
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA computational protocol for the calculation of the standard reduction potential of iron complexes: application to Fe2+/3+-Aβ model systems relevant to Alzheimer's disease†
Adrián L. Orjuelaa,
Francisco Núñez-Zarur*b and
Jorge Alí-Torres *a
*a
aDepartamento de Química, Universidad Nacional de Colombia-Sede Bogotá, 111321, Colombia. E-mail: jialit@unal.edu.co
bFacultad de Ciencias Básicas, Universidad de Medellín, Carrera 87 No 30-65, 050026 Medellín, Colombia. E-mail: fnunez@udem.edu.co
First published on 7th September 2022
Abstract
Iron complexes play a key role in several biological processes, and they are also related to the development of neurological disorders, such as Alzheimer's and Parkinson's diseases. One of the main properties involved in these processes is the standard reduction potential (SRP) of iron complexes. However, the calculation of this property is challenging, mainly due to problems in the electronic structure description, solvent effects and the thermodynamic cycles used for its calculation. In this work, we proposed a computational protocol for the calculation of SRPs of iron complexes by evaluating a wide range of density functionals for the electronic structure description, two implicit solvent models with varying radii and two thermodynamic cycles. Results show that the M06L density functional in combination with the SMD solvation model and the isodesmic method provides good results compared with SRP experimental values for a set of iron complexes. Finally, this protocol was applied to three Fe2+/3+-Aβ model systems involved in the development of Alzheimer's disease and the obtained SRP values are in good agreement with those reported previously by means of MP2 calculations.
Introduction
Iron is the most abundant transition metal in the human body and in its unbound state plays an important role in neuronal functions. These functions include signal neurotransmission, aerobic respiration in the mitochondria, and myelinization of neurons, among others.1 Experimental studies on brain tissues have found high concentrations of free iron,2 whose unbalance has been related to the development of neurodegenerative disorders, such as Parkinson's2–4 and Alzheimer's disease (AD).5–8The role of iron in AD is related to its ability to form stable complexes with the amyloid beta peptide (Aβ). These complexes could participate in the generation of reactive oxygen species (ROS)9–11 if their standard reduction potentials (SRP) are higher than those of the natural reducing agents and lower than the O2/H2O2 couple.7,12 The presence of ROS has been an important hallmark in the toxicity observed in the brain at several levels (Fig. 1). However, the role of iron in this mechanism is not fully understood in part due to a lack of experimental information.
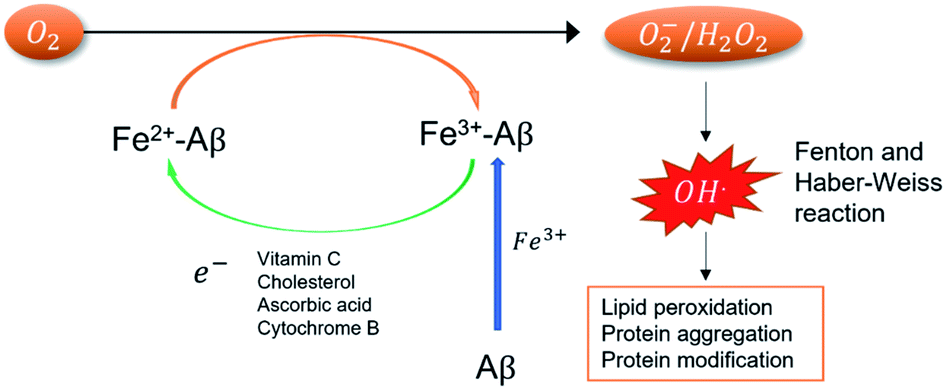 | ||
| Fig. 1 Catalytic cycle of ROS formation mediated by Fe2+/3+-Aβ complexes. | ||
According to Fig. 1, the experimental determination of SRP of iron complexes is crucial to understand the mechanism of ROS formation. However, its experimental determination is difficult to carry out, in part due to the poor solubility of the iron complexes. Computational chemistry must help in this task. Nonetheless, the computation of SRP of iron complexes is challenging due to the subtle electronic effects in the iron reduction process and the description of the solvent effects. Several iron complexes have been computational treated by means of Density Functional theory (DFT) and highly correlated methods.13,14 However, the selection of the computational method and basis set for a correct description of the iron electronic structure is still challenging due to the open-shell nature of these systems and their multiple spin states.15,16 Moreover, other effects like spin crossover are possible to occur.17 The use of highly correlated methods (such as CCSD(T)) provides good results in the energetic and spin description on the ground state.13 Nevertheless, these methods are computational expensive to be applied in large inorganic systems. In this case, DFT offers a good balance between accuracy and computational cost and have been applied in the calculation of SRP of other transition metal complexes.18–21 Recently, Horch used DFT based approaches to study the redox properties of iron metalloenzymes.22
Previously, some of us determined the most favorable structures formed between the Fe2+ and Fe3+ ions and some fragments of amino acids from the Aβ peptide.21 The most stable complexes containing His13-His14 and phenolate of Tyr10 amino acids were the pentacoordinated [Fe2+(O-HisHis)(PhO−)(H2O)]+ and [Fe3+(N-HisHis)(PhO−)(H2O)]+. Moreover, it was found that simultaneous coordination of tyrosine and His13-His14 fragment to Fe2+/3+ is thermodynamically favorable in water at physiological pH. The structures of these complexes are shown in Fig. 2. In that work the calculation of the SRP of Fe-Aβ complexes was done by the direct method with Gibbs energies calculated at the MP2 level for the reduction process Fe3+ → Fe2+. However, this protocol implies to carry out MP2 calculations which are computational expensive.
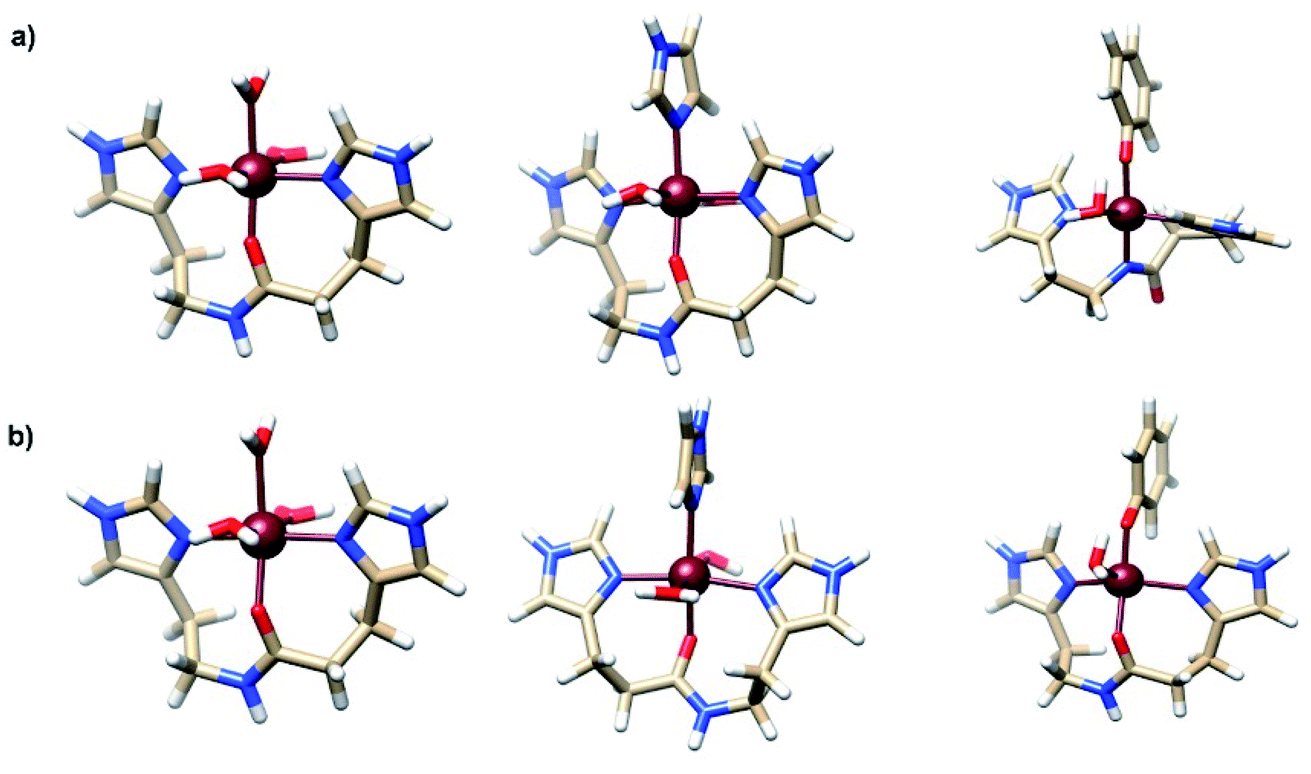 | ||
| Fig. 2 Structure of most stable (a) Fe3+-Aβ and (b) Fe2+-Aβ model systems. Adapted from Alí-Torres et al.21 | ||
In this work, we provide a generalized, reliable method for SRP calculations based on DFT for the calculation of SRP of Fe2+/3+ complexes, which can give accurate results by avoiding the costly MP2 calculations. To do that, we first determine the best combination of functional and basis set for the third ionization energy of Fe, i.e., the reaction Fe2+ → Fe3+, that reproduce the experimental value of 707.42 kcal mol−1.23 We carried out calculations combining 51 density functionals and 38 basis sets to select the combinations with the smallest absolute error. With the best combination of functionals and basis set obtained from the previous step, we calculated the SRPs of 17 iron complexes with reported SRP values (vide infra). Moreover, solvent effects are also accounted for via calibration of cavity and radii parameters of the self-consistent reaction field using the experimentally determined SRP of the [Fe(H2O)6]3+/[Fe(H2O)6]2+ couple.
