
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual determination of nitrite and iron by a single greener sequential injection spectrophotometric system employing a simple single aqueous extract from Areca catechu Linn. serving as a natural reagent†
Kraingkrai Ponhong a,
Watsaka Siriangkhawuta,
Chang Young Leeb,
Norio Teshimac,
Kate Grudpand and
Sam-ang Supharoek*ef
a,
Watsaka Siriangkhawuta,
Chang Young Leeb,
Norio Teshimac,
Kate Grudpand and
Sam-ang Supharoek*ef
aMultidisciplinary Research Unit of Pure and Applied Chemistry, Department of Chemistry and Center of Excellence for Innovation in Chemistry, Faculty of Science, Mahasarakham University, Maha Sarakham 44150, Thailand
bSchool of Energy and Chemical Engineering, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, Republic of Korea
cDepartment of Applied Chemistry, Aichi Institute of Technology, 1247 Yachigusa, Yakusa-cho, Toyota 470-0392, Japan
dDepartment of Chemistry, Faculty of Science and Center of Excellence for Innovation in Analytical Science and Technology for Biodiversity-based Economic and Society, Chiang Mai University, Chiang Mai 50200, Thailand
eDepartment of Medical Science, Mahidol University, Amnatcharoen Campus, Amnat Charoen 37000, Thailand
fDepartment of Chemistry and Center of Excellence for Innovation in Chemistry, Faculty of Science, Mahidol University, Bangkok 10400, Thailand. E-mail: samang.sup@mahidol.ac.th
First published on 11th July 2022
Abstract
Dual determination of nitrite and iron was proposed by using a single greener sequential injection (SI) spectrophotometric system employing a simple single aqueous extract from Areca catechu Linn. The extract served as a natural reagent to replace N-(1-naphthyl)ethylenediamine (NED) of the Griess reagent with nitrite and 1,10-phenanthroline with iron. The color products possessed analytical wavelengths at 430 and 560 nm, respectively. Conditions for the SI procedure were optimized using a univariate experimental design. Calibration ranges were up to 5.0 mg L−1 and 10.0 mg L−1 with limits of detection (LODs) of 0.04 mg L−1 and 0.05 mg L−1 for nitrite and iron(III), respectively, and relative standard deviations (RSDs) being less than 3%. Recoveries of spiked standard nitrite and iron(III) at 0.3 mg L−1 and 0.5 mg L−1 in water samples were 88 to 104% and 84 to 109%, respectively. The developed method successfully achieved dual determination of nitrite and total iron agreeing at a 95% confidence level with the reference methods of the conventional Griess assay and flame atomic absorption spectrometry (FAAS), respectively. The proposed method utilized locally available material from plants and serves the UN-SDGs.
Introduction
Nitrite (NO2−) and iron(III) (Fe3+) are inorganic contaminants in various aquatic environments.1,2 High levels of nitrite can lead to methemoglobinemia in aquatic animals.3 Nitrite also produces carcinogenic N-nitrosamines by reacting with secondary or tertiary amines in the human body,4 while iron(III) is responsible for surface water quality deterioration worldwide.5 Pollution by these substrates is a serious problem because they can enter the human body through food, water, and air. The content of nitrite and iron is an important parameter that is considered when making decisions concerning water quality.Several techniques are available for the detection and investigation of nitrite including electrochemical,6–8 electrochemiluminescence,9,10 chemiluminescence,11–13 chromatographic,14–16 capillary electrophoresis,17–19 spectrofluorimetric,20–22 and spectrophotometric methods.23–28 The most frequently used approach for the spectrophotometric detection of nitrite is the Griess assay, involving a diazo-coupling procedure.24,28
Various methods have been developed to monitor iron such as electrochemical techniques,29,30 chromatography,31,32 atomic absorption spectroscopy,33–36 inductively coupled plasma atomic emission spectroscopy (ICP-AES),37 and spectrophotometry.38–44 The spectrophotometric method is widely used to determine iron in water samples due to its versatility, simplicity, and feasibility. Many reagents are available for iron determination such as 1,10-phenanthroline,28 2-pyridinecarbaldehyde-5-nitro-pyridylhydrazone,39 1-(3′-methoxypropyl)-2-methyl-3-benzyloxy-4-(1H)pyridinone – MRB13,40 and 2-(5-bromo-2-pyridylazo)-5-[N-n-propyl-N-(3-sulfopropyl)amino]aniline.41 However, these reagents are hazardous and/or expensive. They also generate large amounts of chemical waste, which is harmful to human health and causes adverse environmental impacts. To address this issue, natural environmentally friendly reagents are preferred.38,42–45
Spectrophotometric methods can determine nitrite and iron contents using commonly available equipment, thus decreasing the support required to implement a routine task. Reduced reagent consumption and waste generation are also parameters that should be considered when determining whether an analytical method is cost-effective with minimal environmental impacts. An automatic procedure based on sequential injection analysis (SIA)38,41,45 has been proved optimal because microliter volumes of samples and reagents can be aspirated at programmed time intervals by a syringe pump that enables bi-directional discontinuous flow. The SIA setup can be used for different assay protocols without changing the system configuration and is appropriate for simultaneous or multi-determination of nitrite and iron. However, only flow based analysis has previously been presented for simultaneous multi-determination of nitrite and iron using the Griess reaction and 1,10-phenanthroline, respectively.28
Betel nut (Areca catechu Linn.), which is commonly available in tropical areas including Thailand, is composed of phenolics and alkaloids. The phenolics are mainly distributed in the root followed by the fresh unripe fruit, leaf, spike, and vein.46 Betel nut has been reported as a natural reagent for the determination of iron.45 The phenolic compounds include subgroups such as tannins, flavonoids, coumarins, lignans, quinones, stilbenes, and curcuminoids. Tannins, as tannic acid or a gallic acid ester of D-glucose, are polyhydroxyphenols that can act as metal chelators.47 The ortho-position on the benzene ring of tannic acid is unsubstituted and can be coupled with the diazonium ions of nitrite ions with sulfanilamide (SAM), resulting in an azo dye compound.48
To expand the use of the simple extract of betel nut for iron determination previously reported,45 we propose in this study to apply simple aqueous betel nut extract as a natural reagent for the determination of nitrite and iron using a single SI system, with the aim of achieving greener chemical analysis as the simple aqueous betel nut extract replaces the conventional reagents of NED (in the Griess reagent) and 1,10-phenanthroline for nitrite and iron, respectively, for sequential determination using flow procedure with multicommutation as previously reported.28 The developed SI procedure was applied for real surface water samples. The results were validated by comparison with the conventional Griess method49 and flame atomic absorption spectrometry (FAAS).36
Experimental
Chemicals and reagents
All chemicals used were analytical grade and utilized without further purification. Deionized (DI) water (Milli-Q, Millipore, Sweden) was used to prepare all the solutions. Iron(III) (Merck, Germany) standard stock solutions of 1000 mg L−1 were used for all experiments. A standard stock solution of 1000 mg L−1 nitrite was prepared by dissolving 0.1631 g sodium nitrite (Carlo Erba Reagents, French) in 100 mL of DI water while a standard stock solution of iron(III) at a concentration of 100 mg L−1 was prepared by diluting a stock standard of iron(III) in DI water. Working solutions of nitrite and iron(III) were freshly prepared by appropriately diluting the stock solution with DI water. Sulfanilamide (SAM) at 0.5% (w/v) was prepared from 0.50 g SAM in 400 μL of HCl and adjusted with DI water to 100 mL. N-1-Naphthyl ethylene diamine (NED) at 0.3% (w/v) was prepared by weighing 0.30 g of this chemical in 100 mL of DI water; 0.05 M acetate buffer pH 5.5 was prepared from 2.86 g CH3COONa·3H2O and 0.22 mL CH3COOH in a 600 mL beaker. pH values were adjusted using 1.0 mol L−1 HCl or NaOH with final volumes made up to 500 mL with DI water.Extraction of Areca catechu Linn.
Betel nut (Areca catechu Linn.) was collected from a local market in Roi Et Province, Thailand and washed with deionized water. The fresh seeds were peeled, chopped into small pieces, and then dried in an oven at 60 °C for 24 h. Next, the dry seeds were homogenized in a cooking blender (Electrolux, Thailand). The betel nut seeds powder was kept in a polyethylene bottle and stored in a desiccator. Stock solution of the natural reagent was prepared from 0.5 g of betel nut seed powder in 100 mL DI water and then placed in an ultrasonic bath at 60 °C and sonicated for 20 min. The extracted solution was filtered through Whatman No. 1 filter paper and adjusted with DI water to 100 mL in a volumetric flask. The natural reagent was diluted to 100 mL with DI water and acetate buffer at pH 5.5 for nitrite and iron detection by SIA, respectively.Instruments and apparatus
