
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCharacteristic 1H NMR spectra of β-D-ribofuranosides and ribonucleosides: factors driving furanose ring conformations†
Dominik Walczak,
Artur Sikorski,
Daria Grzywacz,
Andrzej Nowacki and
Beata Liberek *
*
Faculty of Chemistry, University of Gdańsk, Wita Stwosza 63, 80-308 Gdańsk, Poland. E-mail: beata.liberek@ug.edu.pl
First published on 13th October 2022
Abstract
A series of β-D-ribofuranosides and ribonucleosides fused with 2,3-O-isopropylidene ring was synthesized and studied in terms of their conformational preferences. Based on the 1H NMR spectra, DFT calculations, and X-ray analysis the E0-like and E4-like conformations adopted by these furanosides are identified. The 3E-like and 2E-like conformations are assigned to ribonucleosides without the 2,3-O-isopropylidene group. The studies are supported by analysis of the structural data of β-D-ribofuranosides and ribonucleosides deposited in the Cambridge Crystallographic Data Center (CCDC) database.† Finally, the factors influencing the conformational preferences of the furanose ring with the β-D-ribo configuration are indicated. These are the unfavorable ecliptic orientation of the 2-OH and 3-OH groups, the 1,3-pseudodiaxial interaction of the aglycone and terminal hydroxymethyl group and the endo-anomeric effect. It is also proved that the exo-anomeric effect acts in β-D-ribofuranosides.
1. Introduction
β-D-Ribofuranosides are commonly found in living organisms both in the form of N- and O-glycosides. Ribonucleosides, are perhaps the best known representatives of this group of compounds because they play essential functions in living organisms. In the form of 5′-phosphates (nucleotides) they are involved in intermediary metabolism, cell signaling, and the biosynthesis of macromolecules. Importantly, nucleotides serve as monomeric units of RNA, a macromolecule essential for all life forms on Earth. Thus, ribonucleosides are responsible for encoding, transmitting, and expressing genetic information in living organisms. Therefore, ribonucleosides and their derivatives are profoundly explored in medicine as anticancer,1–3 antibacterial,4–6 and antiviral7–9 agents. Molnupiravir, a ribonucleoside (Fig. 1A) with proven activity against a number of RNA viruses is currently tested against SARS-CoV-2.10,11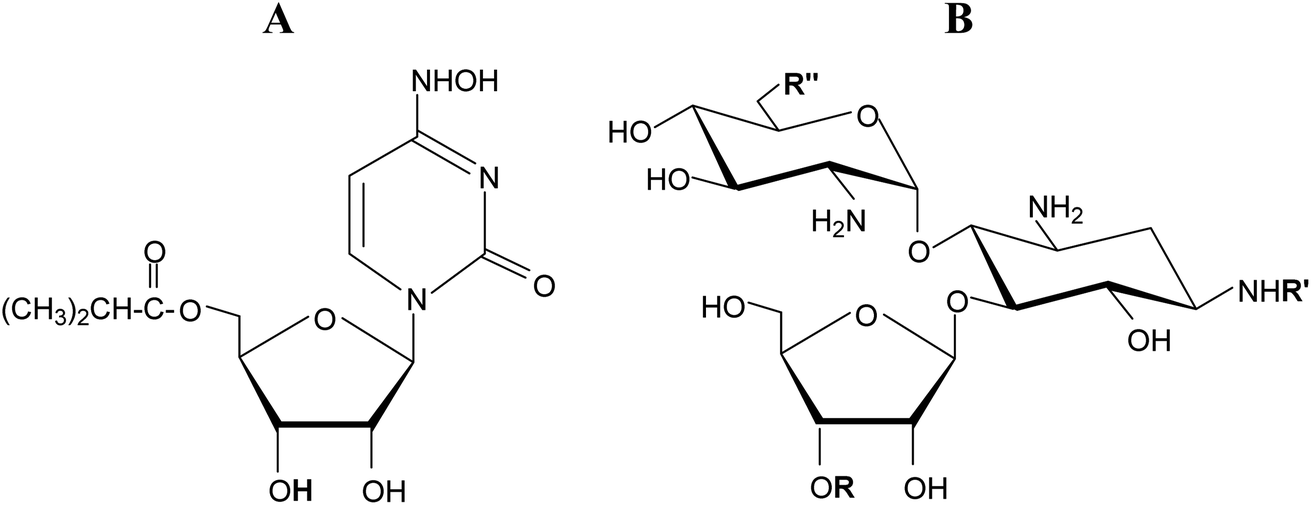 | ||
| Fig. 1 (A) Molnupiravir. (B) Common sugar skeleton of ribostamycin, butirosin B, neomycin, and paromomycin. | ||
β-D-Ribofuranosides, which are not as common as their nitrogen analogues, are also found in natural compounds. Some aminoglycoside-aminocyclitol antibiotics, namely ribostamycin,12 butirosin B,13 neomycin, and paromomycin contain an O-β-D-ribofuranoside ring in their structure (Fig. 1B). It has been suggested that the presence of the ribofuranose ring in these antibiotics may improve the bacterial ribosome selectivity.12 Benzyl β-D-ribofuranoside was isolated from Euphorbia humifusa Willd and its ability to inhibit the LPS-induced NO and TNF-α production in RAW 264.7 cells was demonstrated.14 ADP-ribosylation, the post-translational modifications of proteins involved in many cellular processes, relies on the creation of an O-glycosidic bond between D-ribose and L-serine or L-threonine.15 Phosphorylated glycoconjugates of β-D-ribofuranoside were achieved synthetically.16 Simple β-D-ribofuranosides are often used as intermediate products in the syntheses of more sophisticated ribose derivatives.17–19 β-D-Ribofuranosides possessing phenolic aglycones were synthesized and found to exhibit the Src kinase inhibitory activity.20 Generally, glycosidation of D-ribose is of interest to organic chemists.21
A good understanding of the biological processes in which β-D-ribofuranosides and ribonucleosides participate requires an understanding of their conformational preferences. There are reports indicating that the interactions of nucleosides and their analogues with the target site depend on the furanose ring conformation.22–25 In order to require a specific conformation of the furanose ring in nucleosides, its structure is often modified at the 2′-position26,27 or bridged with an additional ring.27–29 The investigation of the conformationally restricted nucleosides led to the discovery of potent antiviral agents.30,31 It was also demonstrated that changes in the conformation of ribose have an impact on the nucleic acid conformation and function.32 From the chemical point of view, it has to be mentioned that the sugar ring conformation of nucleosides influences their reactivity.33
To avoid the torsion strains present in the flat furanose ring, this adopts one of ten possible envelope (E) or ten possible twist (T) conformations. The descriptors used at the conformation symbol indicate which atoms are placed below (subscript) or above (superscript) the plane formed by the remaining ring atoms (Fig. 2A). The pseudorotation phase angle P was introduced to precisely indicate the conformational state of the furanose ring.34 This parameter is defined to be zero for the 3T2 conformation. The transition from P = 0° to P = 360° exhausts all possible conformational states of the furanose ring, which is illustrated by the pseudorotational wheel (Fig. 2B).
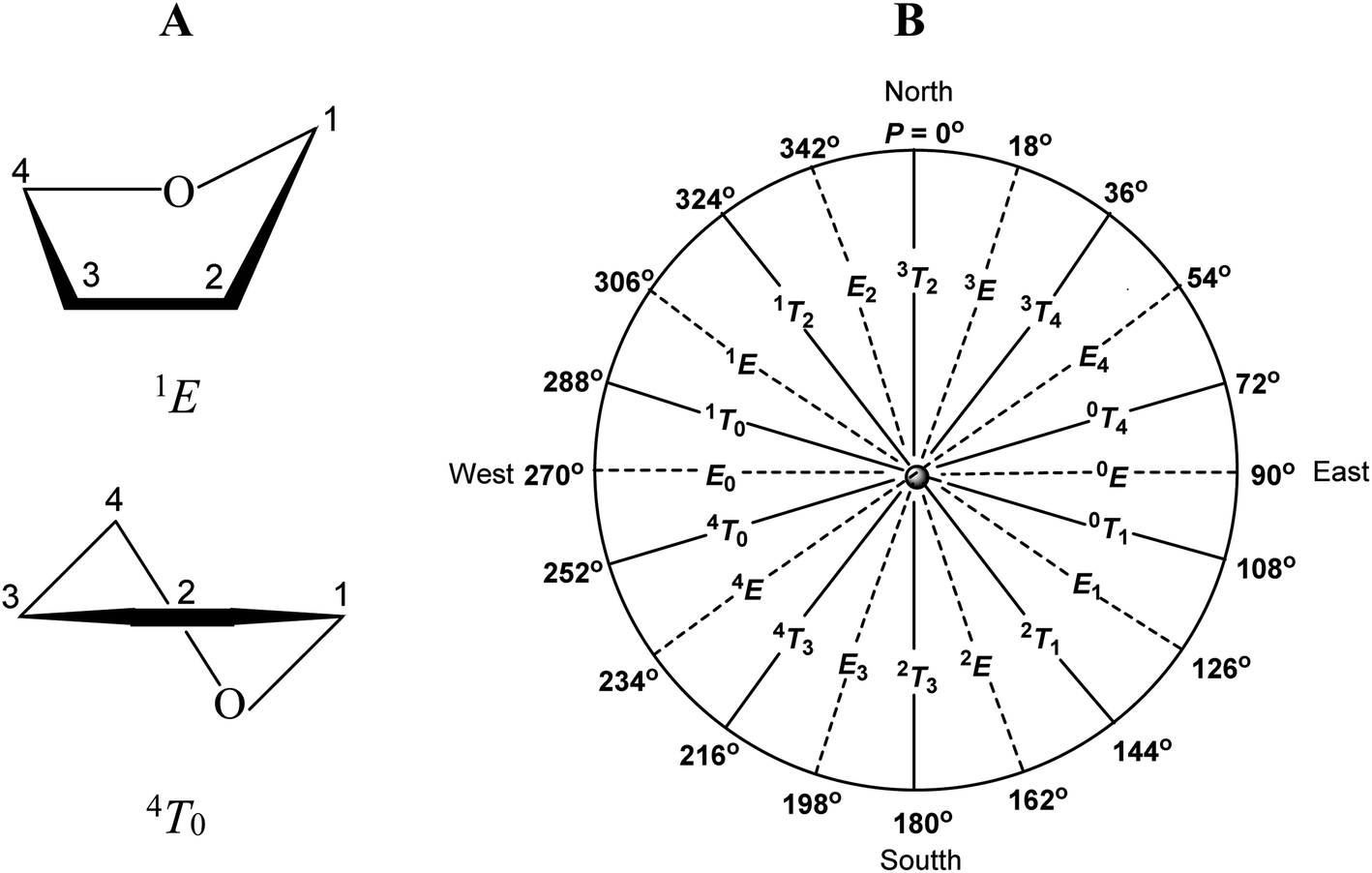 | ||
| Fig. 2 (A) Examples of furanose ring conformations. (B) Pseudorotational wheel for a furanose ring. | ||
The conformation of the furanose ring is difficult to recognize because the five-membered ring is relatively labile. Despite such an inconvenience, much attention has been paid to the conformational analysis of the furanose ring. Respective studies have been carried out based on the X-ray crystallography, NMR data, quantum mechanical calculations, and other techniques.35–43 It does not change the fact that inferring the sugar ring conformation from the 1H NMR data, which is rather simple in the case of pyranosides, is still challenging in the case of furanosides.
In our previous studies, it was demonstrated that a bicyclic structure of D-glucofuranurono-6,3-lactones and their glycosides causes the furanose ring to adopt specific conformations both in the crystal lattice and solution. In the case of β-D-glucofuranurono-6,3-lactones and their O-glycosides it was the 1T2-like conformation.44 In the case of the N-glycoside of α-D-glucofuranurono-6,3-lactone it was the 3T2/3E conformation.45 Thereby, it was proved that the characteristic 1H NMR spectra recorded for β-D-glucofuranurono-6,3-lactones and their O-glycosides are indicative of the 1T2-like conformation for all furanoses both with the β-D-gluco configuration as well as with the β-D-xylo and α-L-ido configurations. In turn, the characteristic 1H NMR spectrum recorded for N-(α-D-glucofuranurono-6,3-lactone) is indicative of the 3T2/3E conformation for all furanoses with the α-D-gluco, α-D-xylo, and β-L-ido configurations.
In this paper we demonstrate how the 2,3-isopropylidene protecting group influences the conformational preferences of β-D-ribofuranosides and ribonucleosides. For the group of 2,3-O-isopropylidene-β-D-ribofuranosides and 2,3-O-isopropylideneribonucleosides, the 1H NMR spectra are presented. These are found to be very distinctive. Based on these spectra the specific conformations of the furanose ring are recognized. We opt for the specific conformation of the presented furanoses, instead of the usually postulated state of the conformational equilibrium, because the recorded 1H NMR signals are sharp. Furthermore, the recorded coupling constants indicative of a given conformation are not sensitive to the change in both the aglycone and the solvent. The conformations diagnosed based on the 1H NMR spectra are proved to be the most stable in the density functional theory (DFT) optimizations. Additionally, the search of β-D-ribofuranosides and ribonucleosides, with and without the 2,3-O-isopropylidene group, from the CCDC database is presented. Finally, analysis of the factors influencing the conformational preferences of the furanose ring with the β-D-ribo configuration is carried out.
2. Experimental
2.1 NMR measurements
The 1H and 13C NMR spectra were recorded on a Varian Mercury 400 (400.49/100.70 MHz) or Bruker AVANCE III 500 (500.13/125.76 MHz) instruments, using standard experimental conditions in CDCl3 or DMSO-d6 with internal Me4Si.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 112.79 (CMe2), 109.64 (C1), 85.43 (C2), 84.48 (C4), 82.12 (C3), 64.87 (C5), 55.14 (OCH3), 26.65, 25.22 (2 × CH3), 21.01 (CH3Ac).O), 112.74 (CMe2), 108.18 (C1), 85.58 (C2), 84.34 (C4), 82.19 (C3), 64.98 (C5), 63.36 (CH2), 26.66, 25.20 (2 × CH3), 21.03 (CH3Ac), 15.06 (CH3).O), 112.73 (CMe2), 108.50 (C1), 85.54 (C2), 84.28 (C4), 82.21 (C3), 69.72 (CH2), 64.99 (C5), 26.64, 25.19 (2 × CH3), 22.81 (CH2), 21.02 (CH3Ac), 10.77 (CH3).O), 112.67 (CMe2), 106.35 (C1), 85.92 (C2), 84.22 (C4), 82.35 (C3), 69.44 (CH), 65.15 (C5), 26.66, 25.19 (2 × CH3), 23.35, 21.29 (2 × CH3), 21.03 (CH3Ac).O), 112.72 (CMe2), 108.52 (C1), 85.54 (C2), 84.28 (C4), 82.21 (C3), 67.84 (CH2), 64.99 (C5), 31.64 (CH2), 26.64, 25.19 (2 × CH3), 21.01 (CH3Ac), 19.47 (CH2), 14.00 (CH3).
O), 112.79 (CMe2), 109.64 (C1), 85.43 (C2), 84.48 (C4), 82.12 (C3), 64.87 (C5), 55.14 (OCH3), 26.65, 25.22 (2 × CH3), 21.01 (CH3Ac).O), 112.74 (CMe2), 108.18 (C1), 85.58 (C2), 84.34 (C4), 82.19 (C3), 64.98 (C5), 63.36 (CH2), 26.66, 25.20 (2 × CH3), 21.03 (CH3Ac), 15.06 (CH3).O), 112.73 (CMe2), 108.50 (C1), 85.54 (C2), 84.28 (C4), 82.21 (C3), 69.72 (CH2), 64.99 (C5), 26.64, 25.19 (2 × CH3), 22.81 (CH2), 21.02 (CH3Ac), 10.77 (CH3).O), 112.67 (CMe2), 106.35 (C1), 85.92 (C2), 84.22 (C4), 82.35 (C3), 69.44 (CH), 65.15 (C5), 26.66, 25.19 (2 × CH3), 23.35, 21.29 (2 × CH3), 21.03 (CH3Ac).O), 112.72 (CMe2), 108.52 (C1), 85.54 (C2), 84.28 (C4), 82.21 (C3), 67.84 (CH2), 64.99 (C5), 31.64 (CH2), 26.64, 25.19 (2 × CH3), 21.01 (CH3Ac), 19.47 (CH2), 14.00 (CH3).2.2 X-Ray measurement
Diffraction data were collected on an Oxford Diffraction Gemini R ULTRA Ruby CCD diffractometer with MoKα (λ = 0.71073 Å) radiation at T = 295(2) K. The lattice parameters were obtained by least-squares fit to the optimized setting angles of the reflections collected by means of CrysAlis CCD.46 Data were reduced using CrysAlis RED software46 and applying multi-scan absorption corrections. The structure was solved with direct methods that carried out refinements by full-matrix least-squares on F2 using the SHELXL-2017/1 program.47All H-atoms bound to N/O/C-atoms were located on a difference Fourier map and refined freely (H-atoms from the methyl group were positioned geometrically and refined using a riding model, with C–H = 0.96 Å and Uiso(H) = 1.5Ueq(C)). All interactions were calculated using the PLATON program (ver. 181115).48 The following programs were used to prepare the molecular graphics: ORTEPII,49 PLUTO-78,50 and Mercury (ver. 2020.2.0).51
Full crystallographic details for title compound have been deposited.†
2.3 DFT calculations
The Molden program52 was used for preparation of all the initial geometries for calculations which were done under default conditions with the aid of the Gaussian 09 program.53 The B3LYP functional (Becke's three-parameter hybrid exchange functional involving the gradient-corrected correlation functional of Lee, Yang and Parr)54,55 combined with the 6-311+G** basis set was used to perform unconstrained geometry optimization of all prepared geometries. No imaginary frequencies could be found for optimized structures as revealed form the Hessians analysis that was done at the same level of theory. Zero-point vibrational energies, molecular entropies as well as thermal energy contributions were obtained form the Hessian calculations, according to statistical thermodynamics formulae which were used to estimate the contribution of each rotamer in equilibrium. The population of rotamers was calculated using the following equation: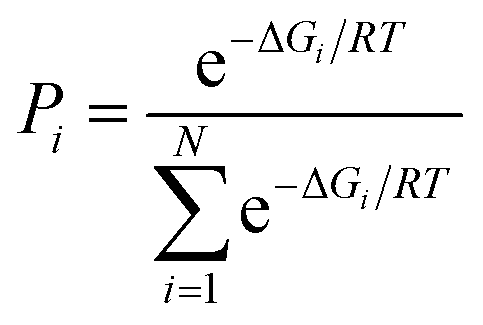
3. Results and discussion
A series of 2,3-O-isopropylidene derivatives of β-D-ribofuranosides (1–10) and ribonucleosides (15–18) was synthesized (Fig. 3). These furanosides constitute fused, bicyclic structures that have a limited freedom of rotation, particularly with regard to adopting the 3T2 and 2T3 conformations, which demand a C2–C3 bond twist. Therefore, 2,3-O-isopropylidene derivatives of ribofuranosides provide a good model for studying the furanose ring conformations other than 3T2 and 2T3.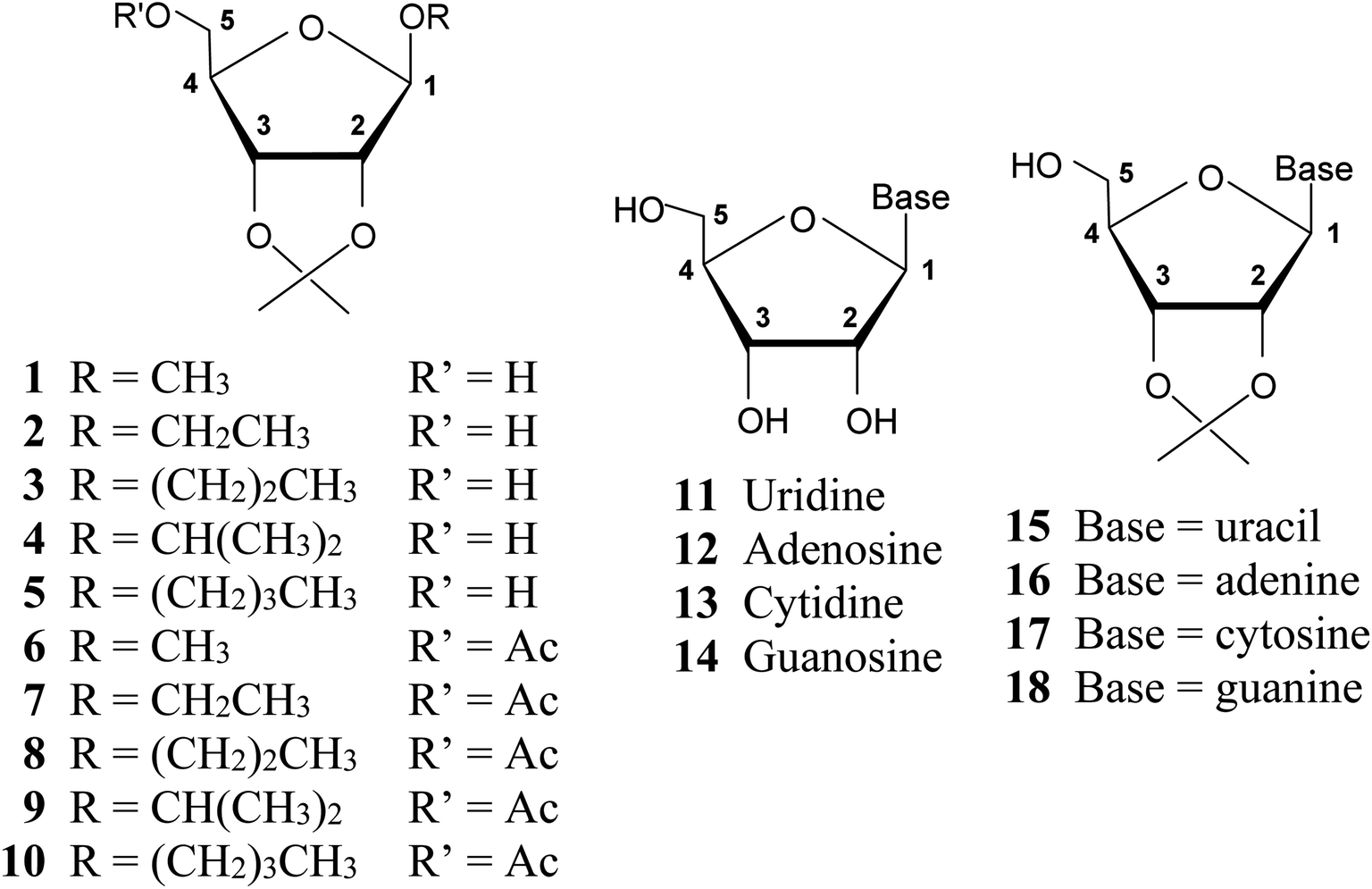 | ||
| Fig. 3 Structures of β-D-ribofuranosides (1–10) and ribonucleosides (11–18) under consideration and the furanose ring atom numbering system used. | ||
Alkyl 2,3-O-isopropylidene-β-D-ribofuranosides (1–5) were synthesized by the reaction of D-ribose with the respective alcohol, carried out in acetone with the addition of SnCl2. Their acetylation provided derivatives 6–10. 2,3-O-Isopropylidenenucleosides (15–18) were synthesized by the reaction of the respective nucleoside (11–14) with 2,2-dimethoxypropane in anhydrous DMF with the addition of p-toluenesulfonic acid (for experimental details see ESI†).
3.1 Conformational analysis of 1–5 based on the 1H NMR spectra
The 1H NMR spectra of alkyl 2,3-O-isopropylidene-β-D-ribofuranosides (1–5) are identical in terms of the coupling constants of the furanose ring protons (Table 1). These are characterized by the zero coupling constants between the trans-oriented H1 and H2 (J1,2) as well as the trans-oriented H3 and H4 protons (J3,4). The third significant J2,3 coupling constant for all these furanosides is always 5.86 Hz or 6.22 Hz which, within the limits of measurement error, can be considered the same value. The identity of the key coupling constants is indicative of the same conformation of the furanose ring in 1–5.| No. | J1,2 trans H/H | J2,3 cis H/H | J3,4 trans H/H | Conformation |
|---|---|---|---|---|
| 1 | ∼0 | 5.86 | ∼0 | 4T0/E0 |
| 2 | ∼0 | 5.86 or 6.23 | ∼0 | 4T0/E0 |
| 3 | ∼0 | 5.86 | ∼0 | 4T0/E0 |
| 4 | ∼0 | 5.86 | ∼0 | 4T0/E0 |
| 5 | ∼0 | 5.86 or 6.23 | ∼0 | 4T0/E0 |
| 6 | ∼0 | 5.86 | 0.73 or 1.10 | E0/1T0 |
| 7 | ∼0 | 5.86 | 0.73 | E0/1T0 |
| 8 | ∼0 | 5.86 or 6.23 | 0.73 or 1.10 | E0/1T0 |
| 9 | ∼0 | 5.86 or 6.23 | 0.73 or 1.10 | E0/1T0 |
| 10 | ∼0 | 5.86 | 0.73 | E0/1T0 |
The zero coupling constant between vicinal protons is an extremely useful hint in the conformational analysis of a furanose ring based on the 1H NMR spectra, typically recorded by every organic chemist. According to the Karplus curve,56 this zero coupling constant indicates that the torsion angle between vicinal protons comes within the 80–100° range. Such a situation is possible solely for the trans-oriented vicinal furanose ring protons when these come as close as possible to each other in the process of the conformational changes. In the case of furanosides 1–5, both the H1 and H2 as well as the H3 and H4 pairs of protons are trans-oriented, and for both, the respective coupling constant is ∼0 Hz. Using a Dreiding model, one may see that the E0 conformation is the only one that ensures the maximum approximation of both the H1 and H2 as well as the H3 and H4 protons. Previously presented data on the torsion angles in the optimized THF ring,45 confirm this assumption. According to them the H1–C1–C2–H2 torsion angle for the β-D-ribo configuration in the E0 conformation is 101°, whereas the H3–C3–C4–H4 torsion angle is −101°. In the E0 conformation, the cis-oriented H2 and H3 protons for the β-D-ribo configuration form the H2–C2–C3–H3 torsion angle that is equal to 0°. The recorded J2,3 = 5.86 Hz or 6.22 Hz coupling constants are in agreement with this value.