The main goal here is to develop a theoretical predictive model and use this model to properly determine the SRP of Fe2+/3+-Aβ model systems, which may help to understand the role of these complexes in the mechanism of ROS formation and ultimately their relevance in the neurodegenerative AD process.
Computational details
To select the best combination of functionals and basis sets we calculated the third ionization energy of iron by using 51 density functionals and 38 basis sets. To calculate the SRP, we performed geometry optimizations with the B3LYP functional and 6-31+G(d) basis set for all atoms for the set of iron complexes used for the calibration process (shown in Fig. 5 below). In addition, frequency calculations were carried out for all molecules considering the harmonic oscillator approximation at 298.15 K and 1 atm of pressure and the ideal-gas model. Since low vibrational modes in all iron complexes are similar before and after reduction process the effect on the calculated ΔS is negligible. On the B3LYP optimized geometries we carried out single-point energy calculations with the functionals and basis set that best reproduce the third iron ionization energy. In order to verify that not important geometrical changes are generated by the use of other functionals, we reoptimized some representative iron complexes with the functionals used in the single-point calculations and we found small geometrical changes based on the calculated RMSD compared to the B3LYP gas-phase optimized geometries. Therefore, we cannot expect significant influence of the DFT functional on the geometries of the complexes (see Table S5 of ESI†).To include solvent effects, we first calibrate the solvation models by varying the radii cavity and scale factors for the [Fe(H2O)6]2+/3+ complexes. With the best combination of implicit solvation models, solvent radii and scale factors obtained, we carried out single-point calculations for all the complexes in the training set with the best combination of functional and basis set obtained previously. Finally, the SRP calculations were performed by using two methods, i.e., direct thermodynamic cycle and the isodesmic method.
All calculations were carried out with the Gaussian 16 suite of programs24 and in all cases, we explored all the possible spin states of iron to determine the ground states. In all cases we observed that energy gaps between spin states are very large.
Results and discussion
The main goal of this work is to obtain reliable SRP values for a series of Fe2+/3+-Aβ model systems, which may provide some insights into the role of iron complexes on the neurodegenerative process of the Alzheimer disease. However, the calculation of these SRPs requires an appropriate selection of electronic structure method, solvent model, and an adequate methodology for the calculation of the SRP of iron complexes. Therefore, we carried out several calibration processes: (i) the third ionization energy of iron i.e. Fe2+ → Fe3+; (ii) the best parameters to include solvent effects within the continuum models; and (iii) the SRP calibration using a validation test of iron complexes with experimentally determined SRP values. Finally, the computational protocol is applied to a series of Fe2+/3+-Aβ complexes which represent plausible coordination spheres for this metal peptide. The relative energies of all the complexes are presented in Table S1 of the ESI.†Iron third ionization energy
