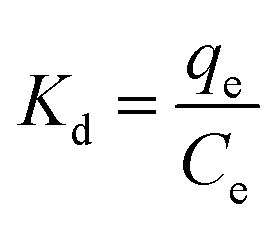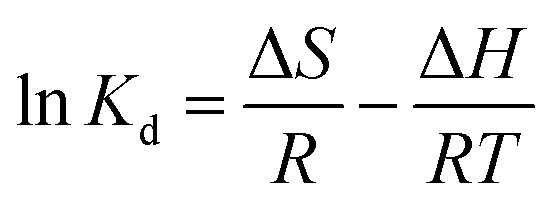DOI:
10.1039/D2RA04323H
(Paper)
RSC Adv., 2022,
12, 28586-28598
Decorated reduced graphene oxide transfer sulfides into sulfur and sulfone in wastewater †
Received
13th July 2022
, Accepted 21st September 2022
First published on 7th October 2022
Abstract
Sulfides cannot be completely removed using oxidation due to the production of sulfate. In this work, a reduced graphene oxide (RGO)/Fe3O4 hybrid material was synthesized via a simple in situ chemical method for sulfide removal. The adsorption capacity of RGO/Fe3O4 was evaluated by sulfide removal from aqueous solution, and different experimental parameters including contact time, solution pH, adsorbent dosage, ion strength and temperature were investigated. The equilibrium data were in accordance with the Langmuir linear isotherm with a maximum uptake capacity of 173 mg g−1. The adsorption of sulfide by the RGO/Fe3O4 hybrid material can be attributed to the synergistic effect of both chemical and physical adsorption according to kinetic, adsorption isotherm and thermodynamic studies. The RGO/Fe3O4 material with oxygenated functional groups could convert sulfides to stable elemental sulfur and sulfone organics. The external magnetic field could easily separate the magnetic RGO/Fe3O4 adsorbent from the liquid. This research provides a novel strategy for the green and low-cost treatment of sulfide-containing wastewater by the RGO/Fe3O4 hybrid material.
1. Introduction
Sulfides (H2S, HS−, S2−) are regarded as one of the most widely existing pollutants in the environment.1 Sulfides in wastewater can react with metals and metal oxides to form metal sulfides, which can corrode metal equipment and metal pipes.2 Sewage leakage induced by pipeline corrosion pollutes the surrounding water, soil and groundwater environment and the corrosion product iron sulfide (FeS) then causes the water body to become black, which worsens the water quality.3 Furthermore, hydrogen sulfide (H2S) gas released from wastewater even at trace levels can cause the water to become obnoxious and is toxic for the human nervous system.4
Sulfides enter the aqueous environment through both natural processes and human activities. Sulfides can be formed spontaneously by the anaerobic decomposition of sulfur-containing organic matter or the reduction of biological sulfates under the action of septic bacteria and sulfate-reducing bacteria, which usually occurs in natural systems such as marine, river and lake sediments as well as in the treatment of municipal and agricultural sewage and waste.5 Moreover, the majority of manufactured dissolved sulfide and gaseous H2S pollutants are discharged into the natural environment. The average sulfide concentrations in wastewater from the petrochemical and leather industries are around 150 g L−1 and 360 mg L−1, respectively.6,7 Dissolved sulfides also have been frequently detected in the municipal wastewater treatment, pulp and paper production, and the manufacturing of coke and steel.1 The removal of sulfides from aqueous solutions is usually done by chemical precipitation, which is defined as sulfides reacting with metal ions such as iron and zinc ions to form insoluble precipitates.8 However, this technology requires a large amount of precipitant input and the precipitate produced is too small to be separated; hence, the comprehensive economic benefits are not good.
Recent studies demonstrated that oxidation and adsorption have been generally adopted for sulfide removal.9 It is well known that sulfides in wastewater mostly exist as reductive S2−, which can be oxidized to elemental sulfur/sulfate by different oxidants such as air, ozone, chlorine, potassium permanganate and hydrogen peroxide.10 However, sulfates are reduced into sulfides again by sulfate-reducing bacteria (SRB), and cannot be completely removed in many aquatic systems.11 Adsorption is a powerful approach that has been widely used for sulfide removal due to its low cost and capability for complete removal.12 To date, various adsorbents (including activated carbon, metal oxides, porous graphitic carbon composite and graphene-based materials) have been employed for sulfide removal. Hariz et al. found that activated carbon calcined at 500 °C could effectively adsorb sulfide in refining wastewater, and its adsorption capacity was 58.82 mg g−1.13 Jacukowicz-Sobala et al. used hybrid polymers containing iron oxides prepared through a carboxyl cation exchanger to remove sulfides from water with an adsorption capacity of 60 mg g−1.14 The sulfide was removed by heterogeneous oxidation and reductive dissolution of iron(III) oxides. Edathil et al. synthesized a graphene-based hybrid adsorbent using desert sand and sucrose, and the maximum adsorption capacity for sulfide was 370 mg g−1.15 The adsorption behavior of sulfide on the adsorbent fitted the Langmuir adsorption isotherm and pseudo-second-order kinetic model well. Sulfide is removed by conversion to elemental sulfur and sulfate.
Among these materials, carbon-based adsorbents have won significant consideration owing to its large specific surface area, abundant adsorption sites and good chemical stability.16,17 In particular, reduced graphene oxide (RGO) exhibits good absorption property.18 RGO hydrogel and aerogels had been used for the removal of toluene, rhodamine 6G and phenol with adsorption capacities of 500 mg g−1, 500 mg g−1 and 150 mg g−1, respectively, and could be reused at least five times.19 Porous 3D RGO showed good adsorption performance for imitated polystyrene microplastics by the strong π–π interaction.20 Moreover, RGO has been also demonstrated to have good catalytic activity in oxidative desulfurization reactions to remove sulfur-containing compounds.21 Therefore, RGO potentially can be used as both adsorbent and catalyst to remove dissolved sulfides from waters through adsorbing and catalytically oxidizing sulfides into stable sulfur-containing organics in cases involving the secondary conversion of sulfates.
However, direct separation of RGO from solution by centrifugation or filtration is a high-cost process after adsorption, which limits its practical application. In addition, the easy aggregation of RGO will reduce the adsorption capacity.22 These problems can be solved well by combining magnetic particles with RGO. Fe3O4 is the most commonly used magnetic particle, which is superparamagnetic and environmentally friendly. Furthermore, it has a large surface area and is a low-cost material.23 RGO loaded with Fe3O4 can prevent the aggregation of RGO platelets, which increases the surface area and improves the adsorption capacity. Simultaneously, the magnetic adsorbent can be easily separated from the solution by an external magnetic field.
In the present study, a reduced graphene oxide/magnetite (RGO/Fe3O4) hybrid material was synthesized by a simple in situ chemical synthesis method. RGO/Fe3O4 was tested to remove sulfide from aqueous solution under different experimental parameters, including contact time, solution pH, adsorbent dosage, ionic strength and temperature. The hybrid material was also characterized by SEM, XRD, FTIR, XPS and BET techniques. Adsorption kinetics, isotherm models and thermodynamics were applied to determine the rate control step, maximum adsorption capacity and adsorption mechanism. Moreover, a corresponding reaction mechanism for the removal of sulfides using the RGO/Fe3O4 hybrid material was put forward.
2. Experimental
2.1 Materials preparation
Graphite powder (99.95%, ≥325 mesh) was purchased from Beijing InnoChem Technology (China). Other reagents and chemicals, including sodium sulfide (Na2S·9H2O), iron(II) chloride tetrahydrate (FeCl2·4H2O), sodium nitrate (NaNO3), potassium permanganate (KMnO4), hydrogen peroxide (H2O2), sulfuric acid (H2SO4, 98%) and hydrochloric acid (HCl, 37%), were obtained from Sinopharm Chemical Reagent (China). All chemicals were of analytically pure grade. Ultrapure water (>18 MΩ cm) was used in all experiments.
2.2 Preparation of the RGO/Fe3O4 hybrid material
The modified Hummers' method24 was used to prepare graphene oxide (GO) from graphite powder (see Text S1†). Synthesis of the RGO/Fe3O4 hybrid material with different weight ratios of GO and FeCl2·4H2O was carried out via a simple in situ chemical synthesis method25 with some modifications. In a typical synthesis of the RGO/Fe3O4 hybrid material, GO (25 mg) was dispersed into ultrapure water (50 mL) by ultrasonication for 45 min. Then, FeCl2·4H2O (2.0281 g) was added to this suspension under nitrogen protection. Sequentially, the mixture was stirred at ambient temperature, and 25% ammonia solution (15 mL) was mixed slowly. Each drop of ammonia solution quickly turned the solution from brown to black, and the stirring of the black mixture was maintained for 15 min. Finally, the resulting mixture was washed repeatedly with ultrapure water and ethanol to obtain a solid sample. The solid sample was freeze-dried to yield the RGO/Fe3O4 hybrid material. The weight ratios of GO and FeCl2·4H2O were set to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1–1:4 (GO, 25 mg; FeCl2·4H2O, 1.0141, 2.0281, 3.0422, and 4.0562 g), where the obtained composites were denoted as RGO/Fe3O4-1, RGO/Fe3O4-2, RGO/Fe3O4-3, RGO/Fe3O4-4, respectively.
:1–1:4 (GO, 25 mg; FeCl2·4H2O, 1.0141, 2.0281, 3.0422, and 4.0562 g), where the obtained composites were denoted as RGO/Fe3O4-1, RGO/Fe3O4-2, RGO/Fe3O4-3, RGO/Fe3O4-4, respectively.
2.3 General characterization
The crystal structures of various composites were determined by X-ray diffraction (XRD) with the X-ray source of the Cu-Kα radiation (Ultima IV, Japan) in the range of 10–80° (2 theta). The surface morphology analysis was carried out by scanning electron microscopy (SEM) (S-4800, Hitachi, Japan). The surface functional groups of the immobilization materials were identified via Fourier-transform infrared spectroscopy (FTIR) (Nicolet iS50, Thermo Fisher, USA). The elemental content and chemical state of various compounds were measured by X-ray photoelectron spectroscopy (XPS) (Thermo ESCALAB 250XI, USA) with an Al Kα X-ray as the excitation source. The specific surface area was determined by the Brunauer–Emmett–Teller (BET) method (ASAP2460, Micromeritics, USA).
2.4 Chemical analysis
Various concentrations of sulfide solution were obtained by diluting the stock solution, which was prepared by dissolving 7.5 g of sodium sulfide with a small amount of water and transferring to a 1000 mL volumetric flask for diluting to line. The concentrations of sulfide before and after adsorption were determined by methylene blue method (See Text S2†). All adsorption experiments were carried out without nitrogen gas protection to mimic the actual wastewater scene.
Batch adsorption experiments were carried out by mixing a specific quantity of RGO/Fe3O4 in 10 mL of determined concentrations of sulfide solution, which was held in different 50 mL stoppered conical flasks. The flasks were placed in a constant temperature oscillator (ZD-85, Yinggong, China), which was kept at room temperature with a fixed speed of 160 rpm for all subsequent experiments. The main experimental parameters for the effect of adsorption, including contact time (0–180 min), pH value (2.0–12.0), adsorbent dosage (0–50.0 mg), initial sulfide concentration (0–350 mg L−1) and temperature (25–45 °C). In addition, this paper explored the effect of ionic strength on sulfide adsorption, which was conducted by changing the concentrations of NaCl and CaCl2 (0–1 M). The detailed experimental procedures are supplied in Text S3.† A volume (1.0 mL) of the dispersion solution was pipetted at the specified moment and filtered quickly with a 0.45 μm filter membrane. Then, the equilibrium concentrations of sulfide were quantified by UV-vis absorbance at 665 nm. All tests were repeated multiple times. The removal rate (%) and uptake capacity (Qe, mg g−1) for sulfide under equilibrium condition of the RGO/Fe3O4 composite were determined from eqn (1) and (2), respectively.
| |
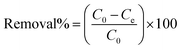 | (1) |
| |
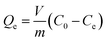 | (2) |
where
C0 (mg L
−1) is the initial concentration of the sulfide solution,
Ce (mg L
−1) is the equilibrium concentration after adsorption,
V (L) is the volume of the sulfide solution, and
m (g) is the weight of the adsorbent added.