3.2 DFT optimizations of methyl 2,3-O-isopropylidene-β-D-ribofuranoside (1)
To verify the conclusions based on the 1H NMR spectra of 1–5, methyl 2,3-O-isopropylidene-β-D-ribofuranoside (1), representing this group of furanosides, was optimized using DFT methods. Taking into account the rotations around the C1–O1, C4–C5, and C5–O5 bonds as well as two (exo and endo) settings of the 2,3-O-isopropylidene group, 54 (2 × 33) rotamers of 1 in the 1E, E1, 3E, and E3 initial conformations, respectively, were prepared. During the optimization, the number of rotamers was significantly reduced, as some of the original structures transformed into the same final geometry. Finally, seven (I–VII) relatively stable structures of 1 were obtained with a population ranging from 56.02% to 1.20%. The geometrical parameters, relative free Gibbs energies, and populations of I–VII in a group of found rotamers of 1 are listed in Table 2. These data indicate that methyl 2,3-O-isopropylidene-β-D-ribofuranoside (1) prefers a really narrow 4T0/E0/1T0 conformational range, which corresponds to all optimized structures I–VII. This conformational range can be limited to the 4T0/E0 conformation with P = ∼260°, which is adopted by six of the seven optimized structures (I–IV, VI, and VII). These structures comprise 97.9% of the population of all optimized structures.| Pa [°] | Conformation | Selected torsion angles [°] | ΔGb [kcal mol−1] | Population [%] | |||||
|---|---|---|---|---|---|---|---|---|---|
| O1–C1–O4–C4 | O4–C1–O1–C6 | H1–C1–C2–H2 | H2–C2–C3–H3 | H3–C3–C4–H4 | |||||
| a P – pseudorotation parameter.b Referred to G = −728.68821 au. | |||||||||
| I | 260.3 | 4T0/E0 | −90.88 | −71.75 | 105.70 | −6.48 | −101.61 | 0.0000 | 56.02 |
| II | 255.9 | 4T0 | −100.01 | −66.53 | 112.18 | −7.01 | −105.13 | 0.7449 | 15.92 |
| III | 262.1 | 4T0/E0 | −90.89 | −71.83 | 104.86 | −5.39 | −102.58 | 0.7455 | 15.91 |
| IV | 272.9 | E0 | −89.19 | −70.70 | 102.02 | 0.15 | −107.17 | 1.1860 | 7.56 |
| V | 278.8 | E0/1T0 | −88.78 | −70.78 | 99.83 | 3.17 | −109.46 | 1.9340 | 2.14 |
| VI | 270.0 | E0 | −87.38 | −68.03 | 101.89 | −1.17 | −103.72 | 2.2497 | 1.25 |
| VII | 266.8 | E0 | −88.33 | −67.97 | 103.42 | −3.15 | −101.35 | 2.2741 | 1.20 |
The results of the optimizations confirm our considerations pertaining to the H1–C1–C2–H2 and H3–C3–C4–H4 torsion angles in 1–5. The mean values of these angles are 104.43° and −104.27°, respectively (Table 2). Importantly, these are almost the same and close to the range of 80–100°, which determines the vicinal coupling constant of ∼0 Hz. Thus, all the presented studies (the 1H NMR spectra and DFT calculations) show that 2,3-O-isopropylidene-β-D-ribofuranoside (1) adopts the 4T0/E0 conformation. Based on the identity of the ring coupling constants (Table 1), it can be concluded that all alkyl 2,3-O-isopropylidene-β-D-ribofuranosides (1–5) adopt the same 4T0/E0 conformation. This means that the set of coupling constants presented in Table 1 is diagnostic of the 4T0/E0 conformation for furanosides not only with the β-D-ribo configuration but also with the β-D-allo, α-L-talo, and β-D-psico configurations. The all mentioned configurations have the same proton arrangement in the furanose ring.
| Pa [°] | Conformation | Selected torsion angles [°] | ΔGb [kcal mol−1] | Population [%] | |||||
|---|---|---|---|---|---|---|---|---|---|
| O1–C1–O4–C4 | O4–C1–O1–C6 | H1–C1–C2–H2 | H2–C2–C3–H3 | H3–C3–C4–H4 | |||||
| a P – pseudorotation parameter.b Related to G = −881.36144 au. | |||||||||
| I | 271.9 | E0 | −86.29 | −68.55 | 100.86 | −0.17 | −103.73 | 0.00 | 33.71 |
| II | 270.7 | E0 | −86.99 | −68.44 | 101.53 | −0.95 | −103.39 | 0.6269 | 11.69 |
| III | 276.0 | E0/1T0 | −86.05 | −68.41 | 99.23 | 2.15 | −105.54 | 0.5447 | 13.44 |
| IV | 274.5 | E0/1T0 | −86.50 | −66.96 | 100.18 | 0.85 | −106.74 | 0.5685 | 12.91 |
| V | 269.5 | E0 | −86.86 | −68.71 | 102.12 | −2.00 | −103.11 | 0.9249 | 7.07 |
| VI | 282.3 | E0/1T0 | −86.17 | −68.76 | 97.88 | 4.96 | −110.67 | 1.0461 | 5.76 |
| VII | 274.5 | E0/1T0 | −86.59 | −69.08 | 100.03 | 1.17 | −105.55 | 1.1960 | 4.47 |
| VIII | 277.7 | E0/1T0 | −86.40 | −67.17 | 98.94 | 2.58 | −107.95 | 1.2406 | 4.15 |
| IX | 271.4 | E0 | −85.65 | −65.42 | 100.72 | −0.77 | −104.18 | 1.3887 | 3.23 |
| X | 275.6 | E0/1T0 | −87.12 | −68.60 | 99.70 | 1.95 | −105.80 | 1.5619 | 2.41 |
| XI | 276.4 | E0/1T0 | −85.58 | −65.68 | 98.79 | 2.20 | −106.63 | 1.9967 | 1.16 |
It has to be mentioned that the lack of a fused 2,3-isopropylidene group clearly changes the conformational preferences of methyl β-D-ribofuranoside. The complex calculations of this compound demonstrated that it adopts the 3T2 conformation from the northern part of the pseudorotational wheel.57
3.3 Conformational analysis of 6–10 based on the 1H NMR spectra
The 1H NMR spectra of alkyl 5-O-acetyl-2,3-O-isopropylidene-β-D-ribofuranosides (6–10) are very distinctive and do not differ substantially from each other. Importantly, with regard to the coupling constants of the furanose ring protons (Table 1), they differ very little from the 1H NMR spectra of their precursors (1–5). Like the latter, they are characterized by the zero coupling constant between the H1 and H2 protons (J1,2) and the same J2,3 coupling constant (5.86 Hz or 6.22 Hz). However, in the case of β-D-ribofuranosides 6–10, a weak J3,4 = 0.73 Hz or 1.10 Hz coupling constant was recorded (again, both values can be considered as the same within the limits of measurement error). There was the zero coupling in the case of β-D-ribofuranosides 1–5, which, as demonstrated above, adopt the 4T0/E0 conformation. The recorded J3,4 coupling constant for 6–10 indicates a slight increase in the H3–C3–C4–H4 torsion angle compared to the situation in 1–5. Such an increase takes place when the E0 conformation approaches the 1T0 conformation. The transition from the 4T0/E0 to the E0/1T0 conformation increases the H3–C3–C4–H4 torsion angle and, at the same time, decreases the H1–C1–C2–H2 torsion angle. The latter angle, however, is still in the range of 80–100°, therefore, zero coupling constant between the H1 and H2 protons is recorded. Adoption of the E0/1T0 conformation does not substantially affect the J2,3 coupling constant, which is still 5.86 Hz or 6.22 Hz, indicating that the H3–C3–C4–H4 torsion angle is close to 0°.3.4 DFT optimizations of methyl 5-O-acetyl-2,3-O-isopropylidene-β-D-ribofuranoside (6)
In order to identify the conformation adopted by β-D-ribofuranosides 6–10, the structure of methyl 5-O-acetyl-2,3-O-isopropylidene-β-D-ribofuranoside (6) was optimized using DFT methods. Taking into account the rotations around the C1–O1, C4–C5, and C5–O5 bonds as well as two (exo and endo) settings of the 2,3-O-isopropylidene ring and two settings of the OAc group 108 (2 × 2 × 33) rotamers of 6 in the 0E, E0, 3E, and E3 conformations, respectively, were prepared. The only two favorable settings of the OAc group were previously demonstrated.58–60 During the optimization, the number of rotamers was significantly reduced. Thus, eleven (I–XI) relatively stable structures of 6 were obtained with a population ranging from 33.71% to 1.16%. The geometrical parameters, relative free Gibbs energies, and populations of I–XI in a group of found rotamers of 6 are listed in Table 3. These data indicate that methyl 5-O-acetyl-2,3-O-isopropylidene-β-D-ribofuranoside (6) prefers the E0 conformation (P = 269.5–271.9°) with a slight deviation towards the 1T0 conformation (P = 274.5–282.3°). The E0 conformation represents 55.70% of the population of all optimized structures, whereas the E0/1T0 conformation represents 44.30%. Thus, it can be stated that methyl 5-O-acetyl-2,3-O-isopropylidene-β-D-ribofuranoside (6) adopts the E0/1T0 conformation, which is really close to the 4T0/E0 conformation adopted by 2,3-O-isopropylidene-β-D-ribofuranoside (1). While the 4T0/E0 conformation is characterized by the zero coupling constants between both the H1 and H2 as well as H3 and H4 protons, the E0/1T0 conformation shows no coupling between the H1 and H2 protons, but a slight coupling (0.73 or 1.10 Hz) between the H3 and H4 is recorded in this conformation. This tenuous difference in the values of the coupling constants is reflected in the calculated torsion angles (Table 3). The H3–C3–C4–H4 torsion angles are slightly, but clearly, greater than the H1–C1–C2–H2 torsion angles. The mean value for the former is 105.75°, whereas it is 100.00° for the latter. Thus, the transition from the 4T0/E0 conformation to the E0/1T0 conformation slightly increases the H3–C3–C4–H4 torsion angle and decreases the H1–C1–C2–H2 torsion angle.The same coupling constants recorded for all alkyl 5-O-acetyl-2,3-O-isopropylidene-β-D-ribofuranosides (6–10) allow for the assumption that all of them adopt the same E0/1T0 conformation. This means that the set of coupling constants recorded for 6–10 (Table 1) is diagnostic of the E0/1T0 conformation for furanosides not only with the β-D-ribo configuration, but also with the β-D-allo, α-L-talo, and β-D-psico configurations.