The calibration of the method and basis set for the calculation of the third ionization energy (3IE) were carried out by using Pople,25 Aldrich,26 Dunning,27 Krauss28 and Los Alamos ECP28 basis sets and different electronic structure methods with diverse types of DFT functionals. This wide set of functionals includes meta-GGA, hyper meta GGA, functionals including dispersion and long-range corrections, PBE correlation, among others. Fig. 4 shows a reduced heatmap for the combination of method and basis set used for the calculation of the 3IE of iron in gas phase with 2% as maximum error. A more comprehensive plot and the ionization energy values with all functionals and basis set combinations can be found in Fig. S1 and Table S1 of ESI.† For all cases we found high spin configurations as ground states (i.e. quintet for Fe2+ and sextet for Fe3+).According to Fig. 3, the best methods are the Minnesota, Half and Half and double hybrid functionals and the best basis set are Pople, Dunning and Aldrich families, which provides the lowest percentage of error when compared with the experimental values. In the Minnesota functionals family we observe that functionals with the lowest percentage of pure Hartree–Fock (HF) exchange exhibit best matches with the experimental 3IE. However, BHandH and BHandHLYP showed a good performance with a 50% HF exchange. Whitin the set of pure functionals VSXC presented the lowest error, 0.73%. In the other hand, the double hybrid functional PBE0DH had an average error of 0.41%. This is in agreement with previous computational works on transition metal systems, where 57 density functionals were tested for the bond dissociation energies of metal–ligand and metal–metal bonds in small compounds with a wide scope of metals.29
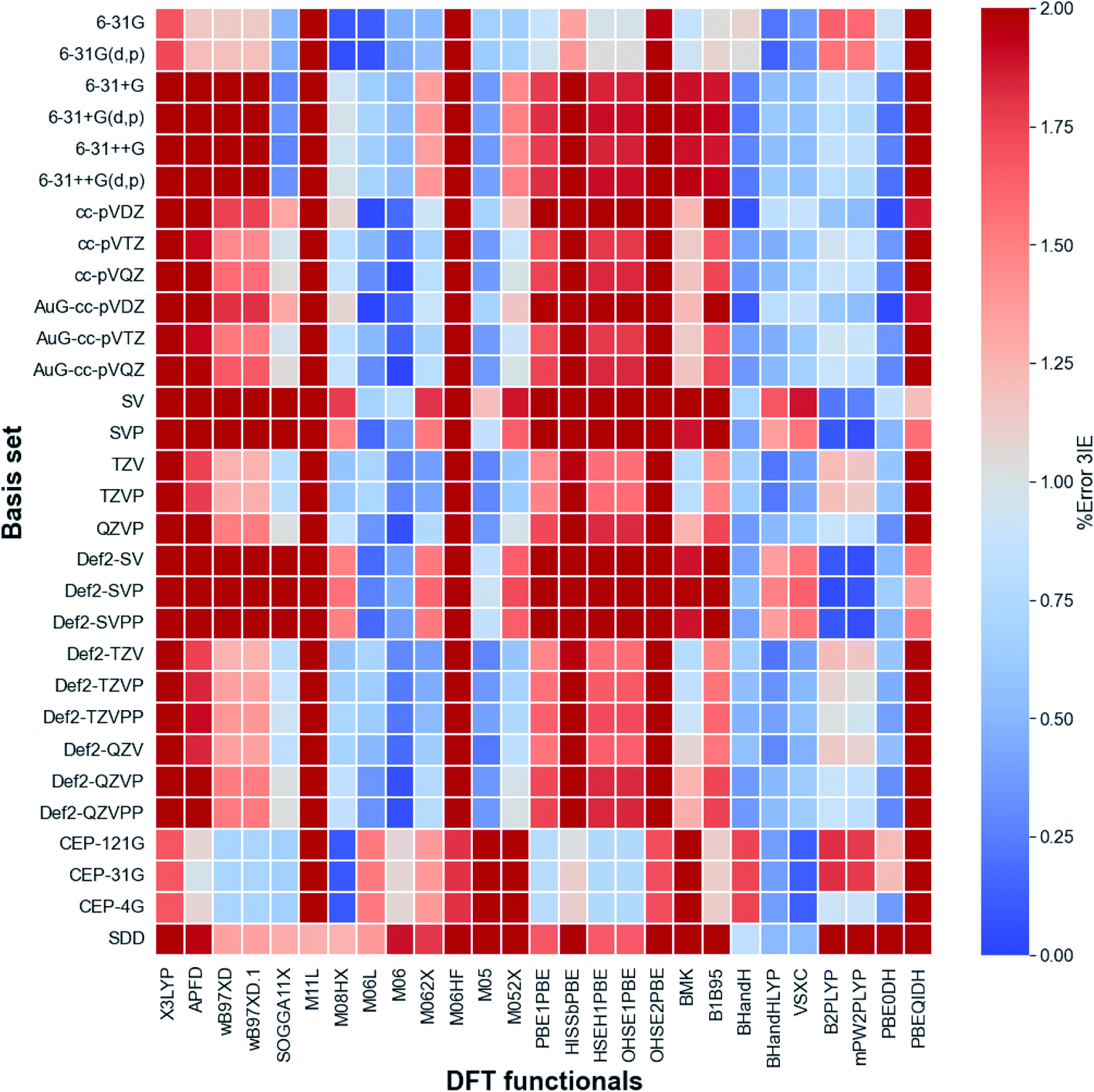 | ||
| Fig. 3 Heatmap for DFT functionals and basis sets. Iron third ionization energy experimental value is 707.42 kcal mol−1.23 | ||
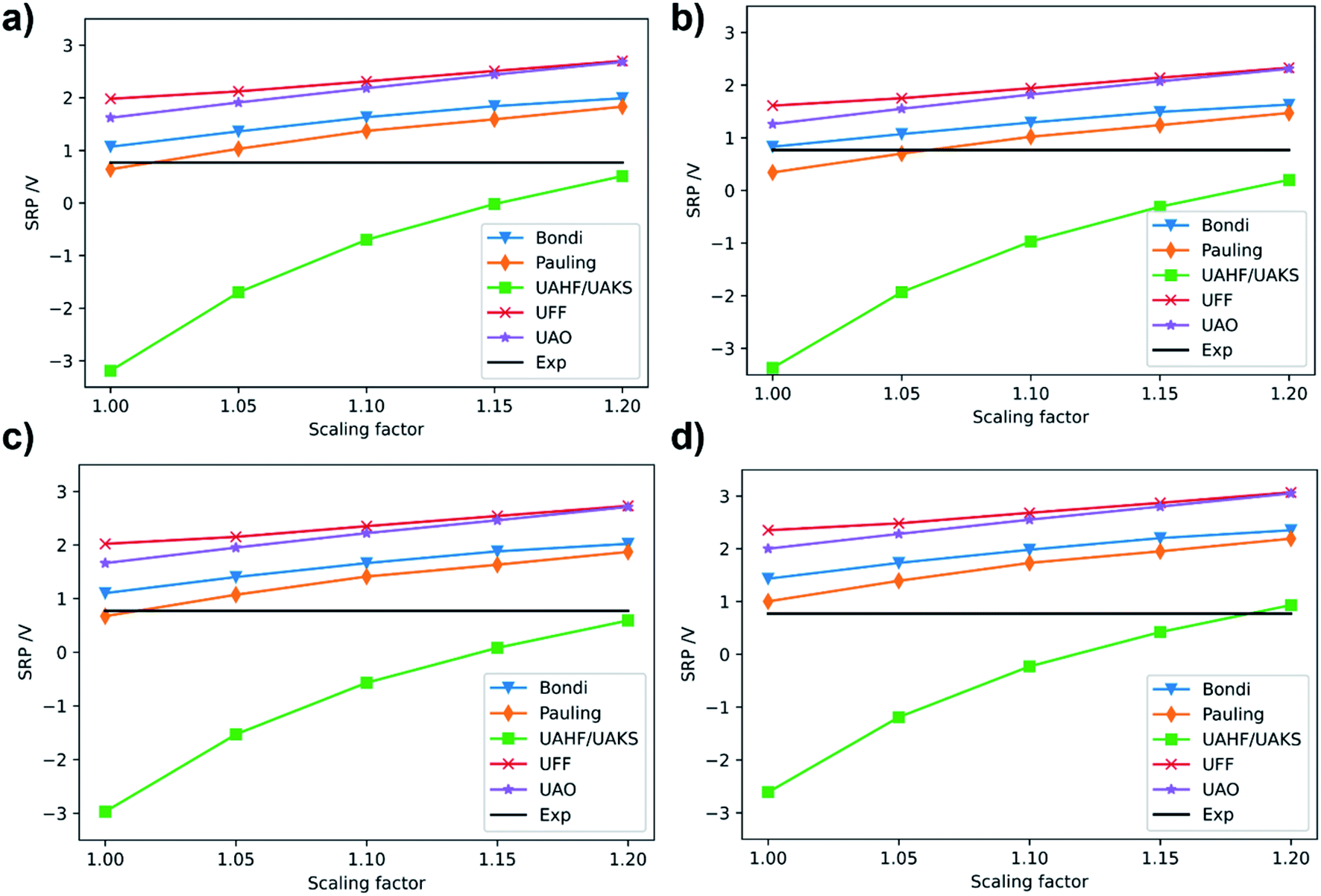 | ||
| Fig. 4 Calculated SRP of [Fe(H2O)6]3+ complex at (a) M06/cc-pVQZ(Fe)-6-31+G(d,p)(O, H); (b) M06L/cc-pVDZ(Fe)-6-31+G(d,p)(O, H); (c) M06/6-31+G(d,p); (d) B3LYP/6-31+G(d,p) levels of theory. Experimental SRP for [Fe(H2O)6]3+ is 0.77 V (solid black line).32 | ||
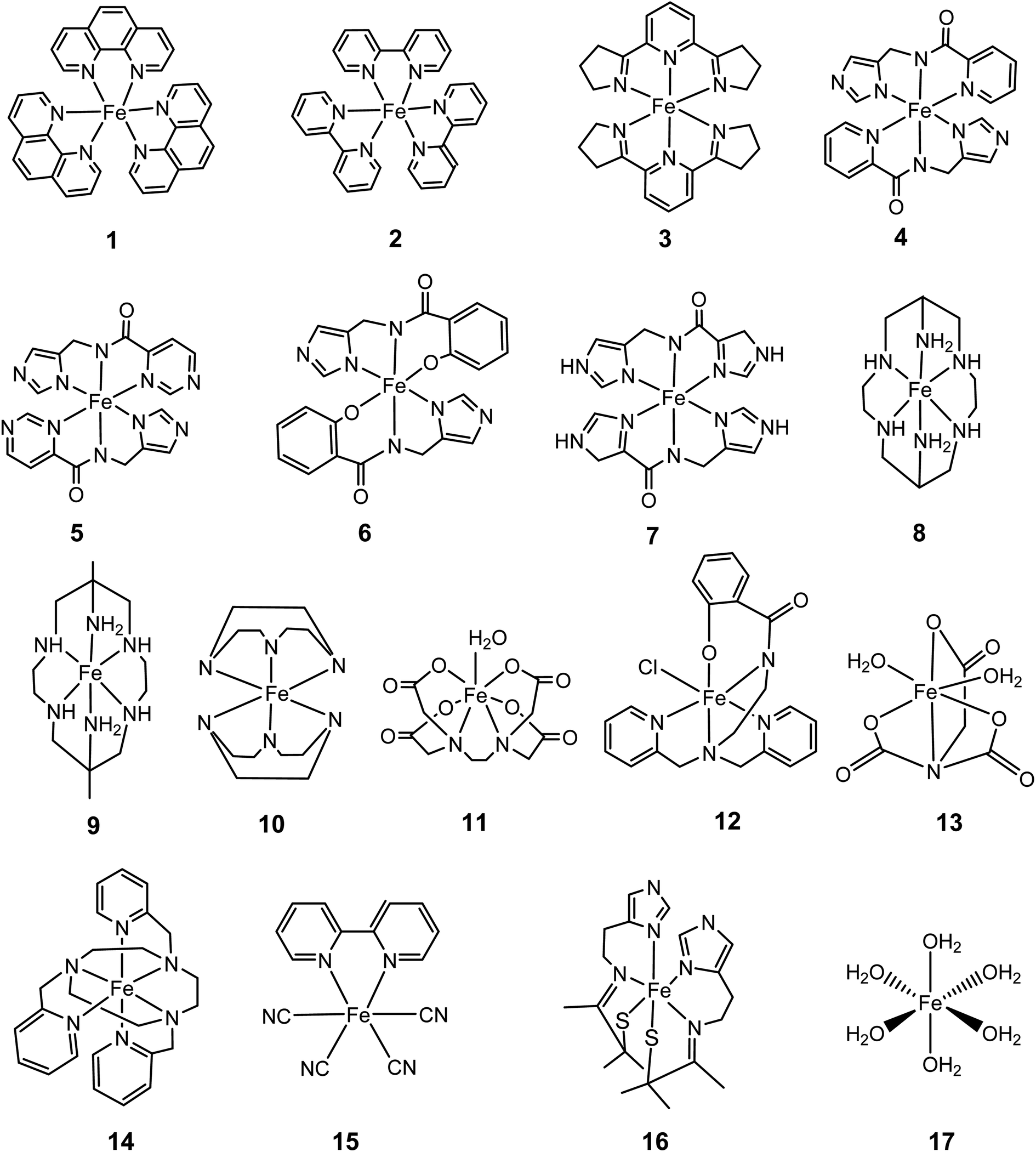 | ||
| Fig. 5 Fe2+/3+ complexes used in the training set for the calibration process of SRP. | ||
Regarding the basis sets performance, we observed that Pople, Dunning and Aldrich basis set give better results with Minnesota, Half-and-Half and double hybrid functionals. In the other hand, Los Alamos basis set family overestimate the 3IE in most cases. Given these results, we selected two different combinations of method/basis set for the subsequent calculations: M06L/cc-pVDZ and M06/cc-pVQZ.
Solvent effects
The Gibbs energy of solvation is an important term in the calculations of the SRP. This contribution can be calculated by using explicit and implicit models. However, the explicit models increase the computational cost due to the quantum treatment of all solvent molecules. The use of polarizable continuum model (PCM)30 simplified the solute–solvent interactions by using a polarizable continuous with similar physicochemical properties. Thus, the Gibbs energy in solution can be calculated as follows:| Gsol = Ges + Gdr + Gcav | (1) |
Once we selected the best combination of method and basis set for the third ionization energy of iron, we turn to calibrate the solvent model parameters, i.e., cavity and scale factors for radii to include the continuum solvent effects into the calculations. For this, we use the [Fe(H2O)6]3+/2+ couple as reference, since its SRP is experimentally available (0.77 V).32 For these calculations, geometry optimizations and frequency calculations were carried out at the B3LYP/6-31+G(d) level in gas phase. This functional and basis set have proven to give proper geometry descriptions in iron