
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUllmann homocoupling of arenediazonium salts in a deep eutectic solvent. Synthetic and mechanistic aspects†
Giovanni Ghigo *a,
Matteo Bonomo*ab,
Achille Antenuccia,
Alessandro Daminab and
Stefano Dughera*a
*a,
Matteo Bonomo*ab,
Achille Antenuccia,
Alessandro Daminab and
Stefano Dughera*a
aDepartment of Chemistry, University of Turin, Via Pietro Giuria 7, 10125 Turin, Italy. E-mail: giovanni.ghigo@unito.it; matteo.bonomo@unito.it; stefano.dughera@unito.it
bNIS Interdepartmental Centre and INSTM Reference Centre, University of Turin, Via Gioacchino Quarello 15/a, 10125 Turin, Italy
First published on 20th September 2022
Abstract
A deep eutectic solvent (DES) based on glycerol and KF is successfully exploited as a solvent medium in Ullmann homocoupling of arenediazonium salts. The reactions were carried out in mild conditions and target products were obtained in fairly good yields. A computational study is presented aiming to understand the reaction mechanism and Raman spectroscopy is employed as an experimental tool to support it.
Introduction
C–C bond formation reactions has always been one of the most explored areas in organic chemistry and an ever-growing number of methods for C–C bond formation have been proposed.1 In fact, the development of improved methods for the efficient construction of C–C bonds is of tremendous interest for both academic and industrial worlds, as attested by Nobel Prize awards in chemistry.2 On these grounds, metal catalysed cross-coupling reactions stand among the most important protocols that allow C–C to be formed.3 In fact, they are highly valuable synthetic tool used for the preparation of a wide variety of organic compounds, from natural and synthetic bioactive compounds to new organic materials, in all fields of chemistry. The oldest (1901) of these metal-catalyzed coupling reactions is certainly the Ullmann reaction,4 an useful protocol for the formation of a new C–C bond between two aryls by the condensation of two molecules of an aromatic halide in the presence of copper. It must be stressed that biaryl compounds are one of the important scaffolds in the field of organic chemistry, and have been extensively applied to pharmaceuticals, functional materials, and agrochemicals.5 However, harsh reaction conditions (generally >200 °C), high copper catalyst loading, poor functional group tolerance and generally low yield of the products had drastically limited the applications of classical Ullmann reaction (Scheme 1). Interestingly, in the last few years with the increasing understanding of its mechanism,6 the use and development of different types of ligands and the application of the green technologies allowed the Ullmann reaction to be carried out under milder conditions with optimal yields and with excellent functional group tolerance.7 These allow broader applications of the Ullmann reaction, including copper-catalyzed nucleophilic aromatic substitution between various nucleophiles with aryl halides, known as Ulmann-type reactions (Scheme 1).8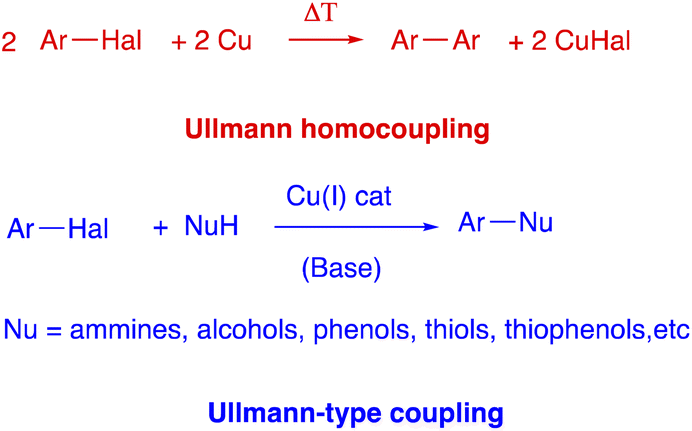 | ||
| Scheme 1 Ullmann homocoupling and Ullmann-type coupling. | ||
In particular, quite recently, Capriati and Perna proposed interesting and improved reinterpretations of Ullmann type reactions, reacting aryl halides with aliphatic amines or polyols in a deep eutectic solvent (DES), in mild conditions and without any ligands.9 It must be stressed that in recent years, the interest in DESs is growing since they are an environmentally benign and sustainable alternative to the conventional organic solvents and, with good reason, they have been defined as “the solvents of the century”.10
Aryl halides are generally the preferred electrophilic partners for Ullmann couplings; other types of compounds have rarely been used. In particular, arenediazonium salts, which in other coupling reactions prove to be a valid alternative to aryl halides,11a,b in Ullmann reactions have been employed sporadically.12 In fact, the literature shows only one significant example in which copper triflate (I) catalyzed homocoupling reactions of arenediazonium salts (bearing either electron donating and electron withdrawing groups) are reported (Scheme 2).12d
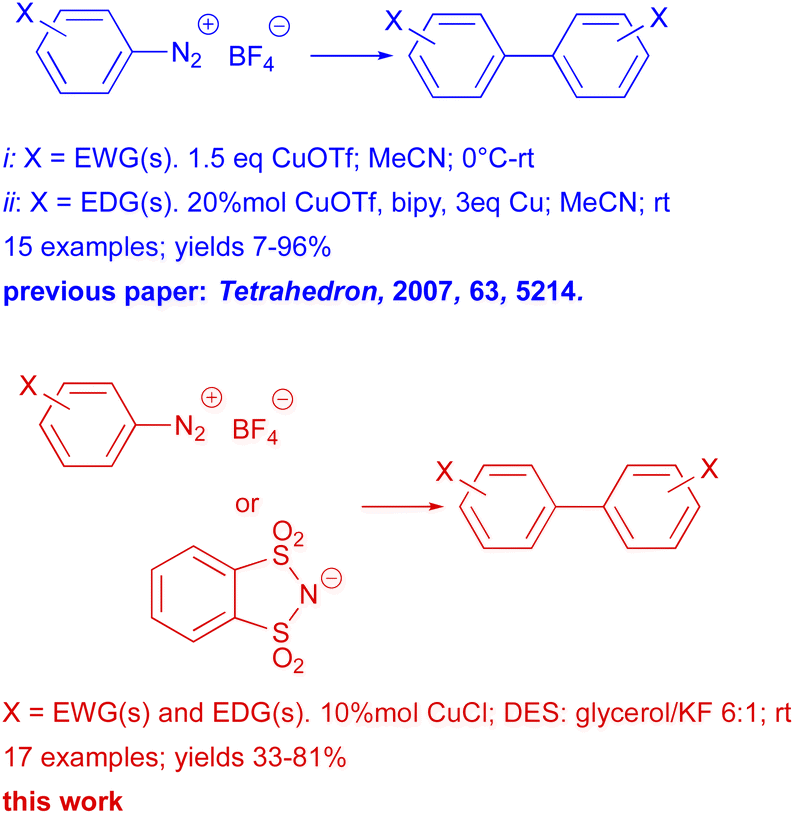 | ||
| Scheme 2 Ullmann homocoupling of arenediazonium salts. | ||
In our previous papers, we carried out a first complete study, both experimental and mechanistic, on the behavior of diazonium salts in deep eutectic solvents (DESs)13a or DESs like solvent system formed by glycerol and potassium halides.13b In fact, despite the increasing use of DESs in organic synthesis they have scarcely utilized in the reactions of diazonium salts. Moreover, it must be stressed that diazonium salt are resourceful building blocks in organic synthesis and the last few years have seen a dramatic resurgence of their chemistry.11c Therefore, in the light of these, in this work we present a thorough study on the Ullmann reactions of arenediazonium salts performed in DESs, in the presence of CuCl as catalyst (Scheme 2).