3.5 2,3-O-Isopropylideneribonucleosides (15–18)
2,3-O-Isopropylideneribonucleosides (15–18) have the same β-D-ribo configuration as 2,3-O-isopropylidene-β-D-furanosides 1–10 discussed above; however, they belong to the class of the N-furanosides. Studies performed on 15–18 well illustrate how a change in the nature of the aglycone affects the conformational preferences of the furanose ring. Because 2,3-O-isopropylideneuridine (15) was obtained in a crystalline form, its geometry is analyzed first.The diffraction data of 2,3-O-isopropylideneuridine (15) are listed in Table 4. The structure of 15 showing the atom numbering scheme and the selected torsion angles are presented in Fig. 4A and B, respectively.
| Compound | 15 |
| Empirical formula | C12H16N2O6 |
| Formula weight | 284.27 |
| Temperature (K) | 295(2) |
| Wavelength (Å) | 0.71073 |
| Crystal system | Orthorhombic |
| Space group | P212121 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|
| Unit cell dimensions | |
| a (Å) | 5.2226(3) |
| b (Å) | 12.7720(7) |
| c (Å) | 19.8559(11) |
| α (Å) | 90° |
| β (°) | 90° |
| γ (°) | 90° |
| V (°) | 1324.45(13) |
| Z | 4 |
| Dcalcd (Mg m−3) | 1.426 |
| Absorption coefficient (mm−1) | 0.115 |
| Absorption correction type | ‘Multi-scan’ |
| F (000) | 600 |
| Crystal size (mm) | 0.38 × 0.24 × 0.11 |
| θ range for data collection (°) | 3.35 ÷ 29.16 |
| Limiting indices | −7 ≤ h ≤ 7, −11 ≤ k ≤ 15, −22 ≤ l ≤ 26 |
| Reflections collected/unique | 9553/3128 [Rint = 0.0345] |
| Completeness 2θ = 25, 24° (%) | 99.7 |
| Refinement method | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 3128/0/216 |
| Goodness-of-fit on F2 | 1.111 |
| Final R indices [I > 2σ(I)] | R1 = 4.80 wR2 = 10.26 |
| R Indices (all data) | R1 = 5.91 wR2 = 10.68 |
| Absolute structure parameter | 0.0(6) |
| Largest diff. peak and hole (eÅ−3) | 0.205 and −0.214 |
| CCDC | 2164567 |
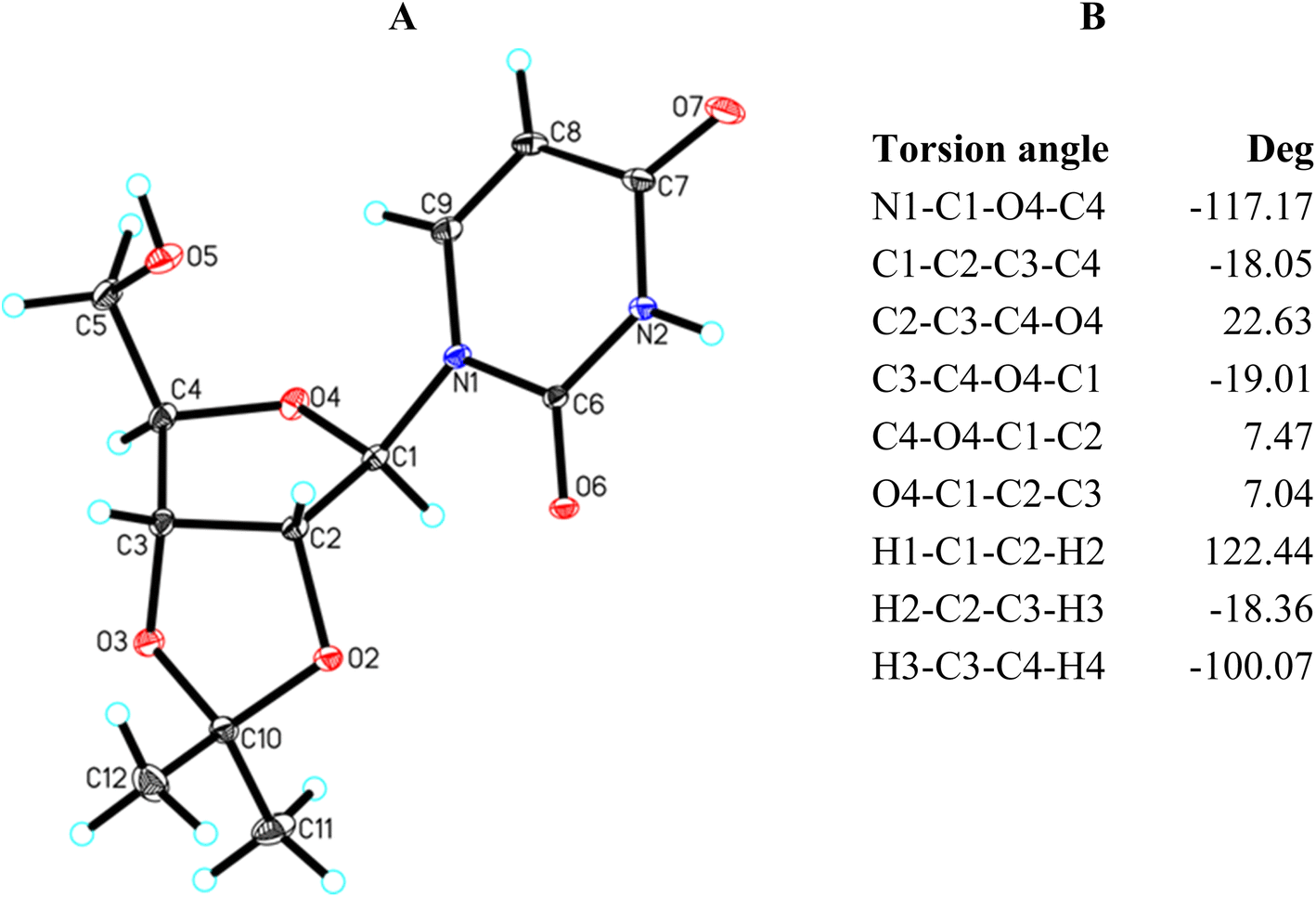 | ||
| Fig. 4 (A) Molecular structure of 15 showing the atom-labelling scheme. Displacement ellipsoids are drawn at the 25% probability level and H atoms are shown as small spheres with an arbitrary radius; (B) selected torsion angles. | ||
X-Ray analysis of 15 reveals that it has a bicyclic structure consisting of fused furanoside (O4/C1/C2/C3/C4) and five-membered 2,3-O-isopropylidene (O2/C2/C3/O3/C10) rings (Fig. 4A). The furanose ring in nucleoside 15 adopts a conformation close to the 4T3 63,64 with ring-puckering parameters65,66 θ = 0.212 Å and ϕ = 124.4(7)°, pseudorotation parameters67 P = 217.0(3)° and τm = 22.8(2)° for the C2–C3 reference bond, and delta parameter68 Δ = 434.0°. The five-membered isopropylidene ring adopts a conformation close to the 3E form63,64 with ring-puckering parameters65,66 θ = 0.300(2) Å and ϕ = 288.4(5)°, pseudorotation parameters67 P = 19.9(2)° and τm = 33.3(1)° for the C3–O3 reference bond, and delta parameter68 Δ = 39.9°. In the crystal of 15, molecules are held together by the N2–H2⋯O7, O5–H5⋯O7 and C1–H1⋯O6 intermolecular interactions to produce a 3D framework (Table 5 and Fig. S1†).
| D–H⋯A | d(D–H) (Å) | d(H⋯A) (Å) | d(D⋯A) (Å) | <D–H⋯A (°) |
|---|---|---|---|---|
| a Symmetry codes: (i) ½ + x, ½ − y, −z; (ii) 1 − x, ½ + y, ½ − z; (iii) −½ + x, ½ − y, −z. | ||||
| N2–H2⋯O6i | 0.90(3) | 2.10(3) | 2.900(3) | 148(2) |
| O5–H5⋯O7ii | 0.82(4) | 1.96(4) | 2.748(3) | 164(4) |
| C1–H1⋯O6iii | 0.97(3) | 2.52(3) | 3.398(3) | 152(2) |
The torsion angles of nucleoside 15 presented in Fig. 4B well illustrate the 4T3 conformation of the furanose ring. In this conformation, the O4, C1, and C2 atoms lay in one plane, whereas the C4 and C3 carbon atoms are located above and below this plane, respectively. Therefore, the torsion angles in which the O4, C1, and C2 atoms are involved are relatively small and amount to 7.47° (C4–O4–C1–C2) and 7.04° (O4–C1–C2–C3), respectively. The remaining torsion angles are larger. These angles formed by two consecutive atoms of those lying in one plane amount to −18.05° (C1–C2–C3–C4) and −19.01° (C3–C4–O4–C1), respectively. The C2–C3–C4–O4 torsion angle is the largest (22.63°) because this is the C3–C4 bond twisted in the 4T3 conformation.
| No. | J1,2 trans H/H | J2,3 cis H/H | J3,4 trans H/H | Conformation | Ref. |
|---|---|---|---|---|---|
| a Not determined. | |||||
| 11 | 5.49 | 5.19 | 3.66 or 3.97 | ∼2E | |
| 12 | 6.11 or 6.41 | 4.88 or 5.19 | 3.05 or 3.36 | ∼2E | |
| D2O | 6.3 | 5.2 | 3.1 | 18% N | 71 |
| DMSO | 6.0 | 4.9 | 3.2 | —a | 70 |
| 13 | 3.97 | a | 4.57 | ∼3E | |
| D2O | 3.9 | 5.2 | 6.2 | 71% N | 71 |
| 14 | 5.80 | 4.89 or 5.18 | 3.36 | ∼2E | |
| D2O | 6.1 | 5.2 | 3.5 | 21% N | 71 |
| 15 | 2.75 | 6.41 | 3.66 or 3.97 | ∼E4 | |
| 16 | 3.05 | 6.10 | 2.44 or 2.75 | ∼E4 | |
| 17 | 2.44 | 6.10 or 6.41 | 3.66 or 3.97 | ∼E4 | |
| 18 | 2.75 | 6.10 | 3.05 or 3.36 | ∼E4 | |
According to the Dreiding model of a furanose ring and the previously reported data on the torsion angles in the optimized THF ring,45 one may see that the trans-oriented vicinal protons can maximally approach each other to form the H–C–C–H torsion angle close to 80–90° (3JH,H ∼0 Hz) or can maximally move away from each other to form the H–C–C–H torsion angle close to 170° (3JH,H ∼8 Hz). These are the extreme arrangements of trans-oriented protons in a furanose ring, and these take place when the trans-oriented protons are attached to the carbon atoms whose bond is twisted. The coupling constants of the trans-oriented H1 and H2 protons in 15–18 are in the narrow range of 2.44–3.05 Hz (2.75 Hz on average). The coupling constants of the trans-oriented H3 and H4 protons are slightly larger and fall within a slightly wider range of 2.44–3.97 Hz (3.36 Hz on average). This may indicate that the H4 proton has more freedom of rotation than the H1 proton. According to the Karplus curve56 as well as Serianni and Barker reports,69 the J1,2 and J3,4 coupling constants recorded for 15–18 are indicative of the respective torsion angles being included in the range of about 120–130°. Simultaneously, the H1–C1–C2–H2 torsion angle has to be slightly smaller than the H3–C3–C4–H4 torsion angle due to the smaller J1,2 coupling constant compared to the J3,4 coupling constant. The 4T3 conformation found in the crystal lattice of 15 with the H1–C1–C2–H2 and H3–C3–C4–H4 torsion angles of 122.27° and −100.59°, respectively, does not meet these requirements. It seems that the best fit is met in the E4-like conformation. This conformation requires the H1 and H2 protons to be ecliptically oriented with the H1–C1–C2–H2 torsion angle of about 120° and allows the H3–C3–C4–H4 torsion angle to be greater than 120°. Such a situation is well illustrated in the crystal structure of 2.3-O-isopropylideneuracil (JAWCEP, Table 8) where the H1–C1–C2–H2 and H3–C3–C4–H4 torsion angles are 113.35° and 129.44°, respectively.
Table 6 also presents the coupling constants recorded for the basic nucleosides (11–14). These show that the furanose ring protons coupling constants of adenosine (12) and guanosine (14) are almost the same, indicating the same conformation. Importantly, the data recorded by us are in accordance with the literature NMR data, both recorded in DMSO70 and in D2O71 (Table 6). The conformation of the structurally non-restricted nucleosides is usually interpreted in terms of a two-state equilibrium between the North and the South conformations.72 Thus, Plavec et al., who recorded very similar coupling constants for 12 and 14 in D2O,71 stated that the equilibrium populations of the North form in 12 and 14 is 18% and 21%, respectively. We would like to propose the statement that the recorded coupling constants for 12 and 14 are indicative of the specific conformation from the southern part of the pseudorotational itinerary (Fig. 2B). According to the Karplus curve56 as well as to the Serianni and Barker reports,69 for the trans-oriented H1 and H2 protons in 12 and 14, the J1,2 coupling constant in the range of 6–6.45 Hz indicates the H1–C1–C2–H2 torsion angle of about 150°. In turn, for the trans-oriented H3 and H4 protons, the J3,4 coupling constant in the range of 3.1–3.58 Hz indicates the H3–C3–C4–H4 torsion angle in the range of 110–120°. The J2,3 coupling constant in the range of 4.83–5.2 Hz for the cis-oriented H2 and H3 protons indicates that the H2–C2–C3–H3 torsion angle reaches a value of 40°. Based on these estimated values of 150°, 40°, and 120° for the H1–C1–C2–H2, H2–C2–C3–H3, and H3–C3–C4–H4 torsion angles, respectively, one may state that adenosine (12) and guanosine (14) adopt the 2E-like conformation (the southern region). Our calculated for the THF ring torsion angles45 as well as the torsion angles found in the CCDC database also confirm that the 2E-like conformation fits well with these estimated angles.
