
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bioorthogonal site-selective conjugation of fluorescent dyes to antibodies: method and potential applications†
Philipp Grossenbacher a,
Maria C. Essersa,
Joël Moserb,
Simon A. Singera,
Stephanie Häuslerc,
Bruno Stiegerc,
Jean-Sébastien Rougiera and
Martin Lochner*a
a,
Maria C. Essersa,
Joël Moserb,
Simon A. Singera,
Stephanie Häuslerc,
Bruno Stiegerc,
Jean-Sébastien Rougiera and
Martin Lochner*a
aInstitute of Biochemistry and Molecular Medicine, University of Bern, Bühlstrasse 28, 3012 Bern, Switzerland. E-mail: martin.lochner@ibmm.unibe.ch
bDepartment of Chemistry, Biochemistry and Pharmaceutical Sciences, University of Bern, Freiestrasse 3, 3012 Bern, Switzerland
cDepartment of Clinical Pharmacology and Toxicology, University Hospital Zürich, University of Zürich, Rämistrasse 100, 8091 Zürich, Switzerland
First published on 5th October 2022
Abstract
Antibodies are immensely useful tools for biochemical research and have found application in numerous protein detection and purification methods. Moreover, monoclonal antibodies are increasingly utilised as therapeutics or, conjugated to active pharmaceutical ingredients, in targeted chemotherapy. Several reagents and protocols are reported to synthesise fluorescent antibodies for protein target detection and immunofluorescence applications. However, most of these protocols lead to non-selective conjugation, over-labelling or in the worst case antigen binding site modification. Here, we have used the antibody disulphide cleavage and re-bridging strategy to introduce bright fluorescent dyes without loss of the antibody function. The resulting fluorescent IgG1 type antibodies were shown to be effective imaging tools in western blot and direct immunofluorescence experiments.
Introduction
The use of antibody drug conjugates (ADCs) has seen a substantial rise in recent times. They offer precise treatment options with fewer side effects, as the payload is guided by the antibody to its specific target.1 In the wake of this development, various techniques have been established, moving from highly heterogenic mixtures to precise homogenous conjugate constructs using monoclonal antibodies with specified drug-to-antibody ratios (DARs) and controlled sites of attachment. For instance, unnatural amino acids can be used to engineer bioorthogonal functional groups into the monoclonal antibody, which subsequently enable site-selective and efficient coupling reactions with a variety of modified drugs.2Traditionally, native antibody modifications have relied on the chemical reactivity of amino acid side chains, and numerous chemical reagents and methods have been developed to conjugate drug molecules to such reactive groups (e.g. the primary amine group of lysine residues).3 Such chemical reagent-based methods often produce heterogeneous mixtures of modified antibodies and involve the risk of over-labelling and altering the antigen binding sites, which results in inactive antibody conjugates. Alternatively, more recent strategies entail the targeting of glycans in native antibodies to guide modifications away from the antigen binding sites.4 Thiols are useful functional groups for protein bioconjugation as they react efficiently with widely used and available maleimides. Thiol-containing antibodies can be obtained through cysteine-engineering5 or generated by full or partial reduction of disulphide bonds, and reacted site-specifically with maleimide reagents.2 A potential drawback of maleimide conjugates is their propensity to undergo retro-Michael addition reactions and transfer their payload onto plasma thiols.6 Another viable approach is to specifically target the interchain disulphide bonds, in particular in IgG1 antibodies. In this subtype, four solvent-accessible disulphide bonds can be cleaved by mild, biocompatible reducing agents (e.g. TCEP or DTT) and then re-bridged using various bis-reactive cysteine reagents that carry the payload or functional groups for subsequent conjugation reactions.2,4 Several re-bridging strategies and agents have been developed, including bissulfones,7,8 divinylsulfonamides,9 arylene dipropiolonitriles (ADPN),10 dibromomethyl heterocycles (C-Lock™),11 dichloroacetone,12 next-generation maleimides,13–15 pyridazinediones16–22 and divinylpyrimidines.23–25
Immunofluorescence applications in general require fluorescently labelled antibodies. Detection of the cellular target can either be achieved directly, using a primary labelled antibody, or indirectly, by using a secondary labelled antibody that recognises the primary (unlabelled) one. Numerous commercial suppliers offer primary or secondary fluorescently labelled antibodies that were raised in different species. The selection of the primary antibody is one of the most important steps to achieve successful experimental outcome in western blot and immunofluorescence applications. Despite the wide variety of commercial antibodies, in many cases antibodies against a specific protein of interest are not available and first have to be generated, validated, and depending on the intended application, fluorescently labelled.
The common workflow in immunofluorescence includes the use of a primary antibody that binds specifically to the cellular target, followed by detection of the primary antibody with a fluorescently labelled secondary antibody. While this method works for most applications, there are limitations that negatively affect experimental outcome (Table 1). Direct immunofluorescence can provide an alternative in such situations; however, it does not come without technical issues either (Table 1).
| Method | Advantages | Disadvantages |
|---|---|---|
| Direct immunofluorescence | • Simplified workflow and shorter sample staining time | • Lower sensitivity for low-abundance targets with limited number of fluorescent dyes that can be attached to primary antibody |
| • No cross-reactivity between secondary antibodies | • Limited number of commercially available fluorescently labelled primary antibodies | |
| • Best solution for specific staining of multiple targets | ||
| Indirect immunofluorescence | • Greater sensitivity for low-abundance targets due to signal amplification by secondary antibodies | • Cross-reactivity between secondary antibodies, in particular when performing multilabel experiments |
| • Numerous inexpensive secondary antibodies commercially available with various dyes and tags attached | • Reaction of secondary antibodies with endogenous immunoglobulins leading to high background fluorescence |
The goal of our study was to provide a practical method to site-specifically label native antibodies with bright fluorescent dyes by targeting the solvent-accessible interchain disulphide bonds. In most examples such conjugations have been shown on monoclonal IgG1 therapeutic antibody trastuzumab (Herceptin®),9,13–21,24 and recently monoclonal IgG4 magacizumab.22 In this work, we explored if the modification approach is more widely applicable to other mono- and polyclonal IgG1 antibodies and validated the fluorescently labelled products in western blot and direct immunofluorescence applications.
Results and discussion
Chemistry
We first synthesised small and versatile re-bridging agent 3, featuring an alkyne clickable handle for fluorescent dye conjugation, by adapting synthetic routes published for very similar dibromopyridazinediones.18,20,26,27 Methylhydrazine was first Boc-protected and then alkylated with propargyl bromide on the remaining free carbamate nitrogen (Scheme 1).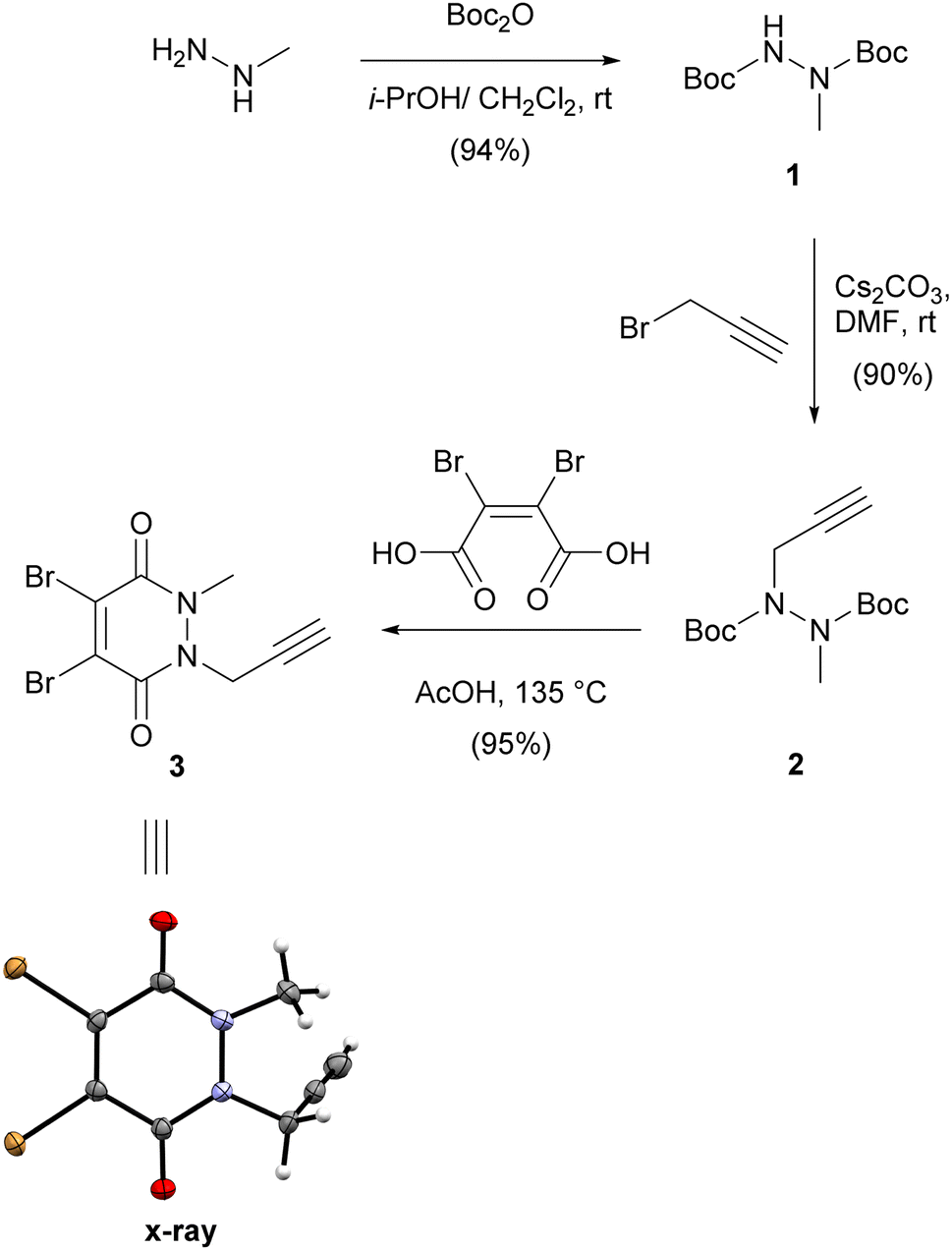 | ||
| Scheme 1 Synthesis of propargyl dibromopyridazinedione 3 and X-ray structure thereof. | ||
This was followed by a one-pot deprotection and cyclisation reaction by heating bis-carbamate 2 with dibromomaleic acid in acetic acid at refluxing temperature.20 Overall, this synthetic route delivered dibromopyridazinedione 3 in 80% yield over three steps as a crystalline solid. We were able to obtain a crystal structure of 3 to confirm the identity of the compound (Scheme 1).
