
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA three dimensional graphdiyne-like porous triptycene network for gas adsorption and separation†
Hui Maa,
Bin-Bin Yanga,
Zhen Wanga,
Kai Wub and
Chun Zhang *a
*a
aCollege of Life Science and Technology, National Engineering Research Center for Nanomedicine, Huazhong University of Science and Technology, Wuhan, 430074, China. E-mail: chunzhang@hust.edu.cn
bTechnology R&D Center, Hubei Tobacco (Group) Co., Ltd, Wuhan, 430070, China. E-mail: 3887253@qq.com
First published on 4th October 2022
Abstract
Graphdiyne, an emerging carbon allotrope, has attracted many researchers devoted to the study of its synthesis and application. The utilization of graphdiyne in gas adsorption and separation has been predicted by computer simulation with many examples. In this work, the triangular basic unit of graphdiyne was introduced into a triptycene-based porous organic polymer to obtain a three dimensional graphdiyne-like porous triptycene network named G-PTN. With high surface area and a microporous structure, G-PTN exhibited convincing application potential for the storage of gas molecules, especially for the selective adsorption of acetylene over ethylene. Computational simulation proved the importance of the triptycene units and three dimensional structure to the selectivity, as well as the potential of graphdiyne units as selective binding sites, suggesting that through judicious design, new three-dimensional porous graphdiyne could be acquired with better gas adsorption and separation performance.
Introduction
Graphdiyne (GDY), a two dimensional (2D) carbon allotrope, is supposed to have excellent mechanical, electronic, optical, and magnetic properties.1–4 Since GDY was proposed in 1997 (ref. 5) and the first GDY film was fabricated on copper foil in 2010,6 many researchers have been dedicated to the study of GDY mainly focussing on the realization of an ideal structure7–9 and the expansion of its application area.10–15 Fundamental building units, consisting of three phenyl rings crosslinked by butadiyne linkages, endow GDY with uniformly distributed pores and an extended π-conjugated system, which were usually considered advantages for gas adsorption and separation as high surface area and selective binding sites. Based on uniform pore size, the superior performance of a monolayer graphdiyne membrane for hydrogen purification from syngas was predicted through computational simulation,16,17 as well as the separation of oxygen from harmful gases.18 The AB stacking of multilayer graphdiyne19 and short interlayer distance resulting from strong interlayer interaction prevented the extension of the pore structure. Recently, Li et al.20,21 adjusted the pore size of graphdiyne through introducing substituted hydrogen or chlorine atoms. Guo et al.22 constructed a series of porous carbon materials through stacking functionalized graphdiyne units in a disordered mode, and proved their outstanding CO2 capture and separation capacity by computation simulation. Concluding from the research of porous polymers, introducing three dimensional rigid building blocks into GDY could reduce the stacking effect between layers and improve its pore performance, so as to expand its application field like gas storage and separation. But so far there have been few relevant reports.Light hydrocarbons comprised of only carbon and hydrogen like ethylene and acetylene were important chemical feedstock for manufacture plastic and other industrial products. However, their similar molecular weight and polarizability led to difficulties in separation of the gas mixture, and furthermore, the utilization of the pure gas. The common industrial separation methods relied on cryogenic distillation – an energy intensive process need high pressure and ultralow temperature – and resulted in tremendous energy cost and carbon emission.23,24 In contrast, adsorptive separation, as an alternative to distillation, did not require phase change of the mixture and possesses improved energy efficiency. Therefore, many researchers have been attracted to the development of new adsorbents with better separation performance in recent years.25–30 For acetylene adsorption and separation from ethylene, it was proved that adjusting the pore size and shape could be an effective way to improve the performance.31–34 However, the precise control of pore size and shape was still a challenge, especially for porous organic polymers. Since the electrostatic potential distribution of acetylene and ethylene were different, introducing proper surface functional groups to obtain selective adsorption sites in porous adsorbents could represent another efficient method. Up to now, some chemical groups like amine groups, imidazolium functional groups, hexafluorosilicate and pigment orange-71 have been demonstrated to exhibit preferential affinity to acetylene.35–38 And more examples were under research and still made sense for providing new design Scheme 1 protocols to obtain adsorbents with better acetylene adsorption and separation performance.
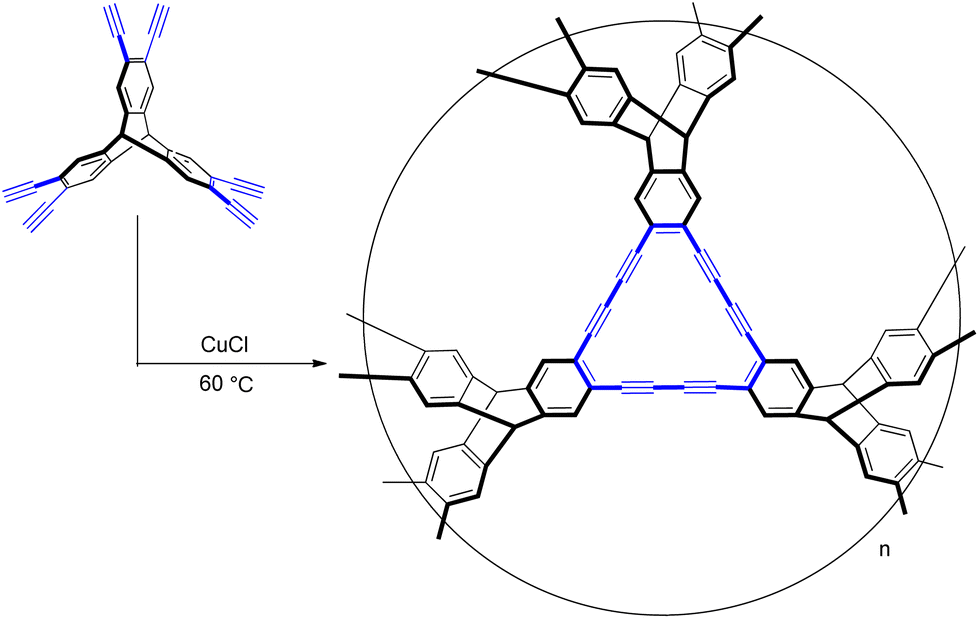 | ||
| Scheme 1 The synthesis of G-PTN. | ||
Triptycene,39–42 with three-dimensional rigid paddlewheel-like structure, has been successfully used as building blocks to construct supramolecular systems,43–48 polymeric materials49,50 and organic porous materials.51–63 In this work, triptycene was introduced into GDY scaffold to prevent the closer packing of two-dimensional layers, so that obtain a three dimensional graphdiyne-like porous triptycene network (G-PTN). Its pore structure and high surface area were characterized, while the selective adsorption capacity toward acetylene over ethylene was also detected and proved by experiments and computation simulation. Which represented a credible way to expand the application of GDY into adsorptive separation of light hydrocarbons, as well as offered a new selective binding site for acetylene over ethylene.
Results and discussion
The synthesis of G-PTN was conducted via a copper-catalyzed cross-coupling reaction of 2,3,6,7,14,15-hexakis[(4-ethynyl-phenyl) ethynyl] triptycene (Scheme S1, Fig. S1–S4†). Fourier-transform infrared (FT-IR) and 13C cross-polarization magic-angle spinning (CP/MAS) NMR experiments were utilized to confirm the formation of diacetylenic linkages. As shown in Fig. S5,† the several signal peaks in NMR spectrum with chemical shifts around 54 ppm, 79 ppm, 94 ppm, 127 ppm and 143 ppm were assigned to the bridgehead carbons of triptycene, diacetylene units and aromatic carbons of triptycene, respectively.20,21 While the oxidation of terminal alkyne could result in the signals around 178 ppm and 194 ppm. In FT-IR spectrum of G-PTN in Fig. S6,† the typical C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C stretching vibration was marked at 2189 cm−1, proving the existence of alkyne groups.
C stretching vibration was marked at 2189 cm−1, proving the existence of alkyne groups.
Fig. S7† exhibited the scanning electron microscope (SEM) and transmission electron microscope (TEM) images of G-PTN, which appeared to be interconnected tiny particles as other amorphous porous organic polymers. The amorphous nature of G-PTN was also demonstrated by a broad peak at 2θ = 20° in powder X-ray diffraction pattern (Fig. S8†). Thermogravimetric analysis (TGA) was employed to test the thermal stability of G-PTN, in Fig. S9,† the material begin to degradation at about 350 °C under nitrogen.
The ideal structure of G-PTN was presented as Fig. 1, which suggested the porous nature of such cage-like material. For verification, nitrogen adsorption isotherm was obtained at 77 K. As shown in Fig. 2a, the curve accorded with type I(b) reversible sorption profile with type H4 hysteresis loop, implying the existence of abundant micropores. The pore size distribution calculated by NLDFT method exhibited two main pore sizes of 0.59 nm and 1.27 nm in Fig. 2b. The Brunauer–Emmett–Teller (BET) surface area (Fig. S10†) of G-PTN was calculated to be 1046 m2 g−1 (Langmuir surface area was 1396 m2 g−1), and the single point adsorption total pore volume of pores less than 320.5 nm diameter at P/P0 = 0.993 was determined to be 0.53 cm3 g−1. The microporous structure of G-PTN was suitable for small gas molecule adsorption. As a typical greenhouse gas, the capture and storage of CO2 was essential in face of global warming and abnormal climate change. The CO2 adsorption capacity of G-PTN was then obtained as 80.7 cm3 g−1 (15.8 wt%) at 273 K and 1 bar and 51.5 cm3 g−1 (10.1 wt%) at 298 K (Fig. S11a and b†), which were comparable with other triptycene-based nitrogen-rich porous organic polymer.60,62
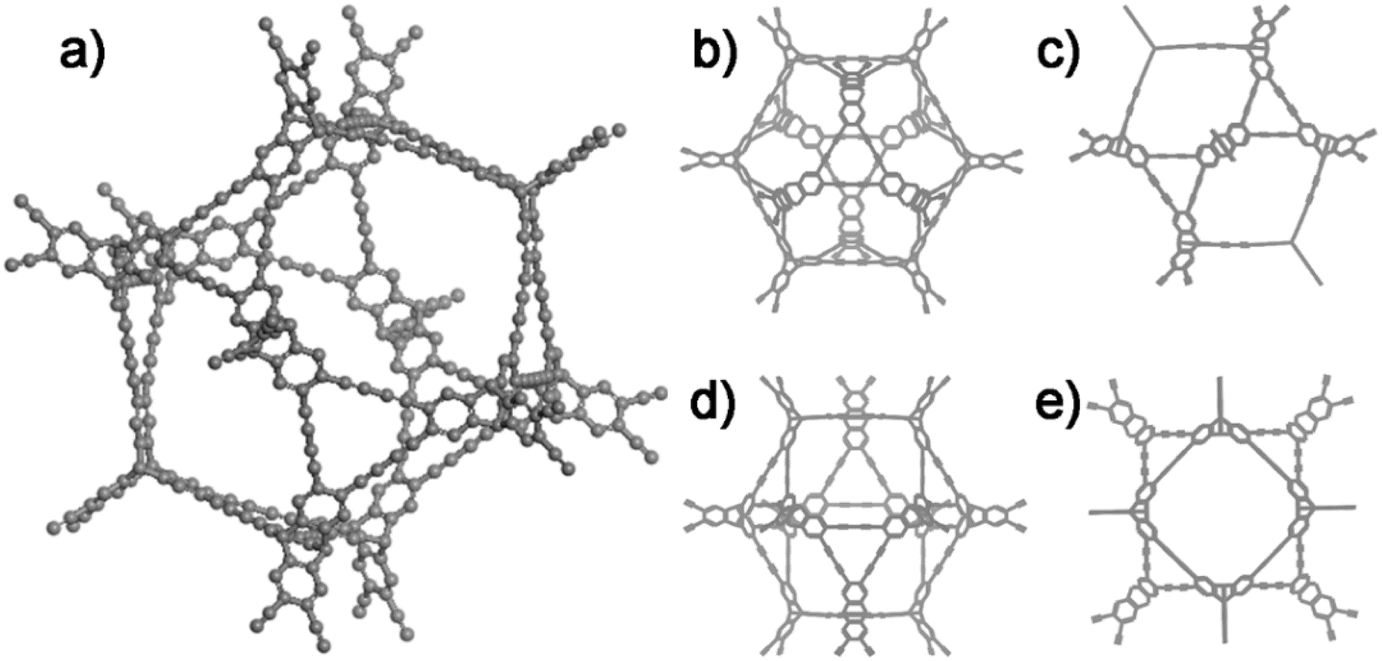 | ||
| Fig. 1 The ideal structure of G-PTN. (a) The three-dimensional structure image of G-PTN; (b) and (c), (d) and (e) the projection drawing of G-PTN from different view angles. | ||
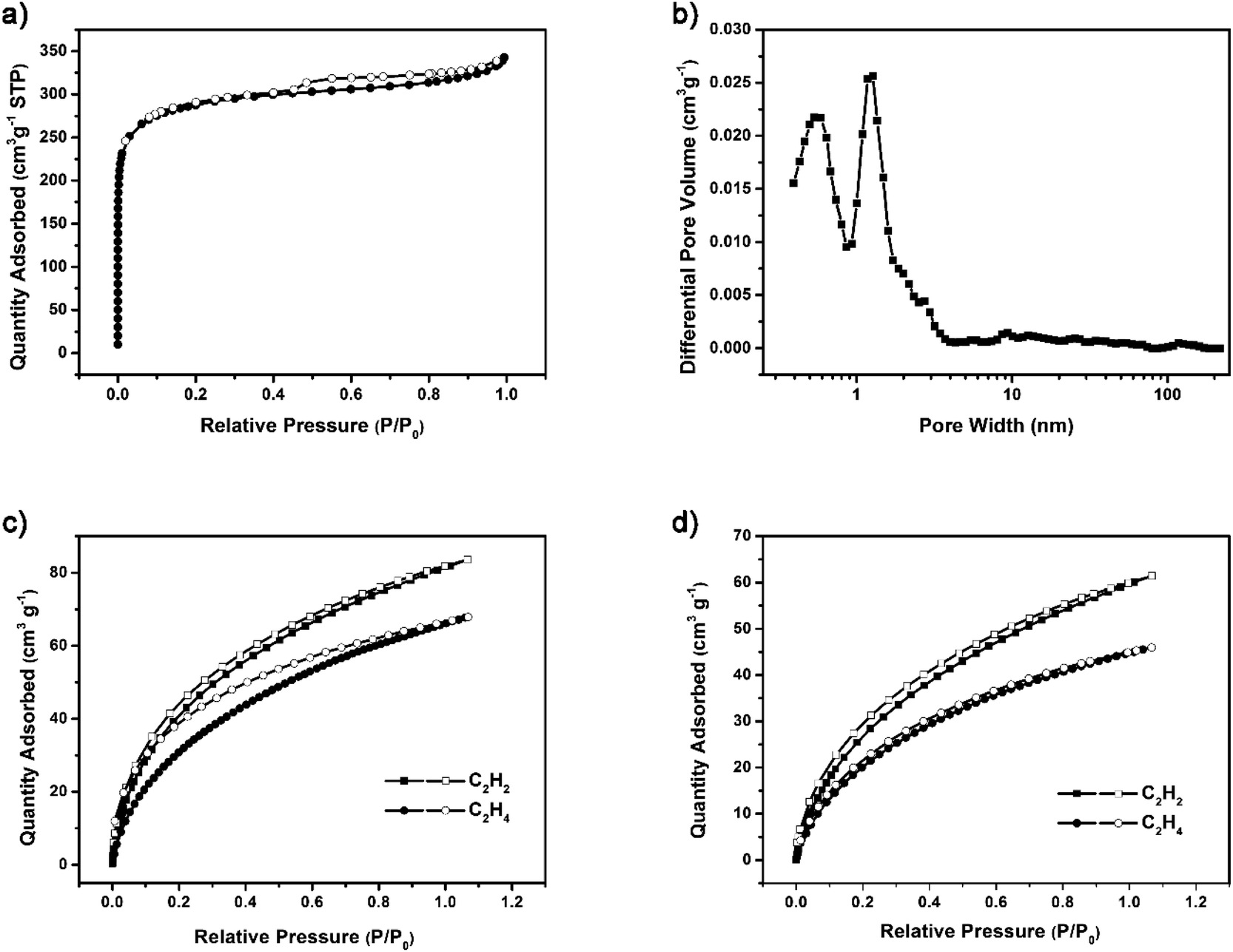 | ||
| Fig. 2 Gas sorption test of G-PTN. (a) Nitrogen sorption and desorption isotherms of G-PTN at 77 K; (b) pore size distribution calculated of G-PTN; C2H2 and C2H4 sorption isotherms of G-PTN at (c) 273 K and (d) 298 K. In (c) and (d), filled symbols denote gas adsorption and empty symbols denote desorption. | ||
The C2H2 and C2H4 adsorption isotherms were collected at 273 K and 298 K to estimate the light hydrocarbons adsorption and separation capacities of G-PTN. As shown in Fig. 2c, the values of 81.1 cm3 g−1 and 66.1 cm3 g−1 were obtained at 273 K and 1 bar for C2H2 and C2H4, respectively, exhibiting manifest acetylene selectivity of G-PTN. Under ambient condition, the gas uptake capacities slightly decreased to 59.6 cm3 g−1 (C2H2) and 44.7 cm3 g−1 (C2H4), approximately remaining the same ratio (Fig. 2d). Since G-PTN hardly exhibited uniform distributed pores, and the primary pore sizes calculated (5.9 Å and 12.7 Å) were wider than both kinetic diameter of C2H2 (3.3 Å) and C2H4 (4.2 Å). It seemed that the acetylene selectivity of G-PTN more likely originated from the stronger affinity of polymer framework toward acetylene.
As further proof, the isosteric heats of adsorption (Qst) for C2H2 and C2H4 were calculated based on the Clausius–Clapeyron equation after fitting the adsorption curves at two different temperature utilizing the dual-site Langmuir model, and the specific values near zero-coverage region were 29.6 kJ mol−1 (C2H2) and 23.9 kJ mol−1 (C2H4) according to Fig. S12a,† which was in support of a stronger interaction between the framework of G-PTN and acetylene. Furthermore, the separation performance of G-PTN for the binary gas mixture of C2H2/C2H4 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :99 v/v) could be predicted via the adsorption isotherms of the pure component gas under the same condition utilizing ideal adsorbed solution theory (IAST).64,65 The data thus obtained were plotted in Fig. S12b,† at 298 K and 1 bar, the value of selectivity was 1.64.
:99 v/v) could be predicted via the adsorption isotherms of the pure component gas under the same condition utilizing ideal adsorbed solution theory (IAST).64,65 The data thus obtained were plotted in Fig. S12b,† at 298 K and 1 bar, the value of selectivity was 1.64.
For further understanding of the selective adsorption, the theoretical calculations were adopted to determine the binding sites and the binding energies when G-PTN was utilized as adsorbent and acetylene or ethylene was utilized as adsorbate. A model molecule termed as M1 consisting the basic triangular unit of graphydine and three triptycene groups (Fig. S13†) was separated out to represent G-PTN in the calculations. As shown in Fig. 3, the van der Waals surface of M1, acetylene and ethylene were coloured according to the value of molecular electrostatic potential (ESP) in BWR color mode and the extreme points were also exhibited. Generally, the intermolecular interactions based on electromagnetic forces could occur between the adsorbents with negative ESP and adsorbates with positive ESP, or the converse. Then two interaction models between acetylene and M1 were screened and optimized, as well as two for ethylene (Fig. S14,† named as M1-C2H21, M1-C2H22, M1-C2H41 and M1-C2H42, respectively), revealing two binding sites of M1 around triptycene units and graphdiyne units. The binding energies were calculated at the PWPB95/def2-QZVPP level, for the more stable binding configurations, the specific values were −23.2 kJ mol−1 for acetylene (M1-C2H21) and −19.3 kJ mol−1 for ethylene (M1-C2H41), which were in accordance with the isosteric heats of adsorption calculated near zero-coverage region previously. The interaction region indicator function (IRI) was employed to visually display interaction regions and chemical bonding regions of the four configurations. In Fig. 4c and d and S15,† the green isosurfaces could represent C–H⋯π interactions and π–π stacking interactions. Given all this, the difference in binding energies between M1-C2H21 and M1-C2H41 could result from three reasons. (1) Hydrogen atoms of acetylene exhibited higher positive ESP (+33.89 kcal mol−1) than that of ethylene (+13.89 kcal mol−1). (2) The linear structure and appropriate length of acetylene allowed its hydrogen atom in intimate contact with the center of phenyl ring (extreme point) to form C–H⋯π interaction, which in Fig. 4d was exhibited as a whole green isosurface. In contrast, in Fig. 4c, the isosurface between ethylene and phenyl ring split into three pieces, representing the off-center positions of hydrogen atoms. (3) For π-conjugated system of benzene rings and butadiyne linkages, the minimum ESP points were around the center of triangular annulus, apparently the hydrogen atom of acetylene was much closer to the points, which could also lead to higher binding energy. To sum up, acetylene molecules presumably preferred to bind with G-PTN at the sites near triptycene units through C–H⋯π interactions and π–π stacking interactions and exhibited higher binding energy than ethylene. In this case, triptycene residues and the three dimensional structure of G-PTN could play an essential role.
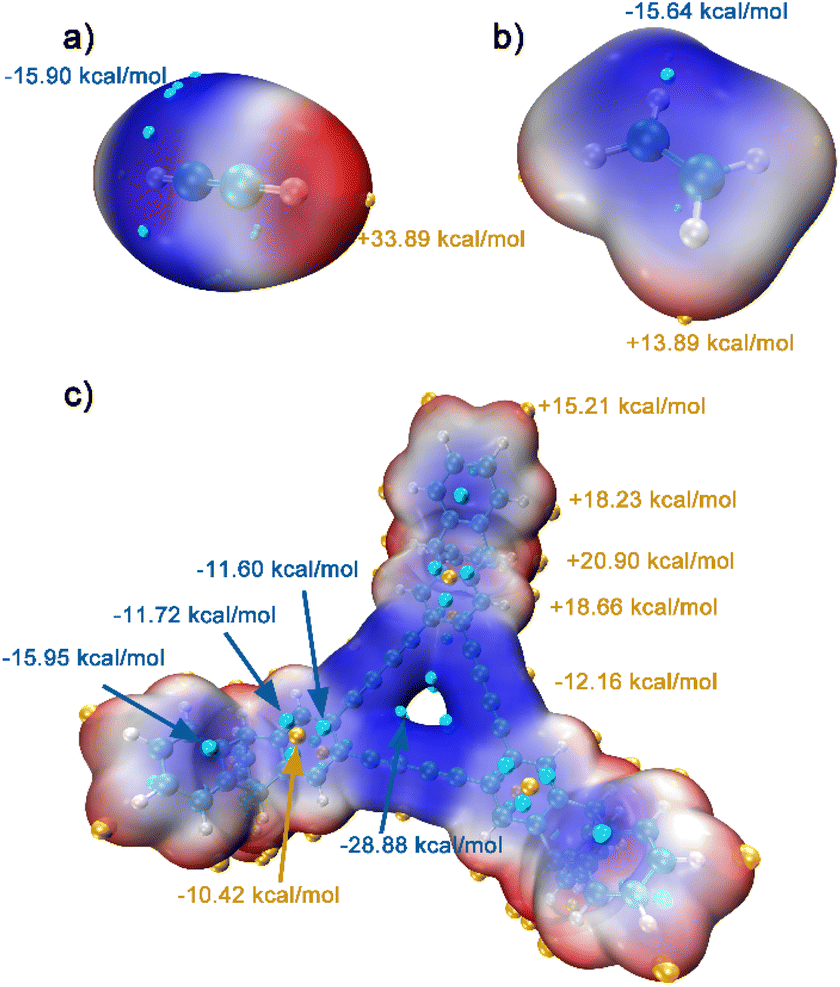 | ||
| Fig. 3 ESP-mapped molecular vdW surface of (a) acetylene, (b) ethylene and (c) model molecule M1. | ||
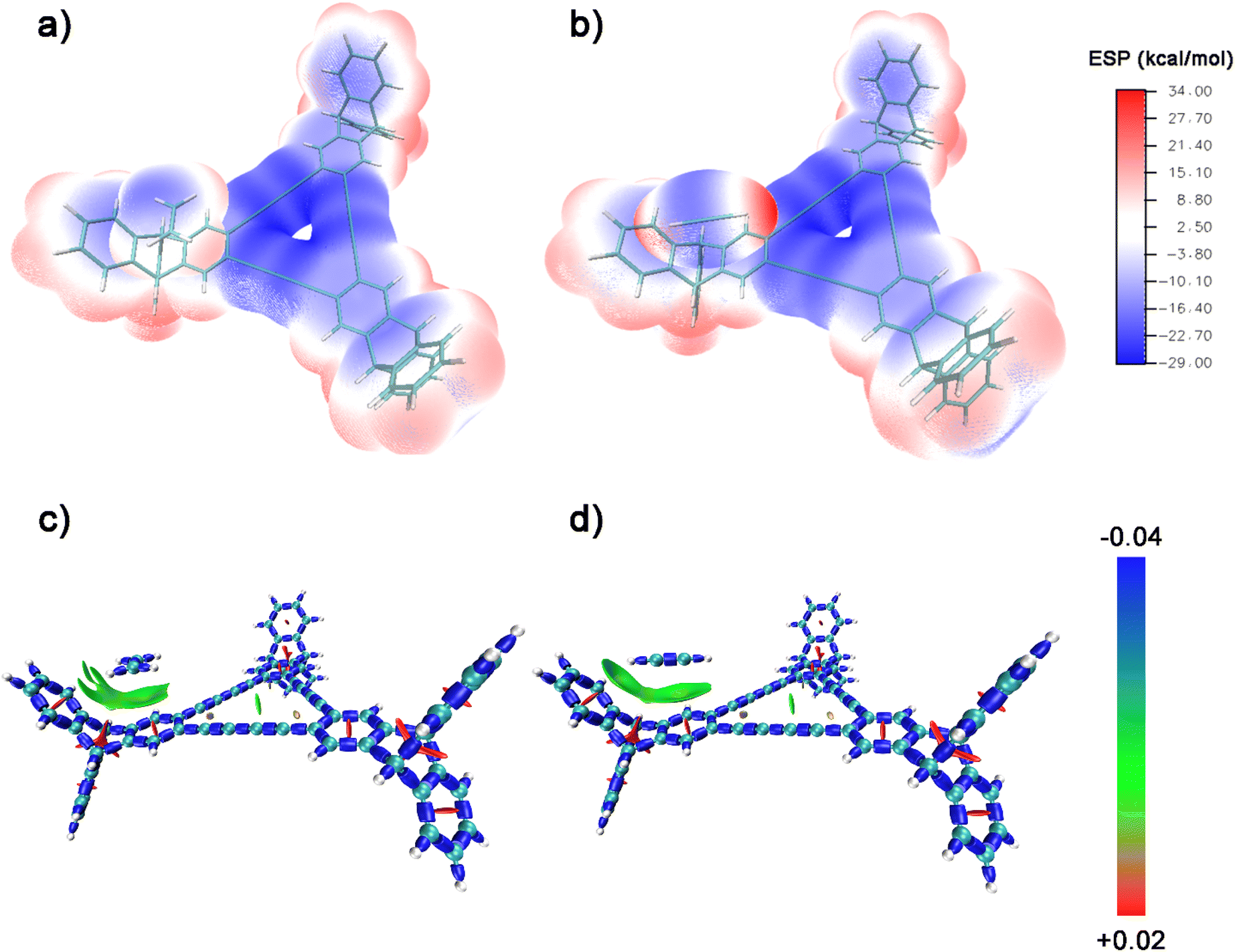 | ||
| Fig. 4 ESP-mapped molecular vdW surface of binding configurations: (a) M1-C2H41 and (b) M1-C2H21. Isosurface maps (IRI = 1.0) of binding configurations: (c) M1-C2H41 and (d) M1-C2H21. In (c) and (d), the isosurface was colored by the value of sign(λ2)ρ in BGR color model, where the blue regions represent notable attractions or chemical bonds, the green regions represent weak interactions like VDW interaction and the red regions represent notable repulsions like steric effect in ring. | ||
In configurations M1-C2H22 and M1-C2H42 (Fig. S14b and d†), the isolated graphdiyne unit could not afford the complete contact with adsorbates, which hindered it from becoming the primary adsorption site (the interaction energies of M1-C2H22 and M1-C2H42 was −13.9 kJ mol−1 and−17.3 kJ mol−1, respectively). For further investigation, two triangular units of graphdiyne and one adsorbate molecule were combined to construct new interaction configurations named as G2-C2H21 and G2-C2H41. The optimized structures were exhibited in Fig. S16.† In the case of acetylene, the triangular units were parallel to each other with a interlayer distance of 3.6 Å and acetylene molecule was perpendicular with the units at the center of the rings. As for G2-C2H41, the dihedral angle of two graphdiyne units was shown as 34.67° to realize the complete contact of the areas with the most positive ESP and the most negative ESP. When the graphdiyne units in G2-C2H41 were froze and the ethylene molecule was replaced with acetylene, a new configuration termed as G2-C2H22 was then obtained through constrained optimization (Fig. S17a†) and the interaction energy was calculated using r2SCAN-3c method as 104.2 kJ mol−1. By contrast with the binding energy of G2-C2H41 (Fig. S17b,† −31.5 kJ mol−1), it could be concluded that in order to realize the complete contact and high binding energy, the ideal three dimensional structures of graphdiyne were disparate for acetylene, ethylene or other gas molecules owing to the slight structural differences. Therefore, by judicious selection of three dimension rigid building blocks participating in the framework of graphdiyne, the selective adsorbents with excellent gas adsorption and separation capacities could be designed and constructed.
Another cage molecule named as M2 was also separated as a model to verify the theoretical calculation results above for the reason that all the phenyl rings of triptycene in G-PTN would participate in the π-conjugated system rather than being isolated in the ideal case. Fig. S18† exhibited the optimized structure of M2, there were two kinds of pores across the molecule with diameters of about 0.66 nm and 1.77 nm, respectively. Taking the atomic volume into account, such sizes were in line with the pore size distribution results calculated from nitrogen adsorption isotherm of G-PTN (5.9 Å and 12.7 Å). The most stable binding configuration of M2 and acetylene (M2-C2H21) was exhibited in Fig. S19a and b† with similar binding mode to M1-C2H21. Employing the same optimized method, M1-C2H2com was obtained for exact comparison with M2-C2H21 (Fig. S19c†). As shown in Fig. S19,† the angles between acetylene and phenyl rings were slightly different in these two configurations, which could result from the electron distribution change during phenyl ring getting involved in π-conjugated system. The binding energies then were calculated using r2SCAN-3c method as −19.1 kJ mol−1 for M2 and −20.0 kJ mol−1 for M1. With extended three dimensional structure, it was clear that triptycene residues could be regarded as cross points of three graphdyine units with dihedral angle of about 120°. For such three dimensional structure, acetylene molecules could more simply realize complete contact with two π-conjugated systems than ethylene, which could result in selective adsorption.
Experimental
The building unit 2,3,6,7,14,15-hexakis [(4-ethynylphenyl) ethynyl]triptycene was synthesized consulting the existing literature66 through the reaction route showed in Scheme S1.† All original reagents were purchased from commercial source without further purification.Synthesis of G-PTN
In a 50 mL pyrex tube, 2,3,6,7,14,15-hexakis[(4-ethynylphenyl) ethynyl]triptycene (310 mg, 0.78 mmol) and cuprous chloride (78 mg, 0.79 mmol) were dissolved in 20 mL pyridine. The mixture was heated to 60 °C, as the reaction processed, brown precipitate was gradually obtained. After 4 days, the reaction system was cooled to room temperature, and the precipitate was filtrated and washed sequentially by dichloromethane, tetrahydrofuran, acetone and ethanol. The product was then obtained after drying in vacuum as brown powder. (303 mg).Computational details
The configurations M1-C2H21, M1-C2H22, M1-C2H41 and M1-C2H42 were screened out through molclus program67 using xtb 6.4.1 (GFN2-xTB method)68 for optimization and energy evaluation. Further constrained optimization were conducted using r2SCAN-3c method69–72 by orca 5.0.3.73–75 The interaction energies were calculated using double-hybrid function PWPB95 in combination with the def2-QZVPP basis set, DFT-D3 dispersion correction and counterpoise correction.76–81 To save computational time, the resolution-of-identity (RI) technique and the auxiliary basis def2/J were used.82The configurations G2-C2H21 and G2-C2H41 were optimized with r2SCAN-3c method in orca 5.0.3. The configuration G2-C2H22 was obtained through replacing the ethylene molecule in G2-C2H41 into acetylene with further constrained optimization freezing all atoms in graphdiyne units by r2SCAN-3c method in orca 5.0.3. The binding energies of G2-C2H22 and G2-C2H41 were both determined at the same calculation level with optimization.
The configurations M2-C2H21 and M1-C2H2com were also screened out through molclus with xtb 6.4.1 (GFN2-xTB method), but did not go through further optimization by density functional theory calculations. The binding energies were evaluated through r2SCAN-3c method in orca 5.0.3.
The ESP colored vdw surfaces were analysed by Multiwfn83–85 and visualised with the VMD visualisation program86 utilizing the wavefunction data obtained from DFT calculations, as well as the IRI isosurface.87
Conclusions
In conclusion, a three dimensional graphdiyne-like porous triptycene network (G-PTN) was synthesized in this work. With high BET surface area of 1046 m2 g−1, its gas sorption capacities for CO2, C2H2 and C2H4 were measured as 51.5 cm3 g−1, 59.6 cm3 g−1 and 44.7 cm3 g−1 at 298 K, respectively, exhibiting selective adsorption capacity toward C2H2 over C2H4. Introducing graphdiyne units into selective adsorbents for C2H2/C2H4 broadened the application potential of graphdiyne, while offered a new design protocol to obtain excellent gas separation materials. Upon adjusting pore size and three-dimensional structure, the separation performance of porous graphdiyne could be further improved and the related work was still under research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (21875079). We thank the Analytical and Testing Center of Huazhong University of Science and Technology for related analysis. We also thank Prof. Yao Yu and Wuhan National High Magnetic Field Center for analysis of solid-state NMR. We thank Dr Zhifang Wang and Prof. Zhenjie Zhang of Nankai University for the acetylene and ethylene adsorption test. The computation is completed in the HPC Platform of Huazhong University of Science and Technology.Notes and references
- Z. Zuo and Y. Li, Joule, 2019, 3, 899–903 CrossRef.
- C. Huang, Y. Li, N. Wang, Y. Xue, Z. Zuo, H. Liu and Y. Li, Chem. Rev., 2018, 118, 7744–7803 CrossRef CAS PubMed.
- X. Gao, H. Liu, D. Wang and J. Zhang, Chem. Soc. Rev., 2019, 48, 908–936 RSC.
- Y. Zhao, L. Chai, X. Yan, W. Huang, T. Fan, O. A. Al-Hartomy, A. Al-Ghamdi, S. Wageh, A. G. Al-Sehemi, Z. Xie and H. Zhang, Mater. Chem. Front., 2022, 6, 528–552 RSC.
- M. M. Haley, S. C. Barand and J. J. Pak, Angew. Chem., Int. Ed. Engl., 1997, 36, 836–838 CrossRef CAS.
- G. Li, Y. Li, H. Liu, Y. Guo, Y. Li and D. Zhu, Chem. Commun., 2010, 46, 3256–3258 RSC.
- Y. Kong, J. Li, S. Zeng, C. Yin, L. Tong and J. Zhang, Chem, 2020, 6, 1933–1951 CAS.
- C. Yin, J. Li, T. Li, Y. Yu, Y. Kong, P. Gao, H. Peng, L. Tong and J. Zhang, Adv. Funct. Mater., 2020, 30, 2001396 CrossRef CAS.
- J. Zhou, Z. Xie, R. Liu, X. Gao, J. Li, Y. Xiong, L. Tong, J. Zhang and Z. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 2632–2637 CrossRef CAS PubMed.
- J. Li, X. Gao, L. Zhu, M. N. Ghazzal, J. Zhang, C.-H. Tung and L.-Z. Wu, Energy Environ. Sci., 2020, 13, 1326–1346 RSC.
- J. He, X. Li, T. Lu, X. Shen, N. Wang and C. Huang, Dalton Trans., 2019, 48, 14566–14574 RSC.
- J. Li, C. Wang, B. Zhang, Z. Wang, W. Yu, Y. Chen, X. Liu, Z. Guo and H. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 49281–49296 CrossRef CAS.
- J. Xu and S. Meng, J. Phys. D: Appl. Phys., 2020, 53, 493003 CrossRef CAS.
- H. Wei, R. Shi, L. Sun, H. Yu, J. Gong, C. Liu, Z. Xu, Y. Ni, J. Xu and W. Xu, Nat. Commun., 2021, 12, 1068 CrossRef CAS PubMed.
- J. Liu, C. Chen and Y. Zhao, Adv. Mater., 2019, 31, 1804386 CrossRef CAS PubMed.
- Y. Jiao, A. Du, M. Hankel, Z. Zhu, V. Rudolph and S. C. Smith, Chem. Commun., 2011, 47, 11843–11845 RSC.
- S. W. Cranford and M. J. Buehler, Nanoscale, 2012, 4, 4587–4593 RSC.
- Z. Meng, X. Zhang, Y. Zhang, H. Gao, Y. Wang, Q. Shi, D. Rao, Y. Liu, K. Deng and R. Lu, ACS Appl. Mater. Interfaces, 2016, 8, 28166–28170 CrossRef CAS PubMed.
- Q. Zheng, G. Luo, Q. Liu, R. Quhe, J. Zheng, K. Tang, Z. Gao, S. Nagase and J. Lu, Nanoscale, 2012, 4, 3990–3996 RSC.
- J. He, N. Wang, Z. Cui, H. Du, L. Fu, C. Huang, Z. Yang, X. Shen, Y. Yi, Z. Tu and Y. Li, Nat. Commun., 2017, 8, 1172 CrossRef PubMed.
- N. Wang, J. He, Z. Tu, Z. Yang, F. Zhao, X. Li, C. Huang, K. Wang, T. Jiu, Y. Yi and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 10740–10745 CrossRef CAS PubMed.
- Y. Dang, W. Guo, L. Zhao and H. Zhu, ACS Appl. Mater. Interfaces, 2017, 9, 30002–30013 CrossRef CAS PubMed.
- D. S. Sholl and R. P. Lively, Nature, 2016, 532, 435–437 CrossRef PubMed.
- C. Y. Chuah, H. Lee and T.-H. Bae, Chem. Eng. J., 2022, 430, 132654 CrossRef CAS.
- Y. He, R. Krishna and B. Chen, Energy Environ. Sci., 2012, 5, 9107–9120 RSC.
- L. Yang, S. Qian, X. Wang, X. Cui, B. Chen and H. Xing, Chem. Soc. Rev., 2020, 49, 5359–5406 RSC.
- E. D. Bloch, W. L. Queen, R. Krishna, J. M. Zadrozny, C. M. Brown and J. R. Long, Science, 2012, 335, 1606–1610 CrossRef CAS PubMed.
- L. Li, R.-B. Lin, R. Krishna, H. Li, S. Xiang, H. Wu, J. Li, W. Zhou and B. Chen, Science, 2018, 362, 443–446 CrossRef CAS.
- Y. Yang, L. Li, R.-B. Lin, Y. Ye, Z. Yao, L. Yang, F. Xiang, S. Chen, Z. Zhang, S. Xiang and B. Chen, Nat. Chem., 2021, 13, 933–939 CrossRef CAS PubMed.
- R.-B. Lin, L. Li, H.-L. Zhou, H. Wu, C. He, S. Li, R. Krishna, J. Li, W. Zhou and B. Chen, Nat. Mater., 2018, 17, 1128–1133 CrossRef CAS.
- B. Zhu, J.-W. Cao, S. Mukherjee, T. Pham, T. Zhang, T. Wang, X. Jiang, K. A. Forrest, M. J. Zaworotko and K.-J. Chen, J. Am. Chem. Soc., 2021, 143, 1485–1492 CrossRef CAS PubMed.
- J. Wang, Y. Zhang, P. Zhang, J. Hu, R.-B. Lin, Q. Deng, Z. Zeng, H. Xing, S. Deng and B. Chen, J. Am. Chem. Soc., 2020, 142, 9744–9751 CAS.
- X. Cui, K. Chen, H. Xing, Q. Yang, R. Krishna, Z. Bao, H. Wu, W. Zhou, X. Dong, Y. Han, B. Li, Q. Ren, M. J. Zaworotko and B. Chen, Science, 2016, 353, 141–144 CrossRef CAS PubMed.
- O. T. Qazvini, R. Babarao and S. G. Telfer, Chem. Mater., 2019, 31, 4919–4926 CrossRef CAS.
- T.-L. Hu, H. Wang, B. Li, R. Krishna, H. Wu, W. Zhou, Y. Zhao, Y. Han, X. Wang, W. Zhu, Z. Yao, S. Xiang and B. Chen, Nat. Commun., 2015, 6, 7328 CrossRef CAS PubMed.
- J. Lee, C. Y. Chuah, J. Kim, Y. Kim, N. Ko, Y. Seo, K. Kim, T. H. Bae and E. Lee, Angew. Chem., Int. Ed., 2018, 57, 7869–7873 CrossRef CAS PubMed.
- Y. Lu, J. He, Y. Chen, H. Wang, Y. Zhao, Y. Han and Y. Ding, Macromol. Rapid Commun., 2018, 39, 1700468 CrossRef PubMed.
- X. Suo, X. Cui, L. Yang, N. Xu, Y. Huang, Y. He, S. Dai and H. Xing, Adv. Mater., 2020, 32, 1907601 CrossRef CAS.
- M.-J. Gu, Y.-F. Wang, Y. Han and C.-F. Chen, Org. Biomol. Chem., 2021, 19, 10047–10067 RSC.
- M. Woźny, A. Mames and T. Ratajczyk, Molecules, 2022, 27, 250 CrossRef PubMed.
- T. Iwata and M. Shindo, Chem. Lett., 2021, 50, 39–51 CrossRef CAS.
- L. Ueberricke and M. Mastalerz, Chem. Rec., 2021, 21, 558–573 CrossRef CAS.
- C.-F. Chen and Y. Han, Acc. Chem. Res., 2018, 51, 2093–2106 CrossRef CAS.
- Y. Gisbert, S. Abid, C. Kammerer and G. Rapenne, Chem.–Eur. J., 2021, 27, 12019–12031 CrossRef CAS PubMed.
- G.-W. Zhang, P.-F. Li, Z. Meng, H.-X. Wang, Y. Han and C.-F. Chen, Angew. Chem., Int. Ed., 2016, 55, 5304–5308 CrossRef CAS.
- Y. Han, Z. Meng, Y. X. Ma and C.-F. Chen, Acc. Chem. Res., 2014, 47, 2026–2040 CrossRef CAS PubMed.
- Z. Meng, Y. Han, L. N. Wang, J. F. Xiang, S. G. He and C.-F. Chen, J. Am. Chem. Soc., 2015, 137, 9739–9745 CrossRef CAS PubMed.
- H. X. Wang, Z. Meng, J. F. Xiang, Y. X. Xia, Y. Sun, S. Z. Hu, H. Chen, J. Yao and C.-F. Chen, Chem. Sci., 2016, 7, 469–474 RSC.
- Y. F. Wang, M. Li, J. M. Teng, H. Y. Zhou, W. L. Zhao and C.-F. Chen, Angew. Chem., Int. Ed., 2021, 60, 23619–23624 CrossRef CAS PubMed.
- K. Kawasumi, T. Wu, T. Zhu, H. S. Chae, T. Van Voorhis, M. A. Baldo and T. M. Swager, J. Am. Chem. Soc., 2015, 137, 11908–11911 CrossRef CAS.
- M. Mastalerz, Acc. Chem. Res., 2018, 51, 2411–2422 CrossRef CAS PubMed.
- M. Mastalerz and I. M. Oppel, Angew. Chem., Int. Ed., 2012, 51, 5252–5255 CrossRef CAS PubMed.
- P. Li, P. Li, M. R. Ryder, Z. Liu, C. L. Stern, O. K. Farha and J. F. Stoddart, Angew. Chem., Int. Ed., 2019, 58, 1664–1669 CrossRef CAS PubMed.
- H. Li, F. Chen, X. Guan, J. Li, C. Li, B. Tang, V. Valtchev, Y. Yan, S. Qiu and Q. Fang, J. Am. Chem. Soc., 2021, 143, 2654–2659 CrossRef CAS PubMed.
- Z. Chen, P. Li, R. Anderson, X. Wang, X. Zhang, L. Robison, L. R. Redfern, S. Moribe, T. Islamoglu, D. A. Gomez-Gualdron, T. Yildirim, J. F. Stoddart and O. K. Farha, Science, 2020, 368, 297–303 CrossRef CAS PubMed.
- J. Mahmood, S.-J. Kim, H.-J. Noh, S.-M. Jung, I. Ahmad, F. Li, J.-M. Seo and J.-B. Baek, Angew. Chem., Int. Ed., 2018, 57, 3415–3420 CrossRef CAS PubMed.
- J. Mahmood, M. A. R. Anjum, S.-H. Shin, I. Ahmad, H.-J. Noh, S.-J. Kim, H. Y. Jeong, J. S. Lee and J.-B. Baek, Adv. Mater., 2018, 30, 1805606 CrossRef PubMed.
- S.-Y. Bae, D. Kim, D. Shin, J. Mahmood, I.-Y. Jeon, S.-M. Jung, S.-H. Shin, S.-J. Kim, N. Park, M. S. Lah and J.-B. Baek, Nat. Commun., 2017, 8, 1599 CrossRef PubMed.
- H.-J. Noh, Y.-K. Im, S.-Y. Yu, J.-M. Seo, J. Mahmood, T. Yildirim and J.-B. Baek, Nat. Commun., 2020, 11, 2021 CrossRef CAS.
- H. Ma, Z. Wang, Y.-H. Zhao, Q. Ou, B. Tan and C. Zhang, Sci. China Mater., 2020, 63, 429–436 CrossRef CAS.
- J.-J. Chen, T.-L. Zhai, Y.-F. Chen, S.-N. Geng, C. Yu, J.-M. Liu, L.-L. Wang, B. Tan and C. Zhang, Polym. Photochem., 2017, 8, 5533–5538 CAS.
- H. Ma, J.-J. Chen, L. Tan, J.-H. Bu, Y. Zhu, B. Tan and C. Zhang, ACS Macro Lett., 2016, 5, 1039–1043 CrossRef CAS PubMed.
- Q.-P. Zhang, Z. Wang, Z.-W. Zhang, T.-L. Zhai, J.-J. Chen, H. Ma, B. Tan and C. Zhang, Angew. Chem., Int. Ed., 2021, 60, 12781–12785 CrossRef CAS PubMed.
- A. L. Myers and J. M. Prausnitz, AIChE J., 1965, 11, 121–127 CrossRef CAS.
- P. Lacomi and P. L. LIewellyn, Adsorption, 2019, 25, 1533–1542 CrossRef.
- C. Moylan, L. Rogers, Y. M. Shaker, M. Davis, H.-G. Eckhardt, R. Eckert, A. A. Ryan and M. O. Senge, Eur. J. Org. Chem., 2016, 185–195 CrossRef CAS.
- T. Lu, Molclus program, Version 1.9.9.6http://www.keinsci.com/research/molclus.html, accessedSep152021 Search PubMed.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- S. Grimme, A. Hansen, S. Ehlert and J.-M. Mewes, J. Chem. Phys., 2021, 154, 064103 CrossRef CAS PubMed.
- H. Kruse and S. Grimme, J. Chem. Phys., 2012, 136, 154101 CrossRef.
- E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer and S. Grimme, J. Chem. Phys., 2017, 147, 034112 CrossRef.
- J. W. Furness, A. D. Kaplan, J. Ning, J. P. Perdew and J. Sun, J. Phys. Chem. Lett., 2020, 11, 8208–8215 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 8, e1327 Search PubMed.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- L. Goerigk and S. Grimme, J. Chem. Theory Comput., 2011, 7, 291–309 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. Hellweg, C. Hattig, S. Hofener and W. Klopper, Theor. Chem. Acc., 2007, 117, 587–597 Search PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 2002, 100, 65–73 CrossRef.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Mol. Graphics Modell., 2012, 38, 314–323 CrossRef CAS PubMed.
- J. Zhang and T. Lu, Phys. Chem. Chem. Phys., 2021, 23, 20323–20328 RSC.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
- T. Lu and Q. Chen, Chem. Methods, 2021, 1, 231–239 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra04031j |
| This journal is © The Royal Society of Chemistry 2022 |