The ring proton coupling constants recorded for uridine (11, Table 6) do not differ much from those recorded for adenosine (12) and guanosine (14). Therefore, we assume that 11, like 12 and 14, adopts a conformation close to the 2E. However, the situation of cytosine (13) is different. The values of the J1,2 and J3,4 coupling constants recorded for 13 are inverted compared to the analogous coupling constants recorded for 11, 12, and 14, whereas the J2,3 coupling constant remained the same. Plavec et al.71 concluded that the equilibrium population of the North form in 13 is 71%. In our opinion, the estimated values of the torsion angles of about 120°, 40°, and 150° for the H1–C1–C2–H2, H2–C2–C3–H3, and H3–C3–C4–H4 torsion angles, respectively, indicate that cytosine (13) adopts the 3E-like conformation (the north region). This is confirmed by the values of the respective torsion angles that we calculated for the THF ring45 and found in the CCDC database (Table 8). As one may see, both the 2E the 3E conformations are typical for the crystal structure of the conformationally non-restricted nucleosides (Table 8).
3.6 Furanoses with β-D-ribo configuration in the CCDC database
To improve presented discussion, the ConQuest search of the Cambridge Structural Database (CSD, Version 5.43, November 2021 update) for the furanose ring with the β-D-ribo-like configuration was done.73,74 Table 7 presents geometric parameters of β-D-ribofuranosides whose conformational freedom is limited due to the fused 2,3-O-isopropylidene ring and β-D-ribofuranosides without such a restriction. Table 8 presents geometric parameters of ribonucleosides whose conformational freedom is limited due to the fused 2,3-O-ketal (isopropylidene or other) and ribonucleosides without such a restriction.| Crystal | Configu-ration | Con for. | O1–C1–O4–C4 [deg] | O4–C1–O1–C6 [deg] | H1–C1–C2–H2 [deg] | H2–C2–C3–H3 [deg] | H3–C3–C4–H4 [deg] |
|---|---|---|---|---|---|---|---|
| a Conformation index numbers refer to consecutive carbon atoms of the furanose ring, with the anomeric carbon atom numbered 1, regardless of whether aldose or ketose is considered.b The D enantiomers of these furanosides adopt the E0 conformation. The respective torsion angles for the D enantiomers have an altered sign. | |||||||
| Without 2,3-O-isopropylidene | |||||||
| ESIHAP | β-D-ribo | 1T2 | −82.35 | −70.10 | 85.63 | 32.59 | −140.27 |
| DEWREC | β-D-ribo | E2 | −87.18 | −85.27 | 74.79 | 38.83 | −147.02 |
| MAQVAB | β-D-ribo | E2 | −91.60 | −69.35 | 89.14 | 44.00 | −152.59 |
| ZOWJAW | β-D-ribo | E2 | −91.98 | −60.49 | 80.66 | 33.05 | −148.56 |
| ZOWJAW | β-D-ribo | E2 | −97.58 | −77.31 | 83.66 | 42.96 | −156.03 |
| GINKOE | β-D-psico | E2 | −90.26 | −44.82 | — | 35.96 | −146.84 |
| UPUKUM | β-D-psico | 3T2 | −105.53 | −52.13 | — | 45.59 | −168.08 |
| UPULAT | β-D-psico | 3T2 | −108.51 | −48.50 | — | 45.88 | −169.45 |
| TEWJUC | β-D-ribo | 2E | −145.49 | −74.80 | 166.23 | −41.50 | −101.25 |
|
|||||||
| With 2,3-O-isopropylidene | |||||||
| ILICUB | β-D-ribo | 4T0 | −92.52 | −71.41 | 110.15 | −11.02 | −97.79 |
| GIVFEW | β-D-ribo | E0 | −89.54 | −70.65 | 104.50 | −7.28 | −89.93 |
| HEWHEZ | β-D-ribo | E0 | −77.65 | −71.79 | 91.58 | 0.92 | −99.68 |
| ILIDAI | β-D-ribo | E0 | −87.27 | −61.82 | 100.79 | 0.43 | −107.75 |
| NUFLOP | β-D-ribo | E0 | −80.03 | −71.52 | 94.85 | 7.65 | −106.45 |
| NUFLUV | β-D-ribo | E0 | −89.43 | −67.53 | 102.43 | −2.92 | −103.53 |
| POGHAU | β-D-ribo | E0 | −82.96 | −70.49 | 92.11 | 8.66 | −109.33 |
| MIPAHY | β-D-allo | E0 | −82.81 | −62.29 | 93.29 | −1.21 | −104.08 |
| POCSUV | β-D-allo | E0 | −85.07 | −76.65 | 96.81 | −0.11 | −102.93 |
| WAVYAU | β-D-psico | E0 | −89.65 | −89.75 | — | −7.94 | −98.55 |
| UDAYAC | β-L-psicob | 0E | 84.26 | 90.18 | — | 1.81 | 99.63 |
| UDAYEG | β-L-psicob | 0E | 89.01 | 90.99 | — | 4.99 | 98.95 |
| DABRON | β-D-ribo | 1T0 | −85.48 | −70.95 | 82.22 | 3.60 | −102.51 |
| ILIDEM | β-D-ribo | 1T0 | −82.74 | −68.12 | 87.21 | 19.02 | −120.09 |
| MUZJID | β-D-ribo | 1T0 | −84.62 | −65.46 | 91.23 | 10.81 | −111.38 |
| VABRUL | β-D-allo | 1T0 | −79.18 | −68.26 | 101.68 | −4.17 | −92.01 |
| Crystal | Nucleobase | Con for. | N1–C1–O4–C4 [deg] | H1–C1–C2–H2 [deg] | H2–C2–C3–H3 [deg] | H3–C3–C4–H4 [deg] |
|---|---|---|---|---|---|---|
| Without 2,3-O-isopropylidene | ||||||
| CEFJUS | Uracil | 3T2 | −116.91 | 94.94 | 36.18 | −161.76 |
| TCYTDH | Cytosine | 3T2 | −116.50 | 100.23 | 49.50 | −166.40 |
| AZCYTD20 | Cytosine | 3E | −121.58 | 97.79 | 43.05 | −166.90 |
| AZURID10 | Uracil | 3E | −116.30 | 94.55 | 36.52 | −153.87 |
| BEZGES | Cytosine | 3E | −116.51 | 89.50 | 38.87 | −165.51 |
| BEURID10 | Uracil | 3E | −110.07 | 87.63 | 42.28 | −163.17 |
| CITXUY10 | Uracil | 3E | −132.33 | 111.80 | 20.16 | −146.66 |
| CYTIDI02 | Cytosine | 3E | −113.15 | 90.06 | 37.43 | −168.85 |
| CYTIDN | Cytosine | 3E | −118.98 | 92.31 | 35.64 | −146.62 |
| DTURID | Uracil | 3E | −119.38 | 94.75 | 40.64 | −166.19 |
| GAKNOV | Adenine | 3E | −117.65 | 93.74 | 42.61 | −162.74 |
| HDTURD10 | Uracil | 3E | −117.06 | 92.00 | 45.69 | −160.57 |
| HICYTM | Cytosine | 3E | −134.00 | 118.73 | 40.67 | −166.75 |
| MEURID | Uracil | 3E | −120.90 | 100.29 | 30.68 | −161.69 |
| MXURID01 | Uracil | 3E | −116.33 | 88.27 | 43.02 | −163.80 |
| TURIDN10 | Uracil | 3E | −116.18 | 88.73 | 43.15 | −169.04 |
| VIKKIK01 | Uracil | 3E | −116.16 | 92.83 | 42.23 | −163.83 |
| CITXOS10 | Uracil | E4 | −153.11 | 129.27 | 20.29 | −158.41 |
| MAQVAB | Uracil | E4 | −146.58 | 121.29 | 26.57 | −163.38 |
| QENGIA | Adenine | 2T1 | −166.93 | 179.95 | −42.01 | −109.69 |
| AMURID | Uracil | 2E | −147.11 | 150.91 | −36.95 | −87.44 |
| BIBXIT02 | Cytosine | 2E | −153.24 | 165.29 | −38.55 | −100.81 |
| BRURID01 | Uracil | 2E | −140.87 | 157.62 | −35.74 | −99.40 |
| CLURID10 | Uracil | 2E | −140.17 | 153.92 | −36.84 | −96.80 |
| CXMURD | Uracil | 2E | −147.99 | 169.16 | −49.63 | −102.43 |
| DAXGEP | Uracil | 2E | −138.38 | 155.06 | −36.90 | −101.68 |
| DAZCYT10 | Cytosine | 2E | −156.66 | 167.86 | −35.18 | −105.19 |
| DMURID | Uracil | 2E | −144.06 | 165.77 | −38.92 | −98.66 |
| DZURID | Uracil | 2E | −145.20 | 158.57 | −26.78 | −100.15 |
| GICMOU | Cytosine | 2E | −136.20 | 148.18 | −46.26 | −105.50 |
| HXURID | Uracil | 2E | −156.73 | 166.33 | −40.07 | −109.97 |
| MEYRID | Thymine | 2E | −145.48 | 160.10 | −38.70 | −96.71 |
| QENGOG | Adenine | 2E | −154.36 | 164.82 | −44.64 | −98.77 |
| ZAYTOI | Hypoxanthine | 2E | −149.28 | 165.78 | −36.72 | −103.84 |
| ZAYTIC | Uracil | 2T3 | −137.08 | 157.77 | −39.85 | −96.14 |
|
||||||
| With 2,3-O-isopropylidene | ||||||
| CAXPEW | Uracil | 3T2 | −119.50 | 104.20 | 14.65 | −125.42 |
| BOYMUX | Uracil | 3E | −125.19 | 107.20 | 19.14 | −141.60 |
| MILDAM | Uracil | 3E | −131.78 | 101.61 | 13.95 | −140.36 |
| AIMCTY | Thymine | 3T4 | −129.82 | 78.91 | 37.25 | −149.06 |
| CICNOR | Uracil | E4 | −140.49 | 102.01 | 34.45 | −146.35 |
| JAWCEP | Uracil | E4 | −139.07 | 113.35 | 12.28 | −129.44 |
| RAGLOB | Adenine | E4 | −138.50 | 112.31 | 24.64 | −149.22 |
| KEXLUU10 | Uracil | 0T1 | −153.93 | 139.55 | −16.13 | −127.59 |
| ELAZUM | Cytosine | E1 | −158.19 | 158.77 | −27.68 | −114.81 |
| JAWCIT | Thymine | E1 | −160.50 | 154.44 | −28.87 | −113.55 |
| RAGLUH | Uracil | E1 | −151.85 | 143.28 | −17.11 | −119.39 |
| WIVZAC | Uracil | E1 | −149.06 | 141.63 | −12.05 | −120.58 |
| BEGKOP | Pyrimidine | 2E | −136.43 | 135.94 | −23.29 | −134.85 |
| EDUJUI | Hypoxanthine | 2T3 | −130.23 | 140.98 | −30.25 | −95.75 |
| GEBRIO | Guanine | E3 | −127.32 | 140.03 | −33.81 | −94.40 |
| 15 | Uracil | 4T3 | −117.33 | 122.27 | −19.10 | −100.59 |
| BOZFEB01 | Uracil | 4T3 | −118.66 | 123.97 | −22.89 | −98.30 |
| ZZZAPA10 | Uracil | 4T3 | −117.53 | 122.92 | −16.30 | −96.22 |
| JUKING | Hypoxanthine | E0 | −83.78 | — | — | — |
| VUYMIL | Guanine | 1E | −107.20 | 107.09 | 4.71 | −117.93 |
|
||||||
| With other 2,3-O-cycloketals | ||||||
| CEZGUJ | Uracil | 0T1 | −159.06 | 146.06 | −4.04 | — |
| WUPBER | Uracil | 2E | −141.04 | 151.81 | −32.70 | −98.22 |
| SUGTUK | Uracil | E3 | −122.33 | 133.96 | −25.81 | −95.07 |
| SUGVAS | Uracil | E3 | −121.56 | 131.62 | −21.04 | −97.95 |
| WUPBIV | Uracil | E3 | −126.05 | 136.20 | −24.15 | −99.35 |
Analysis of the data in Table 7 shows that β-D-ribofuranosides without 2,3-O-isopropylidene fused ring in a crystal structure mainly adopt the E2-like conformation from the northern part of the pseudorotation wheel (red dots in Fig. 5A). The E2 conformation of β-D-ribofuranosides is characterized by a relatively small torsion angle between the H1 and H2 protons, which is in the range of 74.79°–89.14° (82.06° on average). Such a torsion angle is the minimum angle that the trans-oriented protons can achieve in the furanose ring. In turn, the trans-oriented H3 and H4 protons in the E2 conformation of β-D-ribofuranosides move away from each other so that the H3–C3–C4–H4 torsion angle is in the range of 146.84–156.03°. The data in Table 7 clearly show how the H3–C3–C4–H4 torsion angle increases as the conformation changes from the 1T2 (140.27°) through the E2 (150.80° on average) to the 3T2 (169.45°). The last value of the torsion angle is almost the maximum that the trans-oriented protons in the furanose ring can achieve. The H2–C2–C3–H3 torsion angle in the E2 conformation of β-D-ribofuranosides is in the range of 33.05–44.00° (38.96° on average), which is typical of the cis-oriented protons, at least one of which is bound to the carbon laying out of the plane formed by the remaining furanose ring atoms. The maximum torsion angle between the cis-oriented protons is formed when both these protons are attached to the carbons involved in the twist of the furanose ring. This is the case of the H2 and H3 protons of β-D-ribofuranosides in the 3T2 or 2T3 conformations. Therefore, the 3T2 conformation is characterized by the highest values of the H2–C2–C3–H3 torsion angle (45.59° and 45.88°) formed between the cis-oriented H2 and H3 protons. The introduction of 2,3-O-isopropylidene to β-D-ribofuranosides definitively changes their conformational preferences (Table 7). Every 2,3-O-isopropylidene-β-D-ribofuranoside found in the CCDC database adopts the E0-like conformation (orange dots in Fig. 5A). The reasons for this behavior of the furanose ring are discussed in the next paragraph. Typically for the E0 conformation, the values of the H1–C1–C2–H2 torsion angle in the range of 91.58–104.50° (on average 96.99°) and the H3–C3–C4–H4 torsion angle in the range of 89.93–109.33° (on average 101.89°) are similar. The shift towards the 4T0 conformation slightly increases the H1–C1–C2–H2 torsion angle (110.15°) and slightly decreases the H3–C3–C4–H4 torsion angle (97.79°). The shift towards the 1T0 conformation does the opposite and slightly decreases the H1–C1–C2–H2 torsion angle (90.58° on average) and slightly increases the H3–C3–C4–H4 torsion angle (106.50° on average). Also, typically for the E0 conformation, the H2 and H3 protons are eclipsed, which is confirmed by the H2–C2–C3–H3 torsion angle in the range of 0.11–8.66° (4.39° on average). Both the shifts towards the 4T0 and the 1T0 conformations increase this torsion angle to the value of 11.02° and to the averaged value of 9.40°, respectively. All these findings concerning the E0-like conformation of 2,3-O-isopropylidene-β-D-ribofuranosides found in the CCDC database clearly confirm our findings concerning 1–10 based on the 1H NMR spectra and DFT calculations.