complexes.21 On the B3LYP optimized geometries we carried out single-point energy calculations in solution using water as solvent and the PCM,33 C-PCM34 and SMD35 implicit solvation models. For these calculations, we used the best combination of functional and basis sets obtained in the previous step for the metal center (i.e. M06L/cc-pVDZ and M06/cc-pVQZ) and the 6-31+G(d,p) basis set for H and O atoms. We also carried out single point calculations at M06/6-31+G(d,p) and B3LYP/6-31+G(d,p) level for comparison. The calculated SRPs for the [Fe(H2O)6]3+/2+ system with the PCM model using several cavities and scale factors are presented in Fig. 4. The ground states for the iron cations were quintet and sextet for Fe2+ and Fe3+ complexes, respectively.
In general, we observed that the calculated SRP of [Fe(H2O)6]3+/2+ couple shows the same qualitative behavior for all methods, as all SRP values smoothly increase with the scaling factor for the cavity, except for the UAHF/UAKS radii. Thus, our results show that the calculated SRP for the [Fe(H2O)6]3+/2+ couple is dependent on the specific radii and scaling factor, in agreement with previous calculations.31 In all cases the UAHF/UAKS radii underestimate the SRP values, except for a scale factor of 1.2, which provide similar values to the experimental one. In general, PCM with Pauling radii scaled to 1.0 (default value is 1.1) is the option that better reproduces the SRP value for the [Fe(H2O)6]3+/2+ couple with all tested methods. It is worth noting here that the same calculations using the C-PCM model gave the same quantitative trends (Fig. S2 of ESI†).
We also carried out single-point energy calculations in solution on the B3LYP/6-31+G(d) geometries with the aforementioned combinations of functional and basis set by using the SMD model. The results are presented in Table 1. As can be seen, the SMD model reproduces very well the experimental SRP values when using the M06 and M06L functionals and the same basis set used in the calculation of 3IE. The reason for this might be that SMD is parametrized to reproduce experimental solvation energies calculated with the Minnesota functionals family. This is why the SRP of the [Fe(H2O)6]3+/2+ couple calculated at B3LYP/6-31+G(d) gives a highly overestimated value compared to the experimental one (1.43 V). However, the M06/6-31+G(d,p) method also overestimate the experimental SRP and also the SRP calculated at the M06/cc-pVQZ (Fe)-6-31+G(d,p) (O, H) level. This indicates that the 6-31+G(d,p) basis set by itself it is not good enough to reproduce the experimental SRP of [Fe(H2O)6]3+/2+ couple. With all these results in hand, we observe that the M06L/cc-pVDZ (Fe)-6-31+G(d,p) (O, H) methodology well reproduce the SRP of the [Fe(H2O)6]3+/2+ couple.
| M06/cc-pVQZ (Fe)-6-31+G(d,p) (O, H) | M06L/cc-pVDZ (Fe)-6-31+G(d,p) (O, H) | M06/6-31+G(d,p) | B3LYP/6-31+G(d,p) | |
|---|---|---|---|---|
| SRP | 0.71 | 0.74 | 1.10 | 1.43 |
According to these results, SMD and the M06L/cc-pVDZ (Fe)-6-31+G(d,p) (O, H) level represents an appropriate solvent model and method for subsequent calculations.
It is worth mentioning that the calculated SRP may improve with the inclusion of explicit solvent molecules, as have been reported for the aquo–iron complexes.36 Nevertheless, this approach requires an exhaustive conformational search for the position of water molecules in the second coordination shell and therefore it increases significantly the computational cost of such calculations, which is beyond the scope of this work. In our case, using SMD as implicit solvent model reproduces quite well the reported SRP at the M06L/cc-pVDZ (Fe)-6-31+G(d,p) (O, H) level of theory.
Standard reduction potential (SRP) calculations of iron complexes
The main goal of this work is to propose a reliable method for the calculation of the SRP of iron complexes relevant to the Alzheimer disease. However, as we noted before, the SRP are highly sensitive, on one side, to the chemical environment, and on the other side, to the computational protocol used for calculations. Thus, direct calculation of the SRP of iron complexes generates significative errors, and therefore an appropriated protocol to get reliable data to compare to the experiment is needed.The calculation of the SRP can be done by using the direct and isodesmic methods. In the direct method, the SRP calculation implies the calculation of the solvation Gibbs energy and comparison of this value with the Gibbs energy of a known experimental potential (e.g. standard hydrogen electrode, SHE). Then, the SRP can be calculated by using the next equation: E0 = (ΔG0 − ΔGSHE)/nF, where the first term, ΔG0, is the Gibbs energy of the reduction process calculated using electronic structure methods and ΔGSHE is the experimental value of the standard hydrogen electrode (99.9 kcal mol−1).37 For large systems, the optimization including solvation effects is computational expensive, therefore, a Born–Haber cycle can be used, as shown in Scheme 1.18
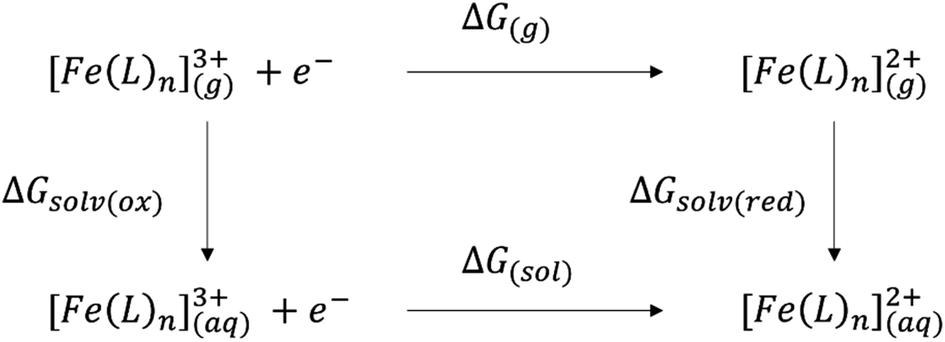 | ||
| Scheme 1 Born–Haber cycle to calculate the SRP of iron complexes. | ||
Then the Gibbs energy in solution is calculated as:
| ΔG(sol) = ΔG(g) + ΔGsolv(red) − ΔGsolv(ox) | (2) |
In the isodesmic method a redox reaction is considered with respect to a reference pair. A proper reference complex must fulfill the following requirements: (i) same coordination sphere; (ii) same numbers and types of bonds; (iii) same charges in the metal centers after and before reduction. The reduction reaction can be express as:
| [Fe(L)n]sol3+ + [Fe(L)n]sol2+,ref → [Fe(L)n]sol2+ + [Fe(L)n]sol3+,ref | (3) |
| E0 = E0,refexp + E0calc − E0,refcalc | (4) |
In this equation E0,refexp is the reported SRP of the reference pair, E0,refcalc is the calculated SRP of the reference pair and E0calc is the calculated SRP for the target system. The isodesmic method have been used to calculate SRP and pKa of cobalt complexes.38,39 Later on Chaparro and Alí-Torres40 showed a good correlation between the experimental and calculated SRP values in a series of 64 copper complexes using the isodesmic method. The advantage of this methodology is the cancelation of errors in the calculation of solvation Gibbs energies. However, the selection of a right reference couple with a known standard potential is one of the main challenges to apply this methodology.
For the present case, we selected a set of 17 Fe2+/3+ complexes (Fig. 5) with known SRP as a calibration set to obtain reliable SRP for Fe-Aβ complexes.41 They all can be used for calculations used the direct method. However, in the case of the isodesmic method only ten of them fits the above requirements on similar coordination shells, oxidation states and number/types of bonds when compared to the model Fe-Aβ complexes used here (Fig. 2).
The calculated standard reduction potentials for this set using SMD and PCM solvation models are presented in Fig. 6 using the direct and the isodesmic method.
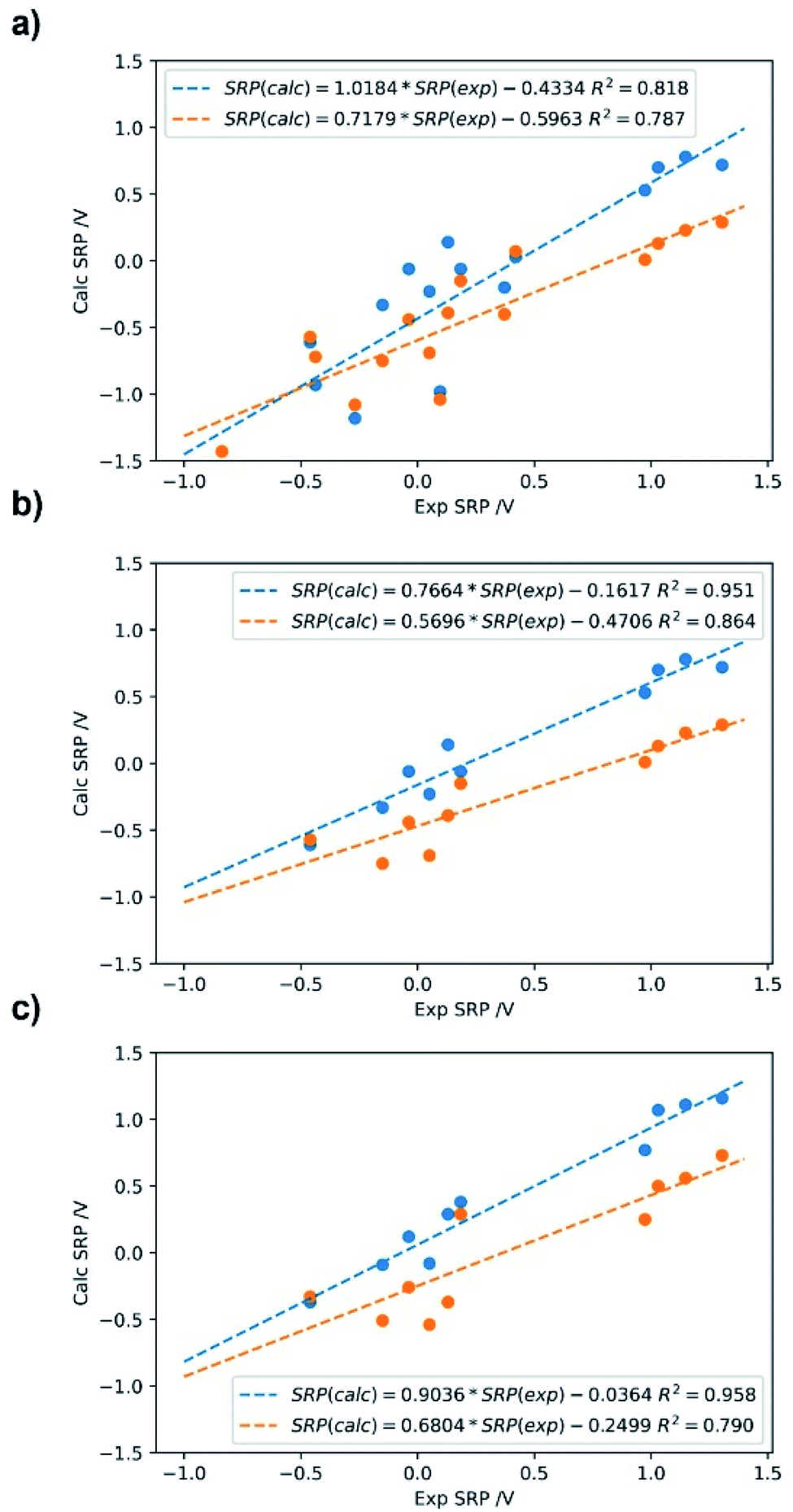 | ||
| Fig. 6 Calculated SRP for the iron complexes in Fig. 5 using M06L/cc-pVDZ(Fe)+6-31+G(d,p) (C,N,H,O); Data in blue: SMD; data in orange: PCM with Pauling radii (scale factor: 1.05). (a) Direct method with 17 molecules; (b) direct method with 10 molecules used in the isodesmic calculations; (c) isodesmic method with 10 molecules. | ||
As can be seen from Fig. 6, good correlations are achieved with both methods and solvation models. In regard to the solvation model, it seems that the SMD model provides better correlation and lower errors than PCM with a Pauling radii scaled to 1.05. Comparing the direct versus the isodesmic method one can infer from Fig. 6a and c with the SMD model that the latter show improved linearity, with R2 = 0.96 versus R2 = 0.82 for the direct method. This is accompanied by a much lower y-intercept value (0.43 versus 0.04 V). In this case, we observed that the isodesmic method is the only one giving an error below the expected experimental uncertainty for electrochemical methods, that is ∼100 mV.