We then attached various azido linkers via activated N-hydroxysuccinimid (NHS) esters to different fluorescent dyes that emit bright light in the red/far-red (Si-rhodamine, SiR)28 or blue region (Pacific Blue, PB), or to biotin (Scheme 2A). Azide 5 with a bright emission in the green region (BODIPY FL, BFL) was purchased from a commercial supplier.
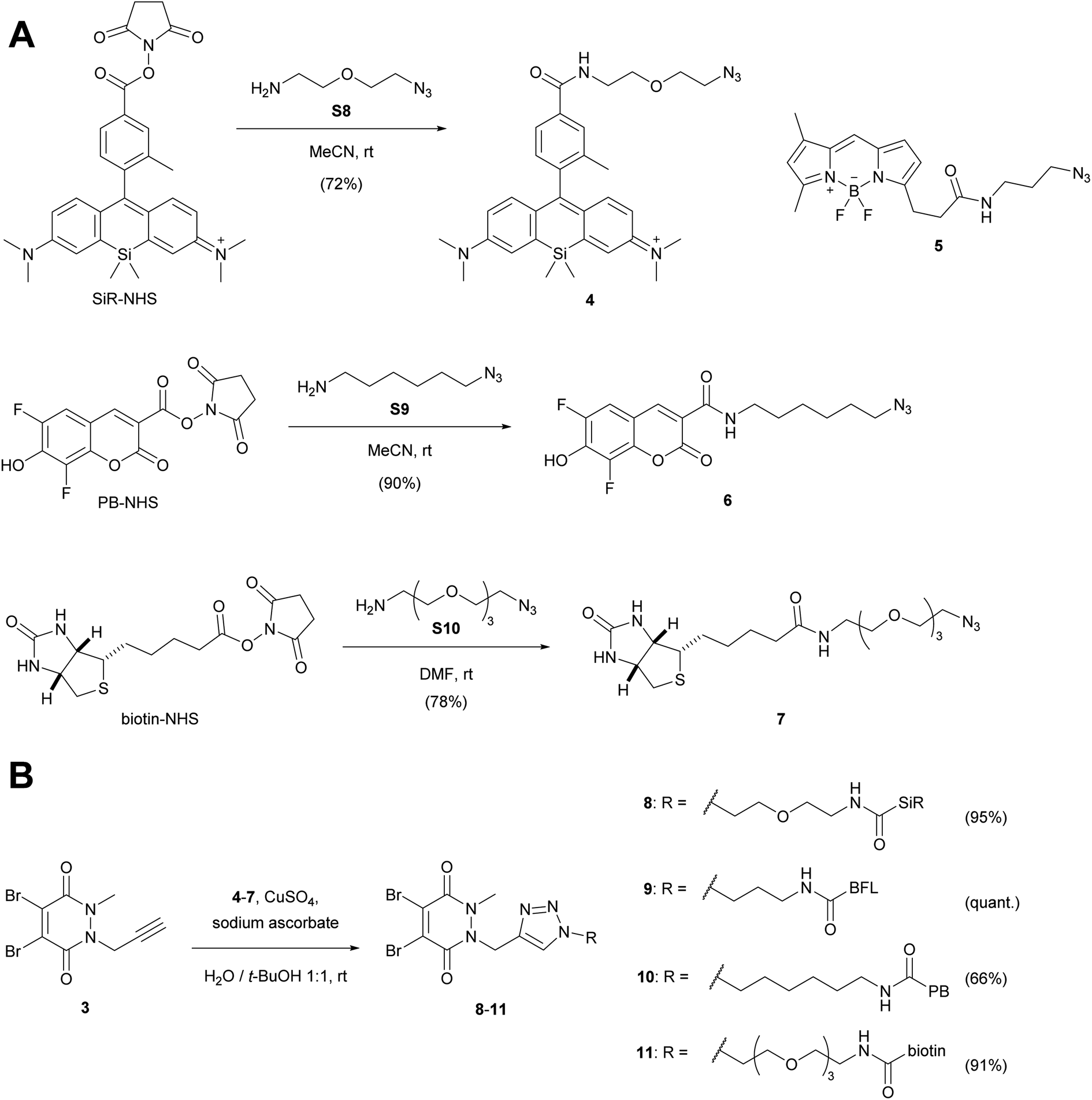 | ||
| Scheme 2 (A) Attachment of linkers to fluorescent dyes and biotin. (B) CuAAC-coupling with alkyne dibromopyridazinedione 3. For the synthesis of SiR-NHS (Scheme S1†), biotin-NHS and the azide linkers S8–S10 see ESI.† PB-NHS was commercially available. SiR, Si-rhodamine; PB, Pacific Blue; BFL, BODIPY FL. | ||
The resulting constructs 4–7 were then coupled to pyridazinedione alkyne 3 using standard Cu-catalysed alkyne–azide cycloaddition (CuAAC) conditions, which gave the final re-bridging agents 8–11 in good to very good yields.
Originally, our synthetic plan was to couple the SiR dye with alternative dibromopyridazinediones via NHS-activated esters, rather than using CuAAC coupling chemistry (Scheme S2, ESI†). Even though the amide forming reactions were successful, one bromine atom was displaced by the NHS leaving group. We therefore speculate that this side reaction might be a potential generic risk when working with dibromopyridazinediones in the presence of NHS-activated esters, although these undesired products might still work as re-bridging agents.
Anti GAPDH antibody modification
Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) is a key enzyme in the glycolytic pathway. It catalyses the conversion of glyceraldehyde-3-phosphate to 1,3-biphopshoglycerate in the presence of NAD+ and inorganic phosphate. In biochemistry labs, GAPDH is commonly used as house-keeping gene in semi-quantitative or quantitative studies to confirm expression or suppression of proteins of interest in proteomic or genetic studies. Apart from its practical utility and important role in carbohydrate metabolism, several studies have shown that GAPDH also binds single-stranded DNA and in some cases acts as a transcription factor.29 Furthermore, GAPDH can initiate apoptosis but can also function as a mediator of cell survival. Other studies thus point to the fact that GAPDH could be an important marker in age-related neurodegenerative diseases, prostate cancer, and viral diseases.30,31For the initial antibody conjugation experiments, we chose a commercially available monoclonal anti GAPDH loading control antibody and followed the reported disulphide re-bridging protocol20 with some slight experimental modifications. The anti GAPDH antibody was incubated with TCEP (10 equiv.) and excess (20 equiv.) dibromopyridazinedione re-bridgers 8–11 in borate buffered saline (BBS) at 4 °C overnight (Fig. 1A). Isolation of the modified antibodies was conveniently achieved by using spin filter columns. Fig. 1B and C show a representative analysis of the SiR-modified anti GAPDH antibody by SDS-PAGE. Two strongly fluorescent bands at 25 kDa and 50 kDa were detected, which correspond to the fluorescently labelled light and heavy chains, respectively (Fig. 1B, lane C-AB).
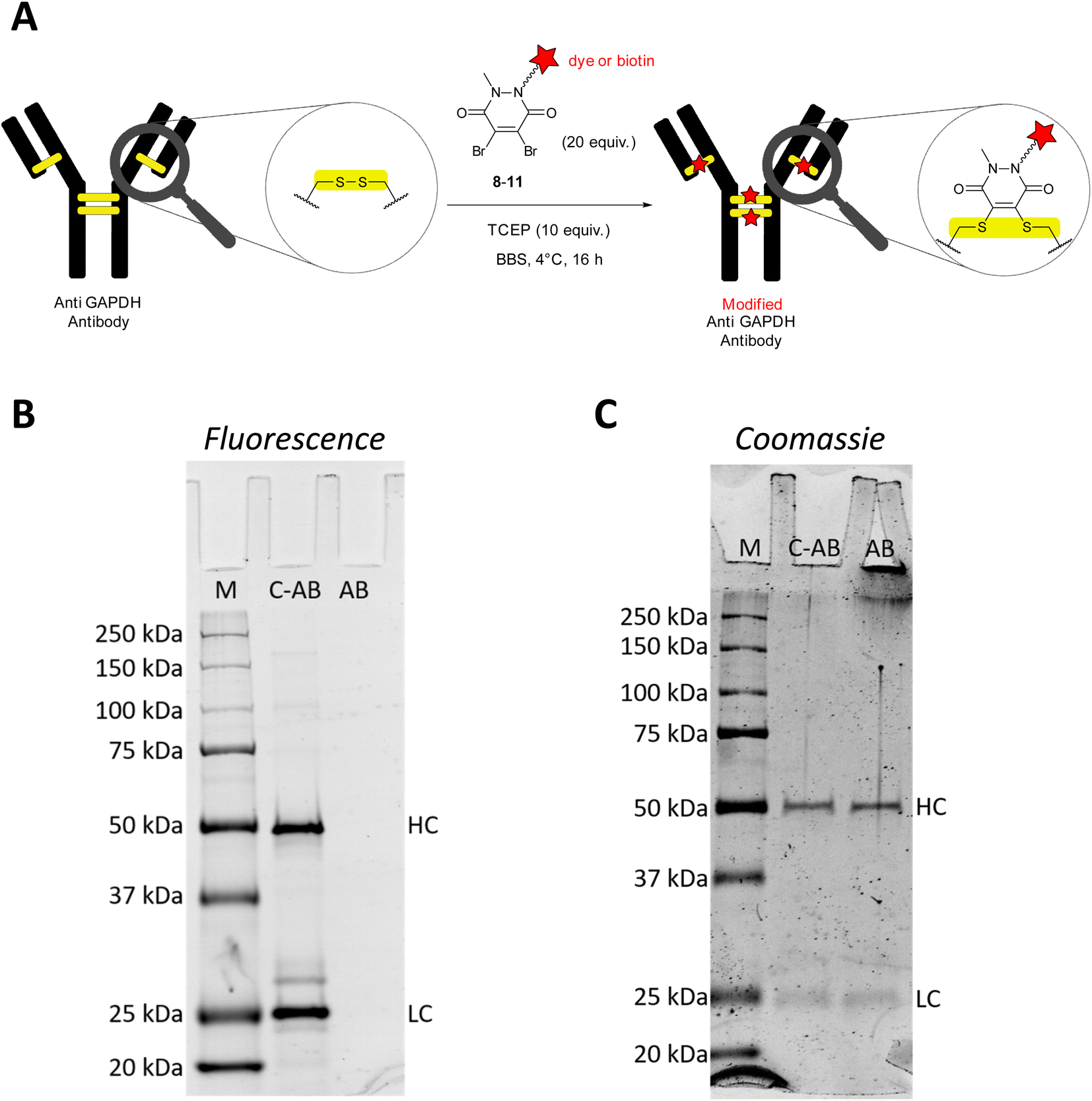 | ||
| Fig. 1 (A) Modification of anti GAPDH antibody by disulphide bond cleavage and re-bridging with dibromopyridazinediones 8–11. (B) Representative SDS-PAGE (reducing conditions) of conjugation reaction of anti GAPDH antibody with SiR-labelled 8. Detection of protein bands by fluorescence (excitation at 635 nm, Cy5 channel). Bands at 50 kDa and 25 kDa correspond to the heavy (HC) and light chains (LC), respectively. (C) SDS-PAGE as in (B), stained with Coomassie blue dye. Loading concentration of C-AB for in-gel fluorescence (B) was tenfold lower than for Coomassie gel (C) to avoid oversaturation of the fluorescence detector. Lanes: M, all Blue Protein Standard; C-AB, SiR-conjugated anti GAPDH antibody; AB, native anti GAPDH antibody. | ||
During the reductive and denaturing SDS-PAGE sample preparation (using 10% mercaptoethanol in the loading buffer and heating), excess thiol presumably leads to disulphide cleavage and antibody fragmentation. The same two bands at 25 kDa and 50 kDa were also detected when a native anti GAPDH antibody sample was prepared analogously for SDS-PAGE and stained with Coomassie (Fig. 1C, lane AB). Omitting the mercaptoethanol in the SDS-PAGE loading buffer for the SiR-conjugated antibody sample resulted in several fluorescent bands at very high molecular weight at around 250 kDa (Fig. S1, ESI†), most probably resulting from incomplete linearisation and aggregation. We therefore deemed the non-reducing SDS-PAGE conditions as less suitable for conjugation reaction control. Due to the very bright fluorescence of the SiR dye, the conjugated antibody SDS-PAGE sample had to be diluted down tenfold to avoid oversaturation of the fluorescence detector (Fig. 1B). It is worth noting that at this low sample concentration Coomassie staining was almost unable to detect the same protein bands.
In addition to SDS-PAGE analysis, we have used size exclusion chromatography (SEC) to characterise the antibody conjugation products. The SEC traces of BFL-modified anti GAPDH antibody after re-bridging with dibromopyridazinedione 9 show the same peaks with very similar retention times as SEC traces of native anti GAPDH antibody (Fig. S2†). This leads us to conclude that the antibody remains intact and a fully re-bridged antibody is obtained. Consequently, it is more likely that the fragmentation of the modified antibodies occurs during the reductive SDS-PAGE sample preparation.
Validation of modified GAPDH antibody and immunofluorescence
To assess the specificity of the modified anti GAPDH antibodies, they were tested in western blot applications, using mouse cardiomyocytes whole cell lysates (Fig. 2). After incubation and blotting, SiR- and BFL-modified antibodies were detected directly by fluorescence emission (Fig. 2A and B), whereas biotin-modified antibody was visualised by complexing with a streptavidin-Cy5 construct (Fig. 2C). In all cases a strong band at around 36 kDa was detected, which is in good agreement with the calculated molecular weight of mouse GAPDH (35.81 kDa).32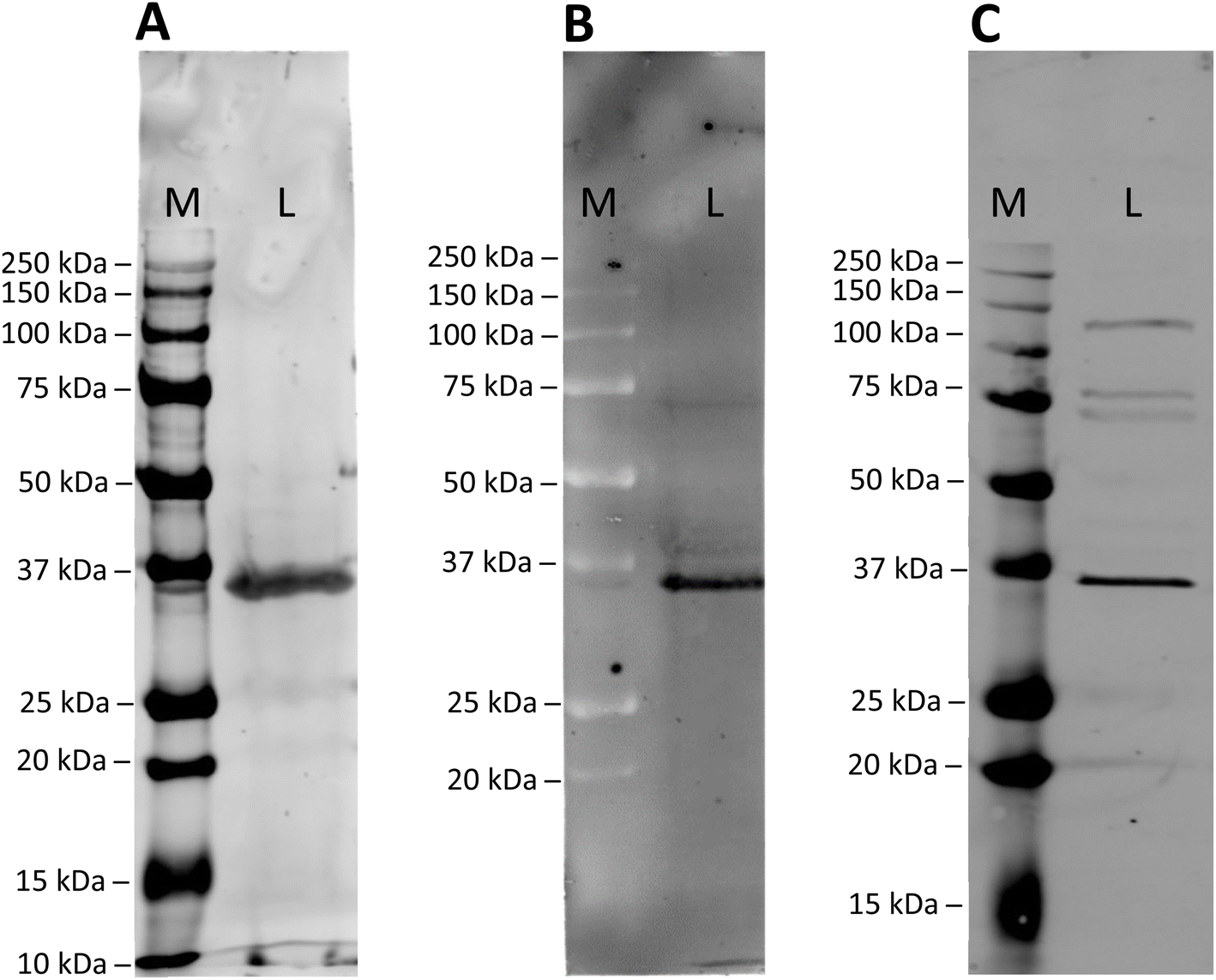 | ||
| Fig. 2 Western blot analysis of mouse cardiomyocyte whole cell lysate using (A) SiR-modified, (B) BFL-modified or (C) biotinylated anti GAPDH antibody. Antibodies were detected directly by fluorescence (A and B; Cy5 and FITC channel, respectively) or indirectly by complexing with streptaviding-Cy5 contstruct (C). The band at 36 kDa corresponds to the molecular weight of mouse GAPDH. Lanes: M, Marker; L, Lysate. | ||
For comparison, we have employed a classical antibody modification method and stochastically modified anti GAPDH antibody with electrophilic, fluorescent reagent SiR-NHS. SDS-PAGE analysis of this directly labelled antibody product revealed the same fluorescent bands for the light and heavy chains (Fig. S3A, ESI†), as for SiR-modified anti GAPDH antibody obtained by re-bridging with dibromopyridazinedione 8 (Fig. 1B). It was noticeable, however, that the fluorescence intensity of the directly labelled antibody was markedly lower than of the re-bridged antibody. More importantly, the directly SiR-labelled antibody failed to detect its antigen GAPDH in whole cell lysate (Fig. S3B, ESI†).
The modified antibodies were then further assessed for their labelling utility in immunofluorescence experiments (Fig. 3). The images show isolated and fixed mouse cardiomyocytes incubated with modified and native anti GAPDH antibodies. Direct immunofluorescence with SiR- and BFL-conjugated antibodies clearly showed the cytosolic localisation of GAPDH in cardiomyocytes (Fig. 3A and D, respectively) compared to control (Fig. 3C). Alternatively, labelling with biotinylated antibody and subsequent incubation with streptavidin-Cy5 fusion construct gave the same staining pattern (Fig. 3B). Direct immunofluorescence staining was very similar to classical indirect immunofluorescence, using primary native anti GAPDH antibody and fluorescently labelled secondary anti mouse antibody (Fig. 3E). In another indirect immunofluorescence control experiment, we have used anti GAPDH antibody, which was re-bridged with alkyne dibromopyridazinedione 3, as primary antibody and a fluorescent secondary anti mouse antibody for staining (Fig. 3F). In this case the staining was again similar, albeit less strong, compared with direct (Fig. 3D) or indirect immunofluorescence (Fig. 3E).
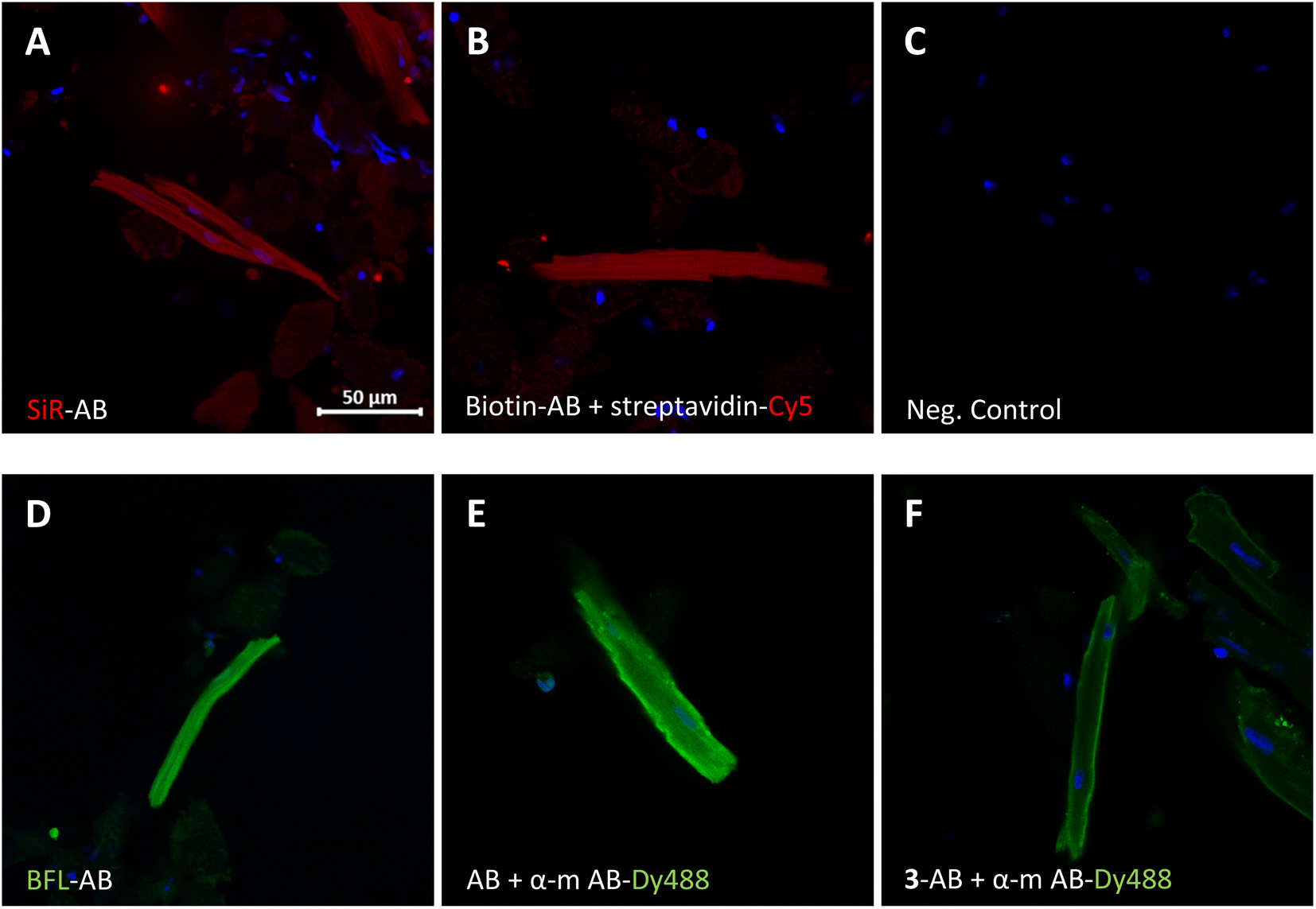 | ||
| Fig. 3 Direct (A and D) and indirect (B, E and F) immunofluorescence with isolated and fixed mouse cardiomyocytes using conjugated and native anti GAPDH antibodies. (A) SiR-modified and (B) biotinylated anti GAPDH antibody. (C) Negative control (DAPI and Cy5 Scan). (D) BFL-modified and (E) native anti GAPDH antibody. (F) Anti GAPDH antibody re-bridged with alkyne dibromopyridazinedione 3. Detection of antibodies by fluorescence (A and D; Cy5 and FITC channel, respectively), by complexing with streptaviding-Cy5 contstruct (B) or by secondary goat anti mouse Dy488-labelled antibodies (E and F). Cell nuclei were stained with DAPI. SiR, Si-rhodamine; BFL, BODIPY FL; Dy488, DyLight® 488; AB, anti GAPDH antibody; α-m AB, anti mouse antibody. Scale bar represents 50 μm. | ||
Modification of other IgG antibodies
We explored the general applicability of the fluorescence modification method with other commercially available or custom-generated IgG antibodies. The targeted antigens included a cytoskeletal protein (β-actin), cardiac ion channels (Nav1.5, Cav1.2, Transient receptor potential cation channel subfamily M member 4 (TRPM4)) and a transmembrane ion pump (Na+/K+-ATPase). The respective IgG antibodies were conjugated with re-bridgers 8 or 9 according to the method shown in Fig. 1A and evaluated in western blot and direct immunofluorescence experiments (Table 2).| Entry | Antigen | Antibody Typea | Tagb | Western Blotc | Direct Immunofluorescencec |
|---|---|---|---|---|---|
| a m, mouse; r, rabbit; mc, monoclonal; pc, polyclonal.b SiR, Si-rhodamine; BFL, BODIPY FL, PB, Pacific Blue.c Y, specific detection or staining of antigen; N, no detection or staining of antigen.d Suitable detection system was not available.e No information available from supplier.f Subtype(s) not known.g Western blot not attempted with SiR-modified antibody, as native antibody did not give signal.h Native antibody did also not give specific staining in indirect immunofluorescence with secondary fluorescent antibody. Experiments in mouse heart sections showed similar inconclusive results for both SiR-modified and native anti TRPM4 antibodies. | |||||
| 1 | GAPDH | IgG1 (m, mc) | SiR | Y | Y |
| 2 | GAPDH | IgG1 (m, mc) | BFL | Y | Y |
| 3 | GAPDH | IgG1 (m, mc) | Biotin | Y | Y |
| 4 | GAPDH | IgG1 (m, mc) | PB | —d | —d |
| 5 | β-Actin | IgG1 (m, mc) | SiR | Y | Y |
| 6 | Nav1.5 | —e | BFL | N | Y |
| 7 | Cav1.2 | IgGf (r, pc) | SiR | —g | Y |
| 8 | Na+/K+-ATPase | IgG1 (m, mc) | SiR | N | Y |
| 9 | TRPM4 | IgGf (r, pc) | SiR | Y | Nh |
In all examples, successful conjugation of fluorescent tags SiR or BFL was evident based on SDS-PAGE of the isolated antibody modification products (Fig. S4, ESI†). For instance, we obtained a SiR-modified anti β-actin antibody that was able to specifically detect its antigen in a cell lysate (entry 5) and gave very clear staining in direct immunofluorescence with mouse cardiomyocytes (Fig. S5 and S6, ESI†). Control indirect immunofluorescence using the native anti β-actin antibody and secondary Dy488-conjugated antibody produced very similar signal. For the studied ion channels and ion pump, the method failed to produce tools to detect Nav1.5, Cav1.2 or Na+/K+-ATPase in cell lysate by western blot (entries 6–8). However, specific direct immunofluorescence signal was clearly obtained with BFL-modified anti Nav1.5, SiR-modified anti Cav1.2 and anti Na+/K+-ATPase antibodies (Fig. S5 and S6, ESI†). It is noteworthy, that the anti Cav1.2 staining tool was obtained from a custom-generated polyclonal antibody population. Conversely, modification of polyclonal anti TRPM4 antibody yielded a product that gave a strong and specific signal in western blot, but direct immunofluorescence was inconclusive (entry 9). In this particular case, TRPM4 detection by indirect immunofluorescence or fluorescence-assisted cell sorting (FACS), using native polyclonal anti TRPM4 antibody and secondary fluorescent antibody, was not conclusive either.
Conclusions
The growing field of ADCs has produced several useful antibody conjugation protocols, some of which selectively target the interchain disulphide bridges. We have adapted the dithiol re-bridging dibromopyridazinedione reagents to introduce bright fluorescent dyes and biotin tags site-selectively into IgG1 type antibodies using a straightforward protocol. A further option, which we have not tested experimentally, would be to re-bridge with general reagent 3 and subsequently “click” the tag of choice to the alkyne-modified antibody.The synthesised anti GAPDH antibody conjugates were shown to give strong and specific signals in western blot and direct immunofluorescence applications. Our produced modified anti GAPDH antibodies could be further evaluated as potential diagnostic tools in various disease models, where GAPDH was proposed as important marker for cell survival.30,31
It appears that the thiol re-bridging modification protocol is not generally transferable to other IgG1 antibodies and the source and quality of the native IgG1 antibody seems to play an important role. It is challenging to generate an imaging antibody tool that works equally well and gives specific labelling in both applications (western blot and direct immunofluorescence), and therefore, modified antibody products must be evaluated carefully for the applications intended. Nonetheless, we have shown that it is possible to start from commercially available monoclonal IgG1 and custom-produced polyclonal IgG antibodies, the latter of which consist of several subtypes having different number and locations of intra-chain disulphide bonds,33,34 and synthetically produce specific ion channel imaging tools for western blot (β-actin, TRPM4) and direct immunofluorescence (β-actin, Nav1.5, Cav1.2, Na+/K+-ATPase) applications.
Experimental section
Chemistry
Commercial reagents and solvents (Acrôs Organics, Fluorochem, Grogg Chemie, Hänseler, Sigma-Aldrich) were used without further purification unless otherwise stated. Pacific Blue NHS ester and BODIPY FL azide 5 were obtained from Lumiprobe GmbH. Dry solvents for reactions were distilled and filtered over columns of dry neutral aluminium oxide under positive argon pressure. Solvents for extraction and flash chromatography were used without further purification.
1H and 13C NMR spectra were recorded on a Bruker AVANCE-300 or 400 spectrometers operating at 300 or 400 MHz for 1H and 75 or 101 MHz for 13C at room temperature unless otherwise stated. Chemical shifts (δ) are reported in parts per million (ppm) relative to tetramethylsilane (TMS) calibrated using residual signals of the solvent or TMS. Coupling constants (J) are reported in Hz. HRMS analyses and accurate mass determinations were performed on a Thermo Scientific LTQ Orbitrap XL mass spectrometer using ESI ionisation and positive or negative mode by the analytical services (mass spectrometry lab of Prof. Dr Stefan Schürch) from the Department of Chemistry, Biochemistry and Pharmaceutical Sciences (DCBP) of the University of Bern, Switzerland. HPLCs were measured on a Thermo-Scientific UltiMate 3000 HPLC with H2O + 0.1% TFA and MeCN + 0.1% TFA as eluents on an Acclaim™ 120 C 18 5 μm 120 Å (4.6 × 150 mm) column.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 to 0:1) to give the desired compound in 94% yield (25.02 g, 101.58 mmol). Physical and spectral data was in accordance with literature.20 Colourless solid: Major rotamer, 1H NMR (300 MHz, CDCl3) δ 6.47 (s, 1H), 3.08 (s, 3H), 1.45 (s, 18H). Minor rotamer, 1H NMR (300 MHz, CDCl3) δ 6.24 (s, 1H), 3.08 (s, 3H), 1.44 (s, 18H). Both rotamers: 13C NMR (75 MHz, CDCl3) δ 155.88, 81.27, 28.33. HRMS (ESI) calculated for [M + H]+ C11H23N2O4+ 247.1652, found 247.1658. Calculated for [M + Na]+ C11H22N2NaO4+ 269.1472, found 269.1477.:0 to 9:1) to give the desired compound in 90% yield (0.53 g, 1.85 mmol). Colourless solid: Mix of rotamers, 1H NMR (300 MHz, DMSO-d6) δ 4.53–3.74 (m, 2H), 3.30–3.18 (m, 1H), 3.11–2.90 (m, 3H), 1.50–1.29 (m, 18H). 13C NMR (75 MHz, DMSO-d6) δ 154.85, 154.16, 153.11, 81.34, 80.72, 80.18, 78.80, 78.65, 74.99, 74.93, 39.39, 38.21, 37.62, 37.46, 36.15, 36.06, 27.81, 27.76, 27.73, 27.69. 1H NMR (400 MHz, at T = 90 °C DMSO-d6) δ 4.36 (d, J = 17.5 Hz, 1H), 3.99 (d, J = 17.9 Hz, 1H), 3.08–2.97 (m, 4H), 1.53–1.32 (m, 18H). HRMS (ESI) calculated for [M + Na]+ C14H24N2NaO4+ 307.1628, found 307.1633.:0 to 1:1) to give the desired compound in 92% yield (0.93 g, 2.91 mmol). A small sample was recrystallised from a mixture of acetone and n-hexane and yielded single crystals of suitable quality for X-ray diffraction analysis (CCDC deposition number: 2171667). Colourless crystals: 1H NMR (300 MHz, CDCl3) δ 4.94 (d, J = 2.5 Hz, 2H), 3.78 (s, 3H), 2.43 (t, J = 2.5 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ 152.87, 152.85, 136.93, 135.16, 75.68, 75.01, 37.17, 34.79. HRMS (ESI) calculated for [M + H]+ C8H7Br2N2O2+ 320.8869, found 320.8872.:0 to 9:1) to give the desired product in quantitative yield (0.030 g, 0.0506 mmol). Purple solid: 1H NMR (300 MHz, DMSO-d6) δ 8.69 (t, J = 5.5 Hz, 1H), 7.93 (s, 1H), 7.87 (dd, J = 7.9, 1.7 Hz, 1H), 7.45 (d, J = 2.4 Hz, 2H), 7.27 (d, J = 7.9 Hz, 1H), 6.88 (d, J = 9.6 Hz, 2H), 6.82 (dd, J = 9.7, 2.5 Hz, 2H), 3.68–3.60 (m, 4H), 3.50 (t, J = 5.7 Hz, 2H), 3.46–3.40 (m, 2H), 3.31 (s, 12H), 2.02 (s, 3H), 0.61 (s, 3H), 0.59 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.22, 165.77, 153.73, 147.16, 141.30, 139.78, 135.30, 134.60, 128.95, 128.90, 126.04, 124.61, 121.56, 114.57, 68.98, 68.68, 49.99, 40.50, 18.85, −1.04, −1.37. HRMS (ESI) calculated for [M-Cl]+ C31H39N6O2Si+ 555.2898, found 555.2878.:1) gradient from 1:0 to 0:1) to give the desired compound in 90% yield (0.049 g, 0.135 mmol). Yellow powder: 1H NMR (300 MHz, CDCl3) δ 9.38 (br. s, 1H), 8.93 (t, J = 5.8 Hz, 1H), 8.80 (d, J = 1.4 Hz, 1H), 7.29–7.20 (m, 1H), 3.48 (q, J = 6.7 Hz, 2H), 3.26 (t, J = 6.8 Hz, 2H), 1.73–1.50 (m, 4H), 1.49–1.32 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 162.16, 160.50, 151.07, 151.06, 148.19, 148.14, 148.10, 147.83, 147.78, 141.03, 141.00, 140.98, 140.95, 140.88, 140.85, 140.61, 140.45, 140.38, 140.21, 137.64, 116.09, 110.30, 110.26, 110.21, 109.99, 110.18, 109.99, 109.94, 51.44, 40.19, 29.20, 28.81, 26.60, 26.45. HRMS (ESI) calculated for [M + H]+ C16H17O4N4F2 367.1212, found 367.1212.:1 gradient from 1:0 to 9:1) to give the desired compound in quantitative yield (0.139 g, 0.313 mmol). Colourless solid: 1H NMR (300 MHz, DMSO-d6) δ 7.82 (t, J = 5.6 Hz, 1H), 6.39 (d, J = 20.1 Hz, 2H), 4.38–4.23 (m, 1H), 4.19–4.05 (m, 1H), 3.68–3.45 (m, 10H), 3.45–3.29 (m, 5H), 3.18 (q, J = 5.9 Hz, 2H), 3.13–3.01 (m, 1H), 2.82 (dd, J = 12.4, 5.0 Hz, 1H), 2.57 (d, J = 12.3 Hz, 1H), 2.06 (t, J = 7.3 Hz, 2H), 1.69–1.38 (m, 4H), 1.36–1.20 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 172.12, 162.73, 69.81, 69.78, 69.71, 69.57, 69.27, 69.18, 61.05, 59.21, 55.43, 50.00, 39.86, 38.45, 35.10, 28.20, 28.04, 25.26. HRMS (ESI) calculated for [M + H]+ C18H33O5N6S 445.2228, found 445.2221.:0 to 9:1) to give the desired compound in 95% yield (0.05 g, 0.05 mmol). Blue solid: 1H NMR (300 MHz, DMSO-d6) δ 8.61 (t, J = 5.4 Hz, 1H), 8.17 (s, 1H), 7.92 (s, 1H), 7.85 (d, J = 7.9 Hz, 1H), 7.45 (d, J = 2.6 Hz, 2H), 7.27 (d, J = 7.9 Hz, 1H), 6.88 (d, J = 9.7 Hz, 2H), 6.81 (dd, J = 9.7, 2.6 Hz, 2H), 5.33 (s, 2H), 4.55 (t, J = 5.1 Hz, 2H), 3.85 (t, J = 5.1 Hz, 2H), 3.63 (s, 3H), 3.56 (t, J = 5.7 Hz, 2H), 3.42 (q, J = 5.5 Hz, 2H), 3.31 (s, 12H), 2.02 (s, 3H), 0.61 (s, 3H), 0.59 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.24, 165.70, 153.72, 152.42, 152.24, 147.16, 141.33, 140.81, 139.80, 135.68, 135.37, 134.70, 134.52, 128.97, 128.92, 126.04, 124.56, 124.47, 121.55, 114.55, 68.66, 68.29, 54.90, 49.51, 42.45, 40.50, 34.85, 18.87, −1.04, −1.37. HRMS (ESI) calculated for [M–Cl]+ C39H45Br2N8O4Si+ 875.1694, found 875.1685.:10:5) gradient from 1:0 to 0:1) to give the desired compound in quantitative yield (0.034 g, 0.049 mmol). Dark orange powder: 1H NMR (300 MHz, DMSO-d6) δ 8.16 (s, 1H), 8.02 (t, J = 5.6 Hz, 1H), 7.69 (s, 1H), 7.08 (d, J = 4.0 Hz, 1H), 6.35 (d, J = 4.0 Hz, 1H), 6.30 (s, 1H), 5.37 (s, 2H), 4.34 (t, J = 7.0 Hz, 2H), 3.64 (s, 3H), 3.07 (dd, J = 9.6, 6.7 Hz, 4H), 2.47 (s, 9H), 2.26 (s, 3H), 1.94 (p, J = 7.0 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 170.92, 159.16, 152.38, 152.25, 140.89, 135.70, 134.72, 132.95, 128.89, 128.19, 125.35, 125.30, 124.14, 120.26, 116.56, 47.35, 42.46, 35.74, 34.81, 33.76, 29.81, 23.95, 14.50, 10.98, 1.14. 19F NMR (376 MHz, DMSO-d6) δ-143.18 (dd, J = 66.5, 33.0 Hz). 11B NMR (96 MHz, DMSO-d6) δ 0.74 (t, J = 32.8 Hz). HRMS (ESI) calculated for [M + Na]+ C25H27O3N8 BBr2F2Na 717.0526, found 717.0523.:1) gradient from 1:0 to 0:1) to give the desired compound in 67% yield (0.071 g, 0.105 mmol). Yellow powder: 1H NMR (300 MHz, DMSO-d6) δ 12.02 (s, 1H), 8.76 (s, 1H), 8.56 (t, J = 5.8 Hz, 1H), 8.16 (s, 1H), 7.74 (d, J = 9.6 Hz, 1H), 5.36 (s, 2H), 4.33 (t, J = 7.1 Hz, 2H), 3.63 (s, 3H), 3.31–3.24 (m, 2H), 1.80 (p, J = 7.1 Hz, 2H), 1.49 (p, J = 6.6 Hz, 2H), 1.32–1.19 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 160.97, 159.66, 152.38, 152.24, 147.00, 140.92, 140.62, 140.59, 140.54, 135.68, 134.73, 123.91, 115.99, 110.53, 110.32, 109.26, 109.18, 49.38, 42.51, 34.80, 29.47, 28.74, 25.68, 25.44. 19F NMR (282 MHz, DMSO-d6) δ-135.17, −154.28. HRMS (ESI) calculated for [M + Na]+ C24H22O6N6Br2F2Na 708.9828, found 708.9854.:0 to 9:1) to give the desired compound in 91% yield (0.175 g, 0.228 mmol). Pale yellow powder: 1H NMR (400 MHz, DMSO-d6) δ 8.12 (s, 1H), 7.80 (t, J = 5.6 Hz, 1H), 6.38 (d, J = 23.7 Hz, 2H), 5.37 (s, 2H), 4.51 (t, J = 5.2 Hz, 2H), 4.30 (dd, J = 7.7, 5.0 Hz, 1H), 4.16–4.08 (m, 1H), 3.80 (t, J = 5.3 Hz, 2H), 3.65 (s, 3H), 3.52–3.43 (m, 8H), 3.38 (t, J = 6.0 Hz, 2H), 3.17 (q, J = 5.9 Hz, 2H), 3.14–3.04 (m, 1H), 2.81 (dd, J = 12.4, 5.1 Hz, 1H), 2.57 (d, J = 12.5 Hz, 1H), 2.06 (t, J = 7.4 Hz, 2H), 1.66–1.54 (m, 1H), 1.56–1.38 (m, 3H), 1.35–1.22 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 172.09, 162.67, 152.39, 152.24, 140.80, 135.69, 134.73, 124.46, 69.67, 69.61, 69.52, 69.14, 68.56, 61.01, 59.17, 55.39, 54.89, 49.50, 42.41, 39.83, 38.41, 35.08, 34.81, 28.17, 28.02, 25.23. HRMS (ESI) calculated for [M + H]+ C26H39O7N8Br2S 765.1024, found 765.1024.
:0 to 0:1) to give the desired compound in 94% yield (25.02 g, 101.58 mmol). Physical and spectral data was in accordance with literature.20 Colourless solid: Major rotamer, 1H NMR (300 MHz, CDCl3) δ 6.47 (s, 1H), 3.08 (s, 3H), 1.45 (s, 18H). Minor rotamer, 1H NMR (300 MHz, CDCl3) δ 6.24 (s, 1H), 3.08 (s, 3H), 1.44 (s, 18H). Both rotamers: 13C NMR (75 MHz, CDCl3) δ 155.88, 81.27, 28.33. HRMS (ESI) calculated for [M + H]+ C11H23N2O4+ 247.1652, found 247.1658. Calculated for [M + Na]+ C11H22N2NaO4+ 269.1472, found 269.1477.:0 to 9:1) to give the desired compound in 90% yield (0.53 g, 1.85 mmol). Colourless solid: Mix of rotamers, 1H NMR (300 MHz, DMSO-d6) δ 4.53–3.74 (m, 2H), 3.30–3.18 (m, 1H), 3.11–2.90 (m, 3H), 1.50–1.29 (m, 18H). 13C NMR (75 MHz, DMSO-d6) δ 154.85, 154.16, 153.11, 81.34, 80.72, 80.18, 78.80, 78.65, 74.99, 74.93, 39.39, 38.21, 37.62, 37.46, 36.15, 36.06, 27.81, 27.76, 27.73, 27.69. 1H NMR (400 MHz, at T = 90 °C DMSO-d6) δ 4.36 (d, J = 17.5 Hz, 1H), 3.99 (d, J = 17.9 Hz, 1H), 3.08–2.97 (m, 4H), 1.53–1.32 (m, 18H). HRMS (ESI) calculated for [M + Na]+ C14H24N2NaO4+ 307.1628, found 307.1633.:0 to 1:1) to give the desired compound in 92% yield (0.93 g, 2.91 mmol). A small sample was recrystallised from a mixture of acetone and n-hexane and yielded single crystals of suitable quality for X-ray diffraction analysis (CCDC deposition number: 2171667). Colourless crystals: 1H NMR (300 MHz, CDCl3) δ 4.94 (d, J = 2.5 Hz, 2H), 3.78 (s, 3H), 2.43 (t, J = 2.5 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ 152.87, 152.85, 136.93, 135.16, 75.68, 75.01, 37.17, 34.79. HRMS (ESI) calculated for [M + H]+ C8H7Br2N2O2+ 320.8869, found 320.8872.:0 to 9:1) to give the desired product in quantitative yield (0.030 g, 0.0506 mmol). Purple solid: 1H NMR (300 MHz, DMSO-d6) δ 8.69 (t, J = 5.5 Hz, 1H), 7.93 (s, 1H), 7.87 (dd, J = 7.9, 1.7 Hz, 1H), 7.45 (d, J = 2.4 Hz, 2H), 7.27 (d, J = 7.9 Hz, 1H), 6.88 (d, J = 9.6 Hz, 2H), 6.82 (dd, J = 9.7, 2.5 Hz, 2H), 3.68–3.60 (m, 4H), 3.50 (t, J = 5.7 Hz, 2H), 3.46–3.40 (m, 2H), 3.31 (s, 12H), 2.02 (s, 3H), 0.61 (s, 3H), 0.59 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.22, 165.77, 153.73, 147.16, 141.30, 139.78, 135.30, 134.60, 128.95, 128.90, 126.04, 124.61, 121.56, 114.57, 68.98, 68.68, 49.99, 40.50, 18.85, −1.04, −1.37. HRMS (ESI) calculated for [M-Cl]+ C31H39N6O2Si+ 555.2898, found 555.2878.:1) gradient from 1:0 to 0:1) to give the desired compound in 90% yield (0.049 g, 0.135 mmol). Yellow powder: 1H NMR (300 MHz, CDCl3) δ 9.38 (br. s, 1H), 8.93 (t, J = 5.8 Hz, 1H), 8.80 (d, J = 1.4 Hz, 1H), 7.29–7.20 (m, 1H), 3.48 (q, J = 6.7 Hz, 2H), 3.26 (t, J = 6.8 Hz, 2H), 1.73–1.50 (m, 4H), 1.49–1.32 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 162.16, 160.50, 151.07, 151.06, 148.19, 148.14, 148.10, 147.83, 147.78, 141.03, 141.00, 140.98, 140.95, 140.88, 140.85, 140.61, 140.45, 140.38, 140.21, 137.64, 116.09, 110.30, 110.26, 110.21, 109.99, 110.18, 109.99, 109.94, 51.44, 40.19, 29.20, 28.81, 26.60, 26.45. HRMS (ESI) calculated for [M + H]+ C16H17O4N4F2 367.1212, found 367.1212.:1 gradient from 1:0 to 9:1) to give the desired compound in quantitative yield (0.139 g, 0.313 mmol). Colourless solid: 1H NMR (300 MHz, DMSO-d6) δ 7.82 (t, J = 5.6 Hz, 1H), 6.39 (d, J = 20.1 Hz, 2H), 4.38–4.23 (m, 1H), 4.19–4.05 (m, 1H), 3.68–3.45 (m, 10H), 3.45–3.29 (m, 5H), 3.18 (q, J = 5.9 Hz, 2H), 3.13–3.01 (m, 1H), 2.82 (dd, J = 12.4, 5.0 Hz, 1H), 2.57 (d, J = 12.3 Hz, 1H), 2.06 (t, J = 7.3 Hz, 2H), 1.69–1.38 (m, 4H), 1.36–1.20 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 172.12, 162.73, 69.81, 69.78, 69.71, 69.57, 69.27, 69.18, 61.05, 59.21, 55.43, 50.00, 39.86, 38.45, 35.10, 28.20, 28.04, 25.26. HRMS (ESI) calculated for [M + H]+ C18H33O5N6S 445.2228, found 445.2221.:0 to 9:1) to give the desired compound in 95% yield (0.05 g, 0.05 mmol). Blue solid: 1H NMR (300 MHz, DMSO-d6) δ 8.61 (t, J = 5.4 Hz, 1H), 8.17 (s, 1H), 7.92 (s, 1H), 7.85 (d, J = 7.9 Hz, 1H), 7.45 (d, J = 2.6 Hz, 2H), 7.27 (d, J = 7.9 Hz, 1H), 6.88 (d, J = 9.7 Hz, 2H), 6.81 (dd, J = 9.7, 2.6 Hz, 2H), 5.33 (s, 2H), 4.55 (t, J = 5.1 Hz, 2H), 3.85 (t, J = 5.1 Hz, 2H), 3.63 (s, 3H), 3.56 (t, J = 5.7 Hz, 2H), 3.42 (q, J = 5.5 Hz, 2H), 3.31 (s, 12H), 2.02 (s, 3H), 0.61 (s, 3H), 0.59 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.24, 165.70, 153.72, 152.42, 152.24, 147.16, 141.33, 140.81, 139.80, 135.68, 135.37, 134.70, 134.52, 128.97, 128.92, 126.04, 124.56, 124.47, 121.55, 114.55, 68.66, 68.29, 54.90, 49.51, 42.45, 40.50, 34.85, 18.87, −1.04, −1.37. HRMS (ESI) calculated for [M–Cl]+ C39H45Br2N8O4Si+ 875.1694, found 875.1685.:10:5) gradient from 1:0 to 0:1) to give the desired compound in quantitative yield (0.034 g, 0.049 mmol). Dark orange powder: 1H NMR (300 MHz, DMSO-d6) δ 8.16 (s, 1H), 8.02 (t, J = 5.6 Hz, 1H), 7.69 (s, 1H), 7.08 (d, J = 4.0 Hz, 1H), 6.35 (d, J = 4.0 Hz, 1H), 6.30 (s, 1H), 5.37 (s, 2H), 4.34 (t, J = 7.0 Hz, 2H), 3.64 (s, 3H), 3.07 (dd, J = 9.6, 6.7 Hz, 4H), 2.47 (s, 9H), 2.26 (s, 3H), 1.94 (p, J = 7.0 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 170.92, 159.16, 152.38, 152.25, 140.89, 135.70, 134.72, 132.95, 128.89, 128.19, 125.35, 125.30, 124.14, 120.26, 116.56, 47.35, 42.46, 35.74, 34.81, 33.76, 29.81, 23.95, 14.50, 10.98, 1.14. 19F NMR (376 MHz, DMSO-d6) δ-143.18 (dd, J = 66.5, 33.0 Hz). 11B NMR (96 MHz, DMSO-d6) δ 0.74 (t, J = 32.8 Hz). HRMS (ESI) calculated for [M + Na]+ C25H27O3N8 BBr2F2Na 717.0526, found 717.0523.:1) gradient from 1:0 to 0:1) to give the desired compound in 67% yield (0.071 g, 0.105 mmol). Yellow powder: 1H NMR (300 MHz, DMSO-d6) δ 12.02 (s, 1H), 8.76 (s, 1H), 8.56 (t, J = 5.8 Hz, 1H), 8.16 (s, 1H), 7.74 (d, J = 9.6 Hz, 1H), 5.36 (s, 2H), 4.33 (t, J = 7.1 Hz, 2H), 3.63 (s, 3H), 3.31–3.24 (m, 2H), 1.80 (p, J = 7.1 Hz, 2H), 1.49 (p, J = 6.6 Hz, 2H), 1.32–1.19 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 160.97, 159.66, 152.38, 152.24, 147.00, 140.92, 140.62, 140.59, 140.54, 135.68, 134.73, 123.91, 115.99, 110.53, 110.32, 109.26, 109.18, 49.38, 42.51, 34.80, 29.47, 28.74, 25.68, 25.44. 19F NMR (282 MHz, DMSO-d6) δ-135.17, −154.28. HRMS (ESI) calculated for [M + Na]+ C24H22O6N6Br2F2Na 708.9828, found 708.9854.:0 to 9:1) to give the desired compound in 91% yield (0.175 g, 0.228 mmol). Pale yellow powder: 1H NMR (400 MHz, DMSO-d6) δ 8.12 (s, 1H), 7.80 (t, J = 5.6 Hz, 1H), 6.38 (d, J = 23.7 Hz, 2H), 5.37 (s, 2H), 4.51 (t, J = 5.2 Hz, 2H), 4.30 (dd, J = 7.7, 5.0 Hz, 1H), 4.16–4.08 (m, 1H), 3.80 (t, J = 5.3 Hz, 2H), 3.65 (s, 3H), 3.52–3.43 (m, 8H), 3.38 (t, J = 6.0 Hz, 2H), 3.17 (q, J = 5.9 Hz, 2H), 3.14–3.04 (m, 1H), 2.81 (dd, J = 12.4, 5.1 Hz, 1H), 2.57 (d, J = 12.5 Hz, 1H), 2.06 (t, J = 7.4 Hz, 2H), 1.66–1.54 (m, 1H), 1.56–1.38 (m, 3H), 1.35–1.22 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 172.09, 162.67, 152.39, 152.24, 140.80, 135.69, 134.73, 124.46, 69.67, 69.61, 69.52, 69.14, 68.56, 61.01, 59.17, 55.39, 54.89, 49.50, 42.41, 39.83, 38.41, 35.08, 34.81, 28.17, 28.02, 25.23. HRMS (ESI) calculated for [M + H]+ C26H39O7N8Br2S 765.1024, found 765.1024.Biology