Results and discussion
Synthetic study
We decided to begin the study by reacting, as a model reaction, 4-nitrobenzenediazonium tetrafluoroborate (1a) in the presence of catalytic amounts (10%) of three different Cu(I) catalysts in several organic solvents and at different temperatures. The results are reported in Table 1. Only in DMSO at 40 °C (in the presence of CuCl, CuI and [(MeCN)4Cu]PF6 as catalysts) we obtained some amount of the target 4,4′-dinitrobiphenyl (2a; Table 1; entries 7,8,9). Few amount of 1-chloro-4-nitrobenzene (3a or 1-iodo-4-nitrobenzene 3b), deriving from collateral Sandmeyer reactions and nitrobenzene (4a) was also recovered (Table 1; entries 7 and 9). We try to add also a ligand such as 1,10-phenanthroline8b without significant increases in yield (Table 1; entry 10, note e). It must be stressed that also Cu(0) in powder, as an alternative to Cu(I), was able to catalyze the reaction, allowing to obtain the target 2a with a yield comparable to that obtained with CuCl, especially using Cu(0) in an amount of 50 mol% (Table 1; entry 11).
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Catalyst | Time (h) | T (°C) | Yields of 2aa (%) |
| a The reactions were carried out with 2 mmol of 1a and 10 mol% of Cu(I) catalysts. Yields refer to pure and isolated 4,4′-dinitrobiphenyl (2a).b The main product was nitrobenzene (4a).c GC-MS analyses of crude residues showed the presence of 2a, 1-chloro-4-nitrobenzene (3a) and 4a. The only isolated product was 2a.d GC-MS analyses of crude residue showed the presence of 2, 1-iodo-4-nitrobenzene (3a′) and 4a. The only isolated product was 2a.e Carrying out the reaction in the presence of 1,10-phenanthroline as a ligand, no significant increases in yield of 2a occurred.f Higher amounts of CuCl did not lead to significant increases in yields. On the contrary, the yields were lower using smaller amounts of CuCl (5%).g The reactions were carried out with 2 mmol of 1a and 50 mol% of Cu catalysts.h Only unreacted 1a was recovered.i The only product was 4a (86% yield).j The reaction was carried out under a nitrogen flow. The only product was 4a (72% yield).k Carrying out the reaction at 40 °C, no significant increases in yield of 2a occurred.l The reaction was carried out under a nitrogen flow. | |||||
| 1 | MeCN | CuCl | 8 | rt | —b |
| 2 | THF | CuCl | 8 | rt | —b |
| 3 | EtOH | CuCl | 8 | rt | —b |
| 4 | Gly | CuCl | 8 | rt | —b |
| 5 | DMSO | CuCl | 4 | rt | 15c |
| 6 | DMSO | CuI | 4 | rt | 12d |
| 7 | DMSO | CuCl | 2 | 40 | 41c |
| 8 | DMSO | [(MeCN)4Cu]PF6 | 2 | 40 | 40 |
| 9 | DMSO | CuI | 2 | 40 | 35d |
| 10 | DMSO | CuCle,f | 2 | 40 | 40c |
| 11 | DMSO | Cu | 3 | 40 | 32g |
| 12 | Gly/K2CO3 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
CuCl | 8 | rt | —h |
| 13 | Gly/Cs2CO3 6:1 |
CuCl | 8 | rt | —h |
| 14 | Gly/KF 6:1 |
— | 5 min | rt | —i |
| 15 | Gly/KF 6:1 |
— | 20 min | rt | —j |
| 16 | Gly/KF 6:1 |
CuCle,f | 1 | rt | 73c,k |
| 17 | Gly/KF 6:1 |
CuCl | 2 | rt | 62c,l |
| 18 | Gly/KF 6:1 |
CuI | 1 | rt | 68d |
| 19 | Gly/KF 10:1 |
CuCl | 1 | rt | 60c |
| 20 | Gly/KF 4:1 |
CuCl | 2 | rt | 45c |

On these basis, we decided, in the light our previous researches,13 to use DES as privileged solvent. Obviously, we have to choose a DES without halide salts as hydrogen bond acceptor to avoid Sandmeyer reactions. We tested DESs in which the hydrogen bond acceptor (HBA) is a salt (i.e. K2CO3 or Cs2CO3) lacking halide anions (Table 1; entries 12 and 13). Unfortunately, no reaction occurred due to the low solubility of the salt in these mixtures. In the light of these results, we resolved to use alkali fluorides as HBAs. In fact, it is well known that it is impossible to obtain aryl fluorides with Sandmeyer reaction. In our previous research, we found that, in this solvent system formed by glycerol and KF in 6:1 ratio, a fast hydrodediazotation reaction occurred, with the formation of nitrobenzene (Table 1; entry 14).13a However, with our delight, adding first CuCl and then diazonium salt to this solvent system, we obtained the target homocoupling adduct 4,4′-dinitrobiphenyl (2a) in good yields. The only by-products were 3a and 4a (Table 1; entry 16). No traces of possible ether, resulting from the nucleophilic attack of the OH groups of glycerol, were never found.
Indeed, there are many advantages associated with diazonium salts. These include their greater reactivity due to the fact that the diazonium group is a better nucleofuge than the halide, which allows the use of mild reaction conditions. Moreover, the reaction do not require a base or additional ligands.
The good yield and simplicity of the procedure encouraged us to explore the generality and scope of this reaction further, using different salts 1a–n. The results are shown in Chart 1. The best results were obtained in the presence of electron-withdrawing groups: 2 were obtained in higher yields and shorter reactions times (about 1 h). Since heteroarenes tetrafluoroborate are often not stable, three heteroarene diazonium o-benzenedisulfonimides 1o–q were alternatively prepared and provided target heterobiaryls 2o–q with satisfactory yields.