Table 2 shows the detailed values of the SRP for the different Fe3+/Fe2+ complexes used for calibration, including the errors respect to the experimental ones with the direct method and the SMD model. It is worth noting that recalculating the SRP values for all complexes in the calibration set using the regression formula SRP (calc) = 1.0184 × SRP (exp) – 0.4334 leads to a lowering of the mean absolute error (MAE) by about 55%.
| Complex (spin multiplicity) | Exp SRP | Direct | Direct linear regression | ||
|---|---|---|---|---|---|
| Calculated SRP | Abs error | Calculated SRP | Abs error | ||
| 1 (2/1) | 1.15 | 0.78 | 0.37 | 1.19 | 0.04 |
| 2 (2/1) | 1.03 | 0.70 | 0.33 | 1.11 | 0.08 |
| 3 (2/1) | 1.30 | 0.72 | 0.58 | 1.13 | 0.17 |
| 4 (2/5) | −0.04 | −0.06 | 0.02 | 0.37 | 0.40 |
| 5 (2/5) | 0.18 | −0.06 | 0.24 | 0.37 | 0.18 |
| 6 (6/5) | −0.84 | −1.55 | 0.71 | −1.10 | 0.26 |
| 7 (2/5) | −0.46 | −0.61 | 0.15 | −0.17 | 0.29 |
| 8 (2/1) | 0.05 | −0.23 | 0.28 | 0.20 | 0.15 |
| 9 (2/1) | −0.15 | −0.33 | 0.18 | 0.10 | 0.25 |
| 10 (2/1) | 0.13 | 0.14 | 0.01 | 0.56 | 0.43 |
| 11 (6/5) | 0.10 | −0.98 | 1.08 | −0.54 | 0.63 |
| 12 (6/5) | −0.27 | −1.18 | 0.91 | −0.73 | 0.47 |
| 13 (6/5) | 0.37 | −0.20 | 0.57 | 0.23 | 0.14 |
| 14 (2/1) | 0.97 | 0.53 | 0.44 | 0.95 | 0.03 |
| 15 (2/1) | 0.54 | 0.03 | 0.51 | 0.46 | 0.09 |
| 16 (2/5) | −0.44 | −0.93 | 0.49 | −0.49 | 0.05 |
| MAE | 0.43 | 0.24 | |||
Focusing on Fig. 6c it can be seen that the isodesmic method provides a good correction in the predicted SRP values. This may be due to the fact that this method cancels out errors due to the electronic structure and solvation description when a complex similar to the complex of interest is used. From Table 3, the regression shows how the use of isodesmic method improve the prediction of the SRP values compared with the direct method. This postulates the isodesmic method as a proper methodology for the SRP calculations of iron complexes. From this data we can see that the absolute errors, both the calculated and the estimated with the linear regression equation, are below the uncertainty provided by the experimental electrochemical methods, and these errors are significantly lower than the obtained by applying the direct method (Table 2). These results confirm that an adequate selection of method, basis set, solvation model and thermodynamic cycles are necessary in order to obtain reliable SRP values.
| Complex (spin multiplicity) | Exp SRP | Direct | Isodesmic | Isodesmic linear regression | Ref pair | ||
|---|---|---|---|---|---|---|---|
| Abs error | Calculated SRP | Abs error | Calculated SRP | Abs error | |||
| 1 (2/1) | 1.15 | 0.37 | 1.11 | 0.04 | 1.18 | 0.03 | 2 |
| 2 (2/1) | 1.03 | 0.33 | 1.07 | 0.04 | 1.14 | 0.11 | 1 |
| 3 (2/1) | 1.30 | 0.58 | 1.16 | 0.14 | 1.24 | 0.06 | 14 |
| 4 (2/5) | −0.04 | 0.02 | 0.09 | 0.13 | 0.06 | 0.10 | 7 |
| 5 (2/5) | 0.18 | 0.24 | −0.04 | 0.22 | −0.08 | 0.26 | 4 |
| 7 (2/5) | −0.46 | 0.15 | −0.37 | 0.09 | −0.44 | 0.02 | 5 |
| 8 (2/1) | 0.05 | 0.28 | −0.05 | 0.10 | −0.10 | 0.15 | 9 |
| 9 (2/1) | −0.15 | 0.18 | −0.05 | 0.10 | −0.10 | 0.05 | 8 |
| 10 (2/1) | 0.13 | 0.01 | 0.32 | 0.19 | 0.31 | 0.18 | 9 |
| 14 (2/1) | 0.97 | 0.44 | 0.90 | 0.07 | 0.95 | 0.02 | 1 |
| MAE | 0.19 | 0.11 | 0.09 | ||||
So far it seems that the isodesmic method outperforms the direct method for the calculation of SRP of iron complexes using the SMD solvation model. However, comparison of adjustments in Fig. 6a and c are not truly fair, since the isodesmic calculation was carried out with only ten iron complexes, the ones with most similar chemical environment to our target Fe2+/3+-Aβ complexes (Fig. 2). Therefore, we develop a third regression for the SRP of iron complexes using the direct method but including only the ten systems used in the isodesmic calculations. The results are shown in Fig. 6b for both SMD and PCM solvation models. As observed, again SMD performs better than PCM, and the adjustment in this case is almost identical to the adjustment in the isodesmic method (R2 = 0.95 versus R2 = 0.96). However, altough the regressions are statistically similar, two factor are against the direct method in comparison to the isodesmic one: (i) a lower slope (0.77 versus 0.91); and (ii) a higher absolute error for the regression, above the experimental accepted value (0.43 V versus 0.11 V). Thus, even comparing the same set of iron complexes with the two methods for calculation of SRP, we noted that the isodesmic method remain superior in performance for the prediction of SRP values of iron complexes.
In order to verify the effect of solvent on the SRP calculations, we reoptimized the geometries of complexes 8 and 9 in solution (water) and calculated their SRPs via direct and isodesmic methods. The results are shown in Table S6 of ESI.† We observed small changes in the calculated SRP, and in fact these effects are lower when using the isodesmic method. This agreement may be attributed to the error cancellation due to the use of a reference pair in the isodesmic method.