Buffers used: PBS (Sigma-Aldrich (P5368) dissolved in 1 L Milli-Q water), BBS (Na2B4O7·10H2O 25 mM, NaCl 25 mM, EDTA 0.5 mM, DMSO 2% in Milli-Q water, pH adjusted with aq. HCl to 8.01), SDS-Running Buffer (25 mM Tris, 190 mM glycine, 0.1% SDS), Transfer Buffer (25 mM Tris, 190 mM glycine, 20% MeOH, 0.1% SDS), TBST (20 mM Tris, pH 7.5, 150 mM NaCl, 0.1 Tween 20), 4x loading buffer (0.2 M Tris pH 6.8, 40% glycerol, 20% 2-mercaptoethanol, 8% SDS, 0.002% bromophenol blue in deionised water).
Antibody conjugation through re-bridging
The buffer, in which the antibody was obtained, was exchanged to Borate Buffered Saline (BBS, pH = 8.0) using spin filter columns (Amicon® Ultra 2 mL, 30′000 Da molecular weight cut-off, 3 × 15 minutes @ 4000 g). The residue was transferred into a PCR reaction tube. The re-bridging compound 8–11 (20 mM in DMSO) was added in 20-fold excess with respect to the antibody. This mixture was incubated for 1 h at 4 °C. Then TCEP (10 mM in water) was added in 10-fold excess with respect to the antibody and the reaction was kept at 4 °C overnight. The reaction residue was transferred into a spin filter column (Amicon® Ultra 2 mL, 30′000 Da molecular weight cut-off) and washed with phosphate buffered saline (PBS, pH = 7.4) (6 × 15 minutes @ 4000 g). After washing, the modified antibody was diluted to the original concentration with PBS, aliquoted and stored at −20 °C.Stochastic antibody fluorescent labelling
The buffer, in which the antibody was obtained, was exchanged to Borate Buffered Saline (BBS, pH = 8.0) using spin filter columns (Amicon® Ultra 2 mL, 30′000 Da molecular weight cut-off, 3 × 15 minutes @ 4000 g). The residue was transferred into a PCR reaction tube. To this a solution of SiR-NHS (10 mM in DMSO) was added in 20-fold excess with respect to the antibody. This mixture was incubated at 4 °C overnight. The reaction residue was transferred into a spin filter column (Amicon® Ultra 2 mL, 30′000 Da molecular weight cut-off) and washed with phosphate buffered saline (PBS, pH = 7.4, 5 × 15 minutes @ 4000 g and 1 × 15 minutes @ 4000 g). After washing, the modified antibody was diluted to the original concentration with PBS, aliquoted and stored at −20 °C.SDS-PAGE
Polyacrylamide gels were cast from 30% acrylamide solution (Carl Roth, A124.1). For a typical SDS-PAGE experiment the modified antibody in PBS was diluted with 4x loading buffer to obtain a loading of 200 ng per lane. In this dilution the mixture was incubated at 92 °C for 30 minutes. A 10% 1 mm polyacrylamide gel was run at 90 V for 15 minutes followed by 120 V up to a total run time of 2.5 h in SDS running buffer. Visualisation was achieved with a Typhoon FLA 9500 fluorescence imager. After the fluorescence analysis, the gel was stained with Coomassie blue staining for 1 h followed by washing with water overnight.Size exclusion chromatography (SEC)
SEC was conducted on an Agilent 1260 infinity II HPLC with 150 mM phosphate buffer + 0.01% NaN3, pH = 7 as eluent, using an Agilent AdvanceBio SEC 2.7 μm 300 Å fully porous column (7.8 × 300 mm) and an Agilent AdvanceBio SEC 2.7 μm 300 Å fully porous guard column (7.8 × 50 mm). Proteins were detected by fluorescence and absorption at 215 nm and 280 nm (Fig. S2, ESI†).Western blotting
In a typical western blot experiment, cardiomyocytes were isolated from mouse heart according to Ozhathil et al.35 and lysed. The lysate was diluted with 4x loading buffer to obtain a loading of 100 μg per lane. In this dilution the mixture was incubated at 37 °C for 30 minutes. A 10% 1 mm polyacrylamide gel was run at 90 V for 15 minutes followed by 120 V up to a total run time of 2.5 h in SDS running buffer. The gel was removed and incubated in transfer buffer for 15 minutes. The gel was sandwiched together with a 0.4 μM nitrocellulose membrane and was wet-blotted at 90 V for 90 minutes in 0 °C transfer buffer. The membrane was then blocked with 3% BSA in TBST for 1 h after which it was rinsed 5x with TBST. Staining of the targets was achieved by incubating the membrane in a solution of the corresponding antibody diluted in TBST + 3% BSA, using a concentration for western blot application recommended by the manufacturer. Visualisation was achieved with a fluorescence imager (GE Healthcare, Typhoon FLA 9500). The biotin-modified antibody was visualised by co-incubating the membrane with streptavidin-Cy5 (Invitrogen™, SA1011) solution.Immunofluorescence
Adult mouse heart cardiomyocytes were isolated as described previously35 and plated onto uncoated PCA microscopy chamber slides (Sartstedt, 94.6140.802). The cells were washed twice with PBS 1x and then fixed with 10% PFA for 15 minutes. The cells were washed twice with PBS 1x, blocked with blocking buffer (PBS 1x + 1% BSA + 0.5% TritonX-100 + 10% Goat Serum) for 30 minutes and subsequently incubated overnight at 4 °C in incubation buffer (PBS 1x + 1% BSA + 0.5% Triton X-100 + 3% Goat Serum). The incubation buffer contained the corresponding antibody in a concentration for immunofluorescence applications, according to the manufacturer's recommendation. The slides were washed 3x with PBS 1x. For indirect immunofluorescence or biotin detection, secondary antibodies (Abcam, ab96879 (goat anti mouse), ab98462 (goat anti rabbit)) or streptavidin-Cy5 was applied, respectively, in the recommended concentration in incubation buffer for 1 h at 22 °C. For direct immunofluorescence using a fluorescent primary antibody, the cells were kept in PBS 1x for 1 h at 22 °C. Subsequently, the slides were washed 3x with PBS 1x, applying DAPI (Sigma-Aldrich, D9542) stain in the second wash. The cover slip was mounted with Fluor Save (Calbiochem, 345789). The microscope used was a Zeiss LSM710 with EC Plan-NeoFluor 40x objective.Author contributions
P. Grossenbacher, B. Stieger and M. Lochner conceived the project. P. Grossenbacher, M. C. Essers, S. A. Singer, B. Stieger, J.-S. Rougier and M. Lochner designed the experiments. P. Grossenbacher, M. C. Essers, J. Moser, S. A. Singer and S. Häusler performed the experiments and data analysis. P. Grossenbacher and M. Lochner wrote the manuscript. All authors edited and approved the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We gratefully acknowledged funding from the Swiss National Science Foundation [CRSII5_180326] to support this work. We thank the Analytical Services from the Department of Chemistry, Biochemistry and Pharmaceutical Sciences, University of Bern, Switzerland, for measuring NMR and MS spectra of synthetic intermediates and final compounds. The X-ray Crystal Structure Determination Service of the same department is acknowledged for measuring, solving, refining, and summarising the structures of compounds 3, S3, S4 and biotin-NHS. The Synergy diffractometer used was funded in part by the Swiss National Science Foundation through a R'Equip programme [206021_177033].References
- P. Khongorzul, C. J. Ling, F. U. Khan, A. U. Ihsan and J. Zhang, Mol. Cancer Res., 2020, 18, 3–19 CrossRef CAS PubMed
.
