wang, Y. Duan, Q. Zhang, A. Komarla, H. Gong, W. Gao and L. Zhang, Small Struct., 2020, 1, 2000018 CrossRef PubMed.
- B. Li, T. Chu, J. Wei, Y. Zhang, F. Qi, Z. Lu, C. Gao, T. Zhang, E. Jiang, J. Xu, J. Xu, S. Li and G. Nie, Nano Lett., 2021, 21, 2588–2595 CrossRef CAS PubMed.
- Y. Zhang, N. Ma, C. Luo, J. Zhu and C. Bao, RSC Adv., 2020, 10, 9378–9386 RSC.
- Z.-J. Liu, K. M. Hoffmeister, Z. Hu, D. E. Mager, S. Ait-Oudhia, M. A. Debrincat, I. Pleines, E. C. Josefsson, B. T. Kile, J. Italiano Jr, H. Ramsey, R. Grozovsky, P. Veng-Pedersen, C. Chavda and M. Sola-Visner, Blood, 2014, 123, 3381–3389 CrossRef CAS.
- H. A. Goubran, J. Stakiw, M. Radosevic and T. Burnouf, Semin. Thromb. Hemostasis, 2014, 40, 296–305 CrossRef CAS PubMed.
- L. Amo, E. Tamayo-Orbegozo, N. Maruri, C. Eguizabal, O. Zenarruzabeitia, M. Riñón, A. Arrieta, S. Santos, J. Monge, M. A. Vesga, F. Borrego and S. Larrucea, Front. Oncol., 2014, 4, 245 Search PubMed.
- C.-M. J. Hu, R. H. Fang, K.-C. Wang, B. T. Luk, S. Thamphiwatana, D. Dehaini, P. Nguyen, P. Angsantikul, C. H. Wen, A. V. Kroll, C. Carpenter, M. Ramesh, V. Qu, S. H. Patel, J. Zhu, W. Shi, F. M. Hofman, T. C. Chen, W. Gao, K. Zhang, S. Chien and L. Zhang, Nature, 2015, 526, 118–121 CrossRef CAS PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- Q. Hu, W. Sun, C. Qian, C. Wang, H. N. Bomba and Z. Gu, Adv. Mater., 2015, 27, 7043 CrossRef CAS PubMed.
- Y. Choi, H. Y. Yoon, J. Kim, S. Yang, J. Lee, J. W. Choi, Y. Moon, J. Kim, S. Lim, M. K. Shim, S. Jeon, I. C. Kwon and K. Kim, Pharmaceutics, 2020, 12, 1165 CrossRef CAS.
- A. D. Garg, L. Galluzzi, L. Apetoh, T. Baert, R. B. Birge, J. M. Bravo-San Pedro, K. Breckpot, D. Brough, R. Chaurio, M. Cirone, A. Coosemans, P. G. Coulie, D. De Ruysscher, L. Dini, P. de Witte, A. M. Dudek-Peric, A. Faggioni, J. Fucikova, U. S. Gaipl, J. Golab, M.-L. Gougeon, M. R. Hamblin, A. Hemminki, M. Herrmann, J. W. Hodge, O. Kepp, G. Kroemer, D. V. Krysko, W. G. Land, F. Madeo, A. A. Manfredi, S. R. Mattarollo, C. Maueroder, N. Merendino, G. Multhoff, T. Pabst, J.-E. Ricci, C. Riganti, E. Romano, N. Rufo, M. J. Smyth, J. Sonnemann, R. Spisek, J. Stagg, E. Vacchelli, P. Vandenabeele, L. Vandenberk, B. J. Van den Eynde, S. Van Gool, F. Velotti, L. Zitvogel and P. Agostinis, Front. Immunol., 2015, 6, 588 Search PubMed.
- G. Passeri, J. Northcote-Smith and K. Suntharalingam, RSC Adv., 2022, 12, 5290–5299 RSC.
- P. Xu, H. Zuo, D. Chen, M. Peng, Y. Jiang, X. Liu, J. Ouyang and B. Chen, RSC Adv., 2017, 7, 33905 RSC.
- D. Kalyane, N. Raval, R. Maheshwari, V. Tambe, K. Kalia and R. K. Tekade, Mater. Sci. Eng., C, 2019, 98, 1252–1276 CrossRef CAS PubMed.
- Y. Kato, S. Ozawa, C. Miyamoto, Y. Maehata, A. Suzuki, T. Maeda and Y. Baba, Cancer Cell Int., 2013, 13, 89 CrossRef CAS PubMed.
- J. Fang, H. Nakamura and H. Maeda, Adv. Drug Delivery Rev., 2011, 63, 136–151 CrossRef CAS PubMed.
- L. Galluzzi, A. Buqué, O. Kepp, L. Zitvogel and G. Kroemer, Nat. Rev. Immunol., 2017, 17, 97–111 CrossRef CAS PubMed.
- H. Huang, Y. Huang, Y. Chen, Z. Luo, Z. Zhang, R. Sun, Z. Wan, J. Sun, B. Lu, L. Zhang, J. Hu and S. Li, Biomaterials, 2021, 270, 120708 CrossRef CAS PubMed.
- S. Ugel, E. Peranzoni, G. Desantis, M. Chioda, S. Walter, T. Weinschenk, J. C. Ochando, A. Cabrelle, S. Mandruzzato and V. Bronte, Cell Rep., 2012, 2, 628–639 CrossRef CAS PubMed.
- M. S. Sasso, G. Lollo, M. Pitorre, S. Solito, L. Pinton, S. Valpione, G. Bastiat, S. Mandruzzato, V. Bronte, I. Marigo and J.-P. Benoit, Biomaterials, 2016, 96, 47–62 CrossRef CAS PubMed.
- B. Mirlekar, SAGE Open Med., 2022, 10, 1–15 Search PubMed.
- H. Zhang, Z.-L. Li, S.-B. Ye, L.-Y. Ouyang, Y.-S. Chen, J. He, H.-Q. Huang, Y.-X. Zeng, X.-S. Zhang and J. Li, Cancer Immunol. Immunother., 2015, 64, 1587–1599 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra04326b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |