
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural diversity of CuZn2InSe4 quaternary chalcogenides: electronic and phonon properties from first principles†
Long Ma a,
Wencong Shi*b and
Lilia M. Woods*a
a,
Wencong Shi*b and
Lilia M. Woods*a
aDepartment of Physics, University of South Florida, Tampa, Florida 33620, USA. E-mail: lmwoods@usf.edu
bSchool of Biological Sciences, Nanyang Technological University, 60 Nanyang Drive. Singapore 637551, Singapore. E-mail: wencong.shi@ntu.edu.sg
First published on 20th September 2022
Abstract
First principles simulations are utilized to calculate the electronic and vibrational properties of several metastable structural phases of the CuZn2InSe4 quaternary chalcogenide, including stanite, kesterite, primitive mixed CuAu, wurtzite-stanite, and wurtzite-kesterite lattices. We find that although each phase is formed by nearest cation-chalcogen bonds, the structural diversity due to cation and polyhedral arrangements has direct consequences in the electronic structure. The simulations further indicate that hybrid functionals are needed to account for the s–p and p–d orbital hybridization that is found around the Fermi level, which leads to much enhanced energy band gaps when compared with standard exchange-correlation approaches. We also find that the thermal conductivities for all phases are relatively low, and the main scattering channel comes from a low frequency optical band hybridized with acoustic phonons. Given that CuZn2InSe4 is a material from a larger class of quaternary chalcogenides, other materials may exhibit similar electronic and vibrational properties, which may be useful for electronic and thermal management applications.
Introduction
Multinary chalcogenides are diverse materials that can accommodate different types of metallic atoms in their structures. Quaternary chalcogenides can exist in a variety of structural phases with tunable properties, which makes them of great interest in many applications, including thermoelectricity,1 superconductivity,2 and nonlinear optics3 among others. Recently, chalcogenide systems containing multiple atoms in their structures have also been suggested as high-entropy compounds for possible thermoelectric applications4,5 Many such materials have a common origin associated with II–VI binary zinc blende structures, and recently a new array of compositions with the chemical formula I–II2–III–VI4 (I = Cu, Ag; II = Cd, Zn, Ca, Co, Fe, V; III = In, Ga, Al, Tl; VI = S, Se, Te) have been synthesized. These new systems have been realized in the laboratory showing that the electric properties can be tuned by stoichiometry variations or doping.6–11Recent experiments have shown that CuZn2InSe4, CuZn2InTe4, CuCd2InTe4, CuMn2InTe4, and AgZn2InTe4 have low thermal conductivity.11–21 These results are consistent with the low measured thermal conductivity for related systems such as Cu2ZnSnS4 and Cu2CdSnSe4, which belong to the widely studied class I2–II–IV–VI4 (I = Cu, Ag; II = Cd, Zn, Ca, Co, Fe, V; IV = Si, Sn, Ge; VI = S, Se, Te).22,23 The close connection between I–II2–III–VI4 and I2–II–IV–VI4 is also evident in the similar structural phases these materials can have. Kesterite, stannite, modified cubic, wurtzite-kesterite, and wurtzite-stannite lattices are possible for both types of materials classes, as shown by experimental and computational studies.11,14–16,24,25
While most computational studies focus on I2–II–IV–VI4, recent investigations of I–II2–III–VI4 representatives report on high through-put calculations for one hundred and fifty structures and their basic electronic properties.14 Simulations for the vibrational and phonon characteristics of selected representatives are also available.26 Given the limited number of such studies, our basic understanding of this emerging class of systems needs further advancement, especially in the context of deeper knowledge of the structure–property relations and transport characteristics.
In this study, using first principles simulations we investigate CuZn2InSe4 and its structural and dynamical stability by simulating five atomic phases previously found for I–II2–III–VI4 materials.14,27 This is a recently synthesized material,28 which has not been investigated in detail from first principles. In-depth calculations for the electronic and vibrational properties together with the lattice thermal conductivity are also performed and analyzed. This investigation helps us develop a better picture for the effects of the phase diversity in a given composition from I–II2–III–VI4 systems, which can be used to translate to other similar materials. The comparison with prior results for Te-based I–II2–III–VI4 systems and other Se-based II2–I–IV–VI4 materials20,29 is especially useful to find commonalities and differences between these related classes.
Methodology
The structural and electronic calculations are based on Density Functional Theory (DFT)30 and are carried out with the Vienna Ab initio Simulations Package (VASP),31 a state-of-the-art code relying on periodic boundary conditions. The exchange-correlation energy is taken into account using the generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) functional.27 The convergence criteria for the atomic relaxation process are 10−8 eV (total energy criteria) and 10−7 eV Å−1 (force criteria), and the energy cut-off is taken at 295.45 eV for all cases. To address the partial occupancies for each orbital, we employed Gaussian smearing method with smearing width of 0.05 eV. We also choose the tetrahedron integration method and Blöchl corrections for the relaxation of the structures with a 12 × 12 × 12 k-mesh.32 The energy band structure and DOS are calculated within the Heyd–Scuseria–Ernzerhof (HSE06) screened Coulomb hybrid density functional.33 This hybrid functional is regarded as a superior method especially for obtaining more accurate energy gaps in semiconductors, in which prominent s–p and p–d hybridization exists. The HSE06 calculations were facilitated by the VASPkit software34 with an optimized 5 × 5 × 2 k and q grids for the KS, PMCA, and ST phases, while 4 × 4 × 4 was used for the WST and WKS k and q grids.The unit cell for each phase is shown in Fig. 1 and it is constructed by following the initial symmetry as described in what follows. After relaxation, the resultant structures are further used to calculate the phonon band structures, phonon density of states (PDOS), and Grüneisen parameters for each CuZn2InSe4 phase by using the PHONOPY code.35,36 The computations were performed based on supercell displacements with central differences within GGA-PBE, by setting energy cutoff values to be 1.3 times the largest default cutoff energy of the atomic pseudopotentials of the CuZn2InSe4 phase (384.08 eV), as recommended by the VASP developer. The atomic displacements are also taken at their default value of 0.03 Å. The phonon properties rely on calculations of the interatomic force constants (IFCs), whose harmonic and anharmonic contributions are obtained by second and third order derivatives of the total energies obtained from VASP in terms of the atomic displacements defined over finite translation groups.37 For the harmonic IFCs, we construct 3 × 3 × 3 supercells with 432 atoms for KS and ST, while the 4 × 4 × 2 supercell are used for the PMCA containing 512 atoms. For the WKS and WST, supercells of 2 × 2 × 4 with 256 atoms and a 3 × 3 × 4 with 576 atoms are constructed, respectively. The third-order anharmonic IFCs are further calculated using the PHONO3PY to find three-phonon interaction matrix elements.35,36
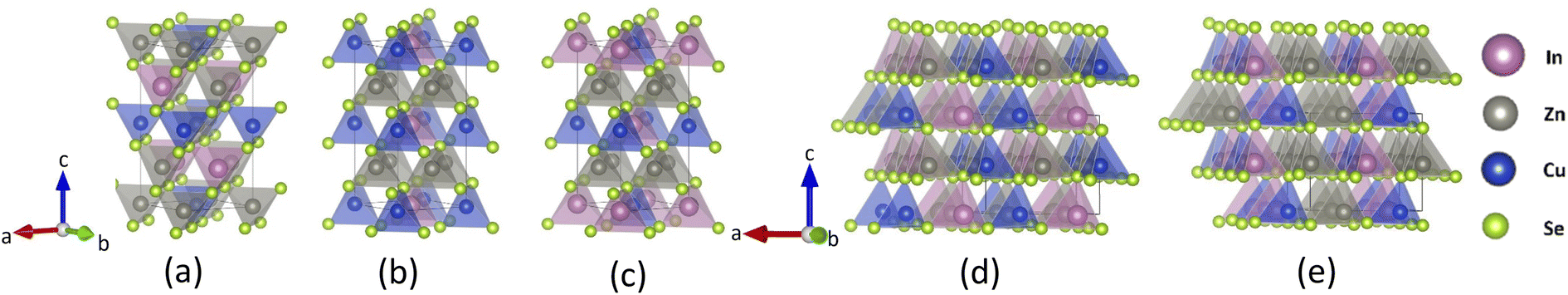 | ||
| Fig. 1 Crystal structures for CuZn2InSe4 with (a) kesterite (KS), (b) primitive mixed CuAu (PMCA), (c) stannite (ST), (d) wurtzite-kesterite (WKS), (e) wurtzite-stannite (WST) phases. | ||
The PHONO3PY code is also used to calculate the lattice thermal conductivity, whose tensor components in α, β directions are
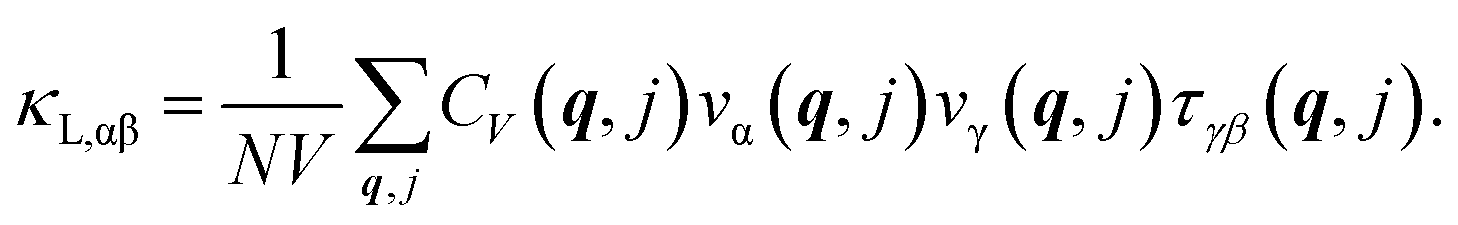
In the above expression, the summation is in the first Brillouin zone over phonon modes labeled by their wave vector q and polarization j. Also, N is the number of sampled points and V is the unit cell volume. The lattice thermal conductivity is determined by the heat capacity CV(q, j), group velocity components vα(q, j), and the transport lifetime tensor τ(q, j) of each vibration mode. Among those, the heat capacity is calculated as CV(q, j) = kBn(q, j)(n(q, j) + 1)(ℏω(q, j)/kBT)2 with ω(q, j) being the frequency and n(q, j) – the Bose–Einstein occupation number at equilibrium of the phonon mode (q, j). The group velocity components are defined as vα(q, j) ≡ ∂ω(q, j)/∂qα. Also, the repeating index α, β, γ in the above expression implies coordinates.
The PHONO3PY code obtains a full solution of the Peierls–Boltzmann transport equation, beyond the relaxation time approximation.38,39 The transport lifetime components follow from the Peierls–Boltzmann equation as they are obtained from the nonequilibrium distribution functions. These depend on transition scattering probabilities, which are found based on perturbation theory by including both normal and Umklapp processes.40 The phonon self-energies needed for the thermal conductivity calculations involve harmonic and anharmonic IFCs that can be obtained via the PHONOPY and PHONO3PY packages. We also consider the phonon-isotope scattering for isotopic mass variations in the atoms. The lattice thermal conductivity is then solved based on the phonon Boltzmann equation with the previously obtained IFCs on a 6 × 6 × 6 q-grid. The anharmonic cutoff-pair distances are taken as 6 Å since larger values do not affect the κL convergency significantly. Convergence tests for the lattice thermal conductivity are given in the ESI† (Fig. S3 and S4†) showing that the chosen size of the q-grid and the magnitude of the cutoff-pair distance result in a good numerical accuracy for the results to follow.
The mode Grüneisen parameter describes the anharmonicity in the lattice. Here it is calculated based on the anharmonic IFCs using the expression γ(q, j) = −(V/ω(q, j))(∂ω(q, j)/∂V), which involves numerical derivatives of the frequency for each phonon mode and can be obtained using central finite difference method that requires the value of ω(q, j) at the equilibrium volume and at slightly larger and smaller than the equilibrium volumes.41 Electron and phonon band structures and densities of states are obtained by using the VASPKIT package.34,35 The crystal structures are visualized with the VESTA software.42
Results and discussions
The quaternary I–II2–III–VI4 group of materials are derivatives of simpler, binary II–VI systems. They are obtained by cation substitution following the chain through binary and ternary compounds II–VI →–I–III–VI2 → I–II2–III–VI4. During the substitution process, charge neutrality and the octet rule are maintained for energetic stability of each new material.43,44 The I–II2–III–VI4 quaternary chalcogenides can be found in five metastable phases Kesterite, KS; primitive mixed CuAu, PMCA; Stannite, ST; wurtzite-kesterite, WKS and wurtzite-stannite, WST.45 To initiate the calculations, the unit cells of these phases are constructed following their associated symmetries as follows: I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) (KS), I2m (ST), P2m (PMCA), Pmn21 (WKS), and Pc (WST).45,46 The lattice parameters of each structural phase after relaxation are summarized in Table 1, showing a marginal difference from the originally prescribed symmetries (a common situation for many complex systems).
(KS), I2m (ST), P2m (PMCA), Pmn21 (WKS), and Pc (WST).45,46 The lattice parameters of each structural phase after relaxation are summarized in Table 1, showing a marginal difference from the originally prescribed symmetries (a common situation for many complex systems).
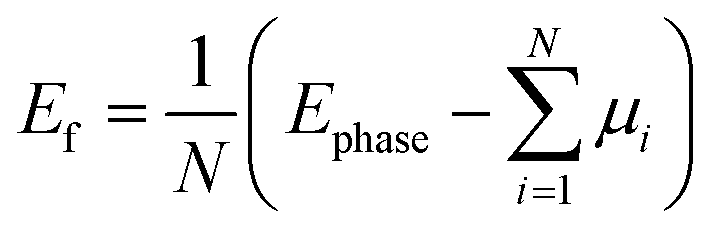 where Ephase is the total energy of the compound in a given structural phase, μi is the chemical potential of each constituent atom, and N is the number of atoms in the unit cells. The energy gaps EG, obtained via PBE and HSE06 approximations, are also given
where Ephase is the total energy of the compound in a given structural phase, μi is the chemical potential of each constituent atom, and N is the number of atoms in the unit cells. The energy gaps EG, obtained via PBE and HSE06 approximations, are also given
| Phases | a (Å) | b (Å) | c (Å) | α (°) | β (°) | γ (°) | ΔE (eV) | Ef (eV) | EPBEG (eV) | EHSEG (eV) |
|---|---|---|---|---|---|---|---|---|---|---|
| KS | 5.517 | 5.517 | 10.998 | 90.000 | 90.000 | 90.000 | 0.000 | −0.523 | 0.756 | 1.887 |
| PMCA | 5.508 | 5.508 | 11.044 | 90.000 | 90.000 | 90.000 | 0.022 | −0.521 | 0.691 | 1.800 |
| ST | 5.506 | 5.506 | 11.052 | 90.000 | 90.000 | 90.000 | 0.001 | −0.523 | 0.816 | 1.932 |
| WKS | 7.785 | 7.789 | 6.402 | 90.079 | 89.961 | 120.020 | 0.206 | −0.510 | 0.449 | 1.538 |
| WST | 7.783 | 7.778 | 6.398 | 90.000 | 90.000 | 119.980 | 0.103 | −0.516 | 0.757 | 1.862 |
The atomic structures for the CuZn2InSe4 quaternary chalcogenide phases are displayed in Fig. 1, where explicit polyhedral arrangements are also given. It shows that the KS and ST lattices have tetragonal symmetry, while the PMCA is very close to an orthorhombic space group, and the wurtzites are hexagonal structures. The lattice parameters for the KS, PMCA, and ST phases share the relation a = b ≈ c/2, while for the WKS and WST, we find that a ≈ b > c. Experimentally, CuZn2InSe4 was recently reported28 in a PMCA-like lattice structure with a = 5.7316 Å, which is about 4% greater than the computational values. Having the lattice parameters, the cell volume for the wurtzites is found as Vwurtzite = a × b × c ≈ 389 Å3, while Vtetra = a × b × c ≈ 333 Å3 for the tetragonal-like symmetries. Given that the atomic volumes in the unit cells are the same for all the phases (Vatoms = 2VCu + 4VZn + 2VIn + 8VSe ≈ 143.9 Å3), the packing density of the KS, PMCA, and the ST phases is ρpacking = Vatoms/Vtetra = 0.43, which is greater than that of the WKS and WST phases ρpacking′ = Vatoms/VWurtzite = 0.37.
For all lattices, the atomic bonds always involve a cation and a Se atom. We further find that within each frame, each cation X (X = Cu, Zn, and In) is bonded with the four adjacent Se atoms to form tetrahedron structures [XSe4]. The distinct features between the different phases describe how the cations are distributed around the chalcogen atoms, which gives some diversity in the relative position of the faces and vertices of the tetrahedra. For instance, Fig. 1a–c, and e illustrate that KS, PMCA, ST, and WST have staggered patterns of [CuSe4] tetrahedra. Specifically, along the [201] direction which covers the adjacent tetrahedra of [CuSe4] in (001) planes, the KS and ST phases line up tetrahedra centralized by different types of cations, such that KS obtains the sequence of [ZnSe4] − [InSe4] − [CuSe4] while ST holds the [InSe4] − [ZnSe4] − [CuSe4] pattern. Similarly, the arrangement in PMCA transforms to [CuSe4] − [ZnSe4] − [CuSe4] with no [CuSe4] tetrahedra touching each other. In contrast, the [CuSe4] tetrahedra in WKS phase share one vertex Se atom along [001] direction with other [CuSe4] tetrahedra, showing a different alternation of the stacking pattern.
The different phases are also studied by examining the total energy difference ΔE and the formation energy per atom Ef. Taking the KS as a reference, we find that the ST phase has practically the same energetic stability. On the other hand, ΔE = 206 meV indicates that the WKS has the least energetic stability. The formation energies of the phases render additional information of how difficult it is to synthesize single crystal CuZn2InSe4 in its different structural phases. We find that Ef for the KS, PMCA, and ST phases is practically the same indicating that these three structures are metastable. The fact that their formation energy values are more negative than the WKS and WST structures shows that the KS, PMCA, and ST lattices are easier to synthesize.
The electronic structure is considered next by analyzing the calculated electronic band structure and density of states (DOS) for each phase, shown in Fig. 2. Standard DFT approximations (GGA and Local-density approximations (LDA) and their variations) are inadequate in finding the energy band gap in many semiconductors that have strong s–p or p–d orbital hybridization around the Fermi level.47–49 Zinc blende derived quaternary chalcogenides fall in that category, thus we perform calculations with the HSE06 hybrid functional (also discussed in Methodology). The band gaps obtained by the standard PBE-GGA approximation are typically 2.5 to 3 times smaller than those found via HSE06 as reported in Table 1. We find that all phases are large gap semiconductors with EHSEG being the largest for the ST phase and smallest for the WKS phase. This is commensurate with the band gap ranges (1.2–1.8 eV) of quaternary compounds from the related I2–II–IV–VI4 family of compounds.50,51
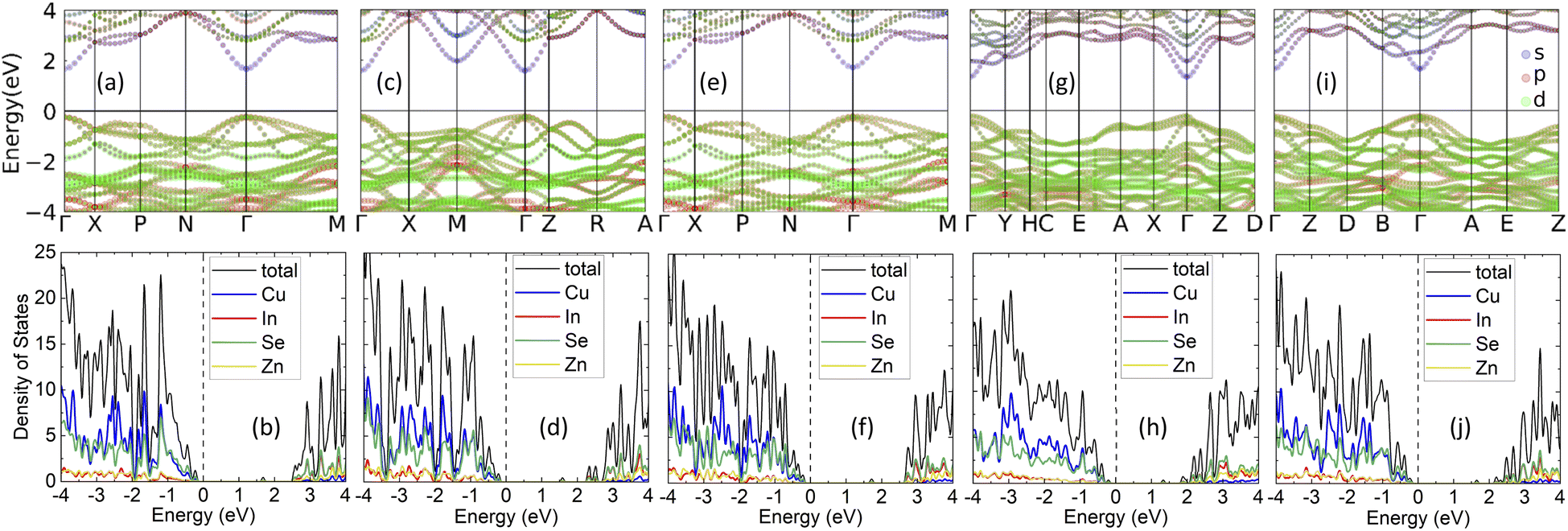 | ||
| Fig. 2 Orbitally projected band structure and atom projected density of states of the CuZn2InSe4 phases: (a) and (b) KS; (c) and (d) PMCA; (e) and (f) ST; (g) and (h) WKS, and (i) and (j) WST. All results are obtained using the HSE06 hybrid functional. | ||
The energy bands, shown in Fig. 2, indicate that all the CuZn2InSe4 phases possess a direct energy gap at the Γ-point. Since the Fermi level is almost at the top of the highest valence band, each phase is a p-type semiconductor. Besides, all phases exhibit one CBM at the Γ point in the Brillouin zone, however, secondary conduction minima are found: for the PMCA at the M-point (higher by 0.39 eV than its peak at Γ), for the WKS at the Y-point (higher by 0.59 eV than its Γ peak), and for the WST at the B-point (higher by 0.61 eV than its Γ peak). These are commensurate with DOS peaks in the conduction band region and are the result of strong s–p orbital hybridization. The conduction regions in the orbitally projected bands and the DOS given in Fig. 2 indicate that the CBM edges mainly contain In-5s and Se-4p orbitals, and that the DOS above the gap is essentially contributed by the Indium and Selenium atoms. The valence bands of all phases mainly contain Se-4p and Cu-3d orbitals in the (−2, 0) eV region as illustrated in Fig. 2. This p–d orbital hybridization not only accounts for the dispersion behaviors at the top of the valence band, but it also shapes the band curves around valence band maximum (VBM) edges. Another perspective of the s–p–d hybridization can be found in Fig. S1 in the ESI,† where the atomic orbitally projected DOS for the Cu, In, and Se atoms is given. The semiconducting behavior of CuZn2InSe4 phases can also be seen in the calculated conductivity at T = 300 K as a function of the chemical potential shown in Fig. S2† (details in the ESI†).
The calculated band structure can also be used to obtain the band effective masses m* for the different phases by using 1/m* = (1/ℏ2)(∂2E/∂ki∂kj) where E is the band energy around the CBM and VBM edges and ki,j are the components of the wave vector k. In Table 2, we show characteristic m* for the different phases (in terms of the electron mass m0) along some directions in the Brillouin zone. Our results show that the effective masses of the holes are larger than the ones of the electrons in magnitude along the same direction indicating greater mobilities for the electrons. For each phase, the effective band masses have similar values in different directions around CBM, while larger variations are found around the VBM, especially for PMCA and WKS. Comparing with the values mh* = 0.6–0.8 m0 of the explored quaternary chalcogenide compounds reported by others,28 the effective masses of the KS and ST phases fall in that range while the PMCA and the wurtzite phases are greater than that benchmark.
| Phase | Direction | me* (m0) | mh* (m0) | vTA1 (m s−1) | vTA2 (m s−1) | vLA (m s−1) |
|---|---|---|---|---|---|---|
| KS | Γ → N | 0.121 | 0.607 | 2284 | 2287 | 3785 |
| KS | Γ → M | 0.130 | 0.910 | 2092 | 2132 | 3998 |
| PMCA | Γ → M | 0.131 | 3.064 | 2279 | 2510 | 4157 |
| PMCA | Γ → Z | 0.125 | 0.560 | 2519 | 2519 | 4022 |
| ST | Γ → N | 0.117 | 0.605 | 2287 | 2254 | 3754 |
| ST | Γ → M | 0.123 | 0.630 | 1952 | 2240 | 4059 |
| WKS | Γ → X | 0.137 | 1.111 | 2135 | 1859 | 4030 |
| WKS | Γ → Z | 0.139 | 0.195 | 1841 | 1850 | 4029 |
| WST | Γ → B | 0.127 | 1.457 | 1809 | 1826 | 4427 |
| WST | Γ → A | 0.127 | 1.391 | 1941 | 2452 | 4046 |
According to Pauling's theory, not only the sharing of the polyhedron elements but also the coordinate number of the polyhedrons influence the lattice stabilities and the electronic properties.43 Therefore, some of the band structure features can be linked with the atomic polyhedral arrangements (Fig. 1), which are typically associated with band edge shifting. Especially, in quaternary chalcogenide compounds such as the CuZn2InSe4, the valence band shifting is mainly attributed to the arrangement of [CuSe4] tetrahedra, while the conduction band shifting comes from the [InSe4] tetrahedra.26,51 For example, given that two nearest [InSe4] tetrahedra in all phases are separated by the similar distance (d ≈ 5.5 Å), the sharing corners of [CuSe4] tetrahedra in WKS result in closer distance between the Cu atoms. This leads to enhanced Coulomb interaction and reduced lattice stability, as also indicated by the WKS smallest total energy, as discussed earlier. As a result, the VBM of WKS phase is pushed up giving a reduced band gap. For the other phases, the separation distance of the nearest [CuSe4] tetrahedra are almost the same (d ≈ 5.5 Å). The staggered patterns of [CuSe4] tetrahedra along [201] shows no touching between adjacent [CuSe4] tetrahedra. However, since the coordination number of [CuSe4] tetrahedra is 6 for the PMCA and WST phases, which is larger than that of the remaining phases (coordination number is 4), the band gaps of PMCA and WST are relatively smaller than KS and ST phases.
Further insight into the type of bonding in each phase can be gained by the calculated Electron Localization Function (ELF), with ELF ∼ 1 representing strong localization and ELF ∼ 0.5 corresponding to an electron gas.52 Fig. 3 shows calculations for ELF with explicitly given projection planes in each case. For all structural phases, the cations are surrounded by the chalcogens within the [XSe4] tetrahedra. The alignment patterns of [XSe4] are also presented by showing the (102) plane in KS, PMCA, and ST phases and (001) plane in WKS and WST phases. Fig. 3 shows that the Cu–Se, Zn–Se bonds exhibit localized electrons around the Se atom, which is indicative of ionic-like type of bonding. For the In–Se bond, on the other hand, the rather large degree of localization in the region between the In and Se atoms shows a covalent-like bonding. The four adjacent ELF basins of Se surrounding the In atom constitute the sp3 hybridization in [InSe4] tetrahedra. This is also consistent with features of the orbitally projected band structure which indicates the conduction band minimum (CBM) edge is mainly composed of In-5s and Se-4p orbitals.
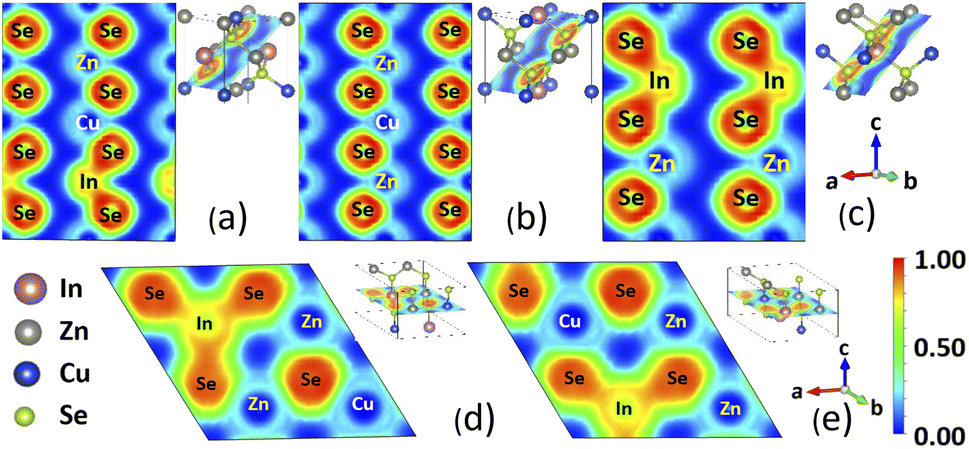 | ||
| Fig. 3 Electron localization function for (a) KS, (b) PMCA, (c) ST, (d) WKS, and (e) WST phases of CuZn2InSe4 compounds. The different projections are shown at the right top corner next to each panel. The atomic composition and the ELF color scale are also displayed. | ||
In addition to the electronic band structure, the vibrational properties are also calculated from first principles, as discussed in Methodology. The results for the phonon band structure and PDOS for the considered CuZn2InSe4 structures are given in Fig. 4. The absence of negative branches in the phonon frequency dispersions indicates that all phases are dynamically stable. In all cases, a dense band of low optical phonons is found in the (1, 2.5) THz region. These types of relatively flat low frequency optical phonons have been obtained for other quaternary chalcogenides in their ST and KS phases, such as the case for Se-based I2–II–IV–VI4 representatives17,18 and Te-based materials from I2–II–IV–VI4 and I–II2–III–VI4 classes.20 The atomically resolved PDOS further show how specific atoms contribute to the phonon dynamics.
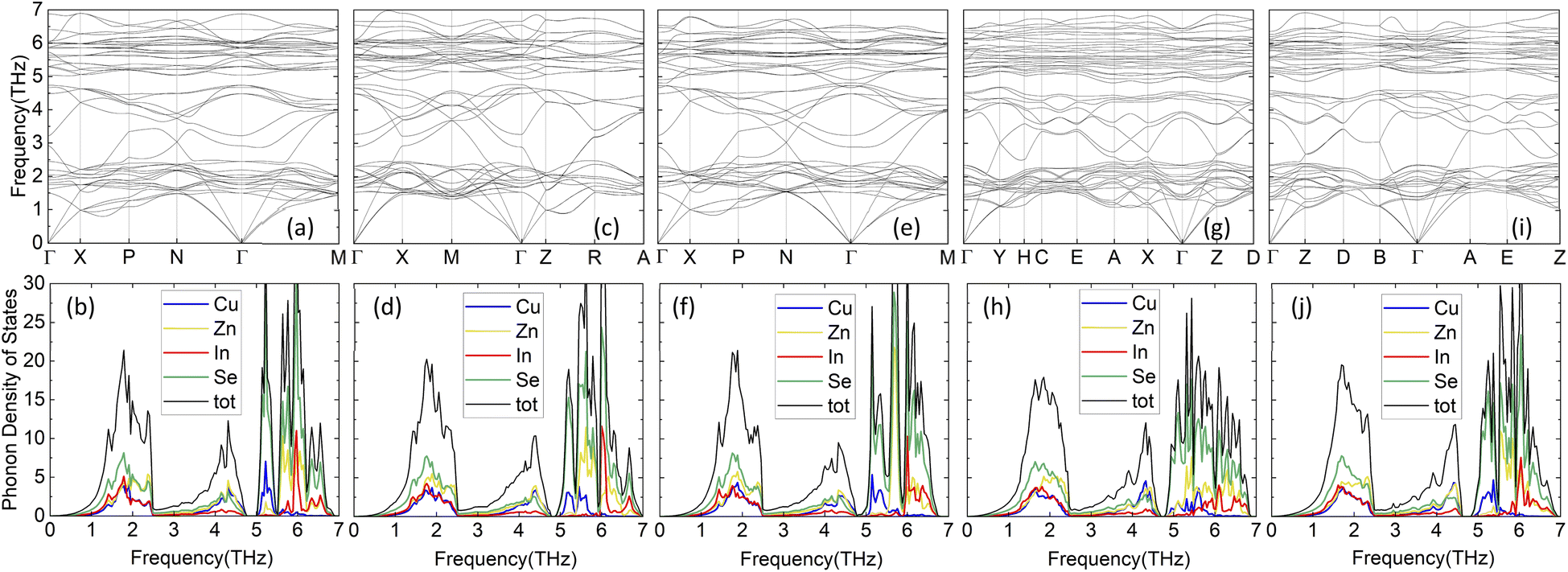 | ||
| Fig. 4 Phonon dispersions and atomically resolved phonon density of states for the considered CuZn2InSe4 phases for CuZn2InSe4: (a) and (b) KS; (c) (d) PMCA; (e) and (f) ST; (g) and (h) WKS, and (i) and (j) WST. | ||
The peak in the centered around ∼1.8 THz comes from vibrations of the Se and all cations, re-enforcing the direct cation-chalcogen bonding in all phases. This feature is associated with the relatively flat dense low-frequency optical phonon bands from the band structure. In the range (2.5, 5) THz, a relatively dispersed phonon band structures stands out.
The PDOS atomic decomposition shows that this frequency range is predominately determined by vibrations involving the Cu, Zn and chalcogen atoms, while the In contributions are rather less important. All phases exhibit a gap at ∼4.8 THz, followed by a frequency range (5, 7) THz primarily again composed of contributions from all atoms. Distinct peaks from Cu–Se at ∼5.3 THz and from In–Se at ∼6.2 THz in that region are found for KS, PMCA, ST, and WST phases. It appears that all structural phases have similar phonon properties with an especially low frequency optical phonon bands but with varying spreads. Among all the phases, the largest spread of this band is WST (1.30–2.35 THz) while the narrowest spread of the band goes to PMCA (1.52–2.32 THz).
From the phonon band structure, we can also find the sound velocities for the transverse acoustic (TA) and longitudinal acoustic (LA) branches around Γ point. Calculated group velocities for the TA and LA branches along certain directions in the Brillouin zone are summarized in Table 2. From these data, vTA = 2519 m s−1 (PMCA phase) is the largest, while vTA = 1809 m s−1 (WST phase) is the smallest. Similarly, the range of the LA group velocities are set by the lower limit of 3754 m s−1 for ST and the upper limit of 4427 m s−1 for WST. We note that the phonon velocities for all CuZn2InSe4 phases compare well with the ones of typical thermoelectric materials, including Bi2Te3 with vTA = 1870 m s−1, vLA = 3390 m s−1 and PbTe with vTA = 1210 m s−1, vLA = 3570 m s−1.21,53
Another important property for the phonon transport is the degree of anharmonicity of the atomic bonds in each lattice. For this purpose, we calculate the Grüneisen parameter γ(q, j) for each mode transition via PHONOPY and the results are shown in Fig. 5. For each CuZn2InSe4 structural phases most of the negative values in the low frequency regime are attributed to transverse acoustic mode scattering, while the higher frequency range with predominantly positive γ(q, j) come from optical phonon mode scattering. One finds a rather constant average γ(q, j) for the ω >3THz region with the KS phase (Fig. 5a) showing largest dispersion. In all cases, the average Grüneisen parameter is found to be 1.8 ± 0.2 for ω >3THz. For comparison, we note that for covalently bonded materials γ ≈ 1.0, while for ionic alkali halides (γ ≈ 2.2).54 The average Grüneisen parameters found here are consistent with mixed ionic-covalent hybridization, as discussed earlier (also, see Fig. 3). Additionally, the magnitude of the average γ in each phase is comparable with the values of other quaternary chalcogenides further re-enforcing the structural connection of these materials.20 The phonon scattering rates are also a measurement of the anharmonicity of the systems. Here these are calculated from three-phonon scattering processes within the relaxation time approximation Γ = 1/τRTA(q, j). The given scattering rate in Fig. 5 display significant similarities for all studied phases. There is a local minimum in the 1−2 THz range, which corresponds to the flat band of low frequency optical phonons (Fig. 4). The maximum ∼4 THz reflects transitioning to higher lying optical phonons followed by Γ around 5 THz, which reflects the corresponding gaps observed in the PDOS for the CuZn2InSe4 phases.
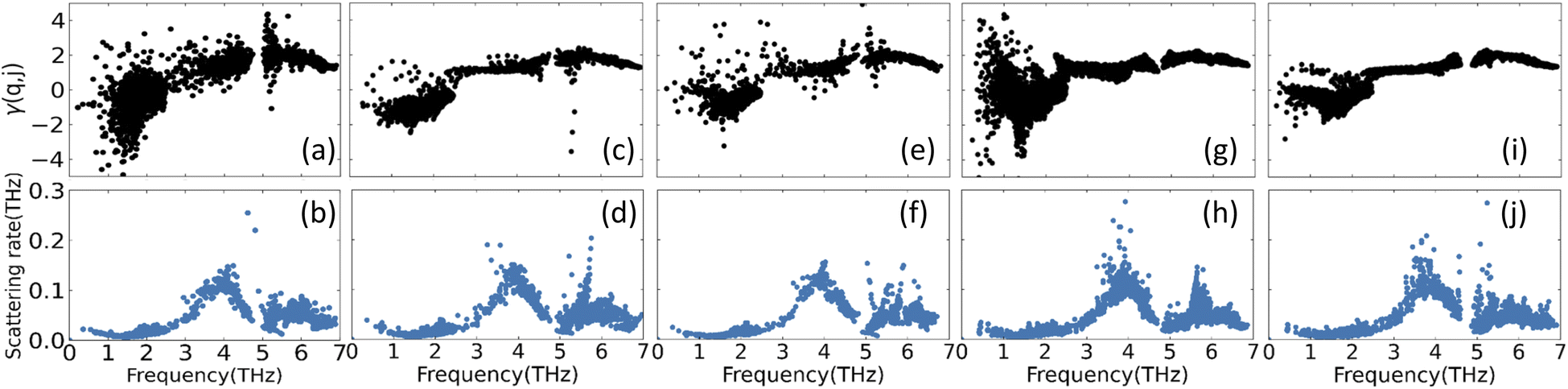 | ||
| Fig. 5 Mode Grüneisen parameters γ(q, j) and room-temperature phonon scattering rates of (a) and (b) KS; (c) and (d) PMCA; (e) and (f) ST; (g) and (h) WKS and (i) and (j) WST of CuZn2InSe4. | ||
The results for the calculated temperature dependent thermal conductivities are presented in Fig. 6. The thermal conduction is anisotropic for the studied latices. Examining the ratio |κxx − κzz|/κxx shows the largest anisotropic degree of 28% for the WST and smallest one of 6% for the ST phases at T = 300 K. It is further interesting to note that from the tetragonal structures, the smallest thermal conductivity overall is obtained for the PMCA lattice (κxx = 1.35, κzz = 1.20 W m−1 K−1 at T = 300 K). From the hexagonal lattices, this distinction belongs to the WKS phase (κxx = 1.24, κzz = 1.36 W m−1 K−1 at T = 300 K). On the other hand, κxx = 1.74, κzz = 1.63 W m−1 K−1 reflect the overall largest thermal conductivity found for the ST phase.
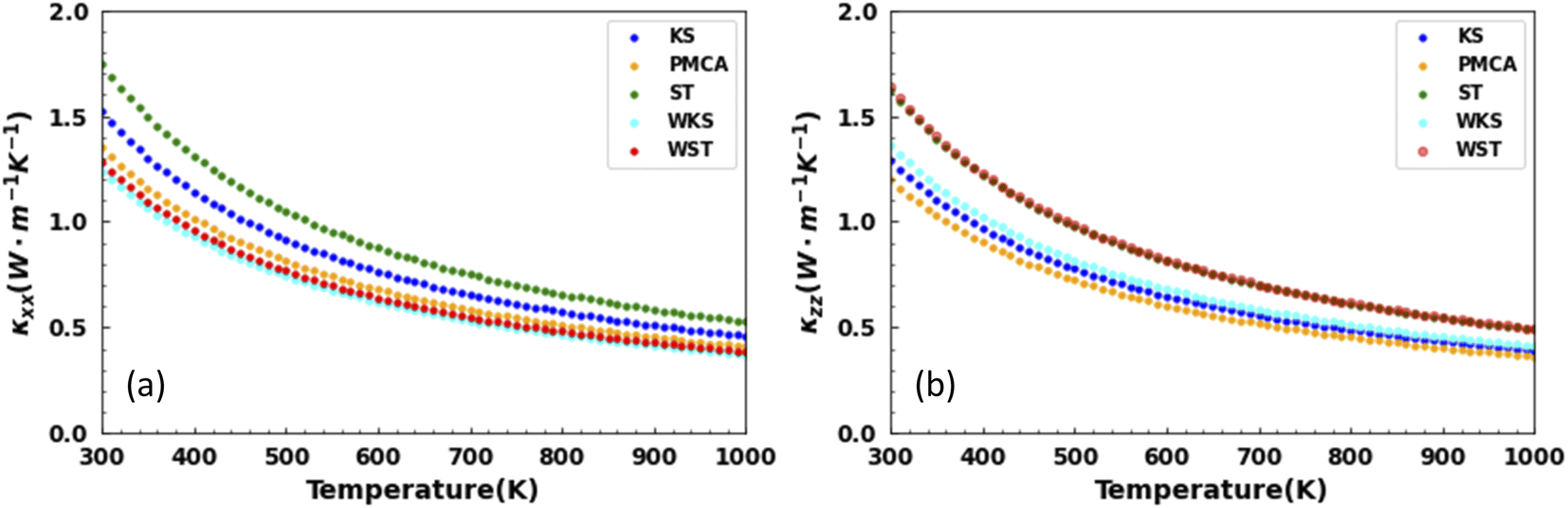 | ||
| Fig. 6 Lattice thermal conductivity components as a function of temperature along the (a) x-axis and (b) z-axis for the lattice phases of CuZn2InSe4. | ||
It is further instructive to compare our results to the thermal conductivity for other related quaternary chalcogenides. Although extensive transport measurements are scarce, existing data for AgZn2InTe4 (κ ∼1.5 W m−1 K−1) and Cu2ZnSnS4 (κ ∼1.69 W m−1 K−1) bulk compounds compare well with the values found for the CuZn2InSe4 phases. Notably, the thermal conductivity for CuZn2InTe4 (κ ∼3.3 W m−1 K−1), Cu2CdSnTe4 (κ ∼2.8 W m−1 K−1), and the Cu2ZnSnSe4 (κ ∼4.44 W m−1 K−1) exhibits higher values than the ones found here.11,17,55,56 It appears that the studied CuZn2InSe4 is characterized by rather low κ, even in the context of other chalcogenides.
More insight into the vibrational properties can be gained by examining the cumulative lattice thermal conductivity calculated as a function of frequency in the high temperature regime using 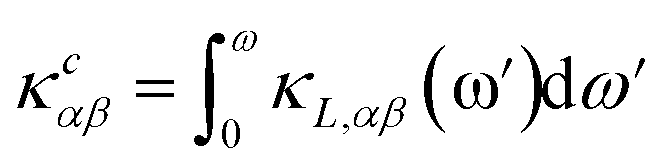 (κL,αβ – defined in Methodology). Fig. 7 shows κcxx and κczz and their derivatives with respect to frequency for all studied phases. The derivatives of κcxx and κczz indicate the density of the phonons that carry heat with respect to ω whose maxima is in the (1, 2) THz range for the different phases. This is also the range with very small scattering rates (Fig. 5) primarily attributed to acoustic phonons. The (2, 4) THz region corresponds to the low frequency optical phonons hybridized to the acoustic modes, whose heat carrying capacity decreases as seen is in the decreasing dκcxx,zz/dω as a function of ω. As the phonon DOS goes to zero (Fig. 4) at ω ∼4 THz the cumulative κcxx,zz reaches a plateau indicating the onset of higher optical phonons with much reduced heat capacity. This also corresponds to the behavior of scattering rates experiencing a maximum at that frequency followed by a gap, as seen in Fig. 5. While these are common features for all structural phases, the various peaks of the derivatives of κcxx and κczz show how different heat carrying phonons contribute to the lattice thermal conductivity for the considered CuZn2InSe4 phases. The cumulative thermal conductivity also gives another qualitative perspective of the anisotropy of the different CuZn2InSe4 structural phases.
(κL,αβ – defined in Methodology). Fig. 7 shows κcxx and κczz and their derivatives with respect to frequency for all studied phases. The derivatives of κcxx and κczz indicate the density of the phonons that carry heat with respect to ω whose maxima is in the (1, 2) THz range for the different phases. This is also the range with very small scattering rates (Fig. 5) primarily attributed to acoustic phonons. The (2, 4) THz region corresponds to the low frequency optical phonons hybridized to the acoustic modes, whose heat carrying capacity decreases as seen is in the decreasing dκcxx,zz/dω as a function of ω. As the phonon DOS goes to zero (Fig. 4) at ω ∼4 THz the cumulative κcxx,zz reaches a plateau indicating the onset of higher optical phonons with much reduced heat capacity. This also corresponds to the behavior of scattering rates experiencing a maximum at that frequency followed by a gap, as seen in Fig. 5. While these are common features for all structural phases, the various peaks of the derivatives of κcxx and κczz show how different heat carrying phonons contribute to the lattice thermal conductivity for the considered CuZn2InSe4 phases. The cumulative thermal conductivity also gives another qualitative perspective of the anisotropy of the different CuZn2InSe4 structural phases.
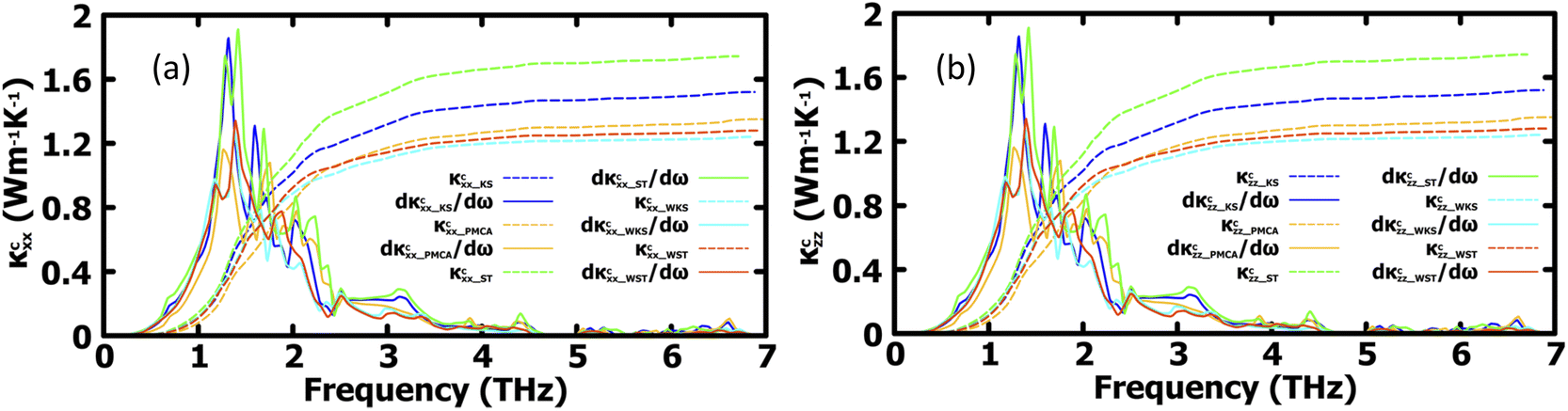 | ||
| Fig. 7 Cumulative lattice thermal conductivity components and the first derivatives as a function of phonon frequency at 300 K along the (a) x-axis and (b) z-axis for the lattice phases of CuZn2InSe4. | ||
Conclusions
The compound CuZn2InSe4 belongs to the I–II2–III–VI4 class of materials, which is similar to I2–II–IV–VI4 quaternary chalcogenides as both types of system are derivatives of the simpler binary II–VI zincblendes. The five structural phases for CuZn2InSe4 can also be found for other representatives of both I–II2–III–VI4 and I2–II–IV–VI4,13,45 although the dynamical stability, electronic and vibrational properties, and associated transport behaviors depend on the particular chemical composition. Here applying first-principles simulations we study in depth the ST, KS, PMCA, WKS, and WST CuZn2InSe4 for their structure–property relations. Drawing similarities with prior studies, the results reported here can serve as a representative example for other quaternary chalcogenides.The hybrid functional calculations yield 2–3 times larger energy gaps as compared with the standard DFT simulations. Experimental measurements report energy gap values consistent with the ones found via the HSE06 hybrid functional.33 This is not surprising since all chalcogenides from both I–II2–III–VI4 and I2–II–IV–VI4 experience strong s–p or p–d orbital hybridization around the Fermi level, which cannot be captured adequately via standard DFT approximations. Thus, in order to obtain electronic transport properties that are in potentially good agreement with experiments, DFT simulations beyond the GGA and LDA methods are needed. Nevertheless, the energy gaps found for CuZn2InSe4 are in the 1.5–1.9 eV range, which is comparable with the band gap ranges (1.2–1.8 eV) of the I2–II–IV–VI4 counterpart family from reports.50,51
Regardless of the chemical composition, for all structures each chalcogen is surrounded by metallic atoms, such that there are only direct metal-chalcogen bonds. At the same time, the specific arrangement of cations gives the specificity for each lattice, which can be seen in terms of polyhedral [XSe4] building block patterns. The shared corners and edges sequence can be related to stability of each lattice as well as conduction and valence band shifts and orbital hybridization which may be unique for each phase of the same material.
Other important results to be noted are the low thermal conductivity values for all CuZn2InSe4 phases, which are similar to several other I–II2–III–VI4 and I2–II–IV–VI4 representatives, as found experimentally and computationally.11,17,55,56 Usually, materials with complex lattices and atom constituents with highly dissimilar masses are characterized with low κ. However, the lattice structure of quaternary chalcogenides is relatively simple given its resemblance to the parent zinc blende, and the constituent atoms come from light mass elements. It appears that the cation-chalcogen direct bonds making up all structural phases may be key to the origin of their low κ. The PDOS and scattering rates show that in the low frequency regime all atoms contribute to the scattering. As a result, the hybridized optical-acoustic phonon band is an effective phonon scattering channel resulting in much reduced κ. We expect that due to the omnipresence of the direct cation-chalcogen bonds such a source of anharmonicity is present regardless of the chemical composition of the I–II2–III–VI4 and I2–II–IV–VI4 materials or their structural phases. Consequently, all such materials are expected to have low thermal conductivity properties.
Author contributions
L. M. and W. S. performed the simulations for all considered phases and their properties. L. M. W. conceived the idea, performed the analysis, and wrote the paper.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We acknowledge financial support from the U.S. National Science Foundation under Grant No. DMR-1748188. Computational resources were provided by USF Research Computing.References
- C. Han, Q. Sun, Z. Li and S. X. Dou, Adv. Energy Mater., 2016, 6, 1600498 CrossRef
.
- J. Ranninger, Z. Phys. B Condens. Matter, 1991, 84(2), 167–178 CrossRef CAS
- I. Chung and M. G. Kanatzidis, Chem. Mater., 2014, 26, 849–869 CrossRef CAS
- R.-Z. Zhang, F. Gucci, H. Zhu, K. Chen and M. J. Reece, Inorg. Chem., 2018, 57, 13027–13033 CrossRef CAS PubMed
- R. Liu, H. Chen, K. Zhao, Y. Qin, B. Jiang, T. Zhang, G. Sha, X. Shi, C. Uher, W. Zhang and L. Chen, Adv. Mater., 2017, 29, 1702712 CrossRef PubMed
- Y. Dong, H. Wang and G. S. Nolas, Phys. Status Solidi RRL, 2014, 8, 61–64 CrossRef CAS
- W. G. Zeier, Y. Pei, G. Pomrehn, T. Day, N. Heinz, C. P. Heinrich, G. J. Snyder and W. Tremel, J. Am. Chem. Soc., 2013, 135, 726–732 CrossRef CAS PubMed
- G. S. Nolas, H. Poddig, W. Shi, L. M. Woods, J. Martin and H. Wang, J. Solid State Chem., 2021, 297, 122058 CrossRef CAS
- G. E. Delgado, J. L. Villegas, P. Silva and V. Sagredo, Chalcogenide Lett., 2009, 6, 293–298 CAS
- D. Hobbis, W. Shi, A. Popescu, K. Wei, R. E. Baumbach, H. Wang, L. M. Woods and G. S. Nolas, Dalton Trans., 2020, 49, 2273–2279 RSC
- D. Hobbis, K. Wei, H. Wang and G. S. Nolas, J. Alloys Compd., 2018, 743, 543–546 CrossRef CAS
- D. Hobbis, W. Shi, A. Popescu, K. Wei, R. E. Baumbach, H. Wang, L. M. Woods and G. S. Nolas, Dalton Trans., 2020, 49, 2273 RSC
- W. Shi, A. R. Khabibullin, D. Hobbis, G. S. Nolas and L. M. Woods, J. Appl. Phys., 2019, 125, 155101 CrossRef
- W. Shi, A. R. Khabibullin and L. M. Woods, Adv. Theory and Simul., 2020, 3, 2000041 CrossRef CAS
- V. L. Bekenev, V. v. Bozhko, O. v. Parasyuk, G. E. Davydyuk, L. v. Bulatetska, A. O. Fedorchuk, I. v. Kityk and O. Y. Khyzhun, J. Electron Spectrosc. Relat. Phenom., 2012, 185, 559–566 CrossRef CAS
- M. v. Jyothirmai, J. Electron. Mater., 2020, 50(4), 1707–1712 CrossRef
- J. M. Skelton, A. J. Jackson, M. Dimitrievska, S. K. Wallace and A. Walsh, APL Mater., 2015, 3, 041102 CrossRef
- T. Gürel, C. Sevik and T. ÇaǧIn, Phys. Rev. B, 2011, 84, 205201 CrossRef
- A. Togo and I. Tanaka, Scr. Mater., 2015, 108, 1–5 CrossRef CAS
- W. Shi, T. Pandey, L. Lindsay and L. M. Woods, Phys. Rev. Mater., 2021, 5, 045401 CrossRef CAS
- X. Gao, M. Zhou, Y. Cheng and G. Ji, Philos. Mag., 2016, 96, 208–222 CrossRef CAS
- F. J. Fan, L. Wu and S. H. Yu, Energy Environ. Sci., 2013, 7, 190–208 RSC
- F. S. Liu, J. X. Zheng, M. J. Huang, L. P. He, W. Q. Ao, F. Pan and J. Q. Li, Sci. Rep., 2014, 4, 1–7 Search PubMed
- Y. Xu, Q. Fu, S. Lei, J. Xiong, S. Sun, Q. Bian, Y. Xiao and B. Cheng, Inorg Chem., 2019, 58, 15283–15290 CrossRef CAS
- A. Shavel, J. Arbiol and A. Cabot, J. Am. Chem. Soc., 2010, 132, 4514–4515 CrossRef CAS
- K. Pal, Y. Xia, J. Shen, J. He, Y. Luo, M. G. Kanatzidis and C. Wolverton, npj Comput. Mater., 2021, 7, 1–13 CrossRef
- J. Hafner, J. Comput. Chem., 2008, 29, 2044–2078 CrossRef CAS PubMed
- G. S. Nolas, M. S. Hassan, Y. Dong and J. Martin, J. Solid State Chem., 2016, 242, 50–54 CrossRef CAS
- C. Wang, S. Chen, J. H. Yang, L. Lang, H. J. Xiang, X. G. Gong, A. Walsh and S. H. Wei, Chem. Mater., 2014, 26, 3411–3417 CrossRef CAS
- R. O. Jones, Rev. Mod. Phys., 2015, 87, 897 CrossRef
- G. Kresse and J. Hafner, Phys. Rev. B, 1993, 47, 558 CrossRef CAS
- P. E. Blöchl, O. Jepsen and O. K. Andersen, Phys. Rev. B, 1994, 49, 16223 CrossRef
- B. G. Janesko, T. M. Henderson and G. E. Scuseria, Phys. Chem. Chem. Phys., 2009, 11, 443–454 RSC
- V. Wang, N. Xu, J. C. Liu, G. Tang and W. T. Geng, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS
- A. Togo, L. Chaput and I. Tanaka, Phys. Rev. B, 2015, 91, 094306 CrossRef
- L. Chaput, Phys. Rev. Lett., 2013, 110, 265506 CrossRef PubMed
- G. Deinzer, G. Birner and D. Strauch, Phys. Rev. B, 2003, 67, 144304 CrossRef
- L. Lindsay, Nanoscale Microscale Thermophys. Eng., 2016, 20, 67–84 CrossRef CAS
- OXFORD CLASSIC TEXTS IN THE PHYSICAL SCIENCES.
- Y. Wang, Z. Lu and X. Ruan, J. Appl. Phys., 2016, 119, 225109 CrossRef
- D. Cuffari and A. Bongiorno, Phys. Rev. Lett., 2020, 124, 215501 CrossRef CAS PubMed
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS
- L. Pauling, J. Am. Chem. Soc., 1929, 51, 1010–1026 CrossRef CAS
- R. J. Gillespie and E. A. Robinson, J. Comput. Chem., 2007, 28, 87–97 CrossRef CAS
- S. Chen, A. Walsh, Y. Luo, J. H. Yang, X. G. Gong and S. H. Wei, Phys. Rev. B, 2010, 82, 195203 CrossRef
- N. Kattan, B. Hou, D. J. Fermín and D. Cherns, Appl. Mater. Today, 2015, 1, 52–59 CrossRef
- S. Chen, X. G. Gong, A. Walsh and S. H. Wei, Phys. Rev. B, 2009, 79, 165211 CrossRef
- R. B. Wexler, G. S. Gautam and E. A. Carter, Phys. Rev. B, 2020, 102, 054101 CrossRef CAS
- S. Botti, D. Kammerlander and M. A. L. Marques, Appl. Phys. Lett., 2011, 98, 241915 CrossRef
- L. Weston and C. Stampfl, Phys. Rev. Mater., 2018, 2, 085407 CrossRef CAS
- A. Hong, Y. Tang and J. Liu, J. Phys. Chem. C, 2021, 125, 24796–24804 CrossRef CAS
- A. Savin, R. Nesper, S. Wengert and T. F. Fässler, Angew Chem. Int. Ed. Engl., 1997, 36, 1808–1832 CrossRef CAS
- Y. Amouyal, Materials, 2017, 10, 386 CrossRef PubMed
- W. F. Sherman, J. Phys. C: Solid State Phys., 1980, 13, 4601 CrossRef CAS
- G. S. Nolas, M. S. Hassan, Y. Dong and J. Martin, J. Solid State Chem., 2016, 242, 50–54 CrossRef CAS
- Y. Dong, A. R. Khabibullin, K. Wei, Z.-H. Ge, J. Martin, J. R. Salvador, L. M. Woods and G. S. Nolas, Appl. Phys. Lett., 2014, 104, 252107 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra04261d |
| This journal is © The Royal Society of Chemistry 2022 |