DOI:
10.1039/D2RA03875G
(Paper)
RSC Adv., 2022,
12, 21548-21557
Cationic palladium(II)-catalyzed synthesis of substituted pyridines from α,β-unsaturated oxime ethers†
Received
23rd June 2022
, Accepted 14th July 2022
First published on 4th August 2022
Abstract
An efficient method for the synthesis of multi-substituted pyridines from β-aryl-substituted α,β-unsaturated oxime ethers and alkenes via Pd-catalyzed C–H activation has been developed. The method, using Pd(OAc)2 and a sterically hindered pyridine ligand, provides access to various multi-substituted pyridines with complete regioselectivity. Mechanistic studies suggest that the pyridine products are formed by Pd-catalyzed electrophilic C–H alkenylation of α,β-unsaturated oxime followed by aza-6π-electrocyclization. The utility of this method is showcased by the synthesis of 4-aryl-substituted pyridine derivatives, which are difficult to synthesize efficiently using previously reported Rh-catalyzed strategies with alkenes.
Introduction
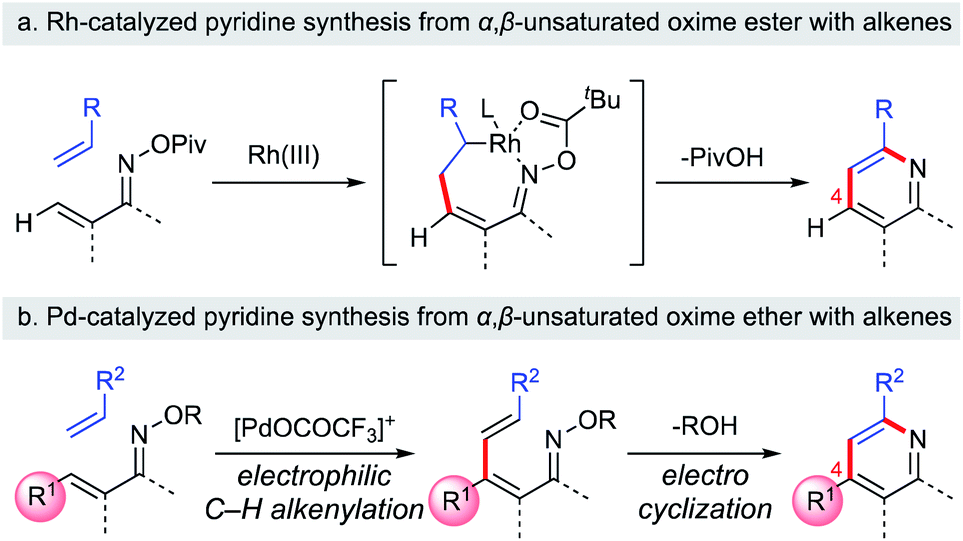
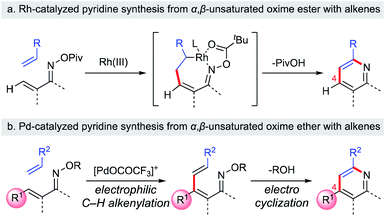
Substituted pyridine scaffolds are ubiquitous in medicinal chemistry. A recent analysis of the ring systems in all FDA-approved pharmaceutical drugs revealed the pyridine ring to be the most prevalent nitrogen-containing heteroaromatic motif.1 In particular, appropriately substituted pyridines exhibit a wide range of biological activities.2 In this context, the development of a versatile approach to build substituted pyridines from readily accessible building blocks is a topic of continuing interest for synthetic chemists. Classical methods to access substituted pyridines generally involve either carbonyl condensation3 or [2 + 2 + 2] cycloaddition.4 However, these methods require harsh reaction conditions and only limited pyridine substitution patterns can be synthesized. Recently, remarkable progress has been reported, with the assembly of the pyridine core via [4 + 2] cycloaddition from 1-azadienes and two-carbon π-components.5 Rh-catalyzed [4 + 2] cycloaddition approaches are particularly useful, and can provide access to multi-substituted pyridines from α,β-unsaturated oximes with alkynes or alkenes.6 For example, the Rovis group reported that the Rh(III)-catalyzed coupling of α,β-unsaturated oximes with activated alkenes was a useful approach to build substituted pyridines with good regioselectivity (Scheme 1a).6e Interestingly, this reaction involves a 7-membered rhodacycle intermediate, which undergoes C–N bond formation/N–O bond cleavage to furnish the pyridine product. Unfortunately, high yields are obtained only with β-unsubstituted α,β-unsaturated oximes. Therefore, efficient strategies to construct 4-substituted pyridines from readily accessible β-substituted α,β-unsaturated oximes with high regioselectivity are still needed.
 |
| | Scheme 1 Synthesis of substituted pyridines from α,β-unsaturated oximes. | |
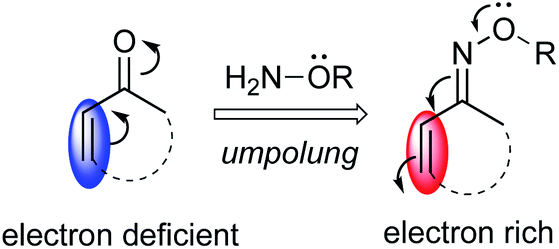
Umpolung reactivity (polarity inversion) is an effective strategy for organic synthesis, as demonstrated by the wealth of examples that have contributed to the development of synthetic chemistry.7 Especially, carbonyl Umpolung using oximes and hydrazones, which have an electron-donating hetero atom in an imine functionality, has been developed to invert the electrophilic reactivity of carbonyl compounds to a nucleophilic character.8,9 However, with regard to α,β-unsaturated oximes, there are only a few reports on extension of this electron-donating ability to conjugated olefins.10 Because of the electron-donating effect of an electron lone pair, conjugated systems of α,β-unsaturated oximes, especially at the β-position, can potentially exhibit nucleophilic reactivity (Fig. 1).
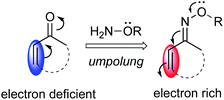 |
| | Fig. 1 Umpolung reactivity of α,β-unsaturated oximes. | |
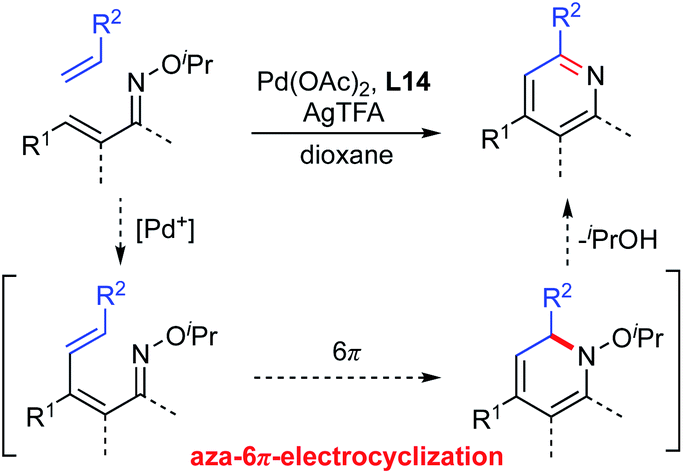
With this unique Umpolung reactivity in mind, we have developed a C–H functionalization of the β-position of α,β-unsaturated oximes using cationic Pd(II) catalysts.11 In this reaction, α,β-unsaturated oximes react with highly electrophilic cationic Pd(II) species, such as Pd(OCOCF3)+, at the nucleophilic β-position via electrophilic C–H activation. Based on this work, we have recently communicated a new method for the synthesis of multi-substituted pyridines from α,β-unsaturated oxime ethers via a two-step process: cationic Pd(II)-catalyzed electrophilic C–H alkenylation of α,β-unsaturated oximes followed by aza-6π-electrocyclization (Scheme 1b).12 The key features of our method are its operational simplicity, broad substrate scope, and the use of a general Pd(OAc)2 catalyst to obtain 4-substituted pyridine derivatives with complete regioselectivity. This work complements previously reported pyridine synthetic methods, since 4-unsubstituted pyridines can already be easily prepared with alkenes by using Rh-catalyzed strategies (Scheme 1a vs. b). In this article, we present a full account of this work, together with additional experimental data to provide insights into the mechanism of the cationic palladium(II)-catalyzed electrophilic C–H activation of α,β-unsaturated oximes.
Results and discussion
Optimization of pyridine ligand
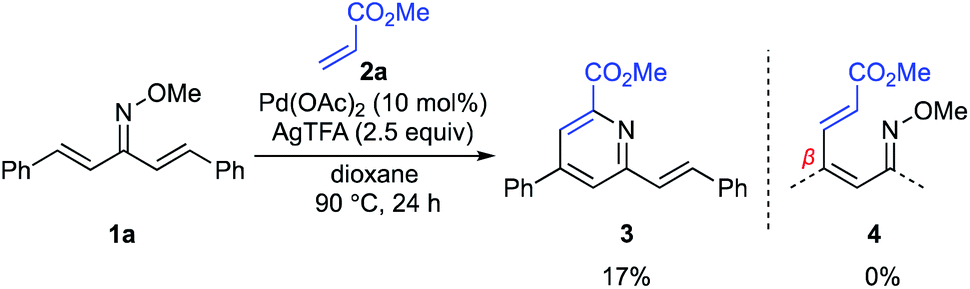
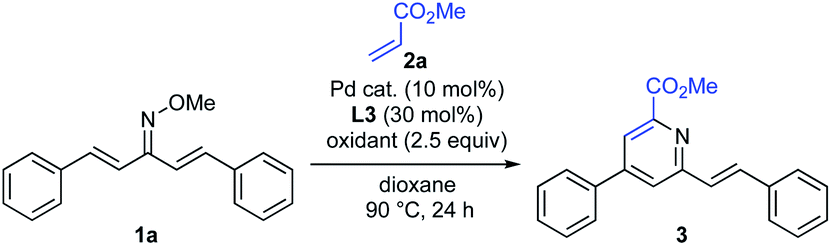
For the examination of this pyridine synthesis, we first chose a symmetrical oxime 1a as a model substrate (Scheme 2). Based on our experience in the development of oxime chemistry,11 oxime ethers of β-substituted α,β-unsaturated ketones readily undergo E/Z isomerism, which might interfere with optimization of the reaction. Thus, our studies began with examination of the reaction of symmetrical α,β-unsaturated O-methyl oxime 1a and methyl acrylate (2a) in the presence of Pd(OAc)2 as a catalyst (Scheme 2). With silver trifluoroacetate (AgTFA) as an oxidant and dioxane as a solvent, the desired pyridine 3 was obtained in 17% yield. It should be noted here that no β-alkenylation product 4 was observed, suggesting that aza-6π-electrocyclization of 4 and subsequent aromatization proceed rapidly to furnish the pyridine 3. Although other solvents such as toluene, 1,2-dichloroethane, DMF, and HFIP were examined, the product yield was not improved. Unfortunately, the high temperature (over 100 °C) reaction caused substrate decomposition.