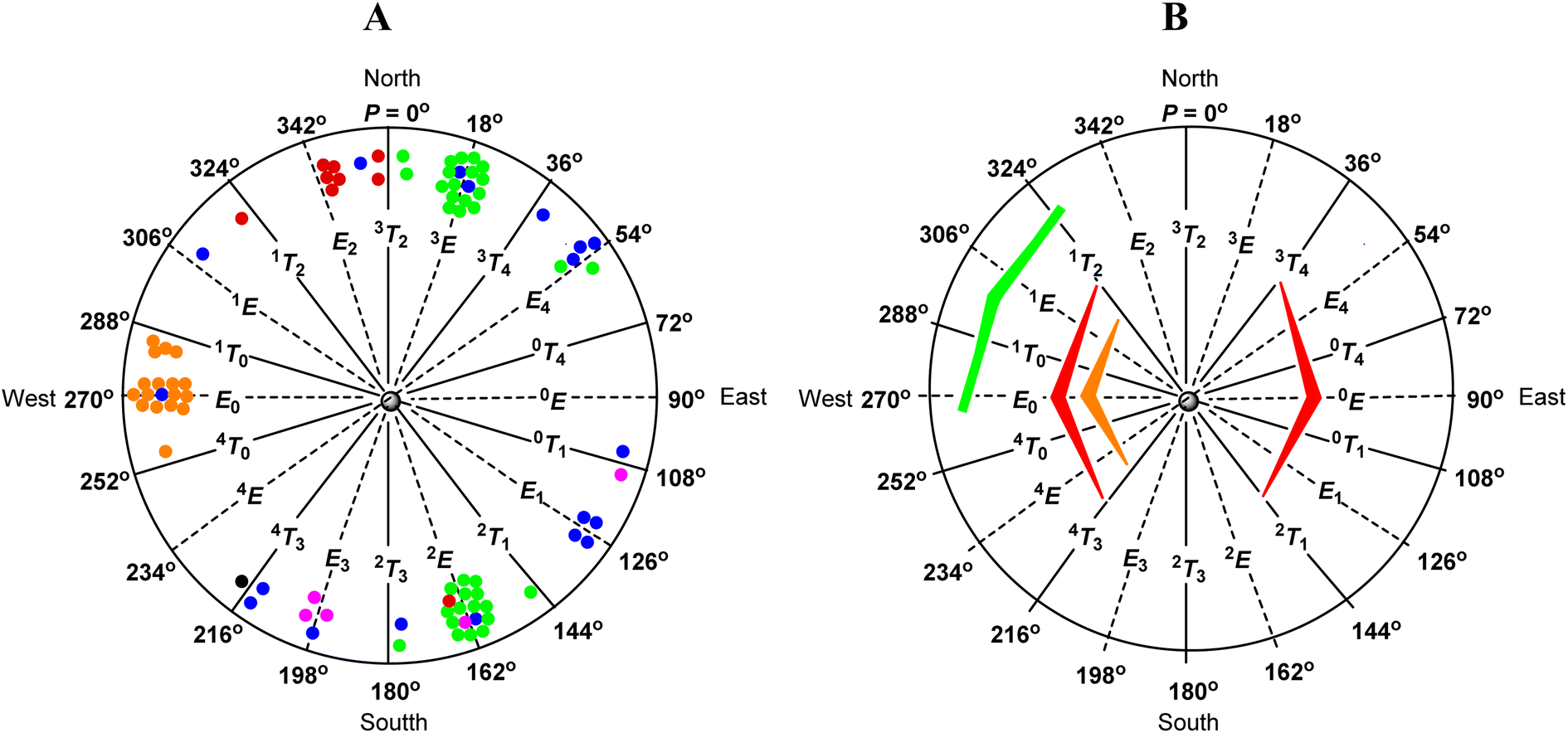 | ||
| Fig. 5 (A) Distribution of sugars with the β-D-ribo configuration of the furanose ring in the CCDC database (red – furanosides; green – nucleosides; orange – 2,3-O-isopropylidenefuranosides; blue – 2,3-O-isopropylidenenucleosides; black – 15; purple – nucleosides with other 2,3-O-ketal rings). (B) Factors influencing the conformational preferences of furanoses with the β-D-ribo configuration (red – unfavorable ecliptic orientation of the 2-OH and 3-OH groups; orange – unfavorable 1,3-pseudodiaxial interaction of the aglycone and terminal hydroxymethyl group; green – favorable anomeric effect). | ||
The CCDC database search shows that the conformational preferences of ribonucleosides (Table 8) are different from that of β-D-ribofuranosides (Table 7). As long as the conformational changes of the furanose ring are not restricted by the 2,3-O-isopropylidene or other 2,3-O-ketal rings, the nucleosides in the crystal adopt the 3E-like or 2E-like conformation, from the North or South regions, respectively, of the pseudorotational itinerary (green dots in Fig. 5A). This finding is fully in agreement with Altona's reports, who indicates these two regions to be the most stable for nucleosides.72 In the 3E-like conformation, the trans-oriented H1 and H2 protons of the ribonucleosides approach each other by an average torsion angle of 95.53°. In turn, the trans-oriented H3 and H4 protons move away from each other by an average torsion angle of 161.75°. The mean value of the H2–C2–C3–H3 torsion angle in this conformation is 38.84°. This is typical of cis-oriented protons, one of which (H3) is bound to the carbon atom (C3) laying outside the plane formed by the other ring atoms. In the case of the 2E-like conformation, the trans-oriented H1 and H2 protons move away from each other by an average torsion angle of 160.67°. In turn, the trans-oriented H3 and H4 protons approach each other by an average torsion angle of 100.52°. The mean value of the H2–C2–C3–H3 torsion angle in this conformation is 38.71°, which again confirms that one of the protons involved in this torsion angle (H2) is bound to the carbon atom (C2) laying outside of the plane formed by the other ring atoms.
The introduction of a 2,3-O-ketal (isopropylidene or other) into a nucleoside hinders their adoption of both the 3E and 2E conformations. However, ribonucleosides with the 2,3-O-ketal fused ring (blue dots for 2,3-O-isopropylidene derivatives, purple dots for other 2,3-O-ketals, and a black dot for 15 in Fig. 5A) clearly behave differently than β-D-ribofuranosides in an analogous situation (orange dots in Fig. 5A). The latter, regardless of their structure, always adopt the E0-like conformation, whereas the former do not have any privileged conformation and can be found throughout the entire range of the pseudorotation wheel.
3.7 Factors influencing the conformational preferences of the β-D-ribofuranose ring
Analyzing the presented results, we found that the first important factor influencing conformational preferences of the furanose ring are unfavourable ecliptic interactions between the substituents of the ring atoms lying in one plane. In the case of the ribofuranose ring, among possible ecliptic interactions, those between the 2-OH and 3-OH groups seem to be particularly unfavorable. In addition to typical torsion strains, these should generate repulsion of dipoles of polar bonds formed between respective carbon and oxygen atoms. From this point of view, the 3T2 and 2T3 conformations of the ribofuranose ring are the most favorable because these conformations allow the dipoles of the polar bonds to be as far apart as possible (Fig. 6A). In turn, the 0E and E0 conformations of the ribofuranose ring, in which the 2-OH and 3-OH groups are ecliptically oriented, are the most unfavorable. These considerations are fully in agreement with the Altona and Sundaralingam reports.72 It should be added that the best stabilization of the 3T2 and 2T3 conformations is often explained by the gauche effect.71,75,76 This effect is not taken into account herein because we are not convinced of its stabilizing action.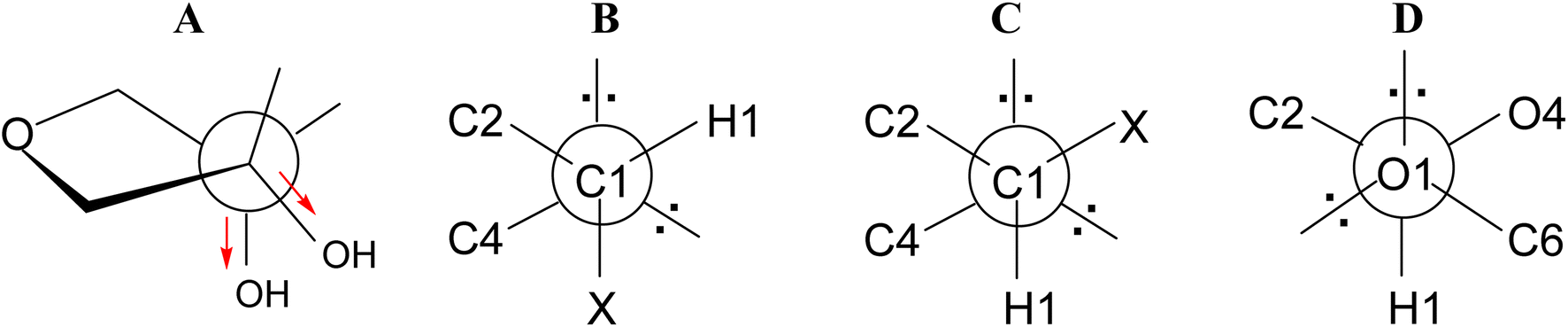 | ||
| Fig. 6 (A) Twist on the C2–C3 bond in the ribofuranose ring; (B) gauche and (C) antiperiplanar orientations of the C4–O4–C1–X atoms; (D) gauche orientation of the O4–C1–O1–C6 atoms. | ||
The second factor influencing the conformational preferences of the furanose ring is an endo-anomeric effect, well known from the pyranose ring. The generalized endo-anomeric effect is defined as the preference of the gauche orientation (Fig. 6B) over the antiperiplanar orientation (Fig. 6C) in the C5–O5–C1–X (pyranose) or C4–O4–C1–X (furanose) atoms arrangement, where the X atom comes from an aglycone.77 This effect increases with increasing electronegativity of the X atom in line with the series: carbon < nitrogen < oxygen < halogen. In the case of a pyranose ring, the anomeric effect implies a preference for the axial orientation of the aglycone.