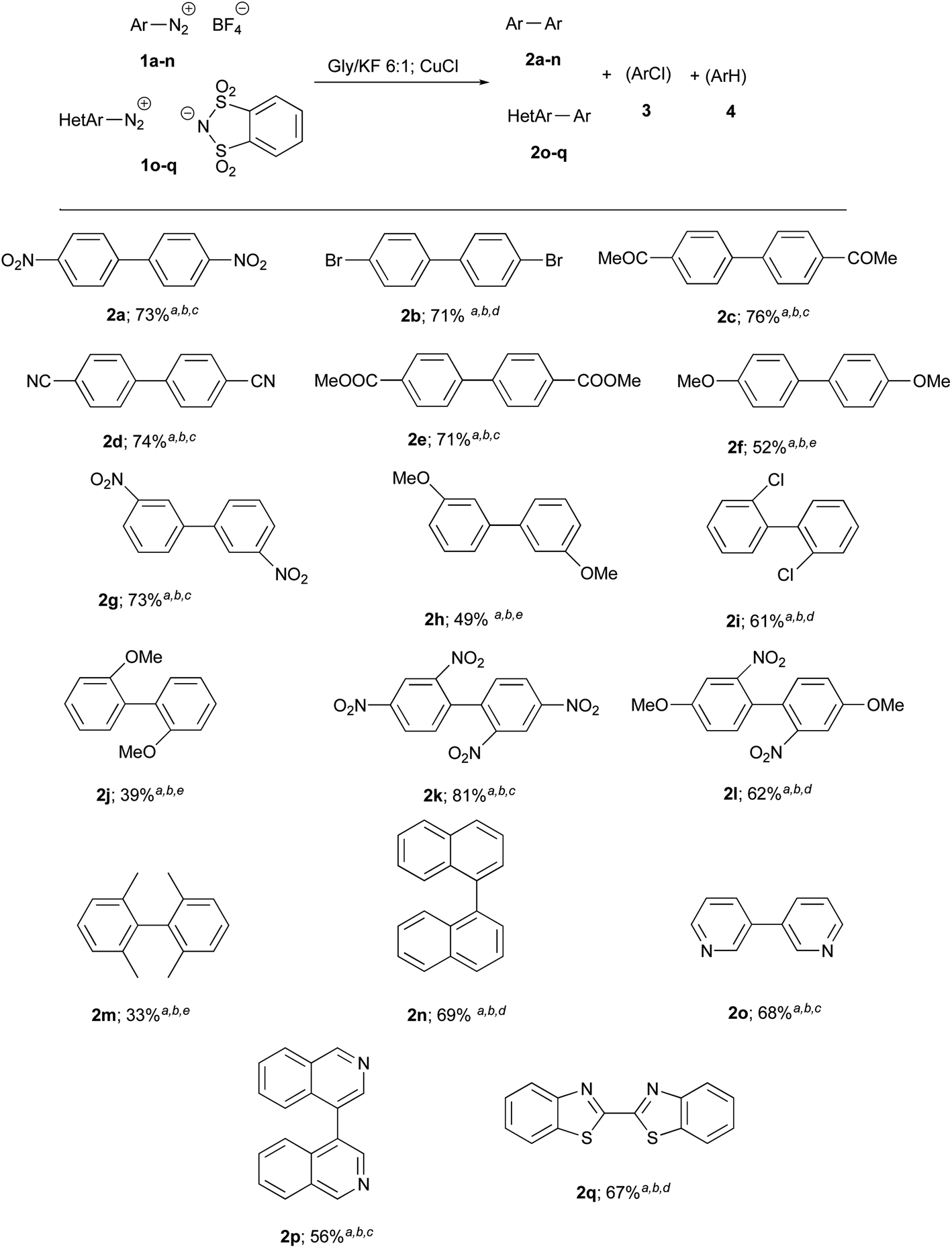 | ||
| Chart 1 Synthesis of biaryl 2a–n or heterobiaryls 2o–q. a The reactions were carried out with 2 mmol of salts 1 and 10 mol% of CuCl. b GC-MS analyses of crude residues showed the presence of biaryls 2, 1-chlorobenzene derivatives 3 and arenes 4. The only isolated products was 2. c The reaction time was about 1 h. d The reaction time was about 2 h. e The reaction times was about 3 h. | ||
Mechanistic study
The mechanism for the coupling of two equivalents of 4-nitrobenzenediazonium salt 1a to 4,4′-dinitrobiphenyl 2a was studied by DFT method (the details are reported in ESI†). We first noticed that the thermodynamics and the large concentration of fluoride could convert the diazonium tetrafluoroborate and the copper chloride in their fluoride derivatives (Scheme 3):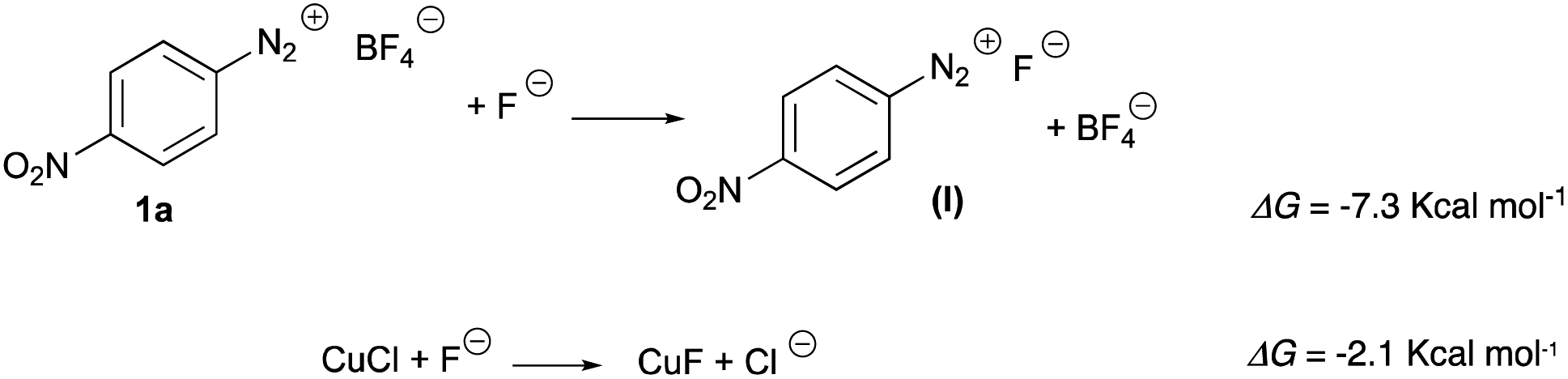 | ||
| Scheme 3 Conversion of 1a and CuCl in fluoride derivatives. | ||
The reaction energy profile and mechanism are shown in Fig. 1; all data and pictures of all structures are reported in ESI.† The diazonium fluoride I forms the complex I-1 with the copper fluoride II, then the fluoride ion moved from the diazonium moiety to the copper to form the complex I-2. The rate determining step of the mechanism is the oxidative addition of the aryl moiety and of the dinitrogen to the copper in TSOA (Fig. 2, left) which is located only 16.2 kcal mol−1 above the reactants in term of free energy and 4.1 kcal mol−1 in term of energy and followed by a downhill sequence of steps from the intermediate I-3. This one loses dinitrogen to form I-4 which is then reduced by the DES through its deprotonated form, the glycerolate III (see also ref. 13a) to form glyceroxyl IV and the intermediate I-5. The free energy barrier (10.8 kcal mol−1 with respect to I-4) is calculated by Marcus theory.14 Then I-5 captures a second diazonium fluoride I yielding the complex I-6. Again the fluoride moves to copper forming I-7 then an internal redox reaction takes place: the copper reduces the aryldiazonium to a aryldiazene radical in I-8. This process is thermodynamically nearly neutral and it is followed by the fast release of dinitrogen through TSNN that forms I-9. After the loss of N2, the intermediate I-10 gives rise to the C–C coupling through TSCC (Fig. 2, right) yielding the complex between the final products P (indeed 2a) and CuIIF3−, I-11 that finally, dissociates.