- S. J. Walsh, J. D. Bargh, F. M. Dannheim, A. R. Hanby, H. Seki, A. J. Counsell, X. Ou, E. Fowler, N. Ashman, Y. Takada, A. Isidro-Llobet, J. S. Parker, J. S. Carroll and D. R. Spring, Chem. Soc. Rev., 2021, 50, 1305–1353 RSC
- G. T. Hermanson, Bioconjugate Techniques, Academic Press, 3rd edn, 2013 Search PubMed
- V. Chudasama, A. Maruani and S. Caddick, Nat. Chem., 2016, 8, 114–119 CrossRef CAS PubMed
- J. R. Junutula, H. Raab, S. Clark, S. Bhakta, D. D. Leipold, S. Weir, Y. Chen, M. Simpson, S. P. Tsai, M. S. Dennis, Y. Lu, Y. G. Meng, C. Ng, J. Yang, C. C. Lee, E. Duenas, J. Gorrell, V. Katta, A. Kim, K. McDorman, K. Flagella, R. Venook, S. Ross, S. D. Spencer, W. Lee Wong, H. B. Lowman, R. Vandlen, M. X. Sliwkowski, R. H. Scheller, P. Polakis and W. Mallet, Nat. Biotechnol., 2008, 26, 925–932 CrossRef CAS PubMed
- B.-Q. Shen, K. Xu, L. Liu, H. Raab, S. Bhakta, M. Kenrick, K. L. Parsons-Reponte, J. Tien, S.-F. Yu, E. Mai, D. Li, J. Tibbitts, J. Baudys, O. M. Saad, S. J. Scales, P. J. McDonald, P. E. Hass, C. Eigenbrot, T. Nguyen, W. A. Solis, R. N. Fuji, K. M. Flagella, D. Patel, S. D. Spencer, L. A. Khawli, A. Ebens, W. L. Wong, R. Vandlen, S. Kaur, M. X. Sliwkowski, R. H. Scheller, P. Polakis and J. R. Junutula, Nat. Biotechnol., 2012, 30, 184–189 CrossRef CAS
- F. A. Liberatore, R. D. Comeau, J. M. McKearin, D. A. Pearson, B. Q. Belonga, S. J. Brocchini, J. Kath, T. Phillips, K. Oswell and R. G. Lawton, Bioconjugate Chem., 1990, 1, 36–50 CrossRef CAS PubMed
- R. B. Del Rosario, R. L. Wahl, S. J. Brocchini, R. G. Lawton and R. H. Smith, Bioconjugate Chem., 1990, 1, 51–59 CrossRef CAS PubMed
- D. Wei, Y. Mao, Z. Xu, J. Chen, J. Li, B. Jiang and H. Chen, Bioorg. Med. Chem., 2021, 51, 116497 CrossRef CAS PubMed
- O. Koniev, I. Dovgan, B. Renoux, A. Ehkirch, J. Eberova, S. Cianférani, S. Kolodych, S. Papot and A. Wagner, Med. Chem. Commun., 2018, 9, 827–830 RSC
- B. Shi, M. Wu, Z. Li, Z. Xie, X. Wei, J. Fan, Y. Xu, D. Ding, S. H. Akash, S. Chen and S. Cao, Cancer Med., 2019, 8, 1793–1805 CrossRef CAS
- Q.-Y. Hu and H. Imase, Methods for making conjugates from disulfide-containing proteins, 2014, p. WO2014083505 Search PubMed
- F. F. Schumacher, J. P. M. Nunes, A. Maruani, V. Chudasama, M. E. B. Smith, K. A. Chester, J. R. Baker and S. Caddick, Org. Biomol. Chem., 2014, 12, 7261–7269 RSC
- J. P. M. Nunes, M. Morais, V. Vassileva, E. Robinson, V. S. Rajkumar, M. E. B. Smith, R. B. Pedley, S. Caddick, J. R. Baker and V. Chudasama, Chem. Commun., 2015, 51, 10624–10627 RSC
- M. Morais, J. P. M. Nunes, K. Karu, N. Forte, I. Benni, M. E. B. Smith, S. Caddick, V. Chudasama and J. R. Baker, Org. Biomol. Chem., 2017, 15, 2947–2952 RSC
- A. Maruani, M. E. B. Smith, E. Miranda, K. A. Chester, V. Chudasama and S. Caddick, Nat. Commun., 2015, 6, 6645 CrossRef CAS
- A. Maruani, H. Savoie, F. Bryden, S. Caddick, R. Boyle and V. Chudasama, Chem. Commun., 2015, 51, 15304–15307 RSC
- M. T. W. Lee, A. Maruani, D. A. Richards, J. R. Baker, S. Caddick and V. Chudasama, Chem. Sci., 2017, 8, 2056–2060 RSC
- E. Robinson, J. P. M. Nunes, V. Vassileva, A. Maruani, J. C. F. Nogueira, M. E. B. Smith, R. B. Pedley, S. Caddick, J. R. Baker and V. Chudasama, RSC Adv., 2017, 7, 9073–9077 RSC
- C. Bahou, D. A. Richards, A. Maruani, E. A. Love, F. Javaid, S. Caddick, J. R. Baker and V. Chudasama, Org. Biomol. Chem., 2018, 16, 1359–1366 RSC
- S. Shao, M.-H. Tsai, J. Lu, T. Yu, J. Jin, D. Xiao, H. Jiang, M. Han, M. Wang and J. Wang, Bioorg. Med. Chem. Lett., 2018, 28, 1363–1370 CrossRef CAS PubMed
- F. Javaid, C. Pilotti, C. Camilli, D. Kallenberg, C. Bahou, J. Blackburn, J. R. Baker, J. Greenwood, S. E. Moss and V. Chudasama, RSC Chem. Biol., 2021, 2, 1206–1220 RSC
- S. J. Walsh, S. Omarjee, W. R. J. D. Galloway, T. T. L. Kwan, H. F. Sore, J. S. Parker, M. Hyvönen, J. S. Carroll and D. R. Spring, Chem. Sci., 2019, 10, 694–700 RSC
- S. J. Walsh, S. Omarjee, F. M. Dannheim, D.-L. Couturier, D. Bexheti, L. Mendil, G. Cronshaw, T. Fewster, C. Gregg, C. Brodie, J. L. Miller, R. Houghton, J. S. Carroll and D. R. Spring, Chem. Commun., 2022, 58, 1962–1965 RSC
- A. R. Hanby, S. J. Walsh, A. J. Counsell, N. Ashman, K. T. Mortensen, J. S. Carroll and D. R. Spring, Chem. Commun., 2022, 58, 9401–9404 RSC
- C. Bahou, E. A. Love, S. Leonard, R. J. Spears, A. Maruani, K. Armour, J. R. Baker and V. Chudasama, Bioconjugate Chem., 2019, 30, 1048–1054 CrossRef CAS PubMed
- M. T. W. Lee, A. Maruani, J. R. Baker, S. Caddick and V. Chudasama, Chem. Sci., 2016, 7, 799–802 RSC
- Y. Koide, Y. Urano, K. Hanaoka, T. Terai and T. Nagano, ACS Chem. Biol., 2011, 6, 600–608 CrossRef CAS PubMed
- C. Nicholls, H. Li and J.-P. Liu, Clin. Exp. Pharmacol. Physiol., 2012, 39, 674–679 CrossRef CAS PubMed
- M. A. Sirover, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1999, 1432, 159–184 CrossRef CAS
- M. A. Sirover, Biochim. Biophys. Acta, Gen. Subj., 2011, 1810, 741–751 CrossRef CAS
- The Universal Protein Resource (UniProt), https://www.uniprot.org/uniprot/P16858, accessed April 2022 Search PubMed.
- H. Liu and K. May, mAbs, 2012, 4, 17–23 CrossRef PubMed
- G. Vidarsson, G. Dekkers and T. Rispens, Front. Immunol., 2014, 5, 520 Search PubMed
- L. C. Ozhathil, J.-S. Rougier, P. Arullampalam, M. C. Essers, D. Ross-Kaschitza and H. Abriel, Int. J. Mol. Sci., 2021, 22, 3401 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2171667, 2171686, 2171687 and 2177612. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2ra05580e |
| This journal is © The Royal Society of Chemistry 2022 |