For kinetic studies, linear pseudo first order, linear pseudo second order and intra-particle diffusion models were used to understand the adsorption mechanism. Equivalently, Langmuir and Freundlich adsorption isotherms in linear and non-linear forms were fitted by the sulfide uptake gained at different initial sulfide concentrations to determine the equilibrium adsorption capacity. Thermodynamic studies were carried out to further analyze the adsorption process.
Furthermore, in order to find an appropriate model to fit the adsorption data, the root mean squared error (RMSE) and χ2 values were used to evaluate the linear and non-linear adsorption isotherm models, combined with the value of the correlation coefficient (R2) from the regressive analysis. The expressions for the above error functions were calculated as follows:
| |
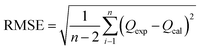 | (3) |
| |
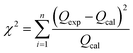 | (4) |
where
Qexp is the adsorption capacity obtained experimentally,
Qcal is the adsorption capacity calculated from the isotherm, and
n is the number of test elements.
3. Results and discussions
3.1 Characterization of the RGO/Fe3O4 hybrid material
3.1.1. Morphology and microstructure analysis. Fig. 1(a–d) shows the morphology of graphite, GO, fresh and used RGO/Fe3O4 by SEM, respectively. It can be seen that flake graphite became thin paper-like graphene oxide, confirming the successful synthesis of GO using the modified Hummers' method.26 The RGO/Fe3O4 hybrid materials showed a stacked sheet-like morphology with some small irregular sheets (Fig. 1(c)). One plausible explanation for this observation is that an electrostatic attraction exists between the Fe2+ and graphene oxide sheets with negatively charged functional groups.27 Fe2+ was oxidized to Fe3+ by the hydroxyl (OH), alkoxy (C–O–C) and carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), and carboxylic acid (COOH), which were reduced simultaneously with the formation of RGO. The detailed redox reaction mechanisms are described in eqn (5)–(7).
O), and carboxylic acid (COOH), which were reduced simultaneously with the formation of RGO. The detailed redox reaction mechanisms are described in eqn (5)–(7).| | |
2Fe2+ + GO → 2Fe3+ + RGO
| (5) |
| | |
2Fe3+ + Fe2+ + 8OH− → Fe3O4 + 4H2O
| (6) |
| | |
3Fe2+ + GO(8OH−) → Fe3O4 + 4H2O + RGO
| (7) |
 |
| | Fig. 1 SEM images of (a) graphite, (b) GO, (c) fresh RGO/Fe3O4 (RGO/Fe3O4-2) and (d) used RGO/Fe3O4 (RGO/Fe3O4-2, C0 = 135.88 mg L−1, T = 298 K, m = 0.050 g, t = 180 min, pH = 11.74). | |
Compared with Fig. 1(c), the morphology variation of the RGO/Fe3O4 hybrid material adsorbed sulfide was not apparent (Fig. 1(d)), which demonstrated that there may be little relationship between the morphology and the adsorption.28
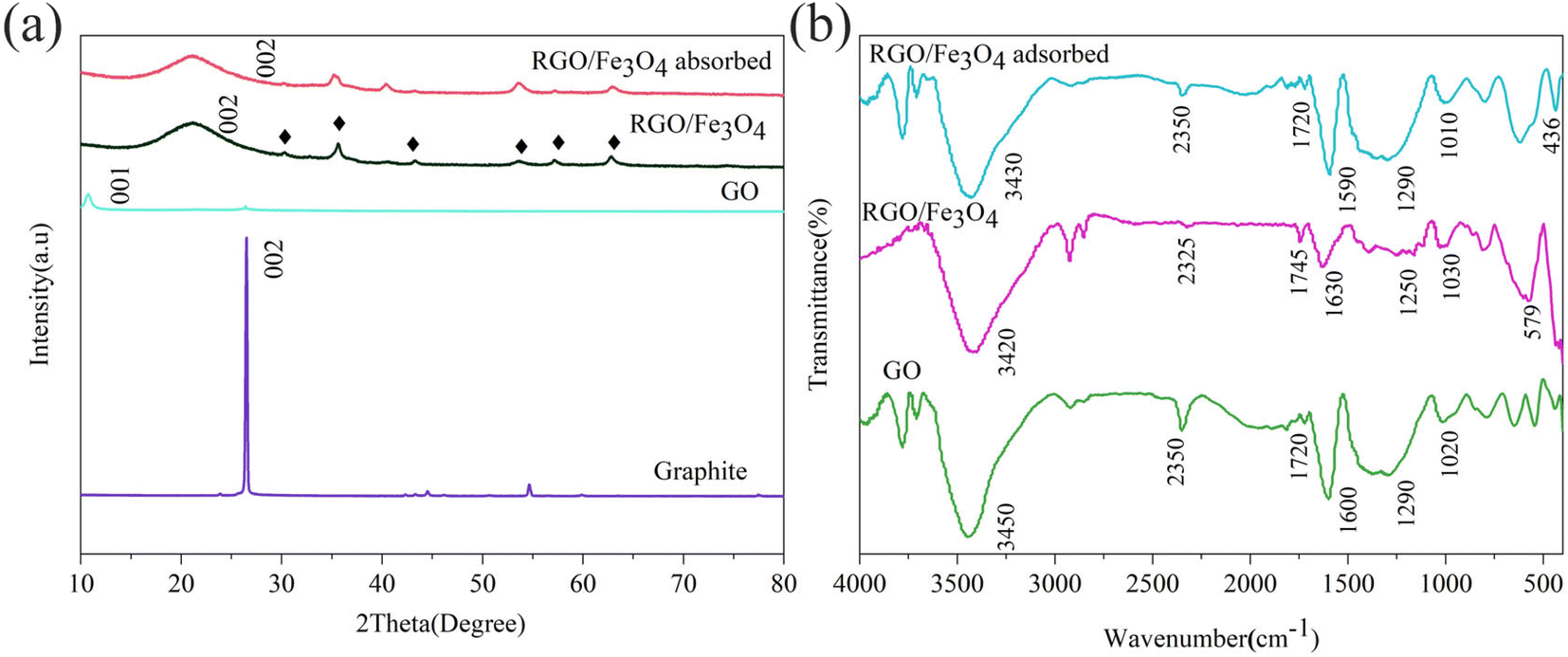
3.1.2. Crystal structure analysis. Fig. 2(a) provides the X-ray diffraction patterns of graphite, GO, fresh and used RGO/Fe3O4. The graphite exhibited a strong diffraction peak at 26.4° (002) with a corresponding interlayer spacing (d-spacing) of 0.3376 nm. From Fig. 2(a), it can be seen that a large peak shifted from 26.4° to 10.7° (001) for the GO with increased interlayer spacing of 0.8252 nm. This could be explained by the presence of oxygen-containing functional groups.29 This result was consistent with the SEM results. The diffraction peaks of the RGO/Fe3O4 hybrid material at 2θ of 30.12°, 35.48°, 43.12°, 53.50°, 57.03° and 62.62° were indexed to the (220), (311), (400), (422), (511), and (440) planes of Fe3O4 (JCPDS#88-0866), respectively.30 Moreover, a strong diffraction peak occurred at 2θ of 21.22° (002) with the interlayer spacing of 0.4184 nm for RGO,31 which was caused by the Fe2+ reduction. The higher interlayer spacing demonstrated that some oxygen-containing functional groups were still retained in RGO.32 These conclusions confirmed that RGO/Fe3O4 was well prepared by in situ chemical method. However, the XRD pattern of RGO/Fe3O4 showed no significant peak change after sulfide adsorption, which might be due to low sulfur content or the amorphous product.
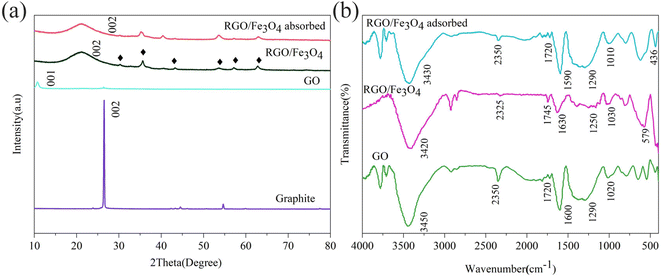 |
| | Fig. 2 XRD diffraction patterns (a) of graphite, GO, fresh and used RGO/Fe3O4 (RGO/Fe3O4-2, C0 = 135.88 mg L−1, T = 298 K, m = 0.050 g, t = 180 min, pH = 11.74) and FTIR spectra (b) of GO, fresh and used RGO/Fe3O4 (RGO/Fe3O4-2, C0 = 135.88 mg L−1, T = 298 K, m = 0.050 g, t = 180 min, pH = 11.74). | |
3.1.3. Surface functional groups analysis. The surface properties and functional groups on the surface of GO, fresh and used RGO/Fe3O4 hybrid material were further studied by FTIR spectroscopy (Fig. 2(b)). In the FTIR spectrum of GO, the peak at 3450 cm−1 was assigned to the O–H stretching vibrations of water molecules that were absorbed on the samples.33 The peak at 1720 cm−1 corresponded to the CO stretching vibration of carboxyl (–COOH) groups or carbonyl (–RCO) groups,34 and the peaks at 1290 and 1020 cm−1 were attributed to the C–O stretching vibration of the epoxy (C–O–C) groups.33 The peak at 1600 cm−1 belonged to the CC bond of the carbon skeleton.33 These data indicated that GO was synthesized well. For RGO/Fe3O4, the above characteristic peaks of oxygen-containing functional groups were weakened, indicating that GO was partially reduced to RGO. Furthermore, the peak at 579 cm−1 was ascribed to the characteristic absorption band of the Fe–O bond of bulk Fe3O4.35 Additionally, from the FTIR spectra of the sulfide-adsorbed RGO/Fe3O4, a new peak at 436 cm−1 was observed, which was attributed to the S–S bond,36 indicating the formation of elemental sulfur and the adsorption of sulfides on RGO/Fe3O4.