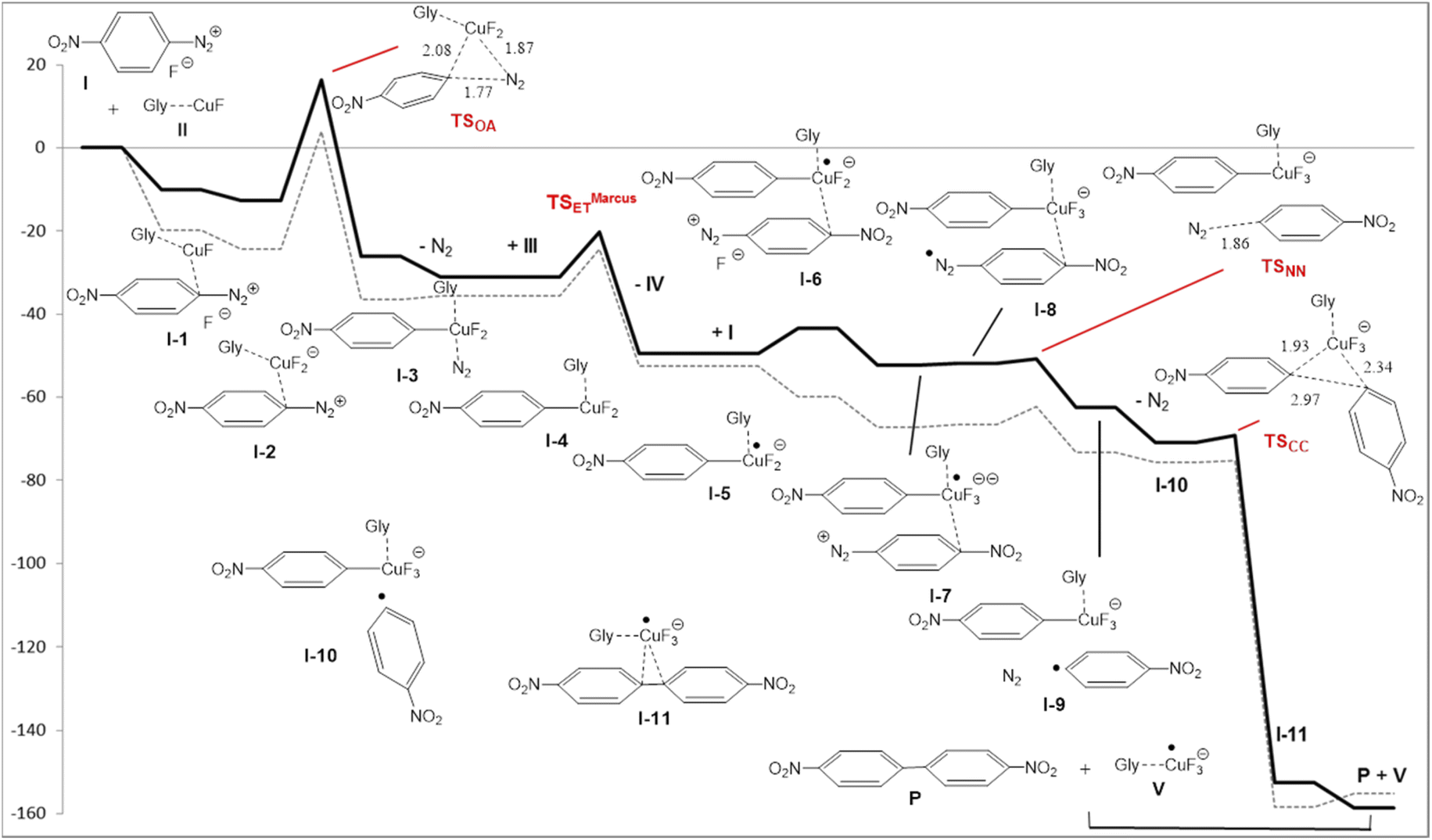 | ||
| Fig. 1 Energy (E + ZPE, dashed line) and free energy (thick line) profiles (in kcal mol−1) for the copper/DES catalyzed coupling of two 4-nitrobenzenediazonium salt 1a to 4,4′-dinitrobiphenyl (2a). | ||
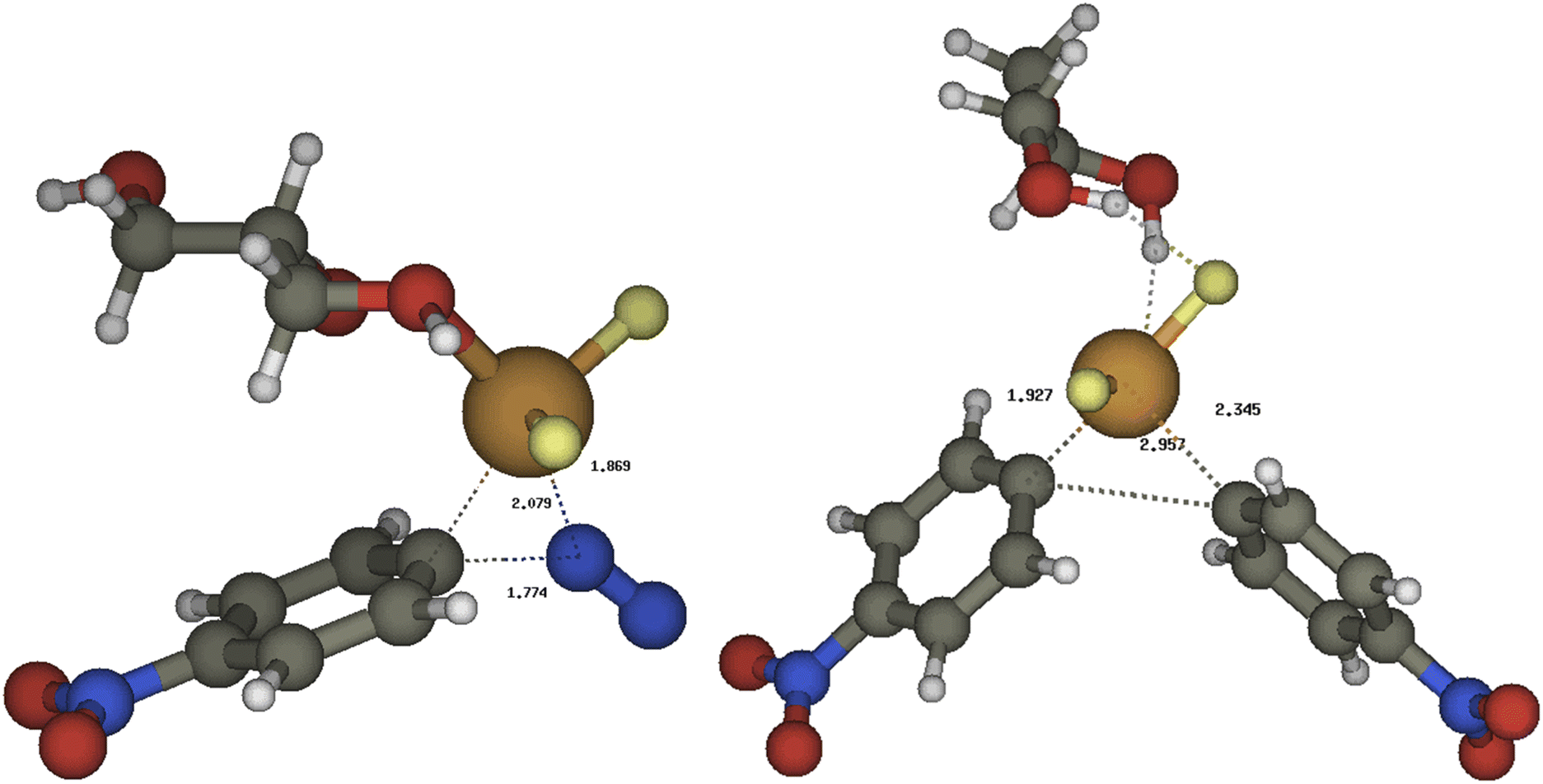 | ||
| Fig. 2 The most relevant transition structures for the copper/DES catalyzed coupling of two 4-nitrobenzenediazonium salt to 2a: left TSOA, right TSCC. | ||
As observed in our previous paper,13a the glyceroxyl IV quickly abstract a hydrogen atom from glycerol generating a new glycerol molecule and the 2-glycerolyl radical VI (or the 1-glycerolyl VI′; Scheme 4):
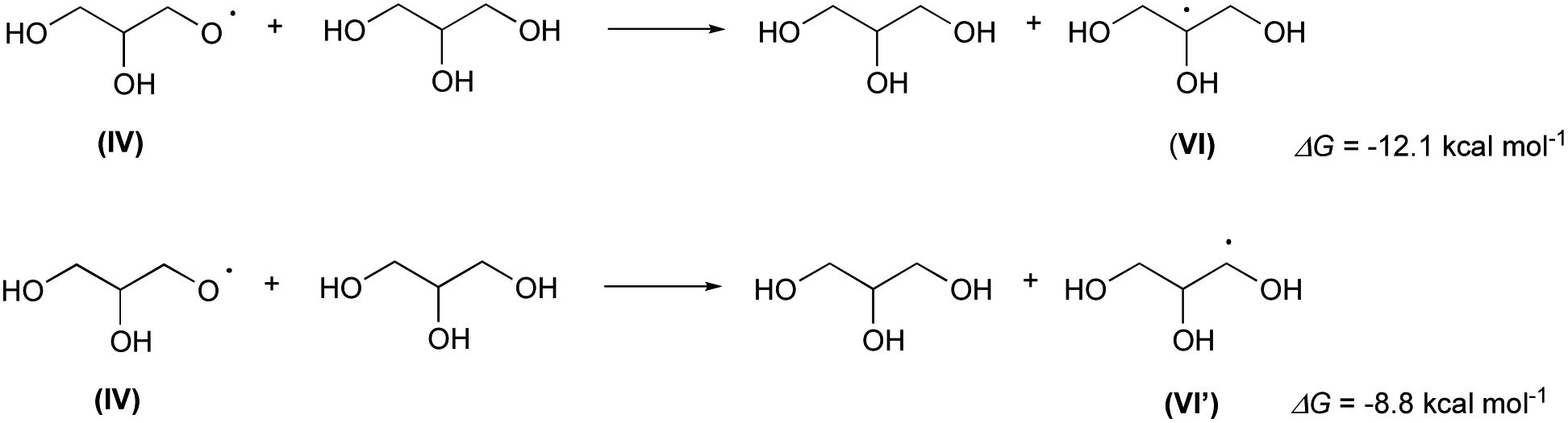 | ||
| Scheme 4 Extraction of H from glycerol by glyceroxyl (IV). | ||
Finally, the radicals VI or VI′ regenerate the catalytic copper(I) yielding as byproducts 1,3-dihydroxypropanone or 1,2-dihydroxypropanale as byproducts (Scheme 5):
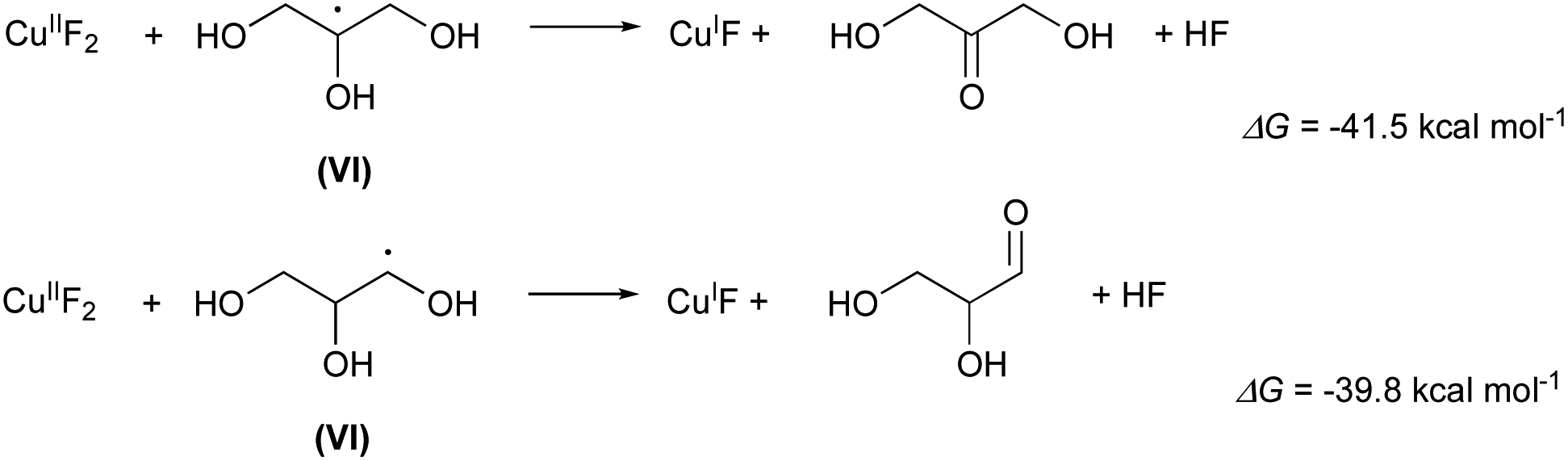 | ||
| Scheme 5 Formation of by-products 1,3-dihydroxypropanone or 1,2-dihydroxypropanale. | ||
The two carbonyl compounds, have both been identified in the NMR spectra of the recovered Gly/KF solvent system (ESI pag. 55†). In fact, the signals are consistent with the presence, in small amounts though, of both dihydroxypropanone (quaternary carbon at 211.97 ppm) and glyceraldehyde (quaternary carbon of the hydrated form at 89.76 ppm, primary carbon at 64.85 ppm, secondary carbon at 74.08 ppm).15 The primary carbon of dihydropropanone is likely hidden by the signal at 62.50 ppm (primary carbon of glycerol).
Therefore, the global reactions are (Scheme 6):
 | ||
| Scheme 6 Global reactions. | ||
Finally, it must be stressed that, in the light of the proposed mechanism, there is an important difference with respect to classic Ullmann protocol, namely the consumption of the less expensive glycerol instead of the metallic copper.
Raman measurements
In order to get further insights into the nature of the species involved in the reaction, we performed some Raman measurements on diazonium salt 1a/DES–CuCl solutions. The results are reported in Fig. 3.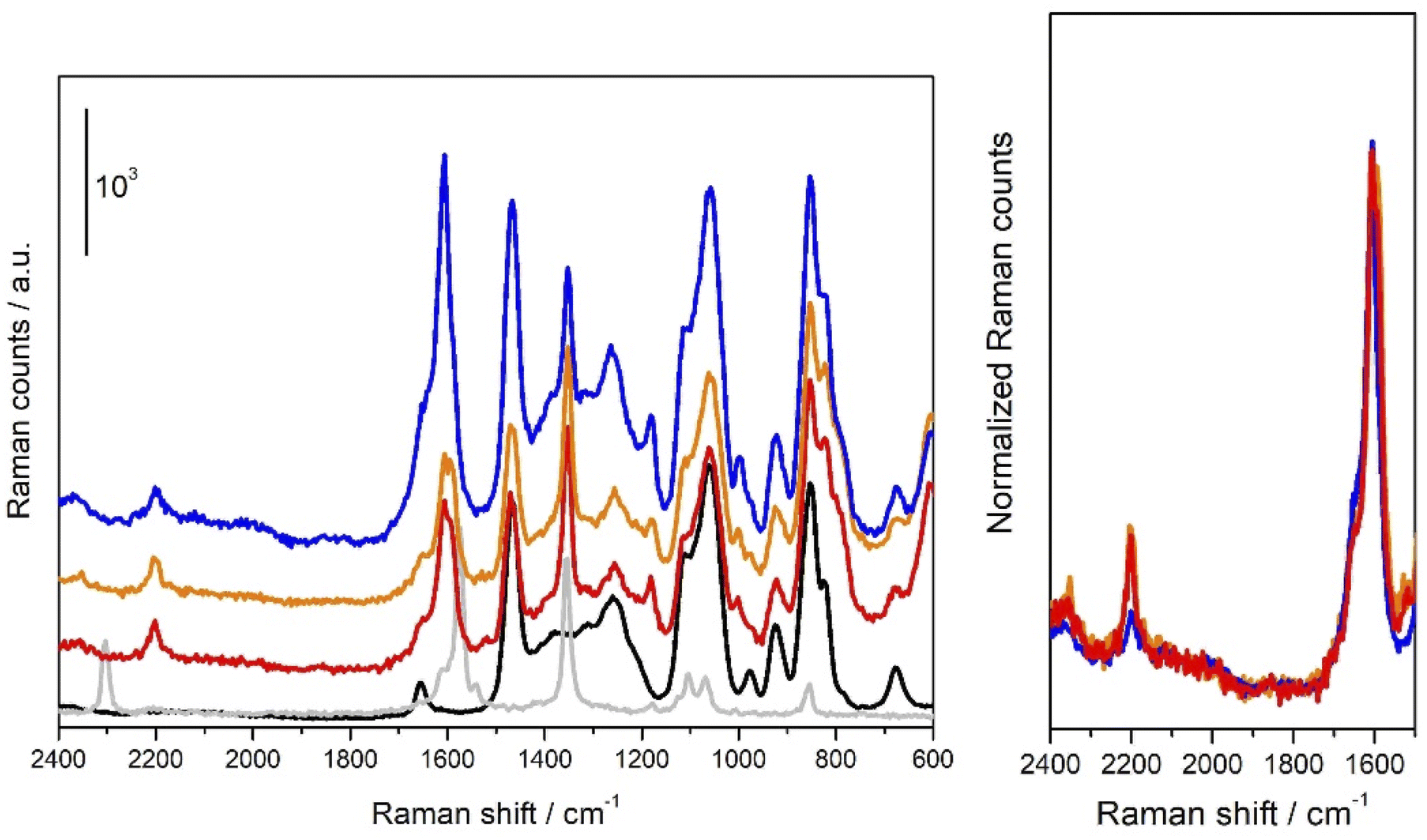 | ||
| Fig. 3 (A) on the left: Raman spectra obtained respectively on solid 1a (grey solid line), DES–CuCl (black solid line), 1a/DES–CuCl solution in closed vessel (red and orange solid lines), 1a/DES–CuCl solution after the removal of cap (solid blue line); (B) on the left: normalized Raman spectra in the 2400–1500 cm−1 Raman shift region (colour code as in the (A) part). | ||
As it can be seen from the comparison between Raman spectrum obtained on 1a/DES–CuCl solutions (red, orange and blue solid lines in Fig. 3A) they significantly differ from those obtained respectively on solid 1a and bare DES–CuCl (light grey and black solid lines in Fig. 3A, respectively). In particular it can be noticed that the band located at 2304 cm−1 in the Raman spectrum of solid 1a assignable to the stretching mode involving N+![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N moiety of 1a, moves to 2204 cm−1 (Dn = −100 cm−1) when 1a/DES–CuCl solutions are concerned.16 Conversely, band at 1354 cm−1 in solid 1a (assignable to NO2 groups involving modes) remains quite unaffected by the interaction with DES–CuCl. The 1620–1560 cm−1 range (typically related to C–C stretching modes of phenyl ring) is characterized by a well-defined band at 1572 cm−1 in solid 1a and undergoes a significant modification after interaction with DES–CuCl, evolving in a complex envelope characterized by two peaks falling respectively at 1606 and 1597 cm−1. It is worth noticing that a close inspection of spectra reported in Fig. 3A as red/orange (recorded in a close vessel) and blue (recorded in the same vessel after removing the cap) solid lines evidenced a further evolution; in particular, as emerging by data reported in Fig. 3B, where the same curves are reported after normalization at the 1606 cm−1 centred peak, a considerable erosion of the 2204 cm−1 band can be observed. This observation let us to hypothesize that red/orange curves contains:
N moiety of 1a, moves to 2204 cm−1 (Dn = −100 cm−1) when 1a/DES–CuCl solutions are concerned.16 Conversely, band at 1354 cm−1 in solid 1a (assignable to NO2 groups involving modes) remains quite unaffected by the interaction with DES–CuCl. The 1620–1560 cm−1 range (typically related to C–C stretching modes of phenyl ring) is characterized by a well-defined band at 1572 cm−1 in solid 1a and undergoes a significant modification after interaction with DES–CuCl, evolving in a complex envelope characterized by two peaks falling respectively at 1606 and 1597 cm−1. It is worth noticing that a close inspection of spectra reported in Fig. 3A as red/orange (recorded in a close vessel) and blue (recorded in the same vessel after removing the cap) solid lines evidenced a further evolution; in particular, as emerging by data reported in Fig. 3B, where the same curves are reported after normalization at the 1606 cm−1 centred peak, a considerable erosion of the 2204 cm−1 band can be observed. This observation let us to hypothesize that red/orange curves contains:
(i) The vibrational features characteristic of 1a molecules interacting with DES–CuCl (testified by the band located a 2204 cm−1) and mainly involving the N+N moiety of 1a molecule, the latter likely being intermediate actors of the reaction more than simply spectators.
(ii) Species coming from reacted 1a molecules and responsible for the band at 1606 cm−1 and possibly for that one located at 1597 cm−1 which could superimpose to signal (located at 1572 cm−1 in the solid) relative to just DES–CuCl perturbed 1a molecules.
As a matter of fact, the peak centred at 1606 cm−1 become predominant in Raman spectrum obtained when 1a/DES–CuCl is exposed to air, reaching an intensity ratio with respect to the 1597 cm−1 as that observed for diphenyl specie 2a.17
It should be noted that it was impossible to record a “zero-time” reaction spectrum (i.e. a spectrum right after the addition of 1a): indeed, the reaction is already partially occurred during the time needed for a complete Raman measurement.
More in details, the red and orange spectra are related to the first and the fourth scan (each scan lasts for 400 seconds), respectively, recorded for a reaction ongoing in a closed vessel: as one can see, after an almost instantaneous reaction as proved by the rising of peaks ascribable to the diphenyl product (vide supra) the latter seems to reach an equilibrium state (orange spectra). This equilibrium state could be easily break only if the cap is removed from the vessel: indeed, the reaction starts again (as proved by the bubbling) and it continues till the peak at 2204 cm−1 completely disappears. One should note that if the reaction is conduced in an open vessel, it proceeds without any equilibrium till the diazonium salt is completely consumed. We tentatively ascribed such a behaviour to a control of the reaction governed by the partial pressure of N2 into the vessel. In fact, the reactions of 1a in Gly/KF 6:1 DES, carried out under nitrogen flow, both in the presence and in the absence of copper, showed a significant slowdown (Table 1; entries 15 and 17).