The manifold of sequential injection analysis (SIA) for the proposed method is shown in Fig. 1a. The system comprised a bidirectional syringe pump (5000 μL, CAVRO, San Jose, CA), 10 selection valve ports (Valco instruments, Houston, TX, USA), a holding coil (250 cm PTFE tubing; OD 1/16′′, ID 0.03′′), one mixing coil (PTFE tubing, ID 0.5 mm, 50 cm long), three 400 μL reaction coils (PTFE tubing, ID 0.5 mm, 205 cm long), and PTFE tubing (ID 0.5 mm). A detection unit including a tungsten lamp as a light source, a spectrophotometer (AvaSpec-3648 StarLine, Avantes, Netherlands), and optic fiber cables (ID 400 nm, 2 m) with a flow through cell (Quartz 10 mm path length, 80 μL internal volume) was utilized for measurement of absorbance at 430 and 560 nm. An avasoft 8.0 Avantes Fiber Optic Spectrometer was used for data acquisition. In-house created software based on Visual Basic 6.0 was used to automatically operate the SI system. A spectrophotometer (UV-1800, Shimadzu, Japan) was used for preliminary screening of tannin compost in various plants and as the standard method to determine nitrite. FAAS (Varian Model AA240FS, USA) measurements were utilized as the standard method for determination of iron. All pH measurements were performed using a 713 pH meter (Metrohm, Herisau, Switzerland).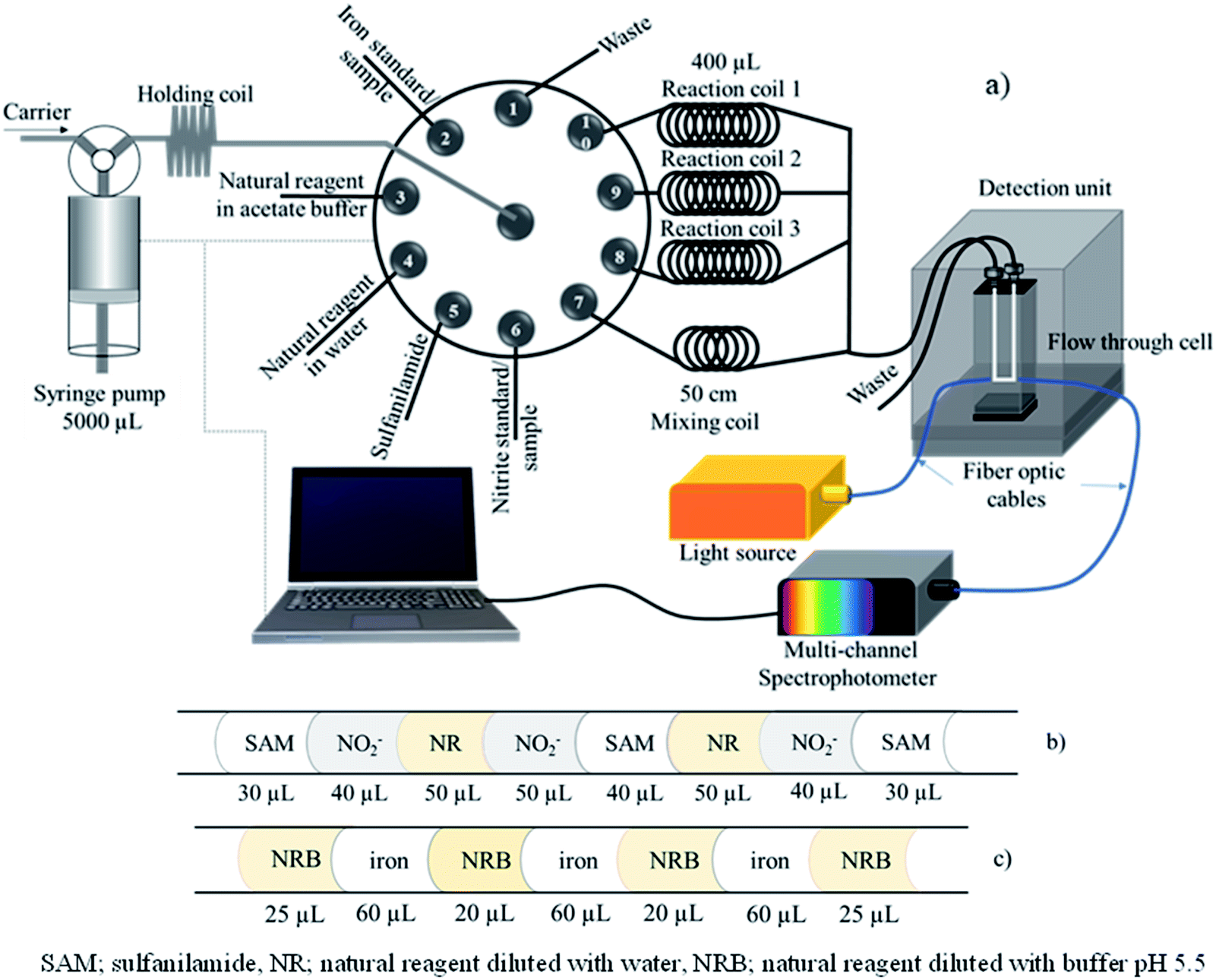 | ||
| Fig. 1 (a) SIA manifold; (b) segment profile for nitrite determination; and (c) segment profile for total iron determination. | ||
SI procedure for determination of nitrite and total iron
The automatic SIA system for the dual determination of nitrite and total iron is illustrated in Fig. 1a with the operational sequence for the developed method shown in Table 1. The holding coil, flow through cell, and PTFE tubing were connected to the rotary 10 port selection valve and initially filled with the carrier (DI water). The tubing connected to the reagents was filled with their respective solution.| Step | Syringe pump | Multi-position valve No. | Operation | Flow rate (μL s−1) | Volume (μL) |
|---|---|---|---|---|---|
| Operational steps for nitrite determination | |||||
| 1 | Out | — | Aspirate carrier into syringe | 100 | 1000 |
| 2 | In | 5 | Aspirate sulfanilamide | 50 | 30 |
| 3 | In | 6 | Aspirate standard nitrite or sample | 50 | 40 |
| 4 | In | 4 | Aspirate natural reagent | 50 | 50 |
| 5 | In | 6 | Aspirate standard nitrite or sample | 50 | 50 |
| 6 | In | 5 | Aspirate sulfanilamide | 50 | 40 |
| 7 | In | 4 | Aspirate natural reagent | 50 | 50 |
| 8 | In | 6 | Aspirate standard nitrite or sample | 50 | 40 |
| 9 | In | 5 | Aspirate sulfanilamide | 50 | 30 |
| 10 | In | 10 | Dispense all solution to reaction coil 1 | 100 | 350 |
| 11 | In | 10 | Delay incubation for 48 s | — | — |
| 12 | Repeat steps 2–9 by the same sequence | ||||
| 13 | In | 9 | Dispense all solution to reaction coil 2 | 100 | 350 |
| 14 | In | 9 | Delay for incubation 48 s | — | — |
| 15 | Repeat steps 2–9 by the same sequence | ||||
| 16 | In | 8 | Dispense all solution to reaction coil 3 | 100 | 350 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||
| Operational steps for iron determination (repeat three times steps 17–25) | |||||
| 17 | Out | — | Aspirate carrier into syringe | 200 | 3000 |
| 18 | In | 3 | Aspirate natural reagent | 50 | 25 |
| 19 | In | 2 | Aspirate iron standard or sample | 50 | 60 |
| 20 | In | 3 | Aspirate natural reagent | 50 | 20 |
| 21 | In | 2 | Aspirate iron standard or sample | 50 | 60 |
| 22 | In | 3 | Aspirate natural reagent | 50 | 20 |
| 23 | In | 2 | Aspirate iron standard or sample | 50 | 60 |
| 24 | In | 3 | Aspirate natural reagent | 50 | 25 |
| 25 | In | 7 | Dispense solution through the flow cell for measurement of the absorbance at 560 nm | 100 | Empty |
| 26 | Out | — | Aspirate carrier solution | 200 | 3000 |
| 27 | In | 10 | Dispense solution in reaction coil 1 through the flow cell for measurement of absorbance at 430 nm | 70 | Empty |
| 28 | Out | — | Aspirate carrier solution | 200 | 3000 |
| 29 | In | 9 | Dispense solution in reaction coil 2 through the flow cell for measurement of absorbance at 430 nm | 70 | Empty |
| 30 | Out | — | Aspirate carrier solution | 200 | 3000 |
| 31 | In | 8 | Dispense solution in reaction coil 3 through the flow cell for measurement of absorbance at 430 nm | 70 | Empty |
Initially, 1000 μL carrier solution as DI water was aspirated into the syringe barrel. Then, SAM, a nitrite standard solution or the sample and the natural reagent diluted with water (crude extracted solution) were aspirated into a holding coil according to the solution sequence as shown in Fig. 1b. After that, 350 μL of aspirated solution and DI water were dispensed to reaction coil 1 via port 10 to incubate the reaction at room temperature and delay 48 s. Reagent solution and nitrite standard were then aspirated by the same sequence following steps 2 to 9 and dispensed to reaction coil 2 to incubate the reaction and delay 48 s. Next, the repetition aspirated solution followed steps 2 to 9 and the solution was delivered to reaction coil 3 for incubation.
During this time, the determination of iron was performed by sequentially aspirating natural reagent in the acetate buffer and iron(III) standard or sample into the holding coil according to the segment profile shown in Fig. 1c. Next, the aspirated solution in the holding coil was delivered through the mixing coil (50 cm) via port 7 and the analyst zone was distributed to the detector unit, detection wavelength set at 560 nm. Quantification of iron was repeated by following steps 17–25 three times.
After the determination of iron, the aspirated zones of nitrite at reaction coils 1, 2, and 3 were subsequently dispensed to the spectrophotometric flow cell for measurement of absorbance at 430 nm. Incubation time for the nitrite assay by this proposed method was approximately 300 s. The total operation time was about 8 min for analysis of nitrite and iron with three repeats of both analyses.
Sample preparation
Surface water samples were collected in 1000 mL polyethylene bottles from different locations in Amnat Charoen Province, Thailand. To preserve the iron metal ions (including of Fe2+ and Fe3+), 2 mL of concentrated nitric acid and 0.5 mL of 30% hydrogen peroxide were added to the water samples for quantification of total iron in terms of iron(III), iron(II) was oxidized to iron(III) by hydrogen peroxide, but not added to the sample for the nitrite assay. All samples were passed through a 0.45 micron nylon filter before analysis by the developed method.Standard method
A standard spectrophotometric method following the Griess reaction49 was utilized for nitrite determination based on the reaction between NED, SAM, and nitrite under acidic medium to produce a pink product with a maximum absorption wavelength of 560 nm. FAAS was used as the standard method for total iron investigation.36Results and discussion
Reaction for nitrite and iron determination exploiting natural reagent
A preliminary study of the reaction between natural reagent extracts, nitrite, and iron(III) was conducted to screen appropriate sources of natural reagent. Four plants including betel nut (Areca catechu Linn.), tummy-wood (Careya sphaerica Roxb.), santol leaves (Sandoricum koetjape Burm. f.), and hairy keruing fruit (Dipterocarpus intricatus) were identified as sources of natural reagent, with extracts from betel nut providing the highest sensitivity for nitrite and total iron determination, as shown in Fig. S1.†Mixtures of the extracted reagent and analyte were studied to identify a suitable pH and wavelength for detection of each complex by UV-vis spectrophotometry. The natural reagent was subjected to reaction with SAM and 5 mg L−1 nitrite for 10 min under acidic medium (∼pH 1.3) and reacted with 10 mg L−1 iron(III) under pH 5.5. Each solution was scanned against the blank reagent to provide absorption spectra in the range 300 to 800 nm. The product colors obtained from the reaction between the natural reagent with nitrite and the natural reagent with iron were yellow and dark purple, respectively. Fig. 2a shows the maximum absorption wavelength of the azo dye compound at 430 nm, while the maximum absorption wavelengths of iron(III)-reagent complexes were achieved at 430 nm and 560 nm, as shown in Fig. 2b. The 560 nm wavelength provided high sensitivity and low background for detection of iron, as illustrated in Fig. S2.† Results presented that the natural reagent could be reacted with nitrite and iron at different pH and wavelength values to determine nitrite and iron(III) contents.
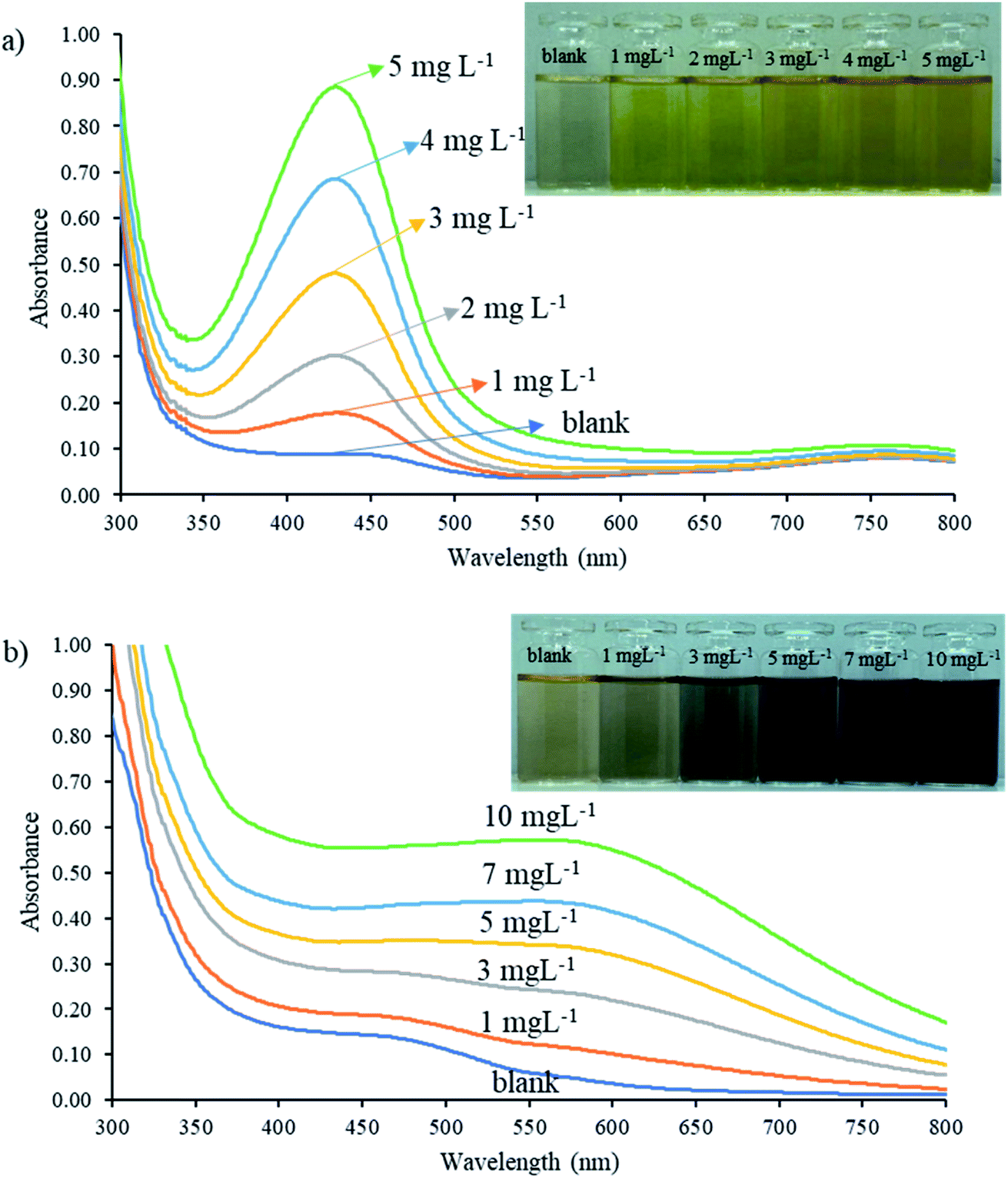 | ||
| Fig. 2 Absorbance spectra of natural reagent extract reactions with (a) nitrite and (b) iron(III) standard solutions from 300 to 800 nm. | ||
The FTIR spectra of the natural reagent showed characteristic bands corresponding to the functional groups of the tannic acid standard such as O–H stretching (3385 cm−1), C–O stretching (1327 cm−1), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching (1611 cm−1), C–H stretching (2925 cm−1) and the presence of aromatic rings indicated by bands at 1520 cm−1, as shown in Fig. S3a.† Characteristic peaks of the azo (NN) band at 1596 to 1497 cm−1 were found after reaction between the natural reagent, SAM, and nitrite, and confirmed it was an azo dye compound (Fig. S3b†). Furthermore, the o-substituted benzene ring absorption at 750 cm−1 proved that the azo group was attached to the ortho position of the benzene ring.50 Characteristic bands of the iron–tannic acid complex showed peaks of the O–H group (3400 cm−1), indicating the presence of polyphenol groups.51 The peak at 1609 indicated that phenolic groups of tannic acid reacted with iron(III) resulting in metal complex formation, as presented in Fig. S3c.† Tannic acid is a natural polyphenol and the hydroxyl group (–OH) in its structure acts as a metal chelator to produce metal–tannic complexes.47 Moreover, the ortho-position on the benzene ring of tannic acid was vacant and could be substituted with diazonium ions of nitrite ions with SAM, resulting in an azo dye complex.48 Therefore, natural reagent extracts from betel nut could be used to determine nitrite and iron contents based on the proposed reaction, as shown in Fig. 3.