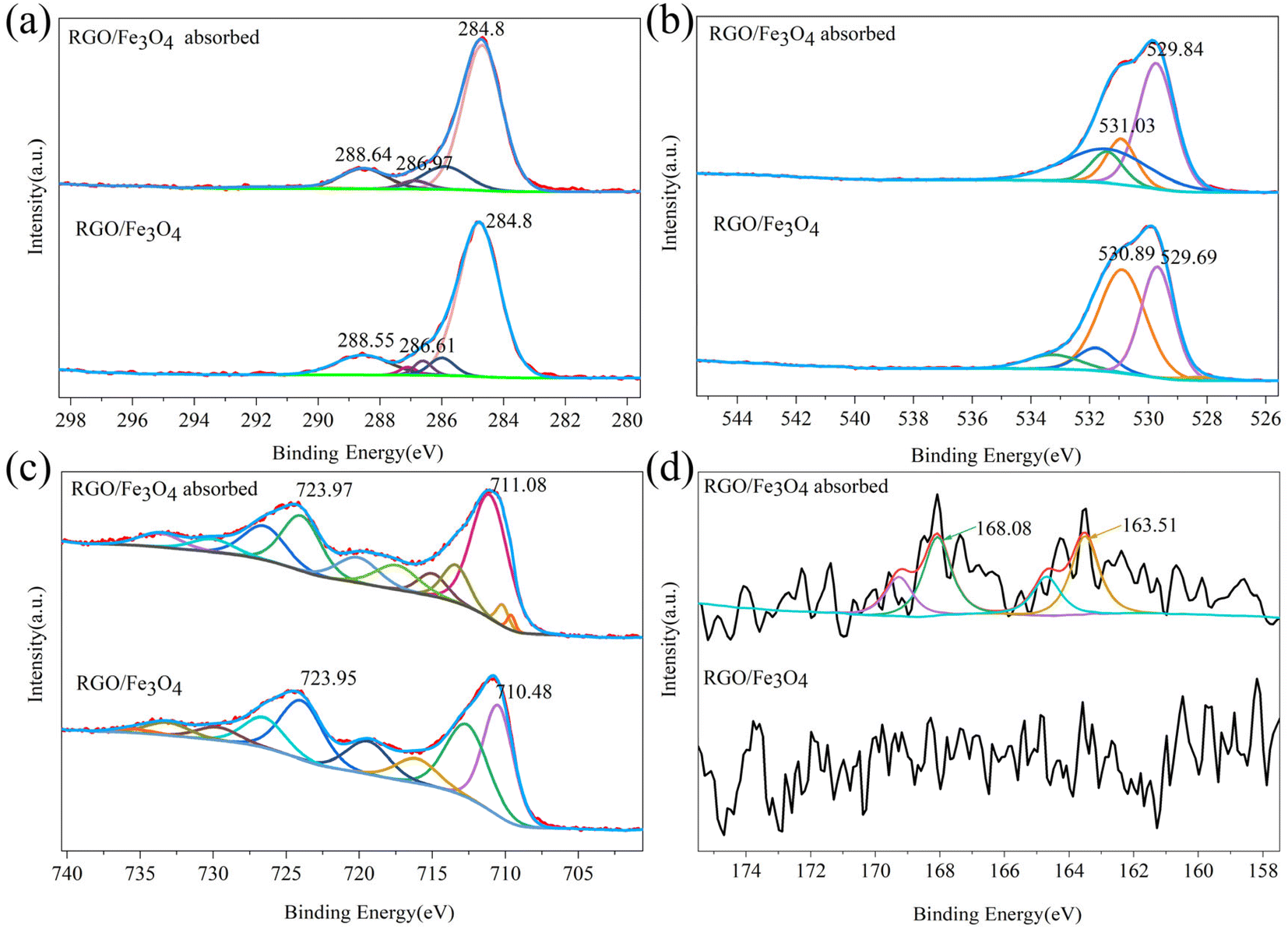
3.1.4. Chemical composition analysis of fresh and used RGO/Fe3O4. XPS spectra were used to further determine the final form of the sulfide adsorbed on RGO/Fe3O4 and their interactions. Fig. S1(a) and (b)† shows the wide spectrum of fresh and used RGO/Fe3O4 hybrid materials, respectively. The characteristic signals of carbon (C 1s at 285 eV), iron (Fe 2p at 711 eV) and oxygen (O 1s at 530 eV) were clearly observed for the fresh RGO/Fe3O4, which confirmed the formation of RGO/Fe3O4.27 The sulfur peak was indistinct on the used RGO/Fe3O4 hybrid material, but the adsorption of sulfides can be determined by combining the previous results and the narrow-spectrum analysis of S 2p. The deconvoluted C 1s for RGO/Fe3O4 before and after the adsorption of sulfide is displayed in Fig. 3(a). The peaks of the fresh RGO/Fe3O4 at 284.8, 286.61 and 288.55 eV were assigned to the C–C and CC bonding hybridization, the C–O bonds and the CO bonds of the RGO,37 respectively, which indicated the presence of oxygen-containing functional groups. The curve-fitted O 1s spectra of the two nanocomposites are shown in Fig. 3(b). The two characteristic peaks of the O 1s spectrum of fresh RGO/Fe3O4 appeared at 529.69 eV and 530.89 eV (Fig. 3(b)). The peak at 529.69 eV was associated with the lattice oxygen of Fe3O4,38 while the peak at 530.89 eV indicated the existence of oxygen-containing groups like the C–O, O–CO and O–H groups.39 Compared to the fresh RGO/Fe3O4, the binding energies in the C 1s and O 1s spectra for the used RGO/Fe3O4 slightly increased. This cans be ascribed to the interaction between the sulfides and oxygen-containing groups on RGO, and their key roles in sulfide removal. As can be seen from Fig. 3(c), the Fe 2p spectrum of RGO/Fe3O4 showed two main peaks located at 710.48 eV and 723.95 eV, which were ascribed to the Fe 2p3/2 and Fe 2p1/2 spin–orbit peaks of Fe3O4, respectively.39 The other peaks corresponded to their satellite peaks. This was similar to the result obtained from the spectra of C 1s and O 1s. The binding energies of Fe 2p also became higher in the used RGO/Fe3O4. This is possibly due to the strong interaction between Fe2+ and RGO through the Fe–O–C bonds.40 These were in agreement with the previous XRD and FTIR characterization. Fig. 3(d) shows the S 2p spectra for the fresh and used RGO/Fe3O4. The S 2p spectra of fresh RGO/Fe3O4 had a larger peak noise and lower intensity than the used RGO/Fe3O4. In addition, elemental sulfur was not introduced in the material preparation process, which indicated that there were no sulfides on the fresh RGO/Fe3O4 composite surface. The S 2p spectra exhibited two prominent peaks at 163.51 and 168.08 eV after sulfide adsorption, corresponding to the elemental sulfur and sulfones, respectively.41,42 These results indicated the successful adsorption and conversion of sulfides on the RGO/Fe3O4 composites.
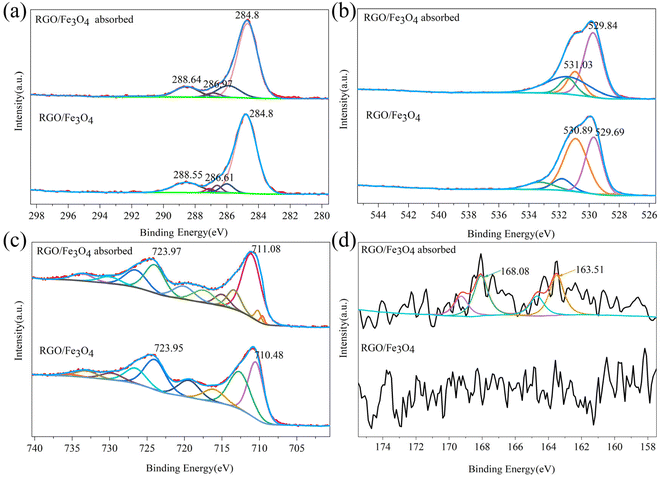 |
| | Fig. 3 XPS spectra of (a) C 1s, (b) O 1s, (c) Fe 2p and (d) S 2p of fresh and used RGO/Fe3O4 (RGO/Fe3O4-2, C0 = 135.88 mg L−1, T = 298 K, m = 0.050 g, t = 180 min, pH = 11.74). | |
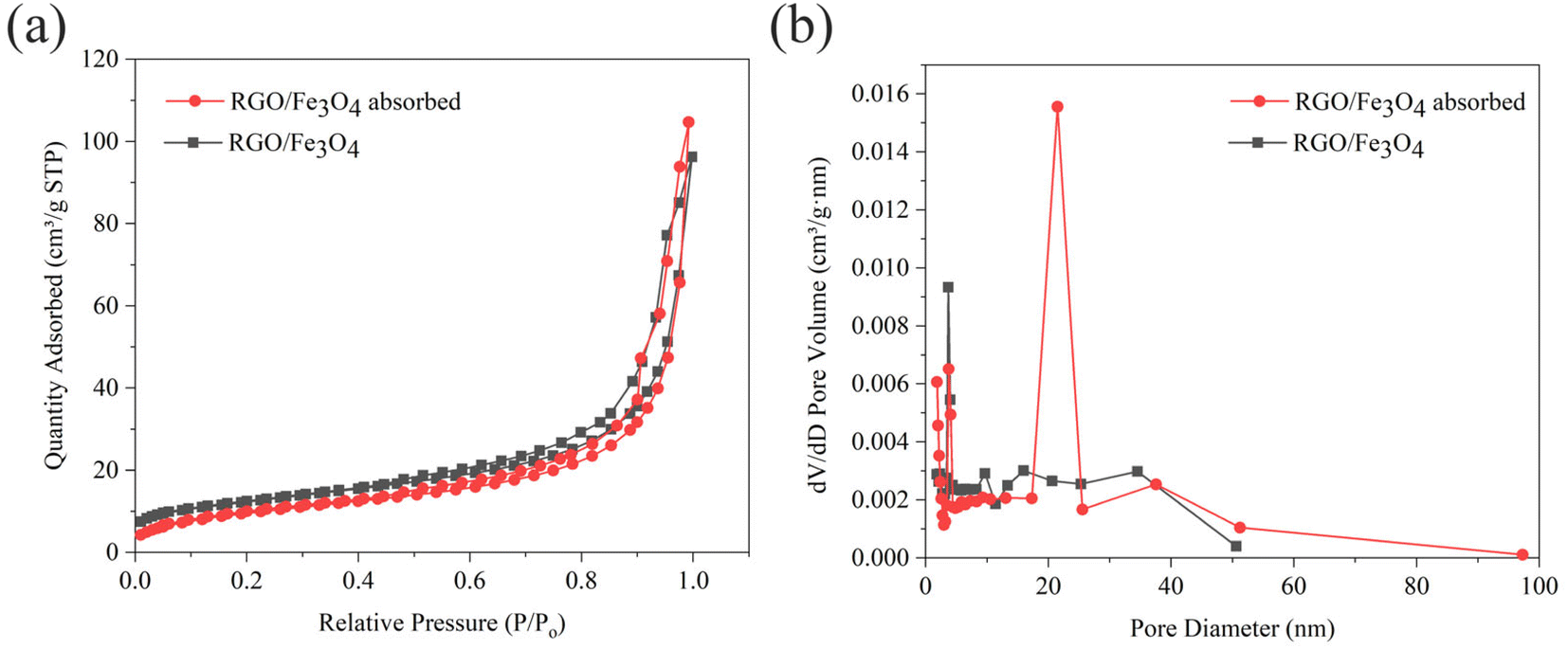
3.1.5. Specific surface area and pore structure analysis of fresh and used RGO/Fe3O4. Fig. 4 shows the N2 adsorption–desorption isotherms and pore size distribution of the fresh and used RGO/Fe3O4. The adsorption isotherms of the two samples both belong to the fourth type isotherm with H3 hysteresis loops according to IUPCA classification,43 which indicate that the two samples are mesoporous materials. It can be seen that the pore size distribution is mainly concentrated at about 10 nm (Fig. 4(b)). Based on the data in Table 1, the specific surface area of RGO/Fe3O4 was higher than that of the RGO/Fe3O4 absorbed. This can be explained by the fact that the oxygen-containing functional groups on the adsorbent reacted with sulfide during the adsorption process, resulting in a decrease in the amount of available oxygen functionalities, which in turn led to a decrease in the specific surface area. This phenomenon is consistent with previous research.44 Moreover, the adsorbed sulfide will cover the surface of the material and block the micropores, which also could reduce the specific surface area.