Conclusions
We have proposed here a mild, easy and efficient revisitation of Ullmann protocol carried out in deep eutectic solvent as green and sustainable solvent media and using diazonium salts as an alternative to classical aryl halides. The key points of the reaction mechanism that we propose consists in the oxidative addition of the first equivalent of arenediazonium salt to the copper catalyst followed by reduction of intermediate by the DES then by the C–C coupling with the second arenediazonium salt equivalent. The reductant property of the DES operates again in the regeneration of the copper(I) catalyst instead of the metallic copper. Furthermore, Raman spectroscopy reveals its interesting role in monitoring the reactive species, proving the remarkably different coordination of the diazonium salt once dissolved in DES compared to solid state; additionally, these measurements prove the kinetic control of the reaction that is driven by the removal of molecular N2 from the reaction environment.Finally, it must be stressed that diazonium salt are resourceful building blocks in organic synthesis and the last few years have seen a dramatic resurgence of their chemistry. The reactions of diazonium salts in deep eutectic solvents is a field so far little explored and can become a valuable tool not only to make their chemistry more sustainable but also useful for finding new types of reactivity.
Experimental
General
All the reactions were carried out in open air glassware. Analytical grade reagents and solvents (purchased from Merck or Alfa-Aesar) were used and reactions were monitored by GC, GC-MS and TLC. Column chromatography and TLC were performed on Merck silica gel 60 (70–230 mesh ASTM) and GF 254, respectively. Petroleum ether refers to the fraction boiling in the range 40–70 °C. Room temperature is 22 °C. Mass spectra were recorded on an HP 5989B mass selective detector connected to an HP 5890 GC with a methyl silicone capillary column. GC analyses were performed on a PerkinElmer AutoSystem XL GC with a methyl silicone capillary column. 1H NMR and 13C NMR spectra were recorded on a Jeol ECZR spectrometer at 600 and 150 MHz respectively. Arenediazonium tetrafluoroborates,18 arenediazonium o-benzenedisulfonimides8h and DES (glycerol/KF 6:1)12a were prepared as reported in the literature. Structures and purity of biaryls 2 were confirmed by their spectral (NMR, MS) and physical data, substantially identical to those reported in the literature. Their NMR spectra are reported in ESI.†
Raman spectra have been collected on an inVia Raman Microscope and adopting a 244 nm exciting LASER line. LASER light/backscattered light have been focused/collected on/from samples through a 15× objective. In the case of solid powders, 5% of the total LASER power has been admitted to reach the samples which were maintained under rotation to avoid/reduce possible decomposition induced by LASER light. For solutions (measured in a Helma QS cuvette and subjected to a magnetic stirring), 10% of the total laser power has been adopted. In any case, stability of samples under the LASER light have been carefully investigated. Backscattered light (after Rayleigh light removal through an edge filter) have been analysed by a 3600 l mm−1 grating and collected through a CCD Peltier cooled detector. For powder, the presented spectrum is resulting from the average of 2 Raman spectra (20 × 20′′ acquisitions) acquired consecutively. For solutions, the presented spectra resulted from 20 × 20′′ acquisitions.
Ullmann homocoupling: typical procedure. Preparation of 4,4′-dinitrobiphenyl (2a)
Copper chloride (19.8 mg; 0.2 mmol; 10% mol) was added to a glycerol/KF 6:1 (5 mL) solvent system. The mixture was stirred at room temperature for 30 min, until the formation of a homogeneous suspension. 4-Nitrobenzenediazonium tetrafluoroborate (1a, 474 mg, 2 mmol) was then added to this mixture which was stirred at room temperature for 1 h; the completion of the reaction was confirmed by the absence of azo coupling with 2-naphthol. Then, the reaction mixture was poured into Et2O/H2O (10 mL, 1:1). The aqueous layer was separated and extracted with Et2O (5 mL). The combined organic extracts were washed with H2O (5 mL), dried with Na2SO4 and evaporated under reduced pressure. GC-MS analyses of the crude residue showed a mixture of 4,4-dinitrobiphenyl (2a), as the major product, MS (EI): m/z = 244 [M]+. Nitrobenzene (4a), MS (EI): m/z = 123 [M]+ and 1-chloro-4-nitrobenzene (3b), MS (EI): m/z = 157 [M]+ were also detected. The crude residue was chromatographed in a short chromatographic column (eluent petroleum ether/ethyl ether 9:1) and pure 2a was obtained (GC, GC-MS, TLC and NMR; 179 mg, 73%). In order to recover the solvent system (Gly/KF 6:1), the aqueous layer was evaporated under reduced pressure, a 13C NMR spectrum (D2O; ESI, page. 55†) was recorded.
Author contributions
G. G.: general organization, conceptualization, writing, review and editing, DFT calculations; M. B.: general organization, conceptualization, writing, review and editing, Raman measurements; A. A.: homocoupling reactions; A. D.: Raman measurements; S. D.: general organization, conceptualization, writing, review and editing, halodediazotation reactions, supervision, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the University of Torino and by Ministero dell'Università e della Ricerca Scientifica.References
- (a) E. J. Corey and X. M. Cheng, The Logic of Chemical Synthesis, John Wiley & Sons: Chichester, 1989 Search PubMed; (b) B. Perry, T. F. Brewer, P. J. Sarver, D. M. Schultz, D. A. Di Rocco and D. W. C. MacMillan, Nature, 2018, 560, 70 CrossRef PubMed.
- C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062 CrossRef CAS PubMed.
- (a) Metal-Catalyzed Cross-Coupling Reactions, ed. A. de Meijere and F. Diederich, Wiley-VCH; Weinheim, Germany, 2nd edn, 2004 Search PubMed; (b) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442 CrossRef CAS PubMed; (c) A. Biffis, P. Centomo, A. Del Zotto and M. Zecca, Chem. Rev., 2018, 118, 2249 CrossRef CAS; (d) L.-C. Campeau and N. Hazari, Organometallics, 2019, 38, 3 CrossRef CAS PubMed.
- (a) F. Ullmann and J. Bielecki, Chem. Ber., 1901, 34, 2174 CrossRef CAS; (b) F. Ullmann, Ber. Dtsch. Chem. Ges., 1903, 36, 238 CrossRef; (c) F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2008, 47, 3096 CrossRef CAS PubMed; (d) C. Sambiagio, S. P. Marsden, A. J. Blacker and P. C. McGowan, Chem. Soc. Rev., 2014, 43, 3525 RSC; (e) S. Mondal, ChemTexts, 2016, 2, 17 CrossRef; (f) M. Lackinger, Chem. Commun., 2017, 53, 7872 RSC; (g) T. Garnier, M. Danel, V. Magnè, A. Pujol, V. Beneteau, P. Pale and S. Chassaing, J. Org. Chem., 2018, 83, 6408 CrossRef CAS PubMed; (h) Q. Yang, Y. Zhao and D. Ma, Org. Process Res. Dev., 2022, 26, 1690 CrossRef CAS.
- (a) T. W. Wallace, Org. Biomol. Chem., 2006, 4, 3197 RSC; (b) S. Yuan, J. Chang and B. Yu, Top. Curr. Chem., 2020, 378, 23 CrossRef CAS.