One of the theories explaining the anomeric effect, known as the hyperconjugation model, assumes that the stabilization of the gauche (axial) conformer is attributed to the delocalization of the antiperiplanar lone-pair of electrons of the ring oxygen atom to the antibonding orbital of the C1–X bond.77,78 In a furanose ring, this delocalization is possible only in a relatively narrow range of conformations, other for α-furanoses and other for β-furanoses. Although, there are some reports that the magnitude of the endo-anomeric effect in furanoses is larger by 2.7 kJ mol−1 (on average) in comparison to analogous pyranoses,79 it seems to us that the tendency is opposite. This is because the delocalization necessary to speak of the anomeric effect in a furanose ring has to be limited compared to the analogous delocalization in the pyranose ring. This limitation results from the fact that the aglycone cannot be fully axially oriented in a furanose ring. In the furanose ring, the bonds can be at most pseudoaxial or pseudoequatorial or have an intermediate character. As shown by the data presented in Tables 7 and 8, the lowest value that the C4–O4–C1–X torsion angle can achieve is ∼80°. This means that in furanoses, the electronically advantageous gauche conformation of the C4–O4–C1–X segment cannot be fully achieved. It does not change the fact that the anomeric effect, although not as strong as in pyranoses, also occurs in furanoses.
The most effective anomeric effect in the case of β-D-furanosides is expected in the 1T2/1E/1T0/E0 conformational range (Fig. 5B), where the aglycone is oriented more or less pseudoaxially and should be the strongest in the 1T0-like conformation. For the same reasons, the most effective anomeric effect in the case of α-D-furanosides is expected in the 2T1/E1/0T1/0E conformational range and should be the strongest in the 0T1 conformation. Consequently, any anomeric effect is expected in the 1T2/1E/1T0/E0 conformational range in the case of α-D-furanosides and in the 2T1/E1/0T1/0E conformational range in the case of β-D-furanosides.
The third factor that influences the conformational preferences of furanoses with the β-D-ribo configuration is the unfavorable 1,3-pseudodiaxial interactions between the aglycone and terminal hydroxymethyl group. These are expected to be the strongest in the 4E/4T0/E0/1T0/1E conformational range (Fig. 5B) and the weakest in the E4/0T4/0E/0T1/E1 conformational range. In the former range of conformations, the substituents of the C1 (aglycone) and C4 (hydroxymethyl group) carbon atoms are closest to each other. In the latter range of conformations, these substituents are furthest to each other. As one may see, the region of action of these unfavorable 1,3-pseudodiaxial interactions overlaps with one of the regions where the unfavorable ecliptic interactions between the 2-OH and 3-OH groups take place (West side, Fig. 5B). In turn, the regions of action of both these unfavorable interactions overlap to some extent with the region of action of the anomeric effect.
The analysis of the data presented herein fully confirms the influence of the above mentioned factors on the conformational preferences of furanosides with the β-D-ribo configuration. As shown by our search of the CCDC database, until the conformational changes of a furanose ring are not restricted by a fused 2,3-O-isopropylidene ring, β-D-ribofuranosides adopt the E2-like conformation, whereas ribonucleosides adopt the 3E-like or 2E-like conformations (Tables 7 and 8 and Fig. 5A). These three conformational spaces are placed in the North or South regions of the pseudorotation wheel where the above indicated unfavorable interactions are minimized (Fig. 5B). However, at least the 3E-like and 2E-like conformations (adopted by ribonucleosides) are not located in the region of action of the anomeric effect. It appears that in the case of ribonucleosides, the anomeric effect is weaker than the cumulative effects of the unfavorable ecliptic arrangement of the 2-OH and 3-OH groups and the 1,3-diaxial interactions of the nucleobase and the hydroxymethyl group. As a result, the 3E-like and 2E-like conformations become the most stable for ribonucleosides. In turn, the anomeric effect influences the conformational preferences of β-D-ribofuranosides (red dots). Their preferred E2-like conformation, compared to the 3E and 2E conformations of nucleosides (green dots), is clearly shifted towards the region where the anomeric effect is active. This is well illustrated by the O1–C1–O4–C4 torsion angle. The anomeric effect acts more efficiently the closer the angle reaches the value of 60° (gauche orientation). As shown by the crystal data of β-D-ribofuranosides without the 2,3-O-isopropylidene fused ring (Table 7), the transition from the 3T2 conformation, through the E2 conformation, to the 1T2 conformation decreases the O1–C1–O4–C4 torsion angle from −108.51°, via −91.98°, to −82.35°. In the case of a furanose ring, the latter value of the O1–C1–O4–C4 torsion angle is optimal for the action of the anomeric effect. Thus, the anomeric effect, stronger in the case of O-furanosides than in the case of N-furanosides, causes β-D-ribofuranosides to adopt the E2 conformation in a crystal lattice.
The action of the anomeric effect is clearly observed in the case of 2,3-O-isopropylidene-β-D-ribofuranosides. The fused 2,3-O-isopropylidene ring hinders the adoption of the 3T2 and 2T3 conformations and favors the conformation in which the ring oxygen atom is placed out of the plane formed by the remaining atoms. However, all 2,3-O-isopropylidene-β-D-ribofuranosides, those that we synthesized (1–10) and those found in the CCDC database (Table 7), adopt solely the E0-like conformation (orange dots, Fig. 5A) and not the 0E-like conformation. The probable reason for this preference is that the E0-like conformation allows the anomeric effect to take place (Fig. 5B). As shown by the crystallographic data (Table 7), in the E0 conformation, the O1–C1–O4–C4 torsion angle is in the range of 77.65–89.65° (85.26° on average), and in the 1T0 conformation, this torsion angle is in the range of 79.18–85.48° (83.00° on average). In the optimized structures, the O1–C1–O4–C4 torsion angle is in the range of 87.38–100.01° (90.78° on average) in the case of 1 (Table 2) and in the range of 85.65–87.12° (86.38° on average) in the case of 6 (Table 3). Such values of the torsion angles mean that the aglycones in these furanosides are oriented pseudoaxially and that the anomeric effect acts optimally in the furanose ring. Our calculations also show that this anomeric effect can act better in the E0/1T0 conformation than in the E0/4T0 conformation because the value of 86.38° is closer to the gauche orientation than the value of 90.78°. This clear preference for the E0-like conformation may indicate that in the case of O-furanosides, the favorable anomeric effect is stronger than the unfavorable 1,3 pseudodiaxial interactions between the aglycone and terminal group.
Regarding the conformational preferences, 2,3-O-ketal derivatives of nucleosides (blue and purple dots, Fig. 5A) behave completely different than their O-furanoside analogues. The 2,3-O-ketal protection group makes it difficult for nucleosides to adopt the 3E or 2E conformations that are most favorable for them. However, 2,3-O-ketalribonucleosides do not adopt the E0-like conformation, characteristic of the analogous β-D-ribofuranosides (orange dots, Fig. 5A). This means that the anomeric effect, in the case of ribonucleosides, is weaker than in the case of β-D-ribofuranosides and it cannot outweigh the unfavorable 1,3-pseudodiaxial interactions of the nucleobase and the terminal hydroxymethyl group. It is also possible that the latter interactions are stronger in the case of ribonucleosides than in the case of β-D-ribofuranosides. As a result, 2,3-O-ketal derivatives of ribonucleosides adopt various conformations in the crystal, which proves that none of them is especially privileged.
It is important to add that the three factors influencing the conformation of the furanose ring listed herein were also noticed in molecular dynamics studies.80
Apart from the endo-anomeric effect, an exo-anomeric effect can also act in the sugar ring. This is caused by the exocyclic oxygen atom from the aglycone and means that the gauche orientation is preferred over the antiperiplanar orientation in the O5–C1–O1–C7 (pyranose) or O4–C1–O1–C6 (furanose) atoms arrangement (Fig. 6D). The exo-anomeric effect does not directly influence the pyranose or the furanose ring conformation. It affects the arrangement of the aglycone carbon atom linked to the glycosidic oxygen atom (C7 in pyranose or C6 in furanose).
The action of the exo-anomeric effect is clearly visible in the β-D-ribofuranosides presented herein, both without and with the 2,3-O-isopropylidene fused ring. In the case of β-D-ribofuranosides without the 2,3-O-isopropylidene found in the CCDC database, the O4–C1–O1–C6 torsion angle (Table 7) is in the range of 44.82–85.27° (64.75° on average). In the case of β-D-ribo and β-D-allofuranosides with the 2,3-O-isopropylidene, this torsion angle is in the range of 61.82–76.65° (68.72° on average). In the case of β-D-ribofuranosides that we optimized, the O4–C1–O1–C6 torsion angle is in the range of 66.53–71.75° (69.65° on average) for 1 (Table 2) and in the range of 65.42–69.08° (67.80° on average) for 6 (Table 3). All these data clearly indicate that the O1–C1–O4–C4 atoms segment adopt the gauche conformation, which means that the exo-anomeric effect acts very well in the presented β-D-ribofuranosides.
4. Summary
Although the furanose ring is inherently labile, there are situations in which a given conformation of this ring is sufficiently stable to recognize it in the 1H NMR spectrum. These are the E0-like conformation adopted by 2,3-O-isopropylidene-β-D-ribofuranosides (1–10), 3E-like and 2E-like conformations adopted by the ribonucleosides (11–14), and the E4-like conformation adopted by 2,3-O-isopropylideneribonucleosides (15–18). The specific 1H NMR data for these four conformations of the furanose ring with the β-D-ribo configuration are presented. Importantly, this data, with regard to the ring proton coupling constants in the indicated conformations, also applies to furanoses with the β-D-allo, β-D-psico and α-L-talo configurations.The most characteristic feature of the presented spectra is the zero coupling between the trans-oriented vicinal protons. This takes place in the case of the H1 and H2 as well as the H3 and H4 pairs of protons in the 4T0/E0 conformation of 1–5 and in the case of the H1 and H2 protons in the E0/1T0 conformation of 6–10. Such a lack of coupling, possible solely for the trans-oriented protons in the furanose ring, indicates that the respective protons maximally approach each other to form the H–C–C–H torsion angle of about 90°. This is also an indication that the furanose ring adopts a specific conformation and does not exist in the conformational equilibrium. Arranged almost ecliptically, the cis-oriented H2 and H3 protons, with the 2,3-O-isopropylidene fused ring in 1–10 and 15–18, form the H2–C2–C3–H3 torsion angle in a range of 0–10°. The respective coupling constant is then in the range of 5.86–6.41 Hz. The same cis-oriented protons not restricted by the 2,3-O-isopropylidene ring can move apart to the torsion angle of about 40°. Then, the J2,3 coupling constant decreases, as in the case of 11–14, where it is in the range of 4.83–5.19 Hz.
The main factor influencing the conformational preferences of the β-D-ribofuranose ring is the unfavorable ecliptic interaction of the 2-OH and 3-OH groups. This is the strongest in the E0 (West side) and 0E (East side) conformations and the weakest in the 3T2 (North side) and 2T3 (South side) conformations. In the case of furanosides with the β-D-ribo configuration on the West side of the pseudorotational wheel, the unfavorable 1,3-pseudodiaxial interaction between the aglycone and terminal hydroxymethyl group also takes place. Not restricted by the 2,3-O-isopropylidene ring ribonucleosides, both 11–14 and those found in the CCDC database, adopt the 3E-like or 2E-like conformations, where these two unfavorable interactions are the most limited. The endo-anomeric effect is too weak to have an influence on the conformational preferences of ribonucleosides. However, the endo-anomeric effect is an important factor in the case of β-D-ribofuranosides. It acts both in β-D-ribofuranosides that are conformationally unrestricted (CCDC database) and restricted by the fused 2,3-O-isopropylidene ring (1–10 plus CCDC database). The endo-anomeric effect causes the former group of O-furanosides to adopt the E2-like conformation and the latter group of O-furanosides to adopt the E0-like conformation. Ribonucleosides with the 2,3-O-ketal fused ring, both 15–18 and those found in the CCDC database, do not adopt the E0-like conformation. This is the best evidence that in their cases, the anomeric effect is not a determinative factor influencing the conformational preferences. In the case of the presented O-furanosides, the action of the exo-anomeric effect is also proved.