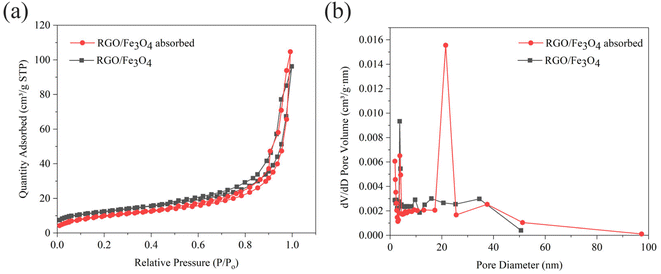 |
| | Fig. 4 (a) N2 adsorption–desorption isotherms, and (b) pore size distribution of fresh and used RGO/Fe3O4 (RGO/Fe3O4-2, C0 = 119.31 mg L−1, T = 298 K, m = 0.4 g, t = 180 min, pH = 11.70). | |
Table 1 The parameters of fresh and used RGO/Fe3O4 (RGO/Fe3O4-2) obtained from BET analysis
| Materials |
BET surface area (m2 g−1) |
Average pore diameter (nm) |
Total pore volume (cm3 g−1) |
| RGO/Fe3O4 |
43.3405 |
13.3843 |
0.076190 |
| RGO/Fe3O4 absorbed |
35.7149 |
17.2854 |
0.161920 |
3.2 Adsorption of sulfide
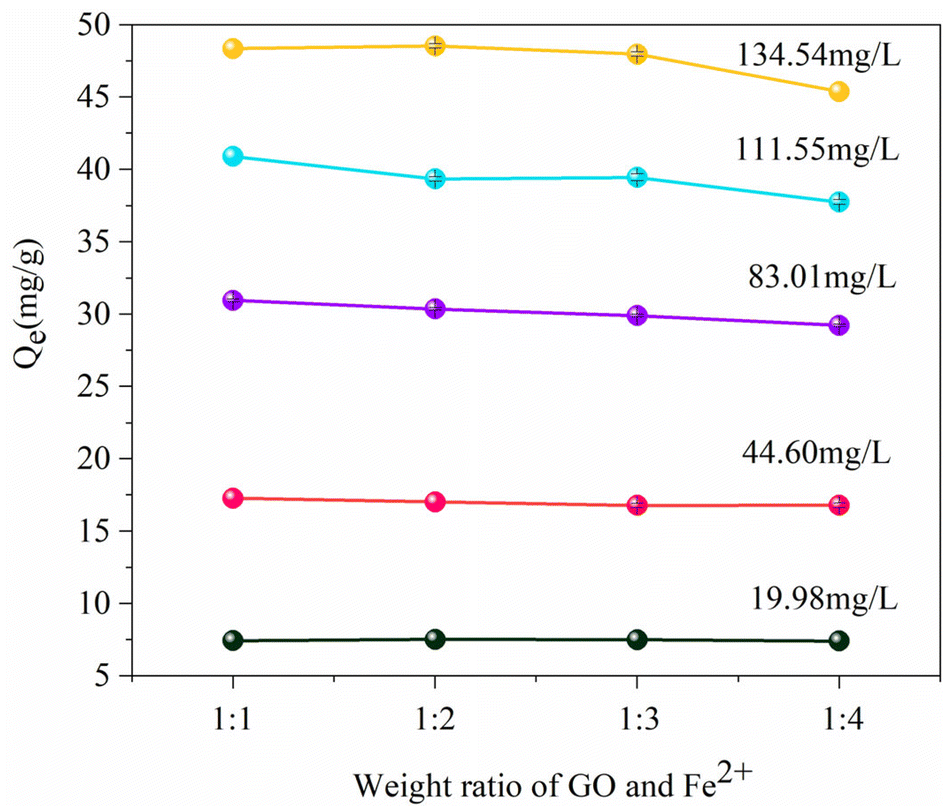
3.2.1. Adsorption effect of various RGO/Fe3O4 materials on sulfide. To investigate the impact of oxygen-containing functional groups on the sulfide removal efficiency and determine the optimal mass ratio of GO to FeCl2·4H2O for sulfide removal, four RGO/Fe3O4 adsorbents with a weight ratio of GO:Fe2+ from 1:1 to 1:4 were fabricated (denoted as RGO/Fe3O4-1, RGO/Fe3O4-2, RGO/Fe3O4-3, RGO/Fe3O4-4). As presented in Fig. 5, different initial sulfide concentrations at 19.98 mg L−1, 44.60 mg L−1, 83.01 mg L−1, 111.55 mg L−1 and 134.54 mg L−1 were used to compare the adsorption capacity. It can be seen that the adsorption capacity of sulfide did not markedly change when the ratio of mGO and mFe2+ increased from 1:1 to 1:3 while fixing the initial concentrations at 19.98, 44.60, 83.01, and 111.55 mg L−1, respectively. As the sulfide concentration was further increased to 134.54 mg L−1, the sulfide sorption capacity of RGO/Fe3O4-2 (48.55 mg g−1) was higher than those of RGO/Fe3O4-1 (48.36 mg g−1), RGO/Fe3O4-3 (47.98 mg g−1) and RGO/Fe3O4-4 (45.38 mg g−1), respectively. Compared with the previous three compounds, the adsorption capacity of RGO/Fe3O4-4 slightly decreased. This is possibly because the oxygen-containing functional groups on GO were more reduced by excess Fe2+ during preparation of RGO/Fe3O4. Therefore, the sulfides cannot be fully bind to the active sites of RGO in high concentration solutions, and further resulted in a decrease in the adsorption capacity. Finally, to balance the removal efficiency and the cost, RGO/Fe3O4-2 was selected for further research.
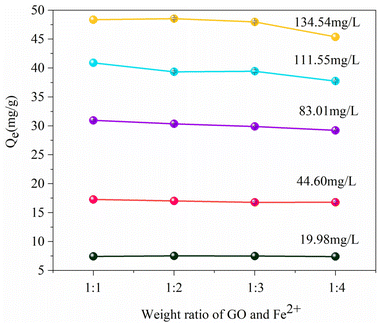 |
| | Fig. 5 The sorption capacities of various RGO/Fe3O4 (T = 298 K, m = 25 mg, t = 180 min). Error bars represent the standard deviation of replicate measurements. | |
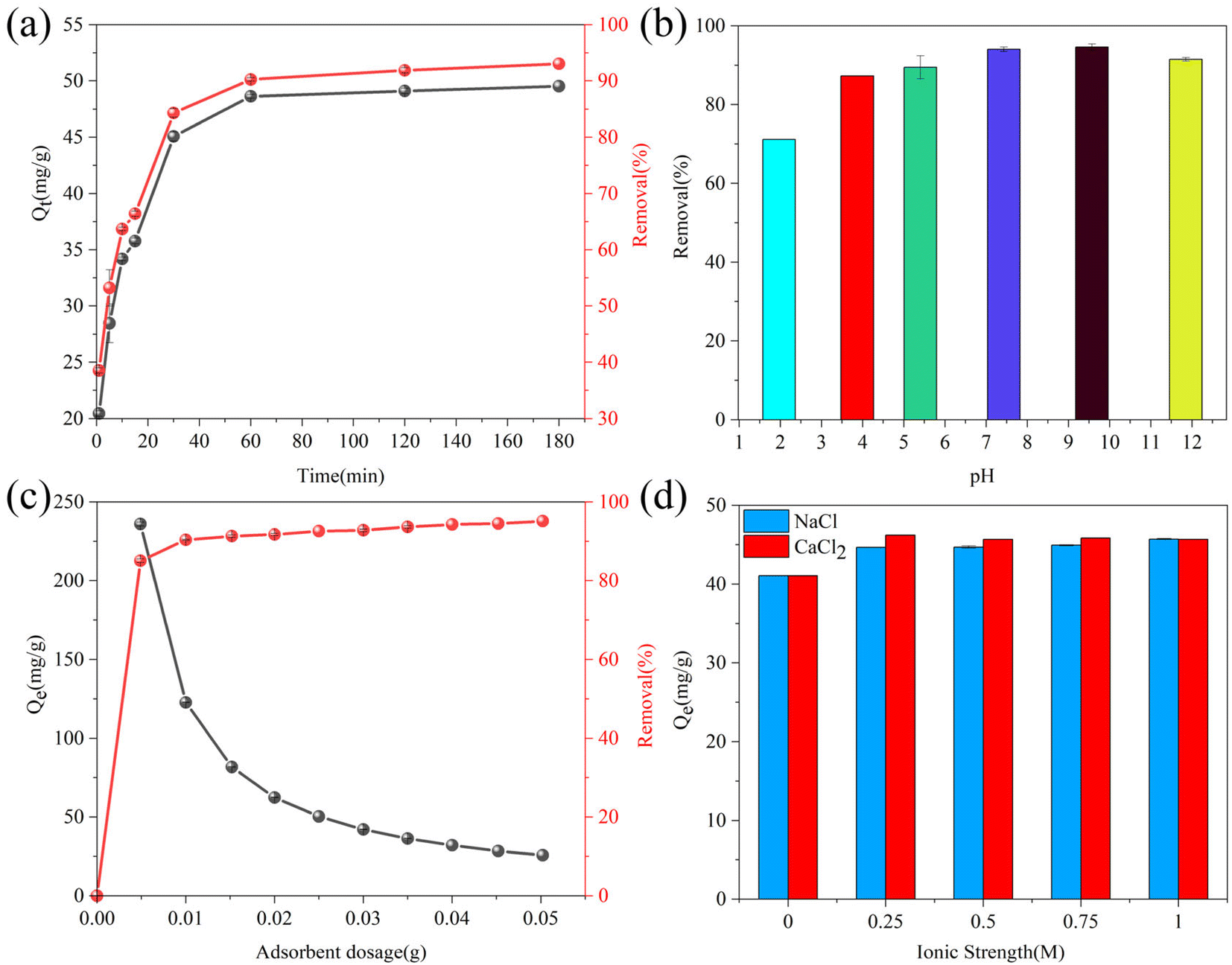
3.2.2. Effect of the contact time. Inceptive adsorption tests were performed to determine the equilibrium contact time of adsorption, and the corresponding experimental results are presented in Fig. 6(a). The sulfide removal efficiency and uptake capacity increased with the contact time. It can be seen that the removal efficiency and adsorption capacity of sulfide increased rapidly in the early stage, which could be ascribed to the presence of sufficient unsaturated active sites on the fresh RGO/Fe3O4 composite. The effect gradually disappeared until it barely increased after 30 min. Subsequently, the adsorption processes gradually reached equilibrium at time intervals ranging from 60 to 120 min. However, upon increasing the contact time to 180 min, the removal efficiency slightly increased because the vacant sites were completely occupied, and thus the equilibrium was established. The sorption equilibrium of RGO/Fe3O4 for sulfide was achieved within 180 min with the adsorption capacity of 49.54 mg g−1. Therefore, 180 min was determined as the contact time for subsequent experiments.
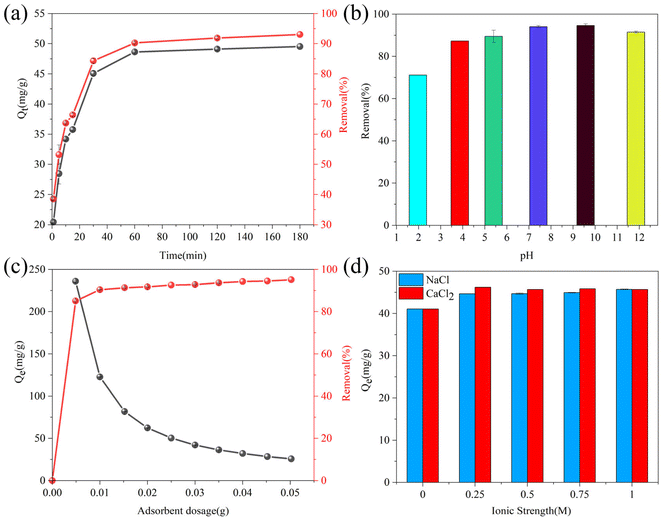 |
| | Fig. 6 (a) Effect of the contact time on sulfide adsorption using RGO/Fe3O4 (C0 = 133.10 mg L−1, T = 298 K, m = 25 mg, pH = 11.72). (b) Effect of pH on the sulfide removal efficiency (C0 = 138.56 mg L−1, T = 298 K, m = 25 mg, t = 180 min). (c) Effect of the adsorbent dosage on sulfide removal using RGO/Fe3O4 (C0 = 135.88 mg L−1, T = 298 K, t = 180 min, pH = 11.74). (d) Effect of the ionic strength on sulfide adsorption using RGO/Fe3O4 (C0 = 116.72 mg L−1, T = 298 K, m = 25 mg, t = 180 min, pH = 11.68). Error bars represent the standard deviation of replicate measurements. | |
3.2.3. Effect of the initial solution pH. The impact of the initial solution pH on the removal efficiency of sulfide by RGO/Fe3O4 was explored at pH 1.97–11.85, and the corresponding results are shown in Fig. 6(b). The sulfide removal performance exhibited obvious fluctuations under acidic and alkaline conditions. Previous research reported that the sulfides mainly existed in the form of H2S (aq.) and HS− under strongly acidic conditions, while the aqueous solution predominantly contains S2− along with HS− under strongly alkaline condition.45 When the solution pH decreased, most S2− would be converted into H2S gas, which was then dispersed from the solution into the air, decreasing the sulfide concentration and causing the low removal efficiency of sulfide according to eqn (1). However, the reaction between the adsorbent and the residual sulfide was not affected by the acidic condition. At alkaline conditions, S2− remained in a relatively stable state. The obtained pH results clearly indicated that the removal of sulfide by RGO/Fe3O4 was not impaired, and the removal efficiency almost retained the same value. The oxygen functional groups in RGO can be classified into acidic functional groups, such as carboxyl, phenol and lactol, and basic functional groups, including ketone, quinone, and carbonyl, according to the acidic or basic nature of the aqueous solution.32 Therefore, the adsorption removal of sulfide by RGO/Fe3O4 was almost unaffected by solution pH, indicating that the synthesized adsorbent could be applied to sulfur-containing wastewater at all pH values. The following batch experiments were conducted at the actual pH of the original solution.