- (a) E. Sperotto, G. P. M. van Klink, G. van Koten and J. G. de Vries, Dalton Trans., 2010, 39, 10338 RSC; (b) X. Ribas and I. Güell, Pure Appl. Chem., 2014, 86, 345 CAS.
- (a) G. Clavè, C. Garel, C. Poullain, B.-L. Renard, T.-K. Olszewski, B. Lange, M. Shutcha, M.-P. Faucon and C. Grison, RSC Adv., 2016, 6, 59550 RSC; (b) F. Ferlin, V. Trombettoni, L. Luciani, S. Fusi, O. Piermatti, S. Santoro and L. Vaccaro, Green Chem., 2018, 20, 1634 RSC; (c) G. Yashwantrao and S. Saha, Tetrahedron, 2021, 97, 132406 CrossRef CAS; (d) Z. Zuo, R. S. Kim and D. A. Watson, J. Am. Chem. Soc., 2021, 143, 1328 CrossRef CAS PubMed.
- (a) Y.-J. Chen and H.-H. Chen, Org. Lett., 2006, 8, 5609 CrossRef CAS PubMed; (b) R. A. Altman, A. Shafir, P. A. Lichtor and S. L. Buchwald, J. Org. Chem., 2008, 73, 284–286 CrossRef CAS PubMed; (c) Y. Zhang, X. Yang, Q. Yao and D. Ma, Org. Lett., 2012, 14, 3056 CrossRef CAS; (d) R. Lv, Y. Wang, C. Zhou, L. Li and R. Wang, ChemCatChem, 2013, 5, 2978 CrossRef CAS; (e) W. Zhou, M. Fan, J. Yin, Y. Jiang and D. Ma, J. Am. Chem. Soc., 2015, 137, 11942 CrossRef CAS PubMed; (f) J. Gao, S. Bhunia, K. Wang, L. Gan, S. Xia and D. Ma, Org. Lett., 2017, 19, 2809 CrossRef CAS PubMed; (g) W. Liu, J. Xu, X. Chen, F. Zhang, Z. Xu, D. Wang, Y. He, X. Xia, X. Zhang and Y. Liang, Org. Lett., 2020, 22, 7486–7490 CrossRef CAS PubMed; (h) A. Antenucci and S. Dughera, Reactions, 2022, 32, 300 CrossRef.
- (a) A. F. Quivelli, P. Vitale, F. M. Perna and V. Capriati, Front. Chem., 2019, 7, 723 CrossRef CAS PubMed; (b) A. F. Quivelli, M. Marinò, P. Vitale, J. Garcia-Alvarez, F. M. Perna and V. Capriati, ChemSusChem, 2022, 15, e202102211 CrossRef CAS; (c) A. F. Quivelli, F. V. Rossi, P. Vitale, J. Garcia-Alvarez, F. M. Perna and V. Capriati, ACS Sustainable Chem. Eng., 2022, 10, 4065 CrossRef CAS.
- (a) A. P. Abbott, D. Boothby, G. Capper, D. L. Davies and R. K. Rasheed, J. Am. Chem. Soc., 2004, 126, 9142 CrossRef CAS PubMed; (b) S. Khandelwal, Y. K. Tailor and M. Kumar, J. Mol. Liq., 2016, 215, 345 CrossRef CAS; (c) D. A. Alonso, A. Baeza, R. Chinchilla, G. Guillena, I. M. Pastor and D. J. Ramón, Eur. J. Org. Chem., 2016, 612 CrossRef CAS; (d) Y. Marcus, Deep Eutectic Solvents, Springer International Publishing, 2019 CrossRef; (e) B. B. Hansen, S. Spittle, B. Chen, D. Poe, Y. Zhang, J. M. Klein, A. Horton, L. Adhikari, T. Zelovich, B. W. Doherty, B. Gurkan, E. J. Maginn, A. Ragauskas, M. Dadmun, T. A. Zawodzinski, G. A. Baker, M. E. Tuckerman, R. F. Savinell and J. R. Sangoro, Chem. Rev., 2020, 121, 1232 CrossRef PubMed; (f) S. E. Hooshmand, R. Afshari, D. J. Ramón and R. S. Varma, Green Chem., 2020, 22, 3668 RSC.
- (a) A. Roglans, A. Pla-Quintana and M. Moreno-Mañas, Chem. Rev., 2006, 106, 4622 CrossRef CAS PubMed; (b) F.-X. Felpin and S. Sengupta, Chem. Soc. Rev., 2019, 48, 1150 RSC; (c) F. Mo, D. Qiu, L. Zhang and J. Wang, Chem. Rev., 2021, 121, 5741 CrossRef CAS PubMed.
- (a) E. R. Atkinson, C. R. Morgan, H. Warren and T. J. Manning, J. Am. Chem. Soc., 1945, 67, 1513 CrossRef CAS; (b) T. Cohen, R. J. Lewarchik and J. Z. Tarino, J. Am. Chem. Soc., 1974, 96, 7753 CrossRef CAS; (c) P. J. Montoya-Pelaez, Y.-S. Uh, C. Lata, M. P. Thompson, R. P. Lemieux and C. M. Crudden, J. Org. Chem., 2006, 71, 5921 CrossRef CAS PubMed; (d) I. Cepanec, M. Litvic, J. Udikovic, I. Pogorelic and M. Lovric, Tetrahedron, 2007, 63, 5614 CrossRef CAS.
- (a) A. Antenucci, M. Bonomo, G. Ghigo, L. Gontrani, C. Barolo and S. Dughera, J. Mol. Liq., 2021, 339, 116743 CrossRef CAS; (b) G. Ghigo, M. Bonomo, A. Antenucci, C. Reviglio and S. Dughera, Molecules, 2022, 9, 1909 CrossRef.
- (a) R. A. Marcus, Angew. Chem., Int. Ed. Engl., 1993, 32, 1111 CrossRef; (b) A. de Aguirre, I. Funes-Ardoiz and F. Maseras, Inorganics, 2019, 7, 32 CrossRef CAS; (c) M. P. Doyle, J. K. Guy, K. C. Brown, S. N. Mahapatro, C. M. VanZyl and J. R. Pladziewicz, J. Am. Chem. Soc., 1987, 109, 1536 CrossRef CAS.
- R. Nguyen, M. Amouroux, A. Duda and Z. Mouloungui, J. Am. Oil Chem. Soc., 2020, 97, 679 CrossRef CAS.
- S. Betelu, I. Tijunelyte, L. Boubekeur-Lecaque, I. Ignatiadis, A. C. Schnepf, E. Guenin, N. Bouchemal, N. Felidj, E. Rinnert and M. Lamy de la Chapelle, J. Org. Inorg. Chem., 2017, 3, 1 Search PubMed.
- I. Gener, G. Buntinx and C. Brémard, Microporous Mesoporous Mater., 2000, 41, 253 CrossRef CAS.
- A. Roe, Preparation of Aromatic Fluorine Compounds from Diazonium Fluoborates in Organic Reactions, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2011; pp. pp. 193–228R Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of computational method, tables of calculated absolute and relative energies, pictures and cartesian coordinates of all optimized structures, NMR spectra of biaryls 2, 13C spectrum of recovered solvent system. See https://doi.org/10.1039/d2ra05272e |
| This journal is © The Royal Society of Chemistry 2022 |