Conflicts of interest
There are no conflicts to declare.References
- V. L. Damaraju, S. J. Damaraju, D. Young, S. A. Baldwin, J. Mackey, M. B. Sawyer and C. E. Cass, Oncogene, 2003, 22, 7524–7536 CrossRef CAS PubMed.
- J. Shelton, X. Lu, J. A. Hollenbaugh, J. H. Cho, F. Amblard and R. F. Schinazi, Chem. Rev., 2016, 116, 14379–14455 CrossRef CAS PubMed.
- M. Guinan, C. Benckendor, M. Smith and G. J. Miller, Molecules, 2020, 25, 2050 CrossRef CAS PubMed.
- S. S. Alneyadi, A. A. Abdulqader, A. A. Salem and I. M. Abdou, Heterocycl. Commun., 2017, 23, 197–203 CrossRef CAS.
- T. D. H. Bugg and R. V. Kerr, J. Antibiot., 2019, 72, 865–876 CrossRef CAS PubMed.
- J. M. Thomson and I. L. Lamont, Front. Microbiol., 2019, 10, 952 CrossRef.
- J. B. Shi, S. Xu, Y. P. Wang, J. J. Li and Q. Z. Yao, Chin. Chem. Lett., 2011, 22, 899–902 CrossRef CAS.
- K. L. Seley-Radtke and M. K. Yates, Antiviral Res., 2018, 154, 66–86 CrossRef CAS.
- M. Dasari, P. Ma, S. C. Pelly, S. K. Sharma and D. C. Liotta, Bioorg. Med. Chem. Lett., 2020, 30, 127539 CrossRef CAS PubMed.
- W. P. Painter, W. Holman, J. A. Bush, F. Almazedi, H. Malik, N. C. J. E. Eraut, M. J. Morin, L. J. Szewczyk and G. R. Painter, Antimicrob. Agents Chemother., 2021, 65, 024288 Search PubMed.
- R. M. Cox, J. D. Wolf and R. K. Plemper, Nat. Microbiol., 2021, 6, 11–18 CrossRef CAS PubMed.
- J. Hu, L. Yang, X. Cheng, Y. Li and Y. Cheng, Adv. Funct. Mater., 2021, 31, 2103718 CrossRef CAS.
- F. Kudo, M. Numakura, H. Tamegai, H. Yamamoto, T. Eguchi and K. Kakinuma, J. Antibiot., 2005, 58, 373–379 CrossRef CAS.
- B. T. T. Luyen, B. H. Tai, N. P. Thao, K. J. Eun, J. Y. Cha, M. J. Xin, Y. M. Lee and Y. H. Kim, Bioorg. Med. Chem. Lett., 2014, 24, 1895–1900 CrossRef CAS PubMed.
- J. O'Sullivan, M. Tedim Ferreira, J.-P. Gagné, A. K. Sharma, M. J. Hendzel, J.-Y. Masson and G. G. Poirier, Nat. Commun., 2019, 10, 1182 CrossRef.
- R. R. Sharipova, M. G. Belenok, I. Y. Strobykina and V. E. Kataev, Russ. J. Org. Chem., 2019, 55, 508–513 CrossRef CAS.
- T. Desai, J. Gigg and R. Gigg, Carbohydr. Res., 1996, 280, 209–221 CrossRef CAS.
- T. Hayashi, Y. Ohishi, H. Abe and M. Inouye, J. Org. Chem., 2020, 85, 1927–1934 CrossRef CAS PubMed.
- A. Das, S. Dasgupta and T. Pathak, Org. Biomol. Chem., 2020, 18, 6340–6356 RSC.
- R. K. Sharma, S. Singh, R. Tiwari, D. Mandal, C. E. Olsen, V. S. Parmar, K. Parang and A. K. Prasad, Bioorg. Med. Chem., 2012, 20, 6821–6830 CrossRef CAS PubMed.
- S. Schmalisch and R. Mahrwald, Org. Lett., 2013, 15, 5854–5857 CrossRef CAS.
- H. Ford, Jr, F. Dai, L. Mu, M. A. Siddiqui, M. C. Nicklaus, L. Anderson, V. E. Marquez and J. J. Barchi, Jr, Biochemistry, 2000, 39, 2581–2592 CrossRef.
- V. E. Marquez, T. Ben-Kasus, J. J. Barchi, K. M. Green, M. C. Nicklaus and R. Agbaria, J. Am. Chem. Soc., 2004, 126, 543–549 CrossRef CAS PubMed.
- M. Raab, S. Kozmon and I. Tvaroška, Carbohydr. Res., 2005, 340, 1051–1057 CrossRef CAS.
- I. A. Il’icheva, K. M. Polyakov and S. N. Mikhailov, Biomolecules, 2020, 10, 552 CrossRef PubMed.
- A. Istrate, M. Medvecky and C. J. Leumann, Org. Lett., 2015, 17, 1950–1953 CrossRef CAS PubMed.
- T. P. Prakash, Chem. Biodiversity, 2011, 8, 1616–1641 CrossRef CAS PubMed.
- H. Thomasen, M. Meldgaard, M. Freitag, M. Petersen, J. Wengel and P. Nielsen, Chem. Commun., 2002, 1888–1889 RSC.
- J. B. Houseknecht and T. L. Lowary, J. Org. Chem., 2002, 67, 4150–4164 CrossRef CAS PubMed.
- V. E. Marquez, S. H. Hughes, S. Sei and R. Agbaria, Antiviral Res., 2006, 71, 268–275 CrossRef CAS PubMed.
- M. Dejmek, M. Šála, H. Hřebabecký, M. Dračínský, E. Prochazkova, D. Chalupská, M. Klima, P. Plačková, M. Hájek, G. Andrei, L. Naesens, P. Leyssen, J. Neyts, J. Balzarini, E. Boura and R. Nencka, Bioorg. Med. Chem., 2015, 23, 184–191 CrossRef CAS.
- M. Evich, A. M. Spring-Connell and M. W. Germann, Heterocycl. Commun., 2017, 23, 155–165 CrossRef CAS.
- K. W. Pankiewicz, J. Krzeminski, L. A. Ciszewski, W.-Y. Ren and K. A. Watanabe, J. Org. Chem., 1992, 57, 553–559 CrossRef CAS.
- C. Altona and M. Sundaralingam, J. Am. Chem. Soc., 1972, 94, 8205–8212 CrossRef CAS PubMed.
- P. Bouř, I. Raich, J. Kaminský, R. Hrabal, J. Čejka and V. Sychrovský, J. Phys. Chem. A, 2004, 108, 6365–6372 CrossRef.
- J. J. Barchi, R. G. Karki, M. C. Nicklaus, M. A. Siddiqui, C. George, I. A. Mikhailopulo and V. E Marquez, J. Am. Chem. Soc., 2008, 130, 9048–9057 CrossRef CAS.
- H. A. Taha, M. R. Richards and T. L. Lowary, Chem. Rev., 2013, 113, 1851–1876 CrossRef CAS.
- A. G. Gerbst, V. B. Krylov, D. Z. Vinnitskiy, A. S. Dmitrenok, A. S. Shashkov and N. E. Nifantiev, Carbohydr. Res., 2015, 417, 1–10 CrossRef CAS PubMed.
- X. Wang and R. J. Woods, J. Biomol. NMR, 2016, 64, 291–305 CrossRef CAS.
- M. B. Patrascu, E. Malek-Adamian, M. J. Damha and N. Moitessier, J. Am. Chem. Soc., 2017, 139, 13620–13623 CrossRef PubMed.
- T. Taniguchi, K. Nakano, R. Baba and K. Monde, Org. Lett., 2017, 19, 404–407 CrossRef CAS PubMed.
- K. Nester, K. Gaweda and W. Plazinski, J. Chem. Theory Comput., 2019, 15, 1168–1186 CrossRef PubMed.
- W. Plazinski, M. U. Roslund, E. Säwén, O. Engström, P. Tähtinen and G. Widmalm, Org. Biomol. Chem., 2021, 19, 7190 RSC.
- B. Liberek, D. Tuwalska, I. do Santos-Zounon, A. Konitz, A. Sikorski and Z. Smiatacz, Carbohydr. Res., 2006, 341, 2275–2285 CrossRef CAS PubMed.
- D. Walczak, A. Nowacki, D. Trzybiński, J. Samaszko-Fiertek, H. Myszka, A. Sikorski and B. Liberek, Carbohydr. Res., 2017, 446–447, 85–92 CrossRef CAS.
- CrysAlis CCD and CrysAlis RED, Version 1.171.36.24, Oxford Diffraction Ltd, Yarnton, England, 2012 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- A. L. Spek, Acta Crystallogr., Sect. D: Struct. Biol., 2009, 65, 148–155 CrossRef CAS PubMed.
- C. K. Johnson, ORTEP II, Report ORNL-5138, Oak Ridge National Laboratory, OakRidge, TN, USA, 1976 Search PubMed.
- S. Motherwell and S. Clegg, PLUTO-78, Program for Drawing and Molecular Structure, University of Cambridge, UK, 1978 Search PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- G. Schaftenaar and J. H. Noordik, J. Comput.-Aided Mol. Des., 2000, 14, 123–134 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03, Revision D.01, Gaussian, Inc., Pittsburgh, PA, USA, 2013 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS.
- H. Günter, NMR Spectroscopy, Basic Principles, Concepts and Applications in Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2013, p. 129 Search PubMed.
- P. Écija, I. Uriarte, L. Spada, B. G. Davis, W. Caminati, F. J. Basterretxea, A. Lesarri and E. J. Cocinero, Chem. Commun., 2016, 52, 6241–6244 RSC.
- D. Tuwalska, A. Sikorski and B. Liberek, Carbohydr. Res., 2008, 343, 404–411 CrossRef CAS PubMed.
- A. Nowacki, D. Walczak and B. Liberek, Carbohydr. Res., 2012, 352, 177–185 CrossRef CAS.
- A. Nowacki and B. Liberek, Carbohydr. Res., 2013, 371, 1–7 CrossRef CAS PubMed.
- M. Satyanarayana, M. A. Viswamitra and P. Ramakrishnan, Curr. Sci., 1976, 45, 826–827 CAS.
- S. K. Katti, T. P. Seshadri and M. A. Viswamitra, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1981, 37, 407–410 CrossRef.
- D. G. Evans and J. C. A. Boeyens, Acta Crystallogr., Sect. B: Struct. Sci., 1989, 45, 581–590 CrossRef.
- W. Saenger, Principles of Nucleic Acid Structure, Springer-Verlag, New York, 1984, p. 19 Search PubMed.
- D. Cremer and J. A. A. Pople, J. Am. Chem. Soc., 1975, 97, 1354–1358 CrossRef CAS.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- S. T. Rao, E. Westhof and M. Sundaralingam, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1981, 37, 421–425 CrossRef.
- C. Altona, H. J. Geise and C. Romers, Tetrahedron, 1968, 24, 13–32 CrossRef CAS.
- A. S. Serianni and R. Barker, J. Org. Chem., 1984, 49, 3292–3300 CrossRef CAS.
- P. H. Wang and S.-S. Lee, J. Chin. Chem. Soc., 1997, 44, 145–149 CrossRef CAS.
- J. Plavec, W. Tong and J. Chattopadhyaya, J. Am. Chem. Soc., 1993, 115, 9734–9746 CrossRef CAS.
- C. Altona and M. Sundaralingam, J. Am. Chem. Soc., 1973, 95, 2333–2344 CrossRef CAS PubMed.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389–397 CrossRef PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- S. Wolfe, Acc. Chem. Res., 1972, 5, 102–111 CrossRef CAS.
- F. D. D'Souza, J. D. Ayers, P. R. McCarren and T. L. Lowary, J. Am. Chem. Soc., 2000, 122, 1251–1260 CrossRef.
- E. Juaristi and G. Cuevas, Tetrahedron, 1992, 48, 5019–5087 CAS.
- I. V. Alabugin, L. Kuhn, M. G. Medvedev, N. V. Krivoshchapov, V. A. Vil’, I. A. Yaremenko, P. Mehaffy, M. Yarie, A. O. Terent’ev and M. A. Zolfigol, Chem. Soc. Rev., 2021, 50, 10253 RSC.
- K. Gaweda and W. Plazinski, Eur. J. Org. Chem., 2020, 674–679 CrossRef CAS.
- K. Nester and W. Plazinski, J. Biomol. Struct. Dyn., 2020, 38, 3359–3370 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2164567. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ra04274f |
| This journal is © The Royal Society of Chemistry 2022 |