3.2.4. Effect of the adsorbent dosage. From Fig. 6(c), we can see the effect of different doses of RGO/Fe3O4 hybrid materials (5–50 mg) on the sulfide removal efficiency and adsorption capacity. The results showed that the sulfide removal efficiency increased as the adsorbent dosage increased, and attained a maximum of 95.11% for 50.0 mg. This phenomenon could be explained as follows: the available active sites for adsorption increased with the increased amount of RGO/Fe3O4, leading to a higher adsorption capacity. Furthermore, there was a decrease in the adsorption capacity from 235.91 to 25.75 mg g−1 when varying the dosage of the adsorbent from 5 to 50 mg, which might be due to the increase of unsaturated adsorption sites. The increase of the adsorbent dosage with constant sulfide solution concentration will increase the amount of unsaturated active sites, which results in the decrease of sulfide content per unit mass of adsorbent, meaning a decrease in the adsorption capacity.46 However, the removal efficiency did not change much when the adsorbent dosing was higher than 25.0 mg because of the absence of sulfide ions bound to the adsorption site. Furthermore, there might be interference between the binding sites when the amount of adsorbent was too much.47 Therefore, considering both results and costs, the adsorbent dosage was selected as 25 mg for all of the subsequent optimization and kinetic studies.
3.2.5. Effect of the ionic strength. Because wastewater usually contains salt, it is important to investigate the effect of the ionic strength in the sulfide adsorption on RGO/Fe3O4. The presence of salts will compete with the sulfide species for available adsorption sites.9 In the present study, NaCl and CaCl2 were used to simulate the ionic strength on the removal of sulfide. Interestingly, the adsorption capacity of sulfide increased when increasing the NaCl and CaCl2 concentrations from 0.25 to 1 M, and the growth extent of CaCl2 was larger (Fig. 6(d)). Previous research reported that increasing the CaCl2 (0–1.0 M) and NaCl (0–0.5 M) concentration could both decrease the adsorption capacity.12 A possible explanation for this might be the diffuse double layer theory. The thickness of the electric double layer can be compressed by the addition of electrolyte, which weakens the electrostatic interaction between the adsorbate and the adsorbent.48 This inconsistency could be due to the existence of electrostatic repulsion between the RGO/Fe3O4 and sulfide, which would be impaired after adding KCl and CaCl2. Therefore, the sulfide adsorption was further enhanced. Furthermore, it is clear that the Ca2+ has a more apparent effect than K+. The reason could be that Ca2+ could form intramolecular bridges such as R–COO−–Ca2+–−OOC–R,49 which further screened the negative charges of the adsorbent and interfered with the electrostatic repulsion.
3.2.6. Adsorption kinetic. Adsorption kinetic analysis was used to investigate the adsorption rate and reaction mechanism of the adsorption process of sulfide by RGO/Fe3O4. In this work, the adsorption kinetics data were fitted using the pseudo-first-order (PFO) model, pseudo-second-order (PSO) model and intraparticle diffusion (IPD) model.50 The corresponding linear plots and parameters of the PFO and PSO models are displayed in Fig. 7(a) and Table 2, respectively. Qe is the uptake capacity at the equilibrium of the RGO/Fe3O4 composite for sulfide, Qt is the uptake capacity at time “t”, kf represents the pseudo-first-order rate constant and ks represents the pseudo-second-order rate constant. It can be seen that the correlation coefficient R2 obtained by the PSO model was larger than that for the PFO model, and the experimentally measured adsorption capacity (Qe(exp) = 49.54 mg g−1) was close to the theoretical value (Qe(cal) = 50.65 mg g−1). This showed that PSO model could better describe the adsorption of sulfide by RGO/Fe3O4. Meanwhile, this further indicated that sulfide adsorption on the RGO/Fe3O4 hybrid material followed a chemisorption mechanism with a rate constant of 0.005193 mg−1 min−1.51 This behavior was in agreement with the characterization results obtained from the used RGO/Fe3O4.
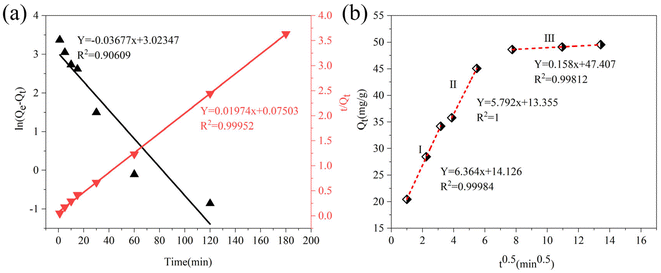 |
| | Fig. 7 Linear plots of (a) PFO and PSO models, and (b) IPD model (C0 = 133.10 mg L−1, T = 298 K, m = 25 mg, pH = 11.72). | |
Table 2 Corresponding equations and parameters of the PFO and PSO kinetic models and IPD model for sulfide adsorption on the RGO/Fe3O4 adsorbent
| Model |
Parameters |
| Pseudo-first order ln(Qe − Qt) = ln(Qe) − kft |
kf (min−1) |
0.03677 |
| Qe(cal) (mg g−1) |
20.56 |
| Qe(exp) (mg g−1) |
49.54 |
| R2 |
0.9061 |
Pseudo-second order 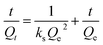 |
ks (mg−1 min−1) |
0.005193 |
| Qe(cal) (mg g−1) |
50.65 |
| Qe(exp) (mg g−1) |
49.54 |
| R2 |
0.9995 |
| Intraparticle diffusion Qe = kit0.5 + Ci |
k1 (mg g−1 min−0.5) |
6.364 |
| C1 |
14.13 |
| R12 |
0.9998 |
| k2 (mg g−1 min−0.5) |
5.792 |
| C2 |
13.36 |
| R22 |
1.0000 |
| k3 (mg g−1 min−0.5) |
0.1580 |
| C3 |
47.41 |
| R32 |
0.9981 |
Fig. 7(b) shows the plot of the IPD model, and the corresponding parameter is presented in Table 2. Ci represents the boundary layer thickness and ki represents the intra-particle diffusion constant, where “i” represents the different stages. The greater the C value, the greater the boundary layer effect.52 As can be seen from Fig. 7(b), the adsorption of sulfide by RGO/Fe3O4 presents a multi-level linear relationship. At the beginning, Qt increased rapidly with time t, then rose slowly, and finally tended to be in dynamic equilibrium, indicating that two or more diffusion steps affecting the rate control were involved in the whole adsorption process. The rapidly rising stage indicated that sulfide diffused from the solution to the outer surface of the adsorbent with a relatively high rate constant (k1 = 6.364 mg g−1 min−0.5). The subsequent ascending phase indicated that sulfide passed through the liquid membrane and onto the RGO/Fe3O4 surface with a smaller rate constant (k2 = 5.792 mg g−1 min−0.5). In the equilibrium stage, the rate constant decreased significantly (k3 = 0.158 mg g−1 min−0.5), which may be due to the reduction of adsorption sites and the electrostatic repulsion between the sulfides on RGO/Fe3O4 and the aqueous solution. It is obvious that none of the three lines goes through the origin, so the adsorption pathways of sulfide on RGO/Fe3O4 may be controlled by other processes besides intra-particle diffusion.53