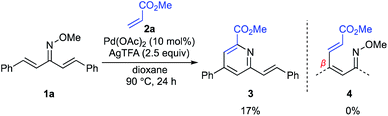 |
| | Scheme 2 Initial experiment. | |
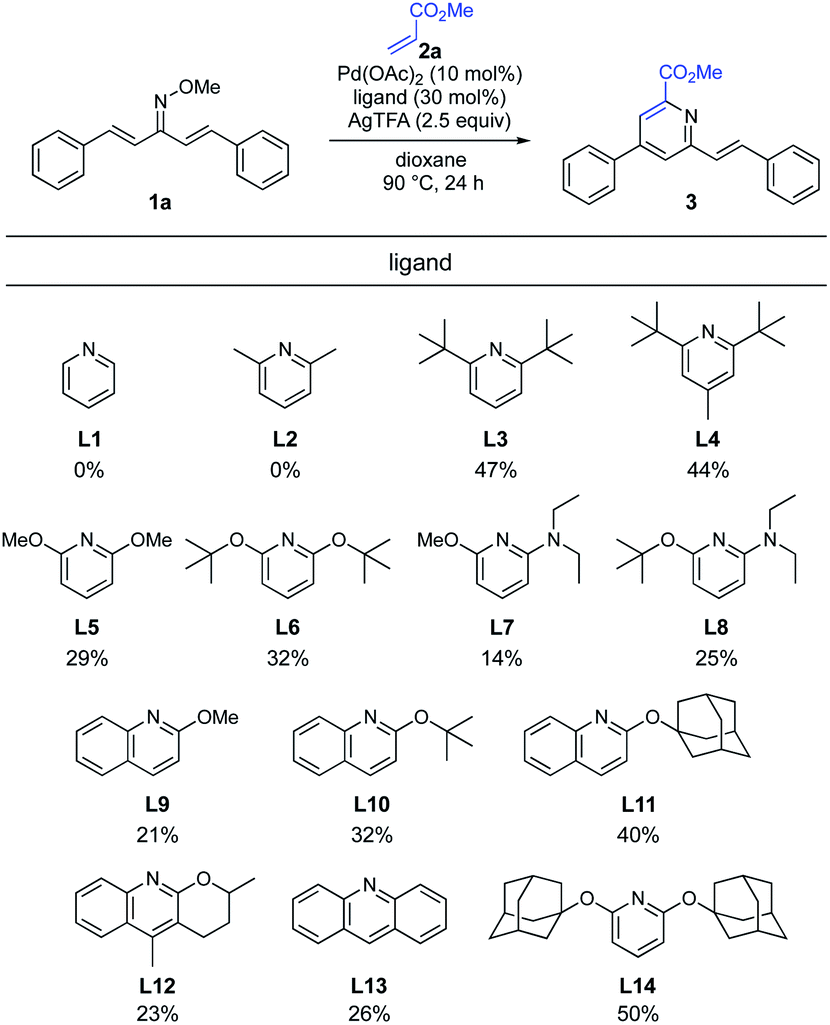
Our experience in the development of Pd-catalyzed β-selective C–H functionalization of α,β-unsaturated oximes showed that the identification of a suitable catalyst ligand would be the key to establish the optimal protocol.11 Recently, Ji and co-workers have reported that the addition of pyridine promoted the Pd-catalyzed C(sp2)–H alkenylation of O-methyl ketoximes.13 With Ji's results in mind, we explored the effect of pyridine derivatives as ligands (Scheme 3). Although the addition of pyridine (L1) and 2,6-lutidine (L2) unexpectedly inhibited the reaction, sterically hindered 2,6-di-tert-butyl pyridine (L3) and 2,6-di-tert-butyl 4-methyl pyridine (L4) significantly increased the reactivity and the desired pyridine 3 was obtained in 47% and 44% yields, respectively. The use of 2,6-dialkoxypyridines L5 and L6, which are more electron-rich ligands than pyridine (L1), afforded the pyridine 3 in 29% and 32% yields, respectively. Unfortunately, the reactions employing pyridines L7 and L8, containing a stronger electron-donating dialkylamino group, decreased the product yield. This was presumably because pyridine with a strong coordination ability deactivates the Pd catalyst. Next, we tested a series of 2-alkoxylquinoline derivatives (L9–L12). Despite the ineffectiveness of 2-methoxyquinoline (L9), the product yield was improved when a quinoline ligand bearing a bulkier side chain, such as tert-butoxy (L10) or 1-adamantyloxy (L11), was used. This substituent effect suggested that the steric hindrance around the nitrogen atom of the quinoline ligand is crucial for high reactivity. Unexpectedly, tricyclic quinoline-derived ligand L12, developed by Yu and co-workers,14 and acridine (L13) led to loss of reactivity. Anticipating a positive effect of a bulkier ligand, we prepared 2,6-diadamantyloxypyridine (L14) in one step from 2,6-difluoropyridine and 1-adamantanol. To our delight, the use of L14 improved the reactivity and afforded 3 in 50% yield.
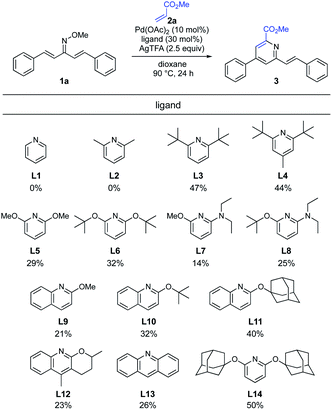 |
| | Scheme 3 Optimization of pyridine ligand. aReaction conditions: 1a (0.2 mmol, 1.0 equiv.), 2a (3.0 equiv.), Pd(OAc)2 (10 mol%), ligand (30 mol%), AgTFA (2.5 equiv.), dioxane (2.0 mL), 90 °C, 24 h. Isolated yield. | |
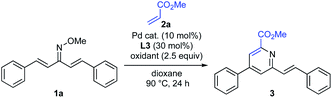
Effect of Pd catalyst and oxidant
Next, the effects of palladium catalyst and oxidizing agent were investigated (Table 1). Without AgTFA, the desired product 3 was not formed (entry 2). When we employed PdCl2 instead of Pd(OAc)2, the reaction proceeded, albeit at the expense of a significantly decreased yield (entry 3). The use of Pd(TFA)2 slightly increased the reactivity and afforded 3 in 53% yield (entry 4). When AgOAc was used as an oxidant, the reaction efficiency was significantly decreased, indicating that the addition of AgTFA is necessary for the C–H alkenylation reaction to proceed (entry 5). These results are consistent with the hypothesis that Pd(OCOCF3)+ generated in situ from Pd(OAc)2 and AgTFA is the reactive species in this transformation (Scheme 1b).15
Table 1 Effect of Pd catalyst and oxidanta
|
| Entry |
Pd cat. |
Oxidant |
Yield |
| Reaction conditions: 1a (0.2 mmol, 1.0 equiv.), 2a (3.0 equiv.), Pd cat. (10 mol%), L3 (30 mol%), oxidant (2.5 equiv.), dioxane (2.0 mL), 90 °C, 24 h. Isolated yield. |
| 1 |
Pd(OAc)2 |
AgTFA |
47% |
| 2 |
Pd(OAc)2 |
None |
0% |
| 3 |
PdCl2 |
AgTFA |
10% |
| 4 |
Pd(TFA)2 |
AgTFA |
53% |
| 5 |
Pd(TFA)2 |
AgOAc |
Trace |
Optimization of oxime ether and its influence on reactivity
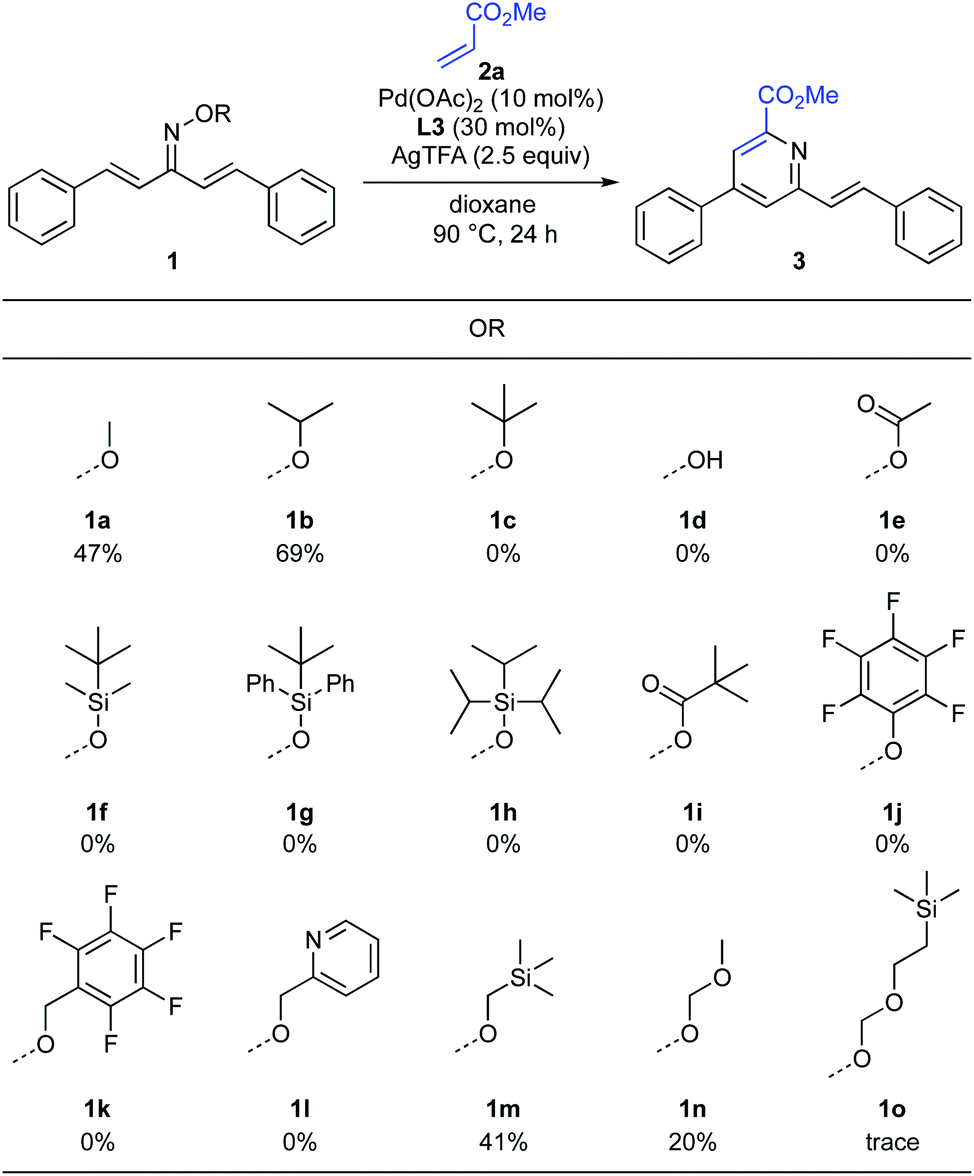
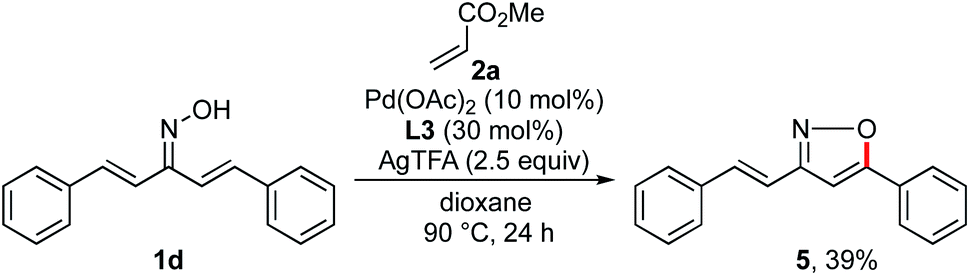
To improve the reaction efficiency, we next focused on modification of the oxime ether moiety (Scheme 4). Tuning the structure of the oxime ether moiety makes it possible to change the electronic state and nucleophilic reactivity of α,β-unsaturated oxime.11 Among the oxime ethers surveyed, O-isopropyl oxime 1b improved the reactivity and the product yield increased to 69%. In the case of the highly sterically hindered O-tert-butyl oxime 1c, the starting material was completely recovered. The parent oxime 1d afforded none of the desired pyridine, instead undergoing intramolecular cyclization to give isoxazole 5 in 39% yield (Scheme 5). Isoxazole 5 was similarly observed with the O-acetyl oxime 1e and O-silyl oximes (1f–1h). Formation of 5 may proceed via deprotection of oxime ester 1e or silyl oximes (1f–1h) to release the parent oxime 1d, followed by intramolecular cyclization.6e Although various oximes bearing O-pivaloyl (1i), O-pentafluorophenyl (1j), O-pentafluorobenzyl (1k), and O-pyridylmethyl (1l) groups were examined, none of them underwent pyridine formation. Heteroatom-containing alkyl oxime ethers such as O-methyl trimethylsilyl oxime 1m, O-methoxymethyl oxime 1n, or O-SEM oxime 1o afforded the desired pyridine, but the product yield was significantly decreased.