C stretching (1611 cm−1), C–H stretching (2925 cm−1) and the presence of aromatic rings indicated by bands at 1520 cm−1, as shown in Fig. S3a.† Characteristic peaks of the azo (NN) band at 1596 to 1497 cm−1 were found after reaction between the natural reagent, SAM, and nitrite, and confirmed it was an azo dye compound (Fig. S3b†). Furthermore, the o-substituted benzene ring absorption at 750 cm−1 proved that the azo group was attached to the ortho position of the benzene ring.50 Characteristic bands of the iron–tannic acid complex showed peaks of the O–H group (3400 cm−1), indicating the presence of polyphenol groups.51 The peak at 1609 indicated that phenolic groups of tannic acid reacted with iron(III) resulting in metal complex formation, as presented in Fig. S3c.† Tannic acid is a natural polyphenol and the hydroxyl group (–OH) in its structure acts as a metal chelator to produce metal–tannic complexes.47 Moreover, the ortho-position on the benzene ring of tannic acid was vacant and could be substituted with diazonium ions of nitrite ions with SAM, resulting in an azo dye complex.48 Therefore, natural reagent extracts from betel nut could be used to determine nitrite and iron contents based on the proposed reaction, as shown in Fig. 3.
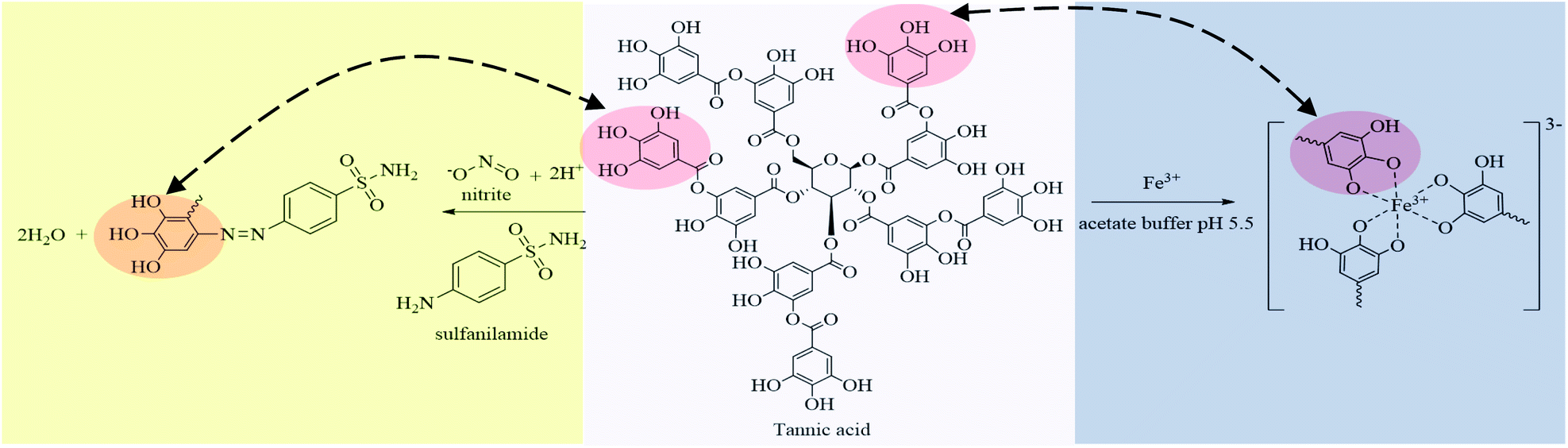 | ||
| Fig. 3 Reaction between the natural reagent with nitrite and iron(III) under different conditions. | ||
Effect of extraction solvent and stability of complexes
Various extraction solvents such as methanol, ethanol, acetonitrile, dichloromethane, water, and warm water (60 °C) were studied. The absorption spectra of complexes with the natural reagent extract from each solvent were monitored in the range 300 to 800 nm. Methanol and ethanol extraction solvents provided the highest absorbance, but solution turbidity was observed, while warm water also gave high absorbance. Therefore, warm water was adopted as the extraction solvent because it provided good absorbance and it was also non-toxic and environmentally friendly. The stability of the reagent after extraction by warm water was studied by monitoring the absorbance at 430 nm and 560 nm hourly for eight hours. Results showed that the absorbance of the complexes did not decrease significantly (2.3% RSD), indicating that the extracted reagent was stable for the intraday SIA experiment.Optimization of SI system for dual determination of nitrite and iron