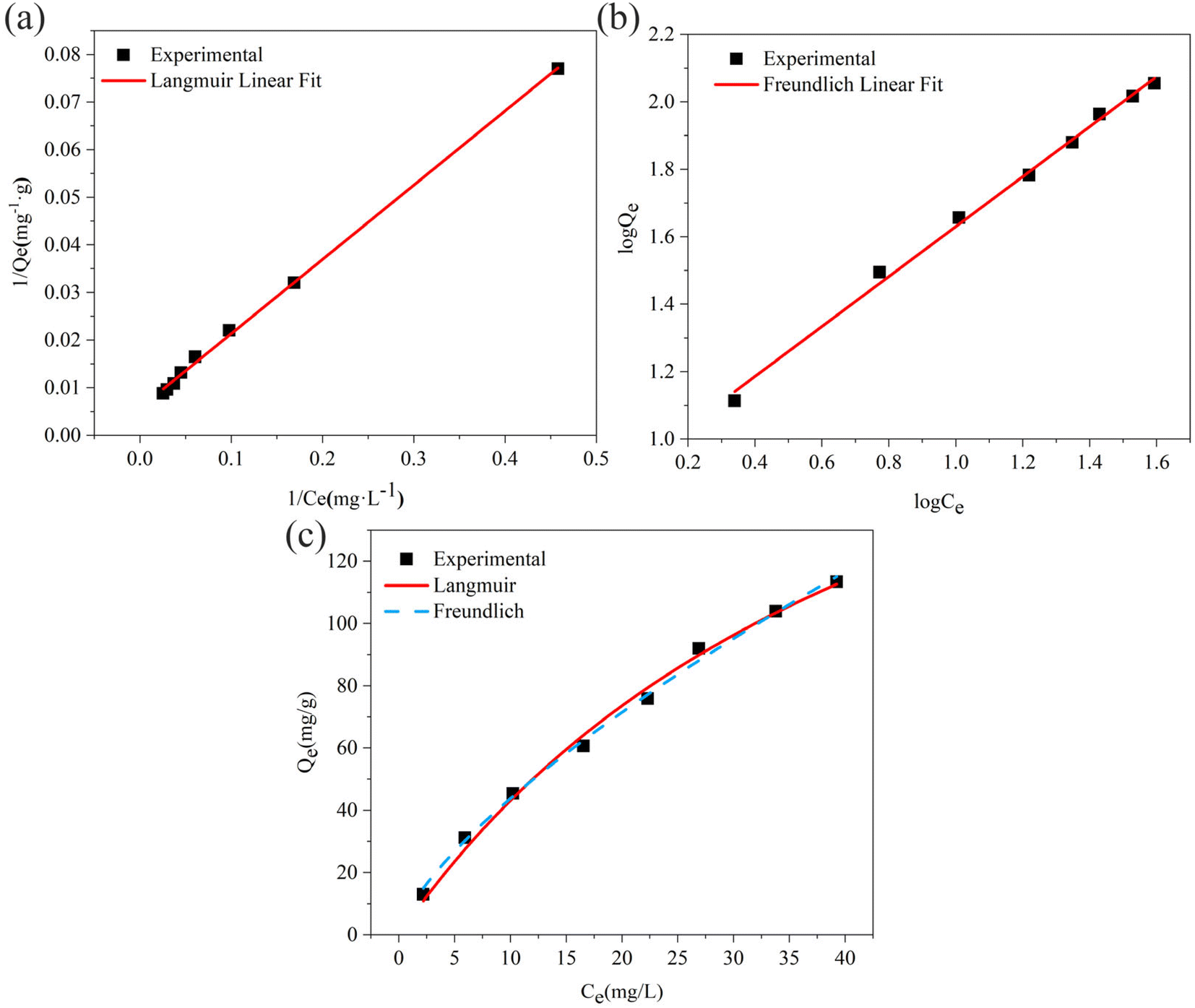
3.2.7. Equilibrium study. To investigate the sulfide adsorption mechanism and the maximum adsorption capacity on the RGO/Fe3O4 hybrid material, linear and nonlinear plots of Freundlich and Langmuir adsorption isotherms were used and the corresponding parameters are shown in Fig. 8 and Table 3, respectively. KL is the Langmuir adsorption constant, KF is the reaction rate constant of the Freundlich linear model, and 1/n (dimensionless) is the affinity of adsorption. According to the R2 of the adsorption isotherm, the Langmuir linear model can fit the experimental data better than the Freundlich linear model with the maximum uptake capacity of 173.01 mg g−1, which indicated that the adsorption of sulfide by RGO/Fe3O4 was a monolayer adsorption. The Freundlich linear isotherm assumed that the adsorption was multilayer and occurred on heterogeneous surfaces. The value of 1/n (0.7406) obtained from the Freundlich linear equation was between 0.1 and 1.0, which indicated that this isotherm was also favorable to describe the adsorption of sulfide on RGO/Fe3O4.54
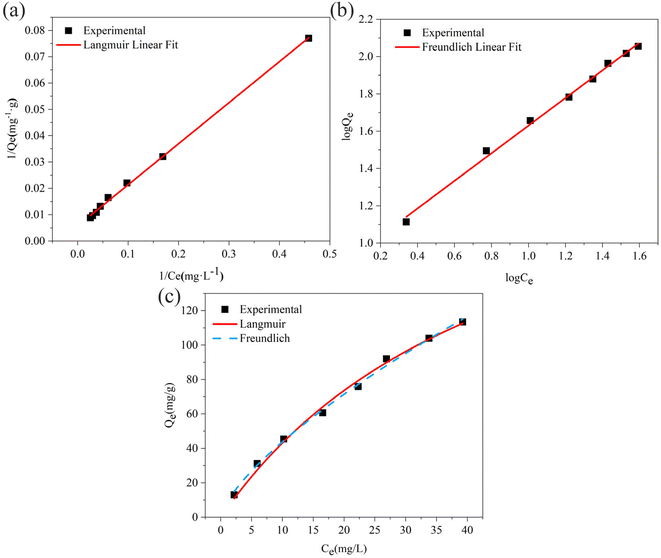 |
| | Fig. 8 (a) Linear Langmuir adsorption isotherm, (b) linear Freundlich adsorption isotherm, (c) non-linear Langmuir and Freundlich adsorption isotherms (C0 = 34.54–325.06 mg L−1, T = 298 K, m = 25 mg, t = 180 min). | |
Table 3 Linear and nonlinear isotherm models and parameters of the Freundlich and Langmuir models for sulfide adsorption on RGO/Fe3O4
| Model |
Parameters |
Langmuir linear isotherm 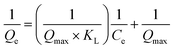 |
qmax (mg g−1) |
173.01 |
| KL |
0.0371 |
| R2 |
0.9987 |
Langmuir non-linear isotherm 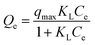 |
qmax (mg g−1) |
251.32 |
| KL |
0.0207 |
| R2 |
0.9940 |
Freundlich linear isotherm 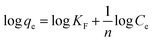 |
1/n |
0.7406 |
| KF |
7.76 |
| R2 |
0.9960 |
Freundlich non-linear isotherm 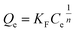 |
1/n |
0.7069 |
| KF |
8.60 |
| R2 |
0.9960 |
To accurately represent the equilibrium data, Langmuir and Freundlich non-linear sorption isotherms were also adopted and are shown in Fig. 8(c). The data revealed that the equilibrium data were well fitted to the Freundlich model with a higher R2 when compared to the Langmuir non-linear isotherm model. The obtained value of 1/n (0.7069) of the isotherm was fine 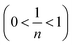 . For the Langmuir non-linear isotherm, R2 (0.9940) was lower than the value obtained from the Langmuir linear equation (0.9987), but qmax (251.32 mg g−1) was higher.
. For the Langmuir non-linear isotherm, R2 (0.9940) was lower than the value obtained from the Langmuir linear equation (0.9987), but qmax (251.32 mg g−1) was higher.
R2, RMSE and χ2 values were used to determine the most suitable adsorption isotherm model.55 As observed from Table 4, the RMSE and χ2 values of the Langmuir linear isotherm model were lower than those of the Freundlich linear isotherm model and the other two non-linear isotherm models, which revealed that the Langmuir linear model was the best fitting isotherm. Therefore, the adsorption process of sulfide on RGO/Fe3O4 was monolayer and the maximum adsorption capacity estimated was 173.01 mg g−1, which was relatively higher than the values for other sulfide adsorbents presented in the literature (Table 5). Consequently, RGO/Fe3O4 could be used as an effective adsorbent for sulfide.
Table 4 RMSE and χ2 values of the linear and nonlinear adsorption isotherm models for sulfide adsorption onto RGO/Fe3O4
| Model |
Parameters |
| RMSE |
χ2 |
| Langmuir |
Linear |
0.0009 |
0.0004 |
| Non-linear |
2.8998 |
1.3837 |
| Freundlich |
Linear |
0.0217 |
0.0019 |
| Non-linear |
2.1782 |
0.5735 |
Table 5 Comparison of the equilibrium time and qmax of sulfide by various materials
| Materials |
Time (min) |
qmax (mg g−1) |
| HIX1 |
180 |
150.00 |
| PGC12 |
180 |
149.25 |
| MAC-500 °C (ref. 13) |
30 |
58.82 |
| AIg/iron oxide-NP56 |
180 |
136.90 |
| GSH composites15 |
180 |
370.40 |
| MnO2-PGC9 |
180 |
526.32 |
| RGO/Fe3O4 (this research) |
180 |
173.01 |
3.2.8. Thermodynamic study. It is not sufficient to determine the adsorption of sulfide on RGO/Fe3O4 as chemisorption by adsorption kinetics. A thermodynamic study is a good complement to further investigate the adsorption process. Fig. 9(a) shows the effect of temperature on the adsorption of sulfide by RGO/Fe3O4. It can be seen from the figure that the adsorption capacity increased with the increase of temperature, which indicated the endothermic nature of the adsorption process. Three thermodynamic parameters, ΔG (Gibbs free energy of adsorption), ΔH (enthalpy change), and ΔS (entropy change) were employed to better understand the adsorption process, which can be obtained through eqn (8)–(10), respectively:57| |
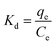 | (8) |
| | |
ΔG = −RTlnKd
| (9) |
| |
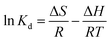 | (10) |
where, Kd (L g−1) is the equilibrium constant value, qe (mg g−1) is the equilibrium concentration of sulfide on RGO/Fe3O4, Ce (mg L−1) is the equilibrium concentration, R is the gas constant, R = 8.314 J mol−1 K−1, and T (K) is the system temperature. ΔS (J mol−1 K−1) and ΔH (kJ mol−1) are obtained from the linear plot of lnKd versus 1/T.
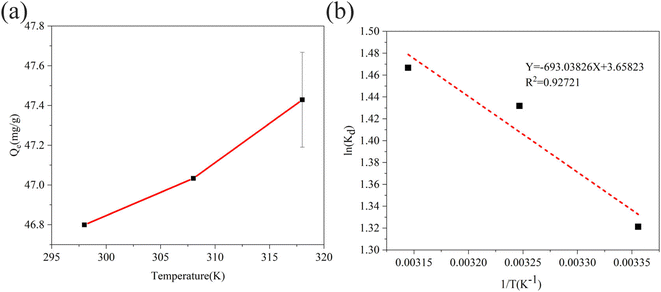 |
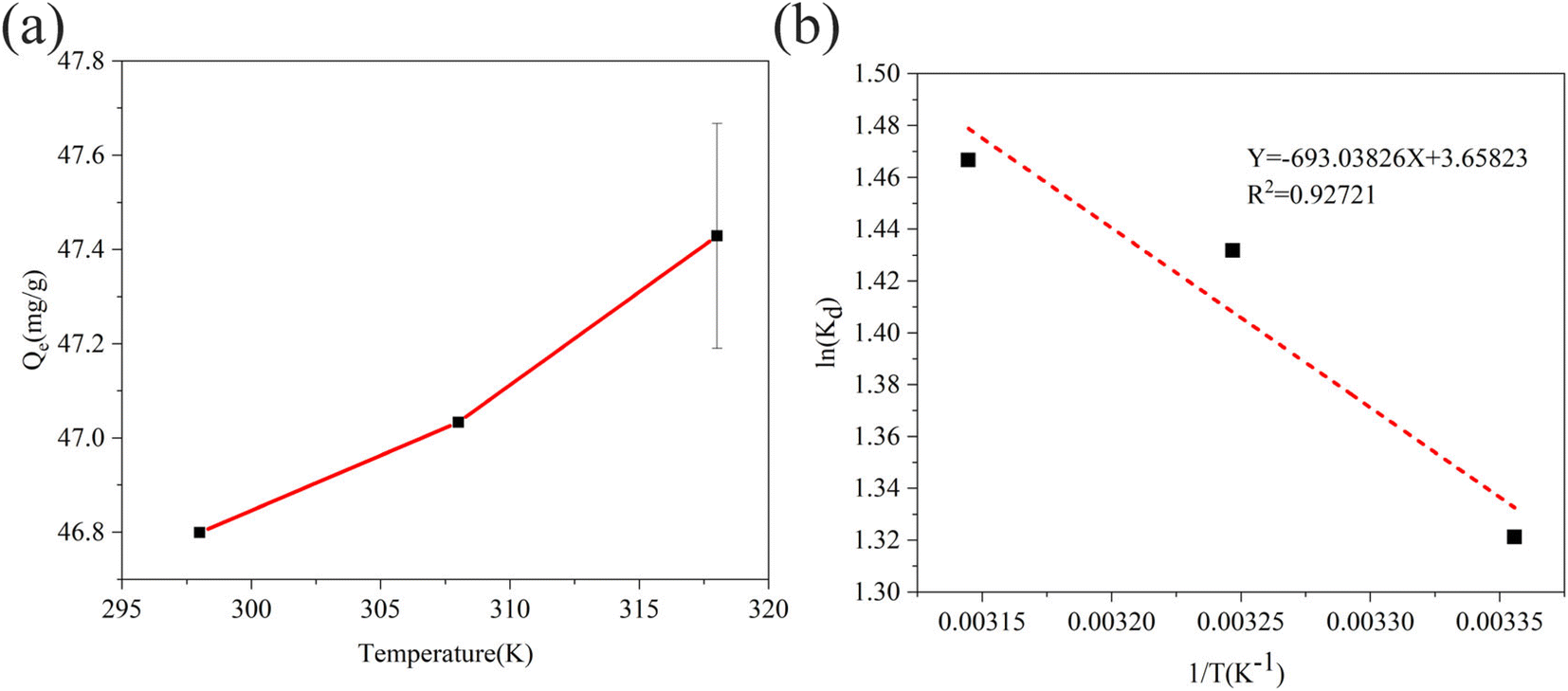
| | Fig. 9 (a) Effect of temperature on sulfide adsorption using RGO/Fe3O4 (C0 = 130.23 mg L−1, m = 25 mg, t = 180 min, pH = 11.72). Error bars represent the standard deviation of replicate measurements. (b) The plot of ln(Kd) vs. 1/T for the sulfide adsorption on RGO/Fe3O4. | |
Fig. 9(b) shows the Van't Hoff plot for sulfide adsorption on RGO/Fe3O4 (eqn. (10)). The thermodynamic parameters of RGO/Fe3O4 adsorption of sulfide are presented in Table 6. The negative ΔG at all temperatures indicates that the adsorption of sulfide by RGO/Fe3O4 is spontaneous. A positive value of ΔH indicates an endothermic adsorption process. ΔS is positive, revealing that the thermodynamic disorder increases during the adsorption of sulfide on RGO/Fe3O4. In addition, the values calculated for the Gibbs free energy of adsorption were −3.2732, −3.6663 and −3.8776 kJ mol−1 at 298 K, 308 K and 318 K, respectively, which was beyond the range of −80 to −400 kJ mol−1, so the adsorption of sulfide by RGO/Fe3O4 is a physical process.58
Table 6 Thermodynamic parameters for the adsorption of sulfide on RGO/Fe3O4
| Temperature (K) |