Fe2+/3+-Aβ complexes
With the conclusion obtained from the previous sections, we used the regression formula SRP(calc) = 0.9036 × SRP(exp) − 0.0364 developed for the isodesmic method in order to predict the SRPs of some representative Fe3+/2+-Aβ model systems relevant to the Alzheimer disease. These complexes were selected since they represent plausible coordination spheres for Fe-Aβ complexes, as have been reported by means of computational21 and Raman experiments.42,43 The geometries for the Fe3+/2+-Aβ complexes were optimized at the B3LYP/6-31+G(d) level. The relative energies of all structures in all possible spin states of the Fe3+/2+ complexes are show in Table S4 of ESI.† These structures as well as their computed SRP are presented in Fig. 7. All calculations were carried out at the M06L/cc-pVDZ(Fe)-6-31+G(d,p) (C, N, H, O) level of theory and by using the SMD model.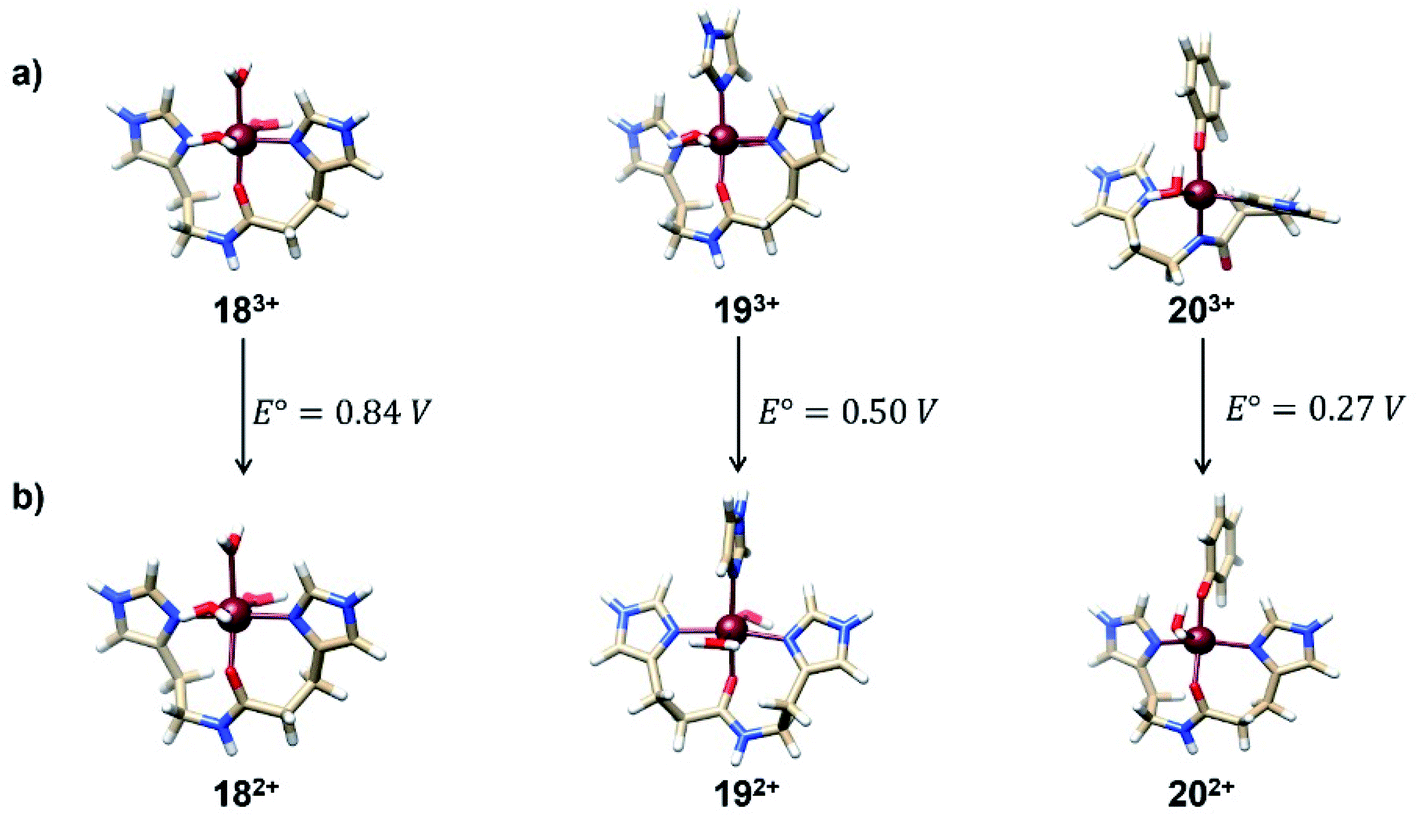 | ||
| Fig. 7 Optimized geometries and calculated SRP values for a series of representative Fe3+/2+-Aβ model systems. For all the Fe3+ complexes the sextet spin-state is the ground state while for Fe2+ is the quintet. | ||
The values obtained from these complexes are in good agreements with previous computational reports which calculated the SRP values of these complexes through MP2 calculations.21 In this work, the use of DFT method decreases the computational effort, which allows the calculation of SRPs of a large set of complexes with a similar performance. The values obtained for the three complexes show that the tyrosine coordination decreased the SRP value allowing the participation of this complex in the ROS formation cycle shown in Fig. 1. This is due to the fact that is the only couple with an SRP lower than the corresponding to the O2/H2O2 couple (0.30 V).44 The proposed protocol showed good performance in the calculation of these SRP values with a good compromise between accuracy and computational cost.
Conclusions
In this work, we propose a computational protocol for the calculation of the SRP of iron complexes based on DFT. The evaluation of 51 density functionals in combination with 38 basis sets showed that the best methods for reproducing the iron third ionization energy were the Minnesota, Half and Half and double hybrid functionals and the best basis set were Pople, Dunning and Aldrich families, since they provided the lowest percentage of error when compared with the experimental value. In the same line, the SMD model was the method that better reproduced the SRP for the [Fe(H2O)6]3+/2+ couple. Similarly, the isodesmic method generated SRP in better agreement with experimental values for a series of iron complexes than the direct method (MAE: 0.43 V for the direct method and 0.11 V for isodesmic). By combining the M06L functional with the SMD implicit solvation model and the isodesmic method we evaluated three iron complexes representing the coordination spheres of Fe2+/3+-Aβ systems. Results showed that this protocol reproduces quite well the results compared with those calculated previously with MP2 method. This confirms this protocol as a feasible tool for the calculation of SRP of iron complexes.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
A. L. O. Thanks UNAL for the teaching assistant scholarship, F. N.-Z. wishes to thank the Universidad de Medellín for financial support (project 1105) and J. A-T. thanks DIB-UNAL for financial support and the Center of Excellence in Scientific Computing (CoE – SciCo) for computational time.References
- R. J. Ward, F. A. Zucca, J. H. Duyn, R. R. Crichton and L. Zecca, The role of iron in brain ageing and neurodegenerative disorders, Lancet Neurol., 2014, 13, 1045–1060 CrossRef CAS PubMed.
- A. A. Belaidi and A. I. Bush, Iron neurochemistry in Alzheimer's disease and Parkinson's disease: targets for therapeutics, J. Neurochem., 2016, 179–197 CrossRef CAS PubMed.
- H. Mochizuki, C. J. Choong and K. Baba, Parkinson's disease and iron, J. Neural Transm., 2020, 127, 181–187 CrossRef PubMed.
- G. Mostile, C. E. Cicero, L. Giuliano, M. Zappia and A. Nicoletti, Iron and Parkinson's disease: a systematic review and meta-analysis, Mol. Med. Rep., 2017, 15, 3383–3389 CrossRef CAS PubMed.
- A. Rauk, The chemistry of Alzheimer's disease, Chem. Soc. Rev., 2009, 38, 2698 RSC.
- J. F. Schenck, E. A. Zimmerman, Z. Li, S. Adak, A. Saha, R. Tandon, K. M. Fish, C. Belden, R. W. Gillen, A. Barba, D. L. Henderson, W. Neil and T. O'Keefe, High-field magnetic resonance imaging of brain iron in Alzheimer disease, Topics in Magnetic Resonance Imaging, 2006, 17, 41–50 CrossRef PubMed.
- D. J. R. Lane, S. Ayton and A. I. Bush, Iron and Alzheimer's Disease: An Update on Emerging Mechanisms, J. Alzheimer's Dis., 2018, 64, S379–S395 CAS.
- J. L. Liu, Y. G. Fan, Z. S. Yang, Z. Y. Wang and C. Guo, Iron and Alzheimer's disease: from pathogenesis to therapeutic implications, Front. Neurosci., 2018, 12, 1–14 CAS.