| Sequence No. | Segment order | Volume (μL) |
|---|---|---|
| a NR: natural reagent diluted with water; SAM: sulfanilamide; NRB: natural reagent diluted with acetate buffer pH 5.5. | ||
| Sequence profiles for nitrite assay | ||
| 1 | NR/SAM/NO2− | 100/100/100 |
| 2 | NR/SAM/NO2−/SAM/NR | 50/50/100/50/50 |
| 3 | SAM/NO2−/NR/NO2−/SAM/NR/NO2−/SAM | 30/30/50/40/40/50/30/30 |
| 4 | SAM/NO2−/NR/SAM/NO2−/NR/SAM/NO2−/NR | 30/30/30/40/40/40/30/30/30 |
|
||
| Sequence profiles for total iron assay | ||
| 5 | NRB/iron/NRB/iron/NRB | 50/75/50/75 |
| 6 | NRB/iron/NRB/iron/NRB/iron/NRB | 35/50/40/50/40/50/35 |
| 7 | NRB/iron/NRB/iron/NRB/iron/NRB/iron/NRB | 30/35/30/40/30/40/30/35/30 |
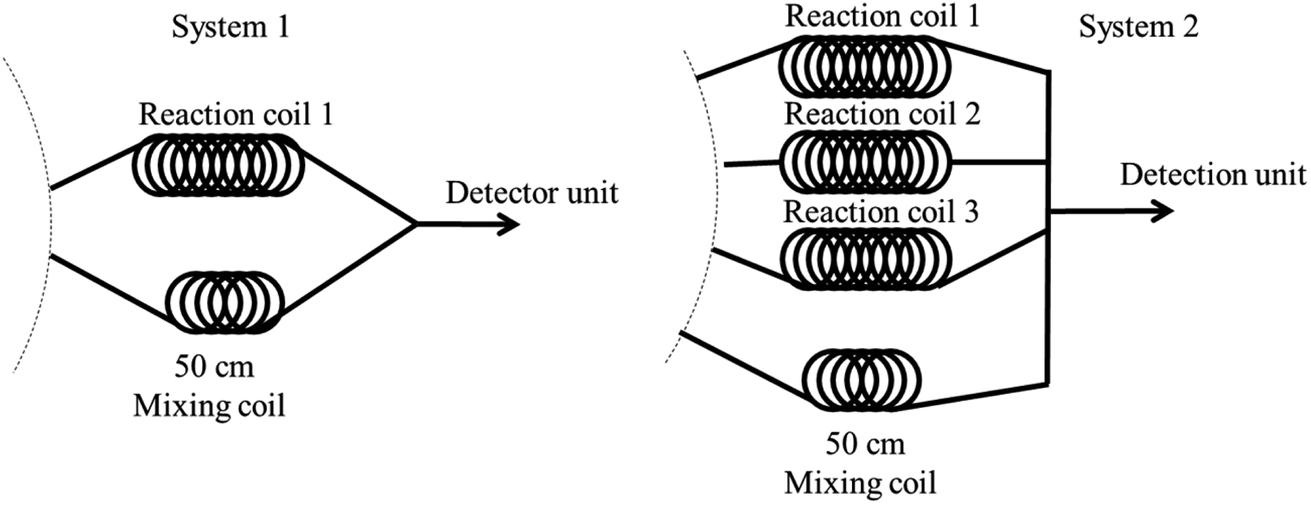 | ||
| Fig. 4 System design sequence for dual determination of nitrite and total iron. | ||
Dual nitrite and total iron determination using system 1 began with aspiration of the mixed nitrite zone that was then dispensed to reaction coil 1 for incubation. Meanwhile, the mixed solution of iron was aspirated and dispensed to another mixing coil through a detector at 560 nm. Analysis time for iron detection was 63 s for each injection. The system was then delayed for 240 s to incubate the nitrite reaction, resulting in enhanced sensitivity (total incubation time for nitrite was approximately 303 s). Finally, the completed reaction zone of nitrite was dispensed through the detector to monitor and record the absorbance value at 430 nm. This operational sequence was repeated three times for both analytes and total analysis time was approximately 16 min.
The second design comprised four reaction coils. Three reaction coils were used for nitrite, while one mixing coil was utilized for iron determination. The operational steps were as follows. First, the mixed solution of nitrite was subsequently aspirated and dispensed into reaction coils 1, 2, and 3 for incubation, respectively. During the nitrite reaction incubation, the mixed zone of iron was analyzed three times, while the mixing solution was dispensed through the 50 cm mixing coil and a detector to record the data. After that, the completed nitrite reactions at reaction coils 1, 2, and 3 were consequently pushed through the detector to monitor absorbance. The second design took 8 min total time for analysis. Therefore, system 2 was selected for dual determination of nitrite and iron.
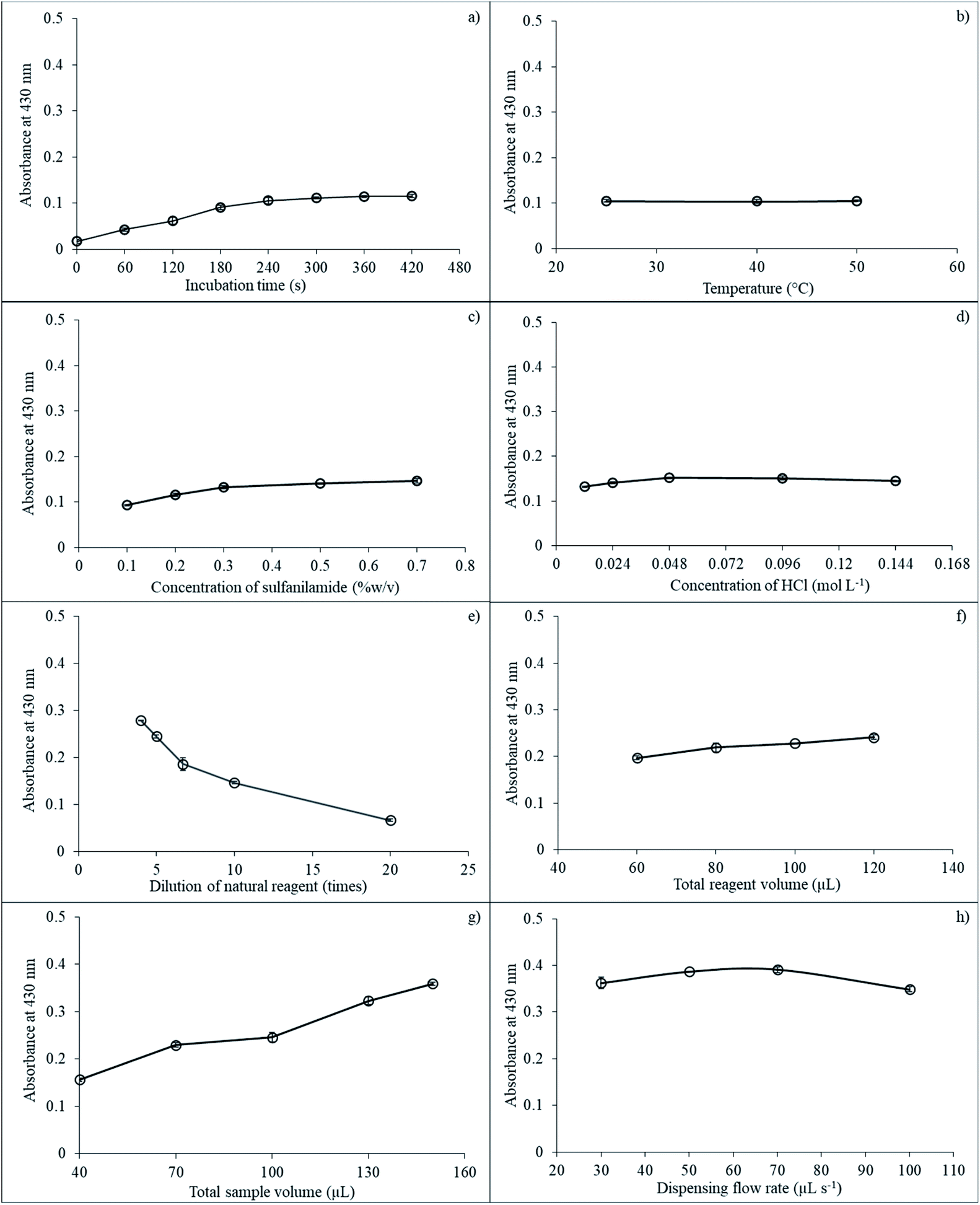 | ||
| Fig. 5 The effect of various parameters on SI for nitrite determination: (a) incubation time; (b) incubation temperature; (c) sulfanilamide concentration; (d) HCl concentration; (e) reagent dilution; (f) total aspirated reagent volume; (g) total aspirated sample volume; and (h) dispensing flow rate. | ||
Next, the effect of temperature on reaction incubation was studied at 25, 40, and 50 °C. Results showed that temperature did not significantly affect absorbance, as presented in Fig. 5b. Therefore, room temperature (25 °C) was used for subsequent experiments.
Concentration of SAM also impacted sensitivity because the SAM reagent reacted directly with nitrite ions under acidic conditions to produce diazonium ions that then reacted with the natural reagent extracts to give a yellow-colored product. Concentration of SAM was varied at 0.1, 0.2, 0.3, 0.5, and 0.7% (w/v). The absorbance showed a slight increase from 0.1 to 0.5% (w/v) and then stabilized at 0.7% (w/v) because the reagent reaction was completed and was sufficient for nitrite detection as presented in Fig. 5c. Optimal SAM concentration was selected at 0.5% (w/v) for nitrite determination.
The reaction between nitrite and natural reagent extracts altered under acidic media. The SAM solution was dissolved in HCl acid to prevent decomposition with the HCl acid concentration varying from 0.012 to 0.144 mol L−1. Absorbance increased from 0.012 to 0.048 mol L−1 and then remained constant, as depicted in Fig. 5d. Therefore, 0.048 mol L−1 HCl was utilized for subsequent experiments.
Total aspirated volume of SAM in HCl was investigated between 40 and 130 μL. The absorbance value negligibility increased from 40 to 100 μL and then remained constant (results not shown). Hence, total aspirated volume of SAM in HCl was employed at 100 μL.
Natural reagent extract dilution is another important parameter. Reagent dilution at 4 to 20 times. Net absorbance decreased slightly from 4 to 5 times and then dropped from 5 to 20 dilution times, as illustrated in Fig. 5e. Therefore, the dilution of the natural reagent extracts of five times was selected for the next procedures.
Total aspirated volumes of the natural reagent extract also impacted detection sensitivity and were studied in the range 60 to 120 μL. Results showed that sensitivity increased slowly from 60 to 80 μL and then leveled off at 120 μL, as shown in Fig. 5f. Thus, 100 μL of total natural reagent aspirated volume was selected for subsequent experiments.
The effect of total aspiration sample volume on the reaction was explored at 40, 70, 100, 130, and 150 μL. Results indicated that sensitivity increased from 40 to 130 μL of total sample volume, while the signal leveled off after 130 μL, as depicted in Fig. 5g. Thus, 130 μL was used for total aspiration sample volume.
The last parameter for nitrite determination by the proposed method was the effect of dispensing flow rate on sensitivity, which was investigated by varying the flow rate in the range 30, 50, 70, and 100 μL s−1. Results showed that the signal increased from 30 to 70 μL s−1. Then, the signal dropped from 70 to 100 μL s−1, as presented in Fig. 5h. Therefore, flow rate at 70 μL s−1 was selected for the proposed method.