ΔG (kJ mol−1) |
ΔH (kJ mol−1) |
ΔS (J mol−1 K−1) |
| 298 |
−3.2732 |
5.762 |
30.4145 |
| 308 |
−3.6663 |
|
|
| 318 |
−3.8776 |
|
|
3.3 Reaction mechanism
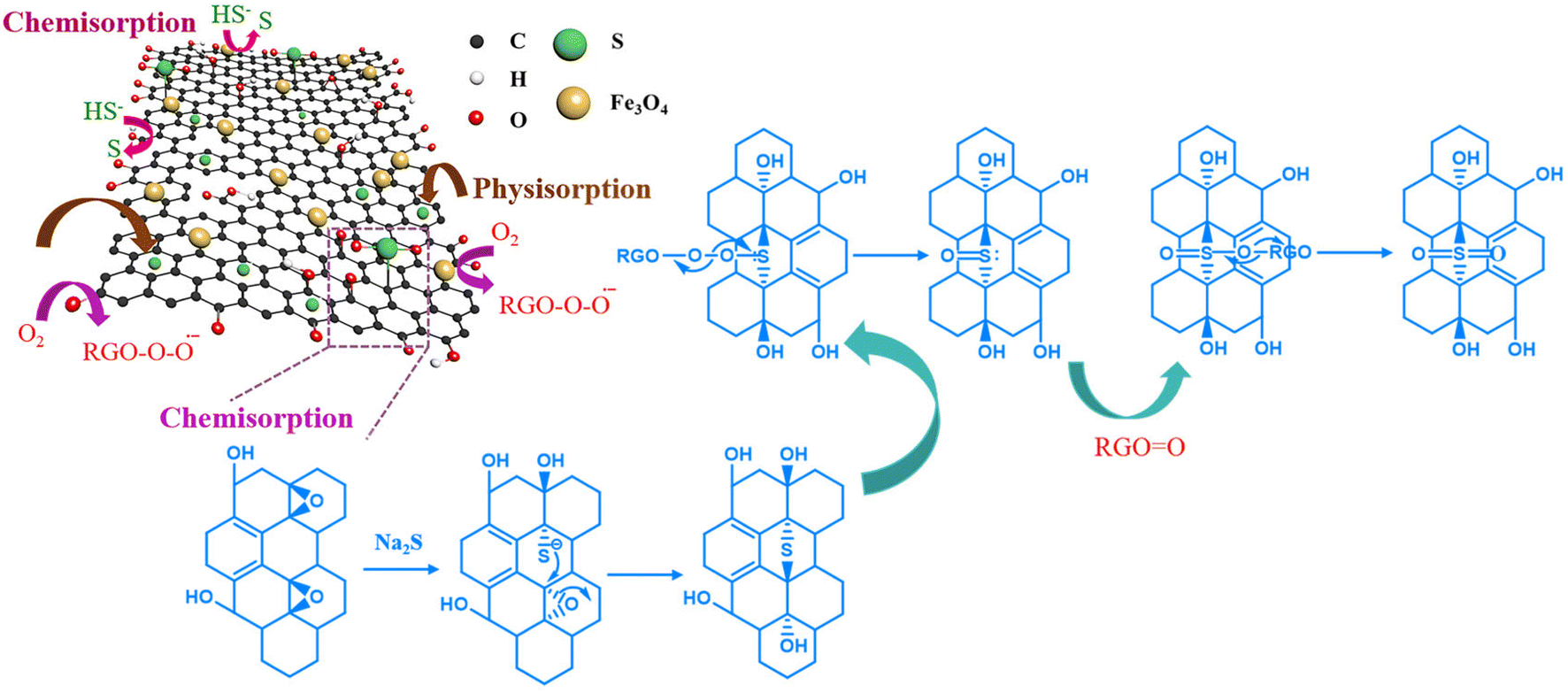
Based on the kinetics, adsorption isotherm and thermodynamic studies, the adsorption of sulfide by the RGO/Fe3O4 hybrid material can be attributed to the synergistic effect from both chemical and physical adsorption.59 According to the analysis of the material characterization and adsorption experiments, the potential mechanism of sulfide removal by the RGO/Fe3O4 hybrid material is proposed and shown in Fig. 10. Sodium sulfide (Na2S⋅9H2O) exists in solution as S2−, HS−, and H2S. On the one hand, sulfide can enter the adsorbent channel easily due to the large average pore diameter of RGO/Fe3O4 (13.3843 nm). The absolute values of ΔH (5.762 KJ mol−1) obtained from the thermodynamic study is in the range of 4–10 kJ mol−1, which indicated that sulfide could be removed through physical adsorption, such as van der Waals force.60 On the other hand, when these compounds contacted with the RGO/Fe3O4 composites, sulfides transferred into elemental sulfur by reacting with hydroxyl (−OH) and carboxyl (−COOH) groups on the RGO/Fe3O4 surface.61 Furthermore, epoxy (C–O–C) groups on RGO reacted with S2− and formed thiols. These thiols then underwent a regioselective epoxy ring-opening dimerization reaction with the adjacent epoxy groups to generate thioethers again because the above reactions could proceed smoothly at room temperature.62 On the other hand, the electronic structures of the contiguous carbon atoms on the RGO surfaces were altered by the carbonyl (CO) groups, which further facilitated the adsorption and activation of dissolved oxygen in water.63 Then, the actively adsorbed oxygen was transformed to O2− radicals (RGO–OO˙−),21 and the oxygen from RGO–OO˙− could be caught by the sulfur atom of thioethers with two lone pair electrons. Then, the O–O bond broke up to form both RGOO and sulfoxide. Subsequently, the sulfoxide was rapidly oxidized to sulfone because sulfoxide cannot be stable in the presence of the highly active species RGOO.64 Lastly, the active sites were gained for next adsorption and activation.21 In addition, the quantitative analysis of elemental sulfur and sulfone accumulated on the RGO/Fe3O4 surface and sulfur compounds in the solution after adsorption need to be further carried out to define the species and content of the products completely.
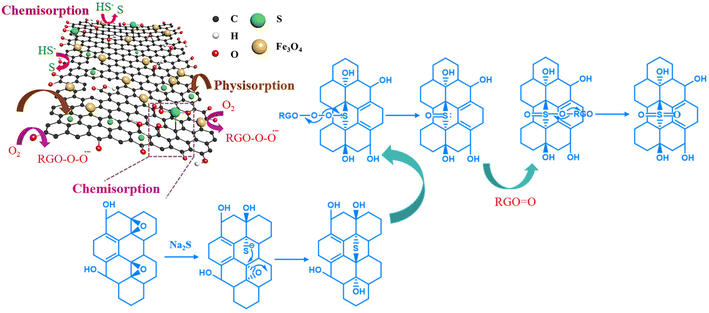 |
| | Fig. 10 Potential mechanism of sulfide removal by the RGO/Fe3O4 hybrid material. | |
4. Conclusion
In this study, a RGO/Fe3O4 adsorbent was synthesized by an in situ chemical method, which was used to remove sulfide from aqueous solution. The adsorption process consists of chemical adsorption and physical adsorption. Through adsorption and oxidation processes, the sulfide was converted into stable elemental sulfur and sulfone, and the secondary conversion of sulfates was thus avoided. The sulfide adsorption by RGO/Fe3O4 reached equilibrium within a contact time of 180 min in a wide pH range. Na+ and Ca2+ improved the sulfide sorption capacity of RGO/Fe3O4 because of the weak electrostatic repulsion. The sulfide adsorption was well fitted to the pseudo-second-order kinetic model. The Langmuir linear isotherm gave a better fit, and the maximum monolayer Langmuir adsorption capacity was estimated to be 173.01 mg g−1. Our thermodynamic study revealed that the adsorption of sulfide by RGO/Fe3O4 was a spontaneous endothermic process. In summary, the RGO/Fe3O4 hybrid material can serve as a potential adsorbent that offers new insight into the treatment of sulfide-containing wastewater.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Key Project of Shandong Provincial Natural Science Foundation (Grant No. ZR2020KE048) and the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDA23050203).
References:
- Ł. J. Wilk, A. Ciechanowska and E. Kociołek-Balawejder, Sep. Purif. Technol., 2020, 231, 115882 CrossRef
 .
. - F. Shi, L. Zhang, J. Yang, M. Lu, J. Ding and H. Li, Corros. Sci., 2016, 102, 103–113 CrossRef CAS .
- J. Cao, Q. Sun, D. Zhao, M. Xue, Q. Shen, D. Wang, Y. Wang and S. Ding, J. Hazard. Mater., 2020, 385, 121511 CrossRef CAS PubMed .
- J. Zhang, Q. Zhu and Z. Xing, J. Hazard. Mater., 2020, 390, 122049 CrossRef CAS PubMed .
- Ł. J. Wilk, A. Ciechanowska and E. Kociołek-Balawejder, Materials, 2020, 13, 1–19 CrossRef PubMed .
- M. Wiemann, H. Schenk and W. Hegemann, Water Res., 1998, 32, 774–780 CrossRef CAS .
- D. Andreides, Z. Varga, D. Pokorna and J. Zabranska, J. Water Process. Eng., 2021, 40, 101834 CrossRef .
- L. Altaş and H. Büyükgüngör, J. Hazard. Mater., 2008, 153, 462–469 CrossRef PubMed .
- P. K. Anjali Achazhiyath Edathil and F. Banat, Environ. Pollut., 2020, 266, 115218 CrossRef PubMed .
- L. Zhang, P. D. Schryver, B. D. Gusseme, W. D. Muynck, N. Boon and W. Verstraete, Water Res., 2008, 42, 1–12 CrossRef CAS PubMed .
- J. Sun, L. Wei, R. Yin, F. Jiang and C. Shang, Water Res., 2020, 171, 115453 CrossRef CAS PubMed .
- A. A. Edathil, P. Pal and F. Banat, J. Environ. Chem. Eng., 2017, 5, 1998–2009 CrossRef CAS .
- I. B. Hariz and L. Monser, Water Sci. Technol., 2014, 4, 264–267 Search PubMed .
- I. Jacukowicz-Sobala, L. J. Wilk, K. Drabent and E. Kociolek-Balawejder, J. Colloid Interface Sci., 2015, 460, 154–163 CrossRef CAS PubMed .
- A. A. Edathil, J. H. Zain, M. A. Haija and F. Banat, New J. Chem., 2019, 43, 3500–3512 RSC .
- Y. Pan, M. Chen, M. Hu, M. Tian, Y. Zhang and D. Long, Appl. Catal., B, 2020, 262, 118266 CrossRef CAS .
- J. H. Yang, Korean J. Chem. Eng., 2021, 38, 674–691 CrossRef CAS .
- M. Khaleghi Abbasabadi, A. Rashidi, J. Safaei-Ghomi, S. Khodabakhshi and R. Rahighi, J. Sulfur Chem., 2015, 36, 660–671 CrossRef CAS .
- R. Castaldo, R. Avolio, M. Cocca, M. E. Errico, M. Lavorgna, J. Šalplachta, C. Santillo and G. Gentile, Chem. Eng. J., 2022, 430, 133162 CrossRef CAS .
- F. Yuan, L. Yue, H. Zhao and H. Wu, Water Sci. Technol., 2020, 81, 2163–2175 CrossRef CAS PubMed .
- Q. Gu, G. Wen, Y. Ding, K.-H. Wu, C. Chen and D. Su, Green Chem., 2017, 19, 1175–1181 RSC .
- P. Benjwal, M. Kumar, P. Chamoli and K. K. Kar, RSC Adv., 2015, 5, 73249–73260 RSC .
- P. Mandal and A. P. Chattopadhyay, Dalton Trans., 2015, 44, 11444–11456 RSC .
- W. Zhang, X. Zou and J. Zhao, J. Mater. Chem. C, 2015, 3, 2788–2791 RSC .
- Z. Feng, C. Zhang, J. Chen, Y. Wang, X. Jin, R. Zhang and J. Hu, RSC Adv., 2013, 3, 4408–4415 RSC .