- M. L. Hegde, P. M. Hegde, K. S. Rao and S. Mitra, Oxidative genome damage and its repair in neurodegenerative diseases: function of transition metals as a double-edged sword, J. Alzheimer's Dis., 2011, 24, 183–198 CAS.
- B. J. Tabner, S. Turnbull, R. Omar, M. A. El-Agnaf and D. Allsop, Production of Reactive Oxygen Species from Aggregating Proteins Implicated in Alzheimers Disease, Parkinsons Disease and Other Neurodegenerative Diseases, Curr. Top. Med. Chem., 2005, 1, 507–517 CrossRef PubMed.
- K. P. Kepp, Bioinorganic Chemistry of Alzheimer's Disease, Chem. Rev., 2012, 112, 5193–5239 CrossRef CAS PubMed.
- C. C. Winterbourn, Toxicity of iron and hydrogen peroxide: the Fenton reaction, Toxicol. Lett., 1995, 82–83, 969–974 CrossRef CAS PubMed.
- M. Radoń, Benchmarking quantum chemistry methods for spin-state energetics of iron complexes against quantitative experimental data, Phys. Chem. Chem. Phys., 2019, 21, 4854–4870 RSC.
- G. M. Sandala, K. H. Hopmann, A. Ghosh and L. Noodleman, Calibration of DFT functionals for the prediction of 57Fe mössbauer spectral parameters in iron-nitrosyl and iron-sulfur complexes: accurate geometries prove essential, J. Chem. Theory Comput., 2011, 7, 3232–3247 CrossRef CAS PubMed.
- S. Song, M.-C. Kim, E. Sim, A. Benali, O. Heinonen and K. Burke, Benchmarks and reliable DFT results for spin-crossover complexes, arXiv: Chemical Physics Search PubMed.
- M. Gruden, S. Stepanovic and M. Swart, Spin state relaxation of iron complexes: the case for OPBE and S12g, J. Serb. Chem. Soc., 2015, 80, 1399–1410 CrossRef CAS.
- J. Conradie and A. Ghosh, DFT calculations on the Spin-crossover complex fe(salen)(NO): a quest for the best functional, J. Phys. Chem. B, 2007, 111, 12621–12624 CrossRef CAS.
- K. Arumugam and U. Becker, Computational redox potential predictions: applications to inorganic and organic aqueous complexes, and complexes adsorbed to mineral surfaces, Minerals, 2014, 4, 345–387 CrossRef.
- L. E. Roy, E. Jakubikova, M. Graham Guthrie and E. R. Batista, Calculation of one-electron redox potentials revisited. Is it possible to calculate accurate potentials with density functional methods?, J. Phys. Chem. A, 2009, 113, 6745–6750 CrossRef CAS PubMed.
- M. A. Rizvi, M. Mane, M. A. Khuroo and G. M. Peerzada, Computational survey of ligand properties on iron(III)–iron(II) redox potential: exploring natural attenuation of nitroaromatic compounds, Monatsh. Chem., 2017, 148, 655–668 CrossRef CAS.
- J. Alí-Torres, L. Rodríguez-Santiago, M. Sodupe and A. Rauk, Structures and Stabilities of Fe2+/3+ Complexes Relevant to Alzheimer's Disease: An ab Initio Study, J. Phys. Chem. A, 2011, 115, 12523–12530 CrossRef PubMed.
- M. Horch, Rational redox tuning of transition metal sites: learning from superoxide reductase, Chem. Commun., 2019, 55, 9148–9151 RSC.
- A. Kramida, Y. Ralchenko, J. Reader, and NIST ASD Team, NIST Atomic Spectra Database (ver. 5.9), National Institute of Standards and Technology, Gaithersburg, MD, 2021, available https://physics.nist.gov/asd [2022, August 19], DOI:10.18434/T4W30F.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision D.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- V. A. Rassolov, J. A. Pople, M. A. Ratner and T. L. Windus, 6-31G* basis set for atoms K through Zn, J. Chem. Phys., 1998, 109, 1223–1229 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- T. H. Dunning, Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- W. J. Stevens, M. Krauss, H. Basch and P. G. Jasien, Relativistic compact effective potentials and efficient, shared-exponent basis sets for the third-, fourth-, and fifth-row atoms, Can. J. Chem., 1992, 70, 612–630 CrossRef CAS.
- N. E. Schultz, Y. Zhao and D. G. Truhlar, Density functional for inorganometallic and organometallic chemistry, J. Phys. Chem. A, 2005, 109, 11127–11143 CrossRef CAS.
- J. Tomasi and M. Persico, Molecular Interactions in Solution: An Overview of Methods Based on Continuous Distributions of the Solvent, Chem. Rev., 1994, 94, 2027–2094 CrossRef CAS.
- K. Hyungjun, P. Joungwon and L. Yoon Sup, A protocol to evaluate one electron redox potential for iron complexes, J. Comput. Chem., 2013, 34, 2233–2241 CrossRef PubMed.
- S. G. Bratsch, Standard Electrode Potentials and Temperature Coefficients in Water at 298.15 K, J. Phys. Chem., 1989, 18, 1–21 CAS.
- B. Mennucci, J. Tomasi, R. Cammi, J. R. Cheeseman, M. J. Frisch, F. J. Devlin, S. Gabriel and P. J. Stephens, Polarizable continuum model (PCM) calculations of solvent effects on optical rotations of chiral molecules, J. Phys. Chem. A, 2002, 106, 6102–6113 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- A. v Marenich, C. J. Cramer and D. G. Truhlar, Universal solvation model based on solute electron density and a continuum model of the solvent defind by the bulk dielectric constant and atomic surface tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef PubMed.
- A. N. Masliy, T. N. Grishaeva and A. M. Kuznetsov, Standard Redox Potentials of Fe(III) Aqua Complexes Included into the Cavities of Cucurbit[n]urils (n = 6–8): A DFT Forecast, J. Phys. Chem. A, 2019, 123, 1341–1346 CrossRef PubMed.
- M. D. Tissandier, K. A. Cowen, W. Y. Feng, E. Gundlach, M. H. Cohen, A. D. Earhart, J. v Coe and T. R. Tuttle, The Proton’s Absolute Aqueous Enthalpy and Gibbs Free Energy of Solvation from Cluster-Ion Solvation Data, J. Phys. Chem. A, 1998, 102(40), 7787–7794 CrossRef CAS.
- B. H. Solis and S. Hammes-Schiffer, Substituent effects on cobalt diglyoxime catalysts for hydrogen evolution, J. Am. Chem. Soc., 2011, 133, 19036–19039 CrossRef CAS PubMed.
- T. Fernández, A. Martínez-Serrano, L. Cussó, M. Desco and M. Ramos-Gómez, Functionalization and Characterization of Magnetic Nanoparticles for the Detection of Ferritin Accumulation in Alzheimer's Disease, ACS Chem. Neurosci., 2018, 9, 912–924 CrossRef PubMed.
- D. Chaparro and J. Alí-Torres, Assessment of the isodesmic method in the calculation of standard reduction potential of copper complexes, J. Mol. Model., 2017, 23, 283 CrossRef.
- A. Galstyan and E. W. Knapp, Accurate redox potentials of mononuclear iron, manganese, and nickel model complexes, J. Comput. Chem., 2009, 30, 203–211 CrossRef CAS PubMed.
- T. Miura, K. Suzuki and H. Takeuchi, Binding of iron(III) to the single tyrosine residue of amyloid β-peptide probed by Raman spectroscopy, J. Mol. Struct., 2001, 598, 79–84 CrossRef CAS.
- T. Miura, K. Suzuki, N. Kohata and H. Takeuchi, Metal binding modes of Alzheimer's amyloid β-peptide in insoluble aggregates and soluble complexes, Biochemistry, 2000, 39, 7024–7031 CrossRef CAS PubMed.
- D. L. Nelson and M. Cox, Lehninger Principles of Biochemistry, W. H Freeman, New York, 2005 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra03907a |
| This journal is © The Royal Society of Chemistry 2022 |