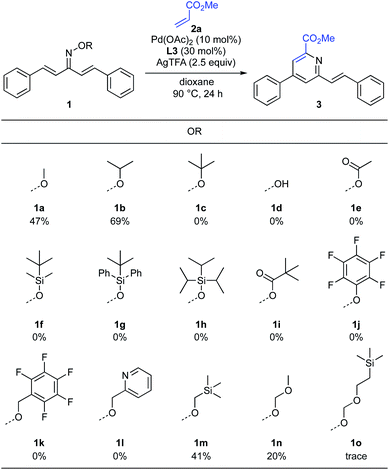 |
| | Scheme 4 Optimization of oxime ether. aReaction conditions: 1 (0.2 mmol, 1.0 equiv.), 2a (3.0 equiv.), Pd(OAc)2 (10 mol%), L3 (30 mol%), AgTFA (2.5 equiv.), dioxane (2.0 mL), 90 °C, 24 h. Isolated yield. | |
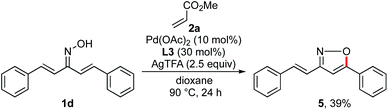 |
| | Scheme 5 Formation of isoxazole 5. | |
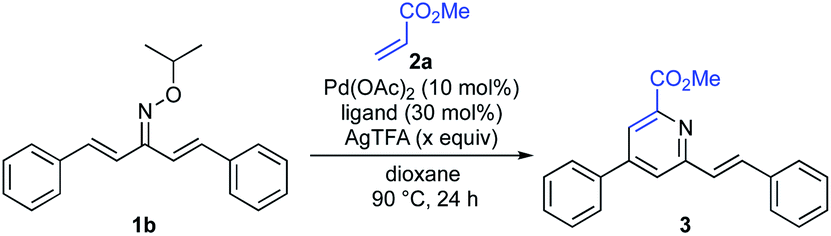
With the optimal oxime ether moiety (isopropyl) in hand, the reaction conditions were further screened (Table 2). Introduction of ligand L14 in place of L3 improved the yield from 69% to 76% (entry 2). After a brief investigation of the amount of oxidant, we found the use of 5.0 equiv. of AgTFA to be optimal, providing the pyridine 3 in 85% yield (entries 3 and 4).
Table 2 Optimization of the reaction conditionsa
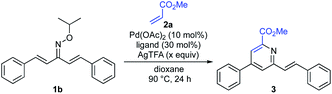
|
| Entry |
Ligand |
AgTFA (x equiv.) |
Yield |
| Reaction conditions: 1b (0.2 mmol, 1.0 equiv.), 2a (3.0 equiv.), Pd(OAc)2 (10 mol%), ligand (30 mol%), AgTFA (x equiv.), dioxane (2.0 mL), 90 °C, 24 h. Isolated yield. |
| 1 |
L3 |
2.5 |
69% |
| 2 |
L14 |
2.5 |
76% |
| 3 |
L14 |
4.0 |
77% |
| 4 |
L14 |
5.0 |
85% |
Substrate scope
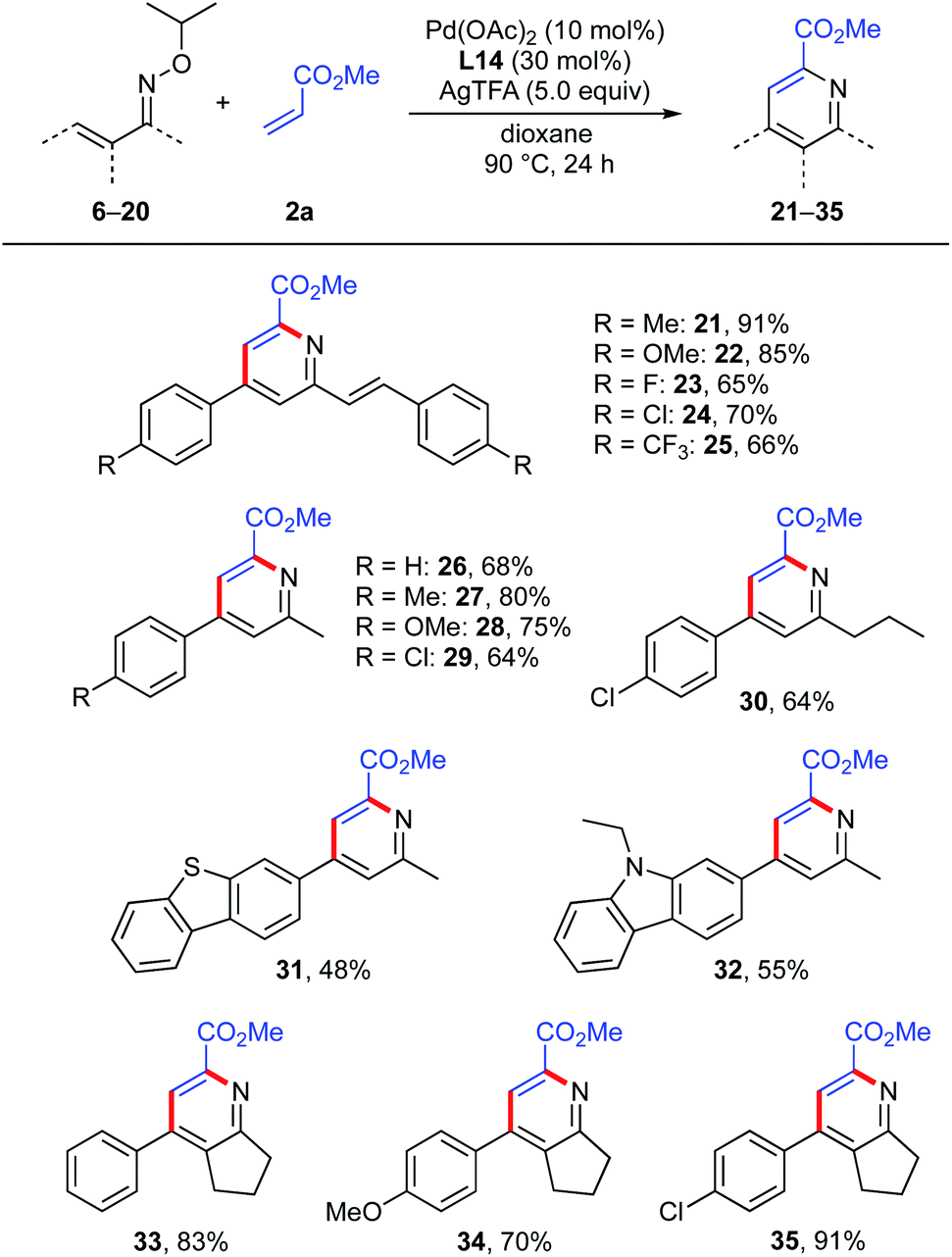
With the optimal oxime moiety and reaction conditions in hand, we surveyed the generality of this multi-substituted pyridine synthesis by examining the reaction of various α,β-unsaturated oximes with methyl acrylate (Scheme 6). Symmetrical oximes bearing an electron-donating group (Me, OMe), halogens (F, Cl) and an electron-withdrawing CF3 group at the para-position of the phenyl ring afforded the expected 2-allyl pyridines (21–25) in good yields (65–91%). Notably, unsymmetrical ketoximes were tolerated, affording the corresponding pyridines (26–30) in good yields (64–80%). Oximes with heteroaromatics such as dibenzothiophene and carbazole were also acceptable, and furnished the desired pyridines 31 and 32 in 48% and 55% yields, respectively. This reaction also worked well with cyclized oximes derived from benzylidene cyclopentanone derivatives, giving the desired 2,3,4,6-tetra-substituted pyridines (33–35) in good yields.
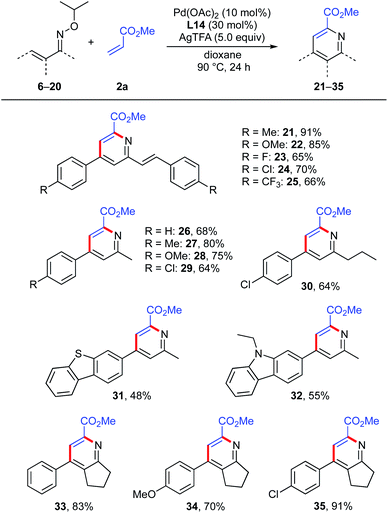 |
| | Scheme 6 Scope of α,β-unsaturated oxime ethers. aReaction conditions: 1 (0.2 mmol, 1.0 equiv.), 2a (3.0 equiv.), Pd(OAc)2 (10 mol%), L14 (30 mol%), AgTFA (5.0 equiv.), dioxane (2.0 mL), 90 °C, 24 h. Isolated yield. | |
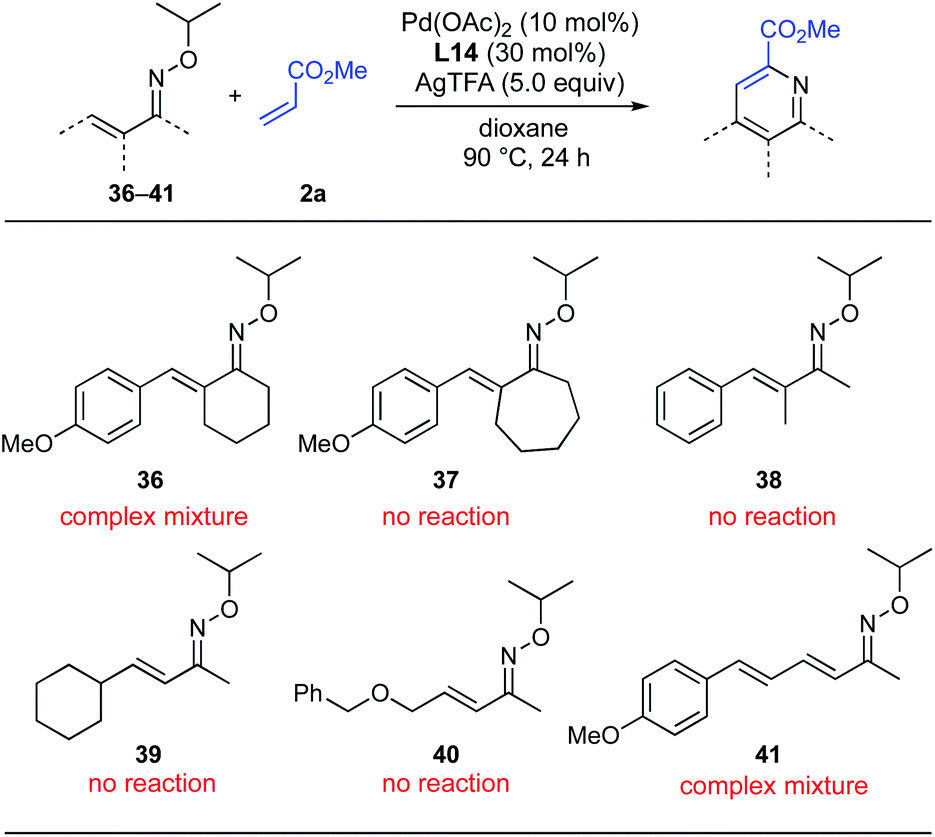
Although the 5-membered cyclized oximes (18–20), giving pyridines 33–35, were found to be suitable substrates for this pyridine synthesis, oximes with 6-membered (36) and 7-membered ring (37) were not tolerated, resulting only in complex mixtures or no reaction (Scheme 7). With α-methyl-substituted acyclic oxime 38, the reaction did not proceed at all and the raw material was completely recovered. Unfortunately, neither a β-alkyl substituent (39 and 40) nor a β-allyl substituent (41) was tolerated, indicating that the substrate scope of this reaction is limited to β-aryl-substituted α,β-unsaturated oximes.
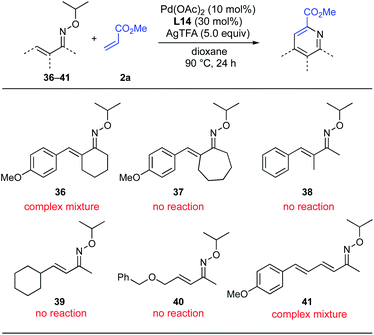 |
| | Scheme 7 Unsuccessful substrates. | |
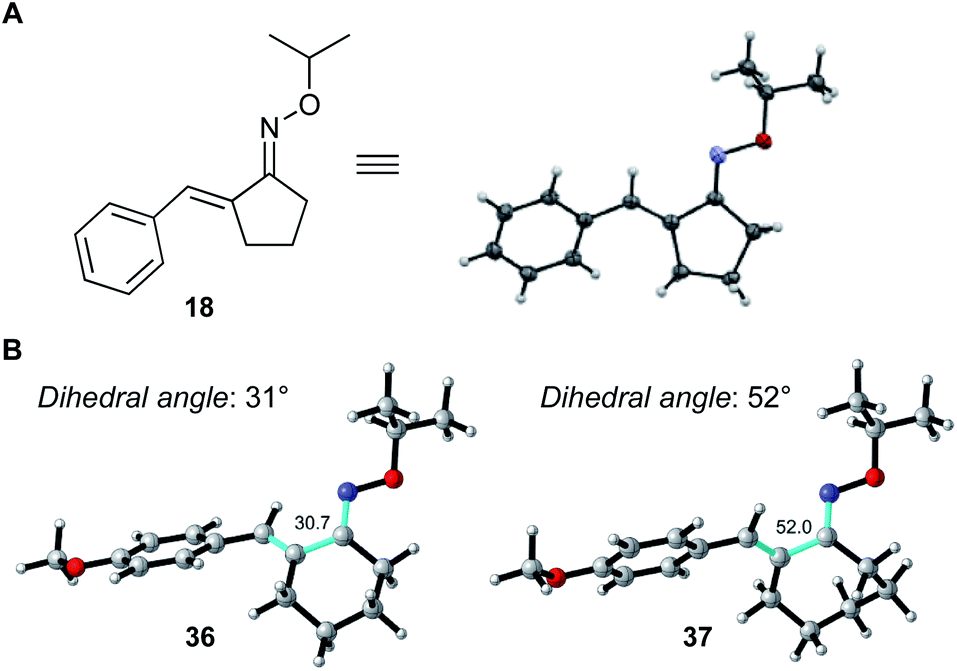
The high reactivities of 2-benzylidenecyclopentenone oximes (18–20) probably originate from the planarity of the conjugated system arising from the 5-membered ring structure, which enables efficient conjugation from oxime–oxygen to carbon–carbon double bond, imparting electron-donating character. As shown in Fig. 2, X-ray crystallographic analysis of α,β-unsaturated oxime 18 confirmed that the conjugated system of 18 is almost planar and the dihedral angle (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–CC) is 6°.16 On the other hand, computational studies with density functional theory (DFT)17,18 revealed that the conjugated systems of 6-membered and 7-membered α,β-unsaturated oximes 36 and 37 are quasi-planar, and the dihedral angles are 31° and 52°, respectively. These results clearly indicate that highly planar conformation of α,β-unsaturated oxime is favorable for the progress of this Pd-catalyzed C–H activation reaction. This also explains the lack of reactivity of 38.
C–CC) is 6°.16 On the other hand, computational studies with density functional theory (DFT)17,18 revealed that the conjugated systems of 6-membered and 7-membered α,β-unsaturated oximes 36 and 37 are quasi-planar, and the dihedral angles are 31° and 52°, respectively. These results clearly indicate that highly planar conformation of α,β-unsaturated oxime is favorable for the progress of this Pd-catalyzed C–H activation reaction. This also explains the lack of reactivity of 38.
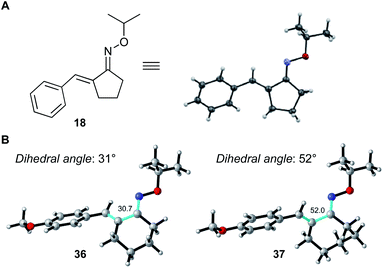 |
| | Fig. 2 (A) ORTEP representation of oxime 18. (B) Calculated structures and NC–CC dihedral angle (highlighted in light green) of oximes 36 and 37.18 | |
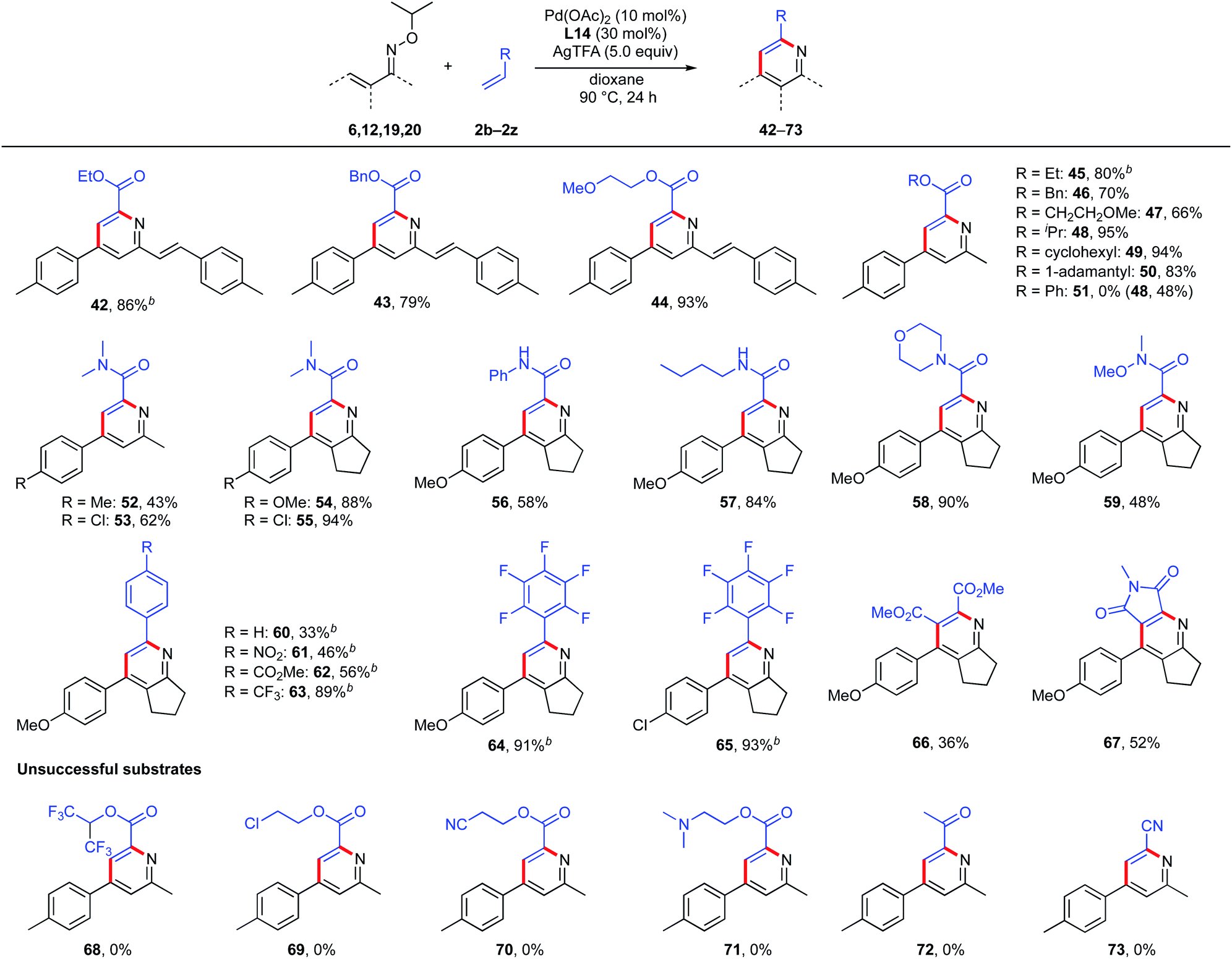
Next, we examined the scope with respect to the partner olefins (Scheme 8). Acrylates bearing ethyl, benzyl, and methoxy ethyl substituents were found to react smoothly, giving the expected 2-phenylethenyl pyridines (42–44) in 86%, 79%, and 93% yields, respectively. Unsymmetrical ketoximes with various acrylates were also compatible and the corresponding pyridines (45–47) were obtained in good yields. Notably, acrylates bearing sterically bulky alkyl groups, such as isopropyl, cyclohexyl, and 1-adamantyl groups worked well, affording the desired products (48–50) in excellent yields. Unexpectedly, the use of phenyl acrylate as a partner olefin gave not pyridine 51 but pyridine, with isopropyl ester 48 as the major product. This product was probably formed by ester exchange reaction between the corresponding pyridine 51 and in situ-generated isopropanol (see below, Mechanistic studies section, Scheme 10). Other acrylate derivatives such as acrylamides were also tolerated and provided the corresponding pyridines 52 and 53 in moderate yields. To our surprise, 5-membered cyclized oximes (19 and 20) reacted efficiently with N,N-dimethyl acrylamide, giving the desired tetra-substituted pyridines 54 and 55 in excellent yields.
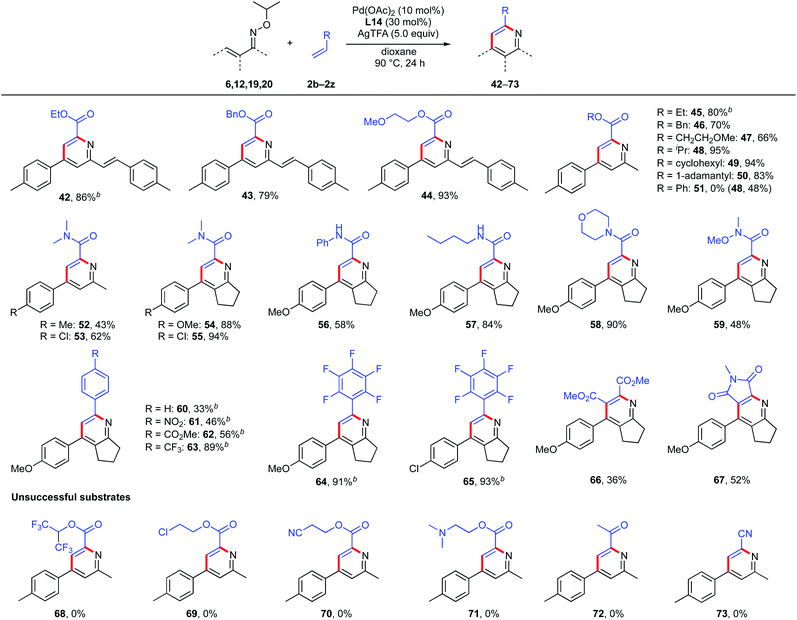 |
| | Scheme 8 Scope of olefins. aReaction conditions: oxime (0.1–0.2 mmol, 1.0 equiv.), alkene (1.5 equiv.), Pd(OAc)2 (10 mol%), L14 (30 mol%), AgTFA (5.0 equiv.), dioxane (2.0 mL), 90 °C, 24 h. Isolated yield. b3.0 equiv. of alkene was used. | |
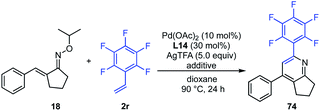
To demonstrate the generality and utility of this multi-substituted pyridine synthesis, the reactions of 5-membered cyclized oximes with other unsaturated olefins were also tested. Various acrylamide derivatives such as phenyl acrylamide, n-butyl acrylamide, and 4-acryloylmorpholine were found to be suitable coupling partners, giving the desired products (56–58) in 58%, 84% and 90% yields, respectively. Weinreb amide-derived acrylamide was also tolerated, giving the corresponding pyridine 59 in moderate yield. Various styrene derivatives containing electron-withdrawing groups such as nitro and ester were also tolerated and afforded the expected pyridines (60–62) in moderate yields. Notably, 4-trifluoromethyl styrene and pentafluorostyrene reacted smoothly with cyclized oximes, providing the corresponding pyridines 63, 64 and 65 in excellent yields. Furthermore, this pyridine synthesis is effective not only for mono-substituted olefins, but also for di-substituted olefins. With dimethyl maleate and N-methyl maleimide, the corresponding penta-substituted pyridines 66 and 67 were obtained in 36% and 52% yields, respectively. Unfortunately, acrylates bearing hexafluoroisopropyl, 2-chloroethyl, 2-cyanoethyl, and 2-dimethylaminoethyl substituents were not tolerated (68–71). With methyl vinyl ketone or acrylonitrile, the reaction did not proceed at all (72 and 73) and the starting material was recovered.
Effect of catalyst loading and the amount of AgTFA
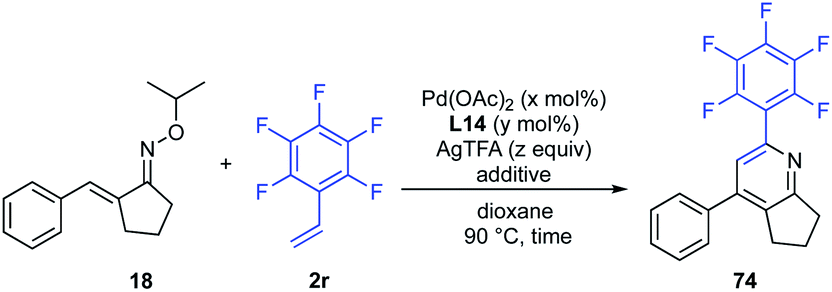
Next, the effect of catalyst loading on the product yield was investigated (Table 3). With decreasing amounts of Pd(OAc)2 to 1 mol% and L14 to 3 mol%, the reaction was clearly slowed down. However, when the reaction time was extended to 80 h, the desired pyridine 74 was obtained in 77% yield (entry 2). Next, the effect of the amount of AgTFA on the product yield was investigated. The use of 1.2 and 2.5 equiv. of AgTFA led to loss of reactivity, and the product yields were decreased to 27% and 44% yields, respectively (entries 3 and 4). However, the use of 10 equiv. of AgTFA improved the reactivity and afforded pyridine 74 in 91% yield (entry 5). These results indicated that the amount of AgTFA is of critical importance to the reaction efficiency.
Table 3 Effect of catalyst loading and the amount of AgTFAa
|
| Entry |
Pd(OAc)2 (x mol%) |
L14 (y mol%) |
AgTFA (z equiv.) |
Time |
Yield |
| Reaction conditions: 18 (0.2 mmol, 1.0 equiv.), 2r (3.0 equiv.), Pd(OAc)2 (x mol%), L14 (y mol%), AgTFA (z equiv.), dioxane (2.0 mL), 90 °C, 24 h. Isolated yield. |
| 1 |
10 |
30 |
5.0 |
24 h |
83% |
| 2 |
1 |
3 |
5.0 |
80 h |
77% |
| 3 |
10 |
30 |
1.2 |
24 h |
27% |
| 4 |
10 |
30 |
2.5 |
24 h |
44% |
| 5 |
10 |
30 |
10 |
24 h |
91% |
Mechanistic studies
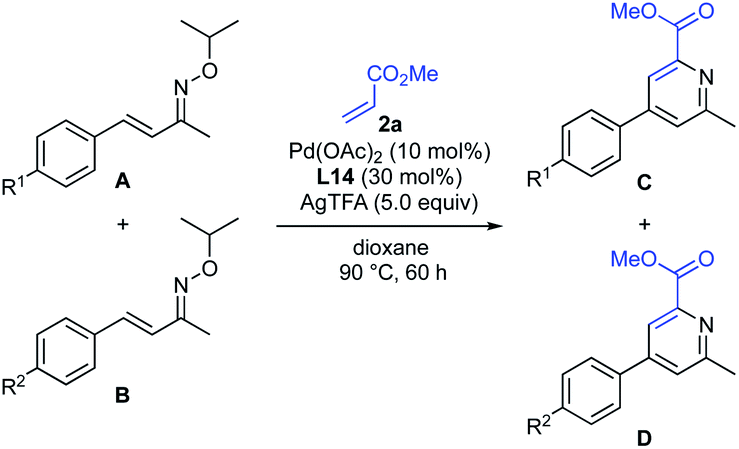
(i) Control experiments with specific substrates and conditions. To gain insight into the reaction mechanism, some competitive reactions were conducted (Table 4). Competitive reaction of methyl acrylate (2a) with α,β-unsaturated oximes, bearing a methyl and a chloro group at the para-position of the phenyl ring, gave the corresponding pyridines in 53% and 27% yields, respectively (entry 1). Similarly, the reactivities of compounds with other substituents (OMe, Me, H, and Cl) were investigated (entries 2–4). The relative reactivity was found to be in order of OMe > Me > H > Cl-substituted α,β-unsaturated oximes, indicating that the reaction could proceed in an electrophilic manner. The results of these competitive experiments were consistent with a mechanism that involves a cationic Pd(II)-catalyzed electrophilic C–H activation15 rather than a standard concerted metalation–deprotonation (CMD) mechanism.19
Table 4 Competitive experimentsa
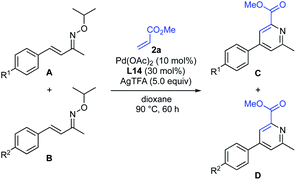
|
| Entry |
Substrates |
Product yield |
| R1 (A, C) |
R2 (B, D) |
C |
D |
C/D |
| Reactions were conducted with 0.2 mmol of oxime A and 0.2 mmol of oxime B. Product yield and the ratio of C and D (C/D) were determined by 1H NMR analysis of the isolated products. |
| 1 |
Me |
Cl |
53% |
27% |
1.9 |
| 2 |
OMe |
Me |
52% |
29% |
1.8 |
| 3 |
Me |
H |
75% |
44% |
1.7 |
| 4 |
H |
Cl |
34% |
26% |
1.3 |
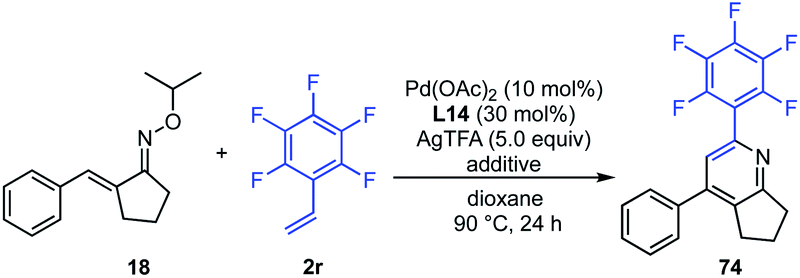
The structure of the oxime ether moiety clearly affected the reactivity, and the O-isopropyl oxime was found to be optimal (Scheme 4). The increased reactivity of O-isopropyl oxime compared to other oxime ethers may be due to the dual role in the C–H alkenylation reaction: (1) the more nucleophilic character of α,β-unsaturated O-isopropyl oxime arising from the strong electron-donating effect of the isopropyloxy group in the imino functionality promotes the electrophilic C–H activation step; (2) in situ-generated 2-propanol, which is generated in the pyridine ring formation step, may have a less adverse effect on the catalytic reactivity. Indeed, with oxime 18 and pentafluorostyrene (2r) as model substrates, the addition of a stoichiometric amount of either methanol or isopropanol to the reaction mixture under standard reaction conditions had a detrimental effect on the reaction efficiency, and the product yields dropped from 83% to 54%, and 66%, respectively (Table 5).20 This result indicates that the in situ-generated alcohol decreases the reaction efficiency, presumably due to deactivation of the Pd catalyst. It should also be noted here that the product yield with the addition of isopropanol was better than that with methanol (entry 2 vs. 3).
Table 5 Control experimentsa
|
| Entry |
Additive |
Yield |
| Reaction conditions: 18 (0.2 mmol, 1.0 equiv.), 2r (3.0 equiv.), Pd(OAc)2 (10 mol%), L14 (30 mol%), AgTFA (5.0 equiv.), additive (1.0 equiv.), dioxane (2.0 mL), 90 °C, 24 h. Isolated yield. |
| 1 |
None |
83% |
| 2 |
MeOH (1.0 equiv.) |
54% |
| 3 |
iPrOH (1.0 equiv.) |
66% |
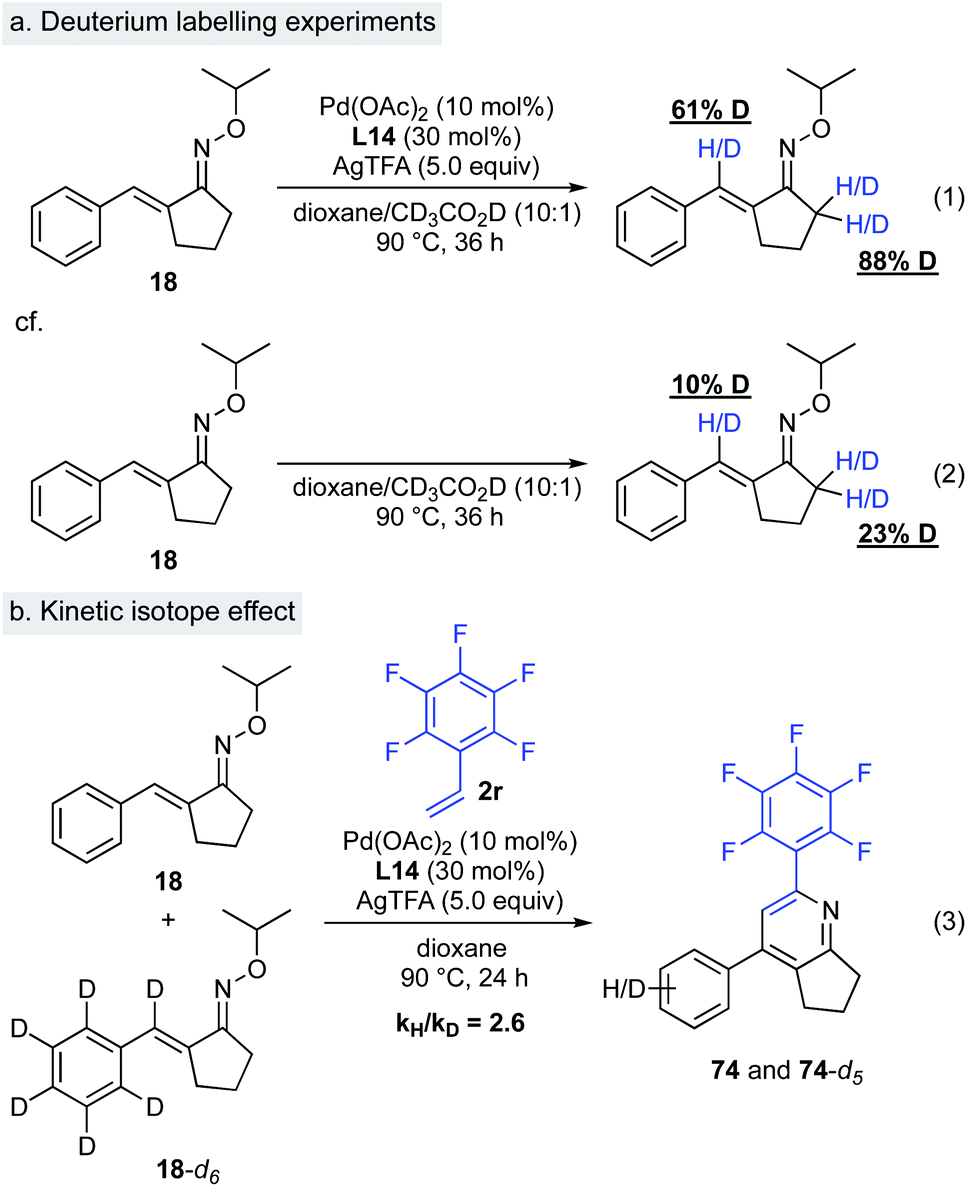
(ii) Deuterium labelling experiments. To probe the nature of this C–H activation reaction, we performed isotope labelling experiments (Scheme 9a). Treatment of α,β-unsaturated oxime 18 with excess amount of acetic acid-d4 in the presence of Pd(OAc)2, L14, and AgTFA resulted in significant deuterium incorporation at the β-position of oxime 18 (Scheme 9, eqn (1)). This result implies that the Pd–C bond formation is reversible. In the same reaction, nearly complete deuterium incorporation at the α-methylene group of oxime 18 occurred, indicating reversible deprotonation at this position under these reaction conditions. In general, deuteration at the α-position of oximes likely takes place with imine–enamine tautomerization. Interestingly, in the absence of catalysts and AgTFA, very little deuterium incorporation at the corresponding sites in oxime 18 was observed (Scheme 9, eqn (2)). A kinetic isotope effect study provided further insight regarding this C–H activation mechanism. A significant isotope effect was observed (kH/kD = 2.6/1), indicating that the C–H activation at the β-position of the α,β-unsaturated oxime could be the rate-limiting step, although further kinetic studies might be needed to confirm this (Scheme 9, eqn (3)).
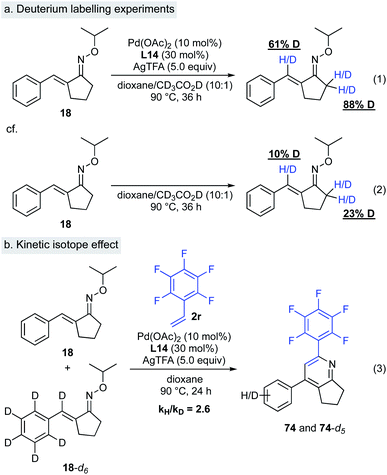 |
| | Scheme 9 Deuterium labelling experiments. | |
(iii) Mechanistic studies on pyridine ring formation. Next, we carried out mechanistic studies on pyridine ring formation. The formation of pyridines from α,β-unsaturated oximes would involve aza-6π-electrocyclization. Thus, Pd-catalyzed electrophilic C–H alkenylation of α,β-unsaturated oxime generates the 1-azatriene intermediate, which undergoes electrocyclization and aromatization with release of isopropanol to furnish the pyridine product (Scheme 10). Such aza-6π-electrocyclization has been demonstrated with various substrates, including oxime ethers.21 However, the expected 1-azatriene intermediate could not be detected in our study, probably because 6π-electrocyclization of the azatriene proceeds very rapidly.
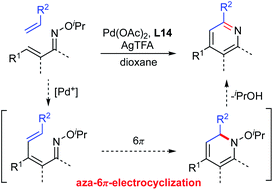 |
| | Scheme 10 Pyridine formation via aza-6π-electrocyclization. | |
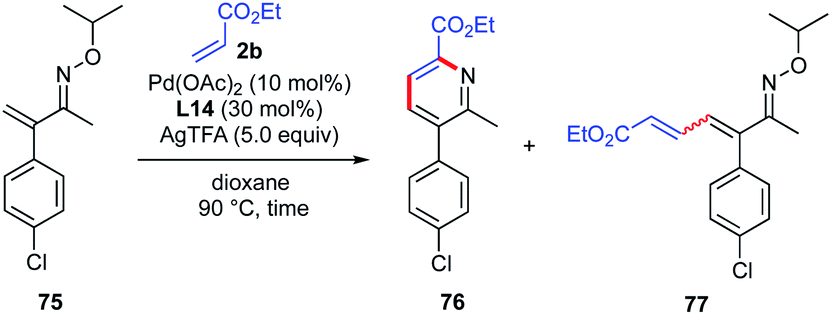
To gain insight into the mechanism of pyridine ring formation, additional experiments were conducted (Table 6). When the reaction was performed with β-unsubstituted oxime 75 and ethyl acrylate (2b) under the same reaction conditions for 4 h, 1-azatriene 77 was obtained in 36% yield as a 1.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of E/Z isomers, but the expected pyridine 76 was not detected (entry 1). However, when the reaction time was extended to 36 h, the pyridine 76 was obtained in 28% yield along with 1-azatriene 77 in 11% yield (entry 2). It should be noted that only (E)-77 was recovered, suggesting that (Z)-77 undergoes much faster electrocyclization than (E)-77.
:1 mixture of E/Z isomers, but the expected pyridine 76 was not detected (entry 1). However, when the reaction time was extended to 36 h, the pyridine 76 was obtained in 28% yield along with 1-azatriene 77 in 11% yield (entry 2). It should be noted that only (E)-77 was recovered, suggesting that (Z)-77 undergoes much faster electrocyclization than (E)-77.
Table 6 Reaction of β-unsubstituted α,β-unsaturated oxime with alkenea
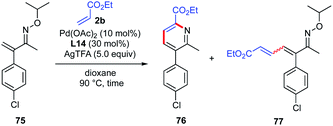
|
| Entry |
Time |
76 |
77 |
| Conditions: see Scheme 6. Isolated yield. The wavy bond of 1-azatriene 77 indicates a mixture of cis and trans configurations of C3–C4 olefins. |
| 1 |
4 h |
0% |
36% (E/Z = 1.3/1) |
| 2 |
36 h |
28% |
11% (E-isomer only) |
To confirm this, (Z)-77 was stirred in dioxane at 90 °C for 36 h. As expected, the corresponding pyridine 76 was obtained in 84% yield and a small amount of E/Z mixture of 77 was recovered (Scheme 11, eqn (1)). In contrast, similar treatment of (E)-77 gave 76 in only 18% yield and most of the (E)-77 was recovered (Scheme 11, eqn (2)). These results clearly indicate that the pyridine product was formed from 1-azatriene intermediates via aza-6π-electrocyclization. Considering both recovery of E/Z mixture of 77 from (Z)-77 (eqn (1)) and formation of a small amount of 76 from (E)-77 (eqn (2)), E/Z isomerization of 1-azatriene appears to proceed to some extent under these reaction conditions.
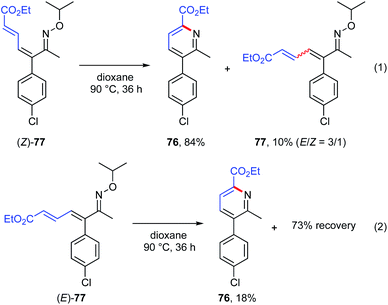 |
| | Scheme 11 Pyridine formation from 1-azatriene via aza-6π-electrocyclization. | |
Proposed mechanism
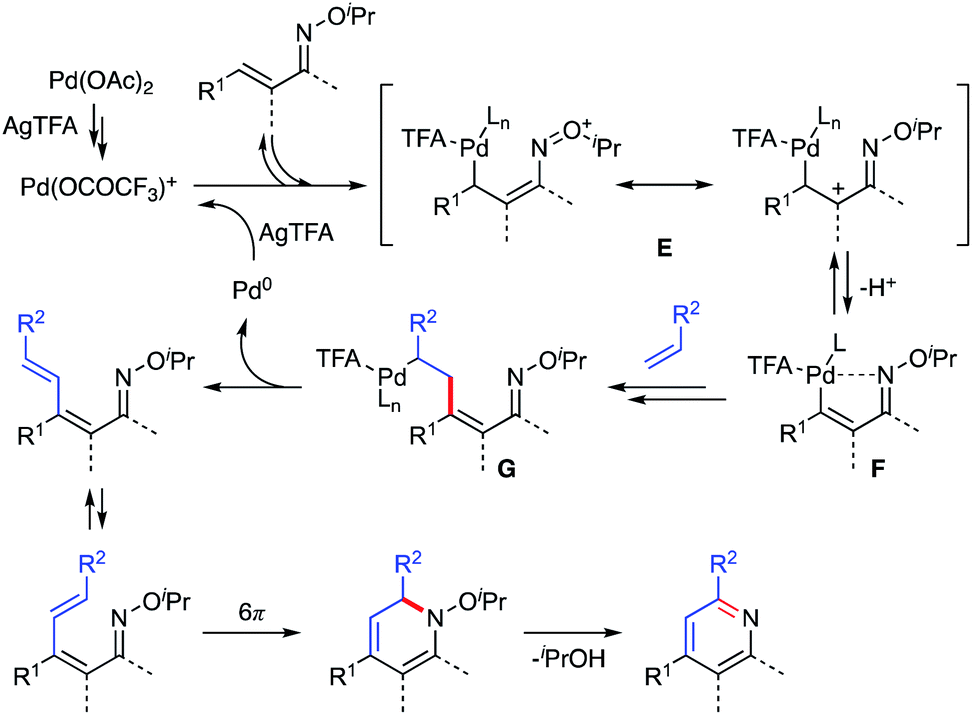
Based on the observations described above, a plausible reaction mechanism is shown in Scheme 12. First, Pd(OCOCF3)+ is formed by the reaction of Pd(OAc)2 and AgTFA.15 Then, the α,β-unsaturated oxime reacts with the electropositive Pd(OCOCF3)+ species at the β-position to generate resonance-stabilized cationic intermediate E, which kinetically releases the β-proton to give vinyl palladium F. The results of the competitive experiments are consistent with this process of electrophilic metalation followed by deprotonation. Moreover, the deuterium labelling experiments indicate that the electrophilic C–H activation process could be reversible. The subsequent Heck-type reaction of complex F with a partner alkene provides intermediate G, which undergoes β-hydride elimination and reductive elimination to afford the 1-azatriene with liberation of Pd(0). The formed Pd(0) can be reoxidized to Pd(OCOCF3)+ by AgTFA to complete the catalytic cycle. The resulting 1-azatriene is immediately converted to a pyridine product via aza-6π-electrocyclization followed by aromatization. An excess amount of AgTFA would promote the formation of the cationic Pd(OCOCF3)+ species.
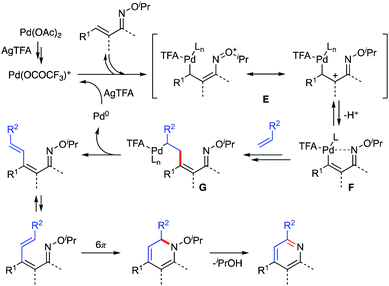 |
| | Scheme 12 Plausible reaction mechanism. | |
Conclusion
We have developed a highly efficient and regioselective synthesis of 4-aryl substituted pyridines from β-aryl-substituted α,β-unsaturated oxime ethers. In the course of reaction optimization, we found that the catalyst ligand significantly affected the reactivity, with a sterically hindered pyridine ligand being optimal. The key features of our approach are broad substrate scope, complete regioselectivity, and high efficiency in producing 4-aryl-substituted pyridines. Mechanistic studies indicated that the Pd(II)-catalyzed C–H activation proceeded in an electrophilic manner, and the pyridine formation proceeded via aza-6π-electrocyclization of β-alkenylated α,β-unsaturated oximes followed by aromatization. Considering the inherent difficulties associated with the synthesis of 4-aryl-substituted pyridines using previously developed Rh-catalyzed approaches with alkenes, we believe this transformation will be useful to enable medicinal chemists to incorporate highly complex pyridines efficiently into bioactive molecules.
Experimental section
General procedure for the synthesis of substituted pyridine derivatives
To a solution of α,β-unsaturated oxime (0.2 mmol, 1.0 equiv.), alkene (0.6 mmol, 3.0 equiv.), AgTFA (1.0 mmol, 5.0 equiv.), and L14 (0.06 mmol, 30 mol%) in dioxane (2.0 mL) was added Pd(OAc)2 (0.02 mmol, 10 mol%). The reaction mixture was stirred at 90 °C (silicone oil bath) for 24 h, then diluted with AcOEt and filtered through a Celite® pad (rinsed with AcOEt). The filtrate was concentrated in vacuo, and the crude product was purified by flash column chromatography on NH2 silica gel to afford the pure product.
Procedure for the synthesis of pyridine 74
To a solution of α,β-unsaturated oxime 18 (46 mg, 0.2 mmol, 1.0 equiv.), pentafluorostyrene (2r, 116 mg, 0.6 mmol, 3.0 equiv.), AgTFA (221 mg, 1.0 mmol, 5.0 equiv.), and L14 (22 mg, 0.06 mmol, 30 mol%) in dioxane (2.0 mL) was added Pd(OAc)2 (4.5 mg, 0.02 mmol, 10 mol%). The reaction mixture was stirred at 90 °C (silicone oil bath) for 24 h, then diluted with AcOEt and filtered through a Celite® pad (rinsed with AcOEt). The filtrate was concentrated in vacuo, and the crude product was purified by flash column chromatography on NH2 silica gel (hexane:AcOEt = 3:1) to afford the desired pyridine 74 (60 mg, 83% yield) as a white solid. mp 102–103 °C; IR (KBr) 1522, 1501, 1219, 990, 772 cm−1; 1H NMR (CDCl3, 300 MHz) δ 7.60–7.30 (m, 5H), 7.26 (s, 1H), 3.25–3.09 (m, 4H), 2.30–2.15 (m, 2H). 13C{1H} NMR (CDCl3, 75 MHz) δ 167.4, 146.3 (m), 146.0, 144.7, 143.0 (m), 139.4 (m), 138.0, 136.1 (m), 135.3, 128.7, 128.6, 128.2, 123.0, 115.8 (m), 34.6, 30.8, 23.4; HRMS (EI-quadrupole) m/z: [M]+ calcd for C20H12F5N, 361.0890; found, 361.0888.
Conflicts of interest
There are no conflicts to declare.
Notes and references
- For selected reviews, see:
(a) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed;
(b) R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed.
- For recent examples, see:
(a) J. J. Cui, M. Tran-Dubé, H. Shen, M. Nambu, P.-P. Kung, M. Pairish, L. Jia, J. Meng, L. Funk, I. Botrous, M. McTigue, N. Grodsky, K. Ryan, E. Padrique, G. Alton, S. Timofeevski, S. Yamazaki, Q. Li, H. Zou, J. Christensen, B. Mroczkowski, S. Bender, R. S. Kania and M. P. Edwards, J. Med. Chem., 2011, 54, 6342–6363 CrossRef CAS PubMed;
(b) R. Singh, E. S. Masuda and D. G. Payan, J. Med. Chem., 2012, 55, 3614–3643 CrossRef CAS PubMed;
(c) J. Felding, M. D. Sørensen, T. D. Poulsen, J. Larsen, C. Andersson, P. Refer, K. Engell, L. G. Ladefoged, T. Thormann, A. M. Vinggaard, P. Hegardt, A. Søhoel and S. F. Nielsen, J. Med. Chem., 2014, 57, 5893–5903 CrossRef CAS PubMed;
(d) T. W. Johnson, P. F. Richardson, S. Bailey, A. Brooun, B. J. Burke, M. R. Collins, J. J. Cui, J. G. Deal, Y.-L. Deng, D. Dinh, L. D. Engstorm, M. He, J. Hoffman, R. L. Hoffman, Q. Huang, R. S. Kania, J. C. Kath, H. Lam, J. L. Lam, P. T. Le, L. Lingardo, W. Liu, M. McTigue, C. L. Palmer, N. W. Sach, T. Smeal, G. L. Smith, A. E. Stewart, S. Timofeevski, H. Zhu, J. Zhu, H. Y. Zou and M. P. Edwards, J. Med. Chem., 2014, 57, 4720–4744 CrossRef CAS PubMed;
(e) J. Popovici-Muller, R. M. Lemieux, E. Artin, J. O. Saunders, F. G. Salituro, J. Travins, G. Cianchetta, Z. Cai, D. Zhou, D. Cui, P. Chen, K. Straley, E. Tobin, F. Wang, M. D. David, V. Penard-Lacronique, C. Quivoron, V. Saada, S. Botton, S. Gross, L. Dang, H. Yang, L. Utley, Y. Chen, H. Kim, S. Jin, Z. Gu, G. Yao, Z. Luo, X. Lv, C. Fang, L. Yan, A. Olaharski, L. Silvermann, S. Biller, S.-S. M. Su and K. Yen, ACS Med. Chem. Lett., 2018, 9, 300–305 CrossRef CAS PubMed;
(f) D. L. Grand, M. Gosling, U. Baettig, P. Bahra, K. Bala, C. Brocklehurst, E. Budd, R. Butler, A. K. Cheung, H. Choudhury, S. P. Collingwood, B. Cox, H. Danahay, L. Edwards, B. Everatt, U. Glaenzel, A.-L. Glotin, P. Groot-Kormelink, E. Hall, J. Hatto, C. Howsham, G. Hughes, A. King, J. Koehler, S. Kulkarni, M. Lightfoot, I. Nicholls, C. Page, G. Pergl-Wilson, M. O. Popa, R. Robinson, D. Rowlands, T. Sharp, M. Spendiff, E. Stanley, O. Steward, R. J. Taylor, P. Tranter, T. Wagner, H. Watson, G. Williams, P. Wright, A. Young and D. A. Sandham, J. Med. Chem., 2021, 64, 7241–7260 CrossRef CAS PubMed.
- For selected reviews, see:
(a) D. M. Stout and A. I. Meyers, Chem. Rev., 1982, 82, 223–243 CrossRef CAS;
(b) U. Eisner and J. Kuthan, Chem. Rev., 1972, 72, 1–42 CrossRef CAS;
(c) M. C. Bagley, C. Glover and E. A. Merritt, Synlett, 2007, 2459–2482 CrossRef CAS.
- For selected reviews, see:
(a) B. Heller and M. Hapke, Chem. Soc. Rev., 2007, 36, 1085–1094 RSC;
(b) J. A. Varela and C. Saá, Chem. Rev., 2003, 103, 3787–3802 CrossRef CAS PubMed.
- For selected reviews, see:
(a) J. M. Neely and T. Rovis, Org. Chem. Front., 2014, 1, 1010–1015 RSC;
(b) M. Behforouz and M. Ahmadian, Tetrahedron, 2000, 56, 5259–5288 CrossRef CAS;
(c) A. V. Gulevich, A. S. Dudnik, N. Chernyak and V. Gevorgyan, Chem. Rev., 2013, 113, 3084–3213 CrossRef CAS PubMed.
- For selected examples, see:
(a) K. Parthasarathy, M. Jeganmohan and C.-H. Cheng, Org. Lett., 2008, 10, 325–328 CrossRef CAS PubMed;
(b) P. C. Too, T. Noji, Y. J. Lim, X. Li and S. Chiba, Synlett, 2011, 2789–2794 CAS;
(c) T. K. Hyster and T. Rovis, Chem. Commun., 2011, 47, 11846–11848 RSC;
(d) D. A. Colby, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2008, 130, 3645–3651 CrossRef CAS PubMed;
(e) J. M. Neely and T. Rovis, J. Am. Chem. Soc., 2013, 135, 66–69 CrossRef CAS PubMed;
(f) J. M. Neely and T. Rovis, J. Am. Chem. Soc., 2014, 136, 2735–2738 CrossRef CAS PubMed.
- For reviews on Umpolung reactivity, see:
(a) X. Bugaut and F. Glorious, Chem. Soc. Rev., 2012, 41, 3511–3522 RSC;
(b) D. Seebach, Angew. Chem., Int. Ed. Engl., 1979, 18, 239–258 CrossRef;
(c) B.-T. Gröbel and D. Seebach, Synthesis, 1977, 357–402 CrossRef.
- For selected examples of the α-cation-stabilizing effect of O-alkyl oximes, see:
(a) X. Creary, Y.-X. Wang and Z. Jiang, J. Am. Chem. Soc., 1995, 117, 3044–3053 CrossRef CAS;
(b) X. Creary and Z. Jiang, J. Org. Chem., 1996, 61, 3482–3489 CrossRef CAS;
(c) X. Creary, E. A. Burtch and Z. Jiang, J. Org. Chem., 2003, 68, 1117–1127 CrossRef CAS PubMed;
(d) M. Schlegel and C. Schneider, Org. Lett., 2018, 20, 3119–3123 CrossRef CAS PubMed;
(e) M. Schlegel and C. Schneider, Chem. Commun., 2018, 54, 11124–11127 RSC;
(f) R. Narayan, R. Fröhlich and E.-U. Würthwein, J. Org. Chem., 2012, 77, 1868–1879 CrossRef CAS PubMed.
- Reviews for Umpolung of carbonyl groups by hydrazone formation, see:
(a) R. Lazny and A. Nodzewska, Chem. Rev., 2010, 110, 1386–1434 CrossRef CAS PubMed;
(b) R. Brehme, D. Enders, R. Fernandez and J. M. Lassaletta, Eur. J. Org. Chem., 2007, 5629–5660 CrossRef CAS; for selected examples of the electrophilic addition at β-position of α,β-unsaturated hydrazones, see:
(c) T. Hashimoto, H. Kimura and K. Maruoka, Angew. Chem., Int. Ed., 2010, 49, 6844–6847 CrossRef CAS PubMed.
- For selected examples of Umpolung reactivity of α,β-unsaturated oximes, see:
(a) Y. Hashimoto, H. Ishiwata, S. Tachikawa, S. Ban, N. Morita and O. Tamura, Chem. Commun., 2017, 53, 2685–2688 RSC;
(b) M. Furugori, K. Yoshida, Y. Hashimoto, N. Morita and O. Tamura, Chem. Pharm. Bull., 2021, 69, 1010–1016 CrossRef PubMed;
(c) N. Umemoto, A. Imayoshi and K. Tsubaki, Tetrahedron Lett., 2020, 61, 152213–152216 CrossRef CAS. For examples of the electrophilic addition at β-position of α,β-unsaturated oximes, see:
(d) A. M. Chibiryaev, A. Y. Denisov, D. V. Pyshnyi and A. V. Tkachev, Russ. Chem. Bull., 2001, 50, 1410–1418 CrossRef CAS;
(e) R. Fernandes, K. Mhaske, R. Balhara, G. Jindal and R. Narayan, Chem.–Asian J., 2022, 17, e202101369 CrossRef CAS PubMed.
- T. Yamada, Y. Hashimoto, K. Tanaka III, N. Morita and O. Tamura, J. Org. Chem., 2020, 85, 12315–12328 CrossRef CAS PubMed.
- T. Yamada, Y. Hashimoto, K. Tanaka III, N. Morita and O. Tamura, Org. Lett., 2021, 23, 1659–1663 CrossRef CAS PubMed.
-
(a) X. Fu, J. Yang, K. Deng, L. Shao, C. Xia and Y. Ji, Org. Lett., 2019, 21, 3505–3509 CrossRef CAS PubMed;
(b) X.-P. Fu, S.-B. Tang, J.-Y. Yang, L.-L. Zhang, C.-C. Xia and Y.-F. Ji, Eur. J. Org. Chem., 2019, 5974–5977 CrossRef CAS.
- J. He, S. Li, Y. Deng, H. Fu, B. N. Laforteza, J. E. Spangler, A. Homs and J.-Q. Yu, Science, 2014, 343, 1216–1220 CrossRef CAS PubMed.
-
(a) C. Jia, D. Piao, J. Oyamada, W. Lu, T. Kitamura and Y. Fujiwara, Science, 2000, 287, 1992–1995 CrossRef CAS PubMed;
(b) W. Lu, Y. Yamaoka, Y. Taniguchi, T. Kitamura, K. Takaki and Y. Fujiwara, J. Organomet. Chem., 1999, 580, 290–294 CrossRef CAS;
(c) C. Jia, W. Lu, J. Oyamada, T. Kitamura, K. Matusda, M. Irie and Y. Fujiwara, J. Am. Chem. Soc., 2000, 122, 7252–7263 CrossRef CAS;
(d) P. K. Hota, A. Jose and S. K. Mandal, Organometallics, 2017, 36, 4422–4431 CrossRef CAS.
- CCDC-2166741 (18) contains the ESI† crystallographic data for this paper.
-
(a) Computations were carried out with the Gaussian 09 series of programs. M. J. Frisch, et al., Gaussian 09, Revision D.01, Gaussian Inc., Wallingford, CT, 2013, see complete reference in the ESI.† All compounds were calculated with ωB97X-D/6-311+G(d,p), and an ultrafine grid was used for geometry optimization;
(b) J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- Structures have been represented in the figures using CLYview. C. Y. Legault, CYLview, 1.0b, Université de Sherbooke, 2009, https://www.cylview.org Search PubMed.
- For selected examples, see:
(a) S. I. Gorelsky, D. Lapointe and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 10848–10849 CrossRef CAS PubMed;
(b) D. Garcia-Cuadrado, P. de Mendoza, A. A. C. Braga, F. Maseras and A. M. Echavaren, J. Am. Chem. Soc., 2007, 129, 6880–6886 CrossRef CAS PubMed;
(c) M. Lafrance, C. N. Rowley, T. K. Woo and K. Gagnou, J. Am. Chem. Soc., 2006, 128, 8754–8756 CrossRef CAS PubMed;
(d) D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118–1126 CrossRef;
(e) D. L. Davies, S. M. A. Donald and S. A. Macgregor, J. Am. Chem. Soc., 2005, 127, 13754–13755 CrossRef CAS PubMed;
(f) S. I. Gorelsky, D. Lapointe and K. Fagou, J. Org. Chem., 2012, 77, 658–668 CrossRef CAS PubMed.
- In the presence of water, hydrolysis of the oximes proceeded preferentially. Therefore, it could not be applied to water.
- For selected examples, see:
(a) N. Kanekiyo, T. Kuwada, T. Choshi, J. Nobuhiro and S. Hibino, J. Org. Chem., 2001, 66, 8793–8798 CrossRef CAS PubMed;
(b) B. M. Trost and A. C. Gutierrez, Org. Lett., 2007, 9, 1473–1476 CrossRef CAS PubMed;
(c) S. Liu and L. S. Liebeskind, J. Am. Chem. Soc., 2008, 130, 6918–6919 CrossRef CAS PubMed;
(d) K. Tanaka, H. Mori, M. Yamamoto and S. Katsumura, J. Org. Chem., 2001, 66, 3099–3110 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Yoshimitsu Hashimoto
,
Yoshimitsu Hashimoto