- X. Zou, Y. Zhou, S. Chen, W. Li, Y. Qiang, B. Xiang, Q. Wu, F. Chen and W. Huang, J. Alloys Compd., 2019, 782, 17–27 CrossRef CAS .
- G. Bharath, R. Madhu, S.-M. Chen, V. Veeramani, D. Mangalaraj and N. Ponpandian, J. Mater. Chem. A, 2015, 3, 15529–15539 RSC .
- N. Wang, J. Feng, J. Chen, J. Wang and W. Yan, Chem. Eng. J., 2017, 316, 33–40 CrossRef CAS .
- K. Krishnamoorthy, M. Veerapandian, K. Yun and S. J. Kim, Carbon, 2013, 53, 38–49 CrossRef CAS .
- J. Xu, H. Yang, W. Fu, K. Du, Y. Sui, J. Chen, Y. Zeng, M. Li and G. Zou, J. Magn. Magn. Mater., 2007, 309, 307–311 CrossRef CAS .
- M. H. Kahsay, N. Belachew, A. Tadesse and K. Basavaiah, RSC Adv., 2020, 10, 34916–34927 RSC .
- Y. J. Oh, J. J. Yoo, Y. I. Kim, J. K. Yoon, H. N. Yoon, J.-H. Kim and S. B. Park, Electrochim. Acta, 2014, 116, 118–128 CrossRef CAS .
- J. Wu, Z. Ye, W. Liu, Z. Liu and J. Chen, Ceram. Int., 2017, 43, 13146–13153 CrossRef CAS .
- L. Zhou, H. Deng, J. Wan, J. Shi and T. Su, Appl. Surf. Sci., 2013, 283, 1024–1031 CrossRef CAS .
- C.-L. Lin, C.-F. Lee and W.-Y. Chiu, J. Colloid Interface Sci., 2005, 291, 411–420 CrossRef CAS PubMed .
- T. Heitmann and C. Blodau, Chem. Geol., 2006, 235, 12–20 CrossRef CAS .
- X. Zhao, Y. Jia and Z. H. Liu, J. Colloid Interface Sci., 2019, 536, 463–473 CrossRef CAS PubMed .
- V. Karthika, M. S. AlSalhi, S. Devanesan, A. A. Kasi Gopinath and M. Govindarajan, Sci. Rep., 2020, 10, 18912 CrossRef PubMed .
- M. Pudukudy, Q. Jia, Y. Dong, Z. Yue and S. Shan, RSC Adv., 2019, 9, 32517–32534 RSC .
- N. A. Zubir, C. Yacou, J. Motuzas, X. Zhang and J. C. D. d. Costa, Sci. Rep., 2014, 4, 4594 CrossRef PubMed .
- N. R. Urban, K. Ernst and S. Bernasconi, Geochim. Cosmochim. Acta, 1999, 63, 837–853 CrossRef CAS .
- R. Pietrzak, T. Grzybek and H. Wachowska, Fuel, 2007, 86, 2616–2624 CrossRef CAS .
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS .
- I. Kondratowicz, M. Nadolska, S. Şahin, M. Łapiński, M. Prześniak-Welenc, M. Sawczak, E. H. Yu, W. Sadowski and K. Żelechowska, Appl. Surf. Sci., 2018, 440, 651–659 CrossRef CAS .
- B.-W. Pang, C.-H. Jiang, M. Yeung, Y. Ouyang and J. Xi, J. Hazard. Mater., 2017, 324, 732–738 CrossRef CAS PubMed .
- A. A. Edathil, P. Kannan, M. A. Haija and F. Banat, Environ. Res., 2021, 196, 110429 CrossRef CAS PubMed .
- G. Chen and L. Shi, RSC Adv., 2017, 7, 43445–43454 RSC .
- J. D. Filius, D. G. Lumsdon, J. C. L. Meeussen, T. Hiemstra and W. H. V. Riemsdijk, Geochim. Cosmochim. Acta, 2000, 64, 51–60 CrossRef CAS .
- K. Vermöhlen, H. Lewandowski, H.-D. Narres and M. J. Schwuger, Colloids Surf., A, 2000, 163, 45–53 CrossRef .
- T. M. Berhanea, J. Levya, M. P. S. Krekelerb and N. D. Danielson, Chemosphere, 2017, 176, 231–242 CrossRef PubMed .
- Y.-S. Ho, J. Hazard. Mater., 2006, 136, 681–689 CrossRef CAS PubMed .
- G. McKay, M. S. Otterburn and J. A. Aga, Water, Air, & Soil Pollution, 1985, 24, 307–322 Search PubMed .
- A. H. Jawad, N. S. A. Mubarak and A. S. Abdulhameed, J. Polym. Environ., 2019, 28, 624–637 CrossRef .
- R. Han, J. Zhang, P. Han, Y. Wang, Z. Zhao and M. Tang, Chem. Eng. J., 2009, 145, 496–504 CrossRef CAS .
- A. R. P. Hidayat, D. O. Sulistiono, I. K. Murwani, B. F. Endrawati, H. Fansuri, L. L. Zulfa and R. Ediati, J. Environ. Chem. Eng., 2021, 9, 106675 CrossRef CAS .
- P. Pal, A. A. Edathil, L. Chaurasia, K. Rambabu and F. Banat, Polym. Bull., 2018, 75, 5455–5475 CrossRef CAS .
- H. V. Tran, L. T. Hoang and C. D. Huynh, Chem. Phys., 2020, 535, 110793 CrossRef CAS .
- M. Hadadian, E. K. Goharshadi, M. M. Fard and H. Ahmadzadeh, Appl. Phys. A, 2018, 124, 239 CrossRef .
- X.-L. Sun, Z. Liu and Z.-L. Cheng, J. Hazard. Mater., 2021, 403, 123851 CrossRef CAS PubMed .
- B. von Oepen, W. Kördel and W. Klein, Chemosphere, 1991, 22, 285–304 CrossRef CAS .
- A. A. Edathil, P. Kannan, M. AbuHaija and F. Banat, Environ. Res., 2021, 196, 110429 CrossRef CAS PubMed .
- K. Tatsuta, Y. Suzuki, T. Toriumi, Y. Furuya and S. Hosokawa, Tetrahedron Lett., 2007, 48, 8018–8021 CrossRef CAS .
- X. Lu, W.-L. Yim, B. H. R. Suryanto and C. Zhao, J. Am. Chem. Soc., 2015, 137, 2901–2907 CrossRef CAS PubMed .
- S. Chen, W. Lu, Y. Yao, H. Chen and W. Chen, React. Kinet., Mech. Catal., 2013, 111, 535–547 CrossRef .
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Hongtao Cui
ab,
Hongtao Cui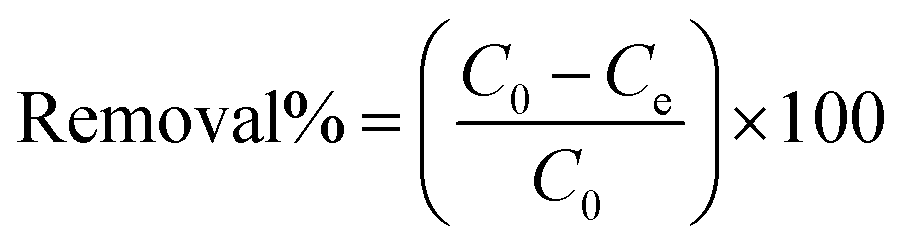
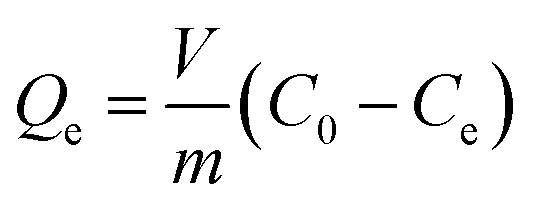
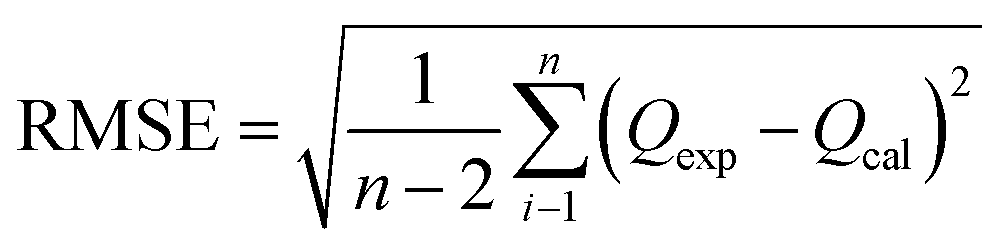
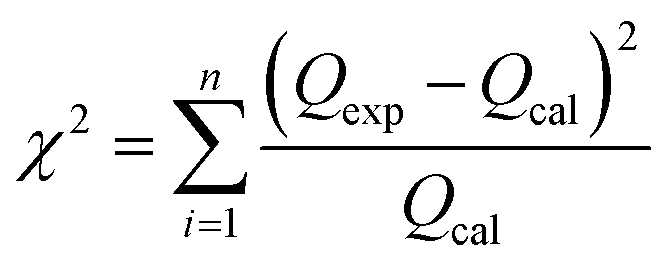


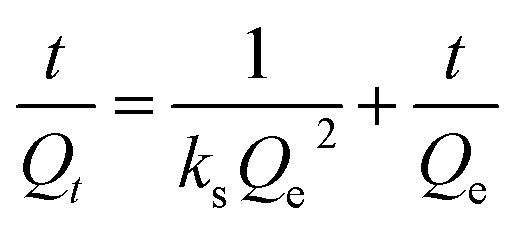
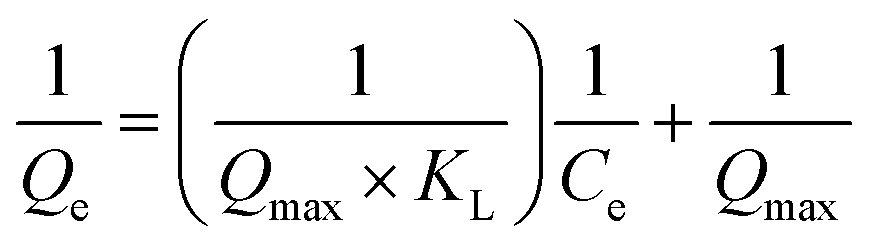
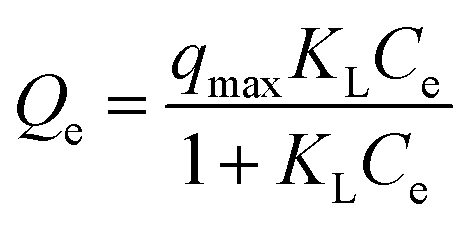
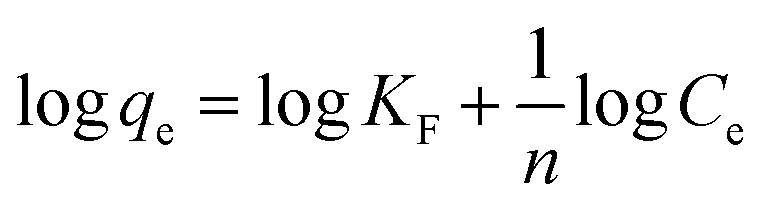
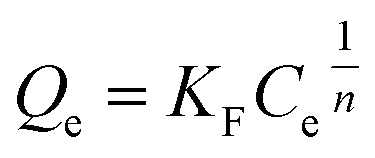
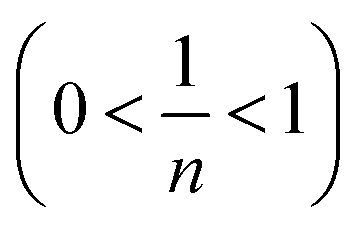 . For the Langmuir non-linear isotherm, R2 (0.9940) was lower than the value obtained from the Langmuir linear equation (0.9987), but qmax (251.32 mg g−1) was higher.
. For the Langmuir non-linear isotherm, R2 (0.9940) was lower than the value obtained from the Langmuir linear equation (0.9987), but qmax (251.32 mg g−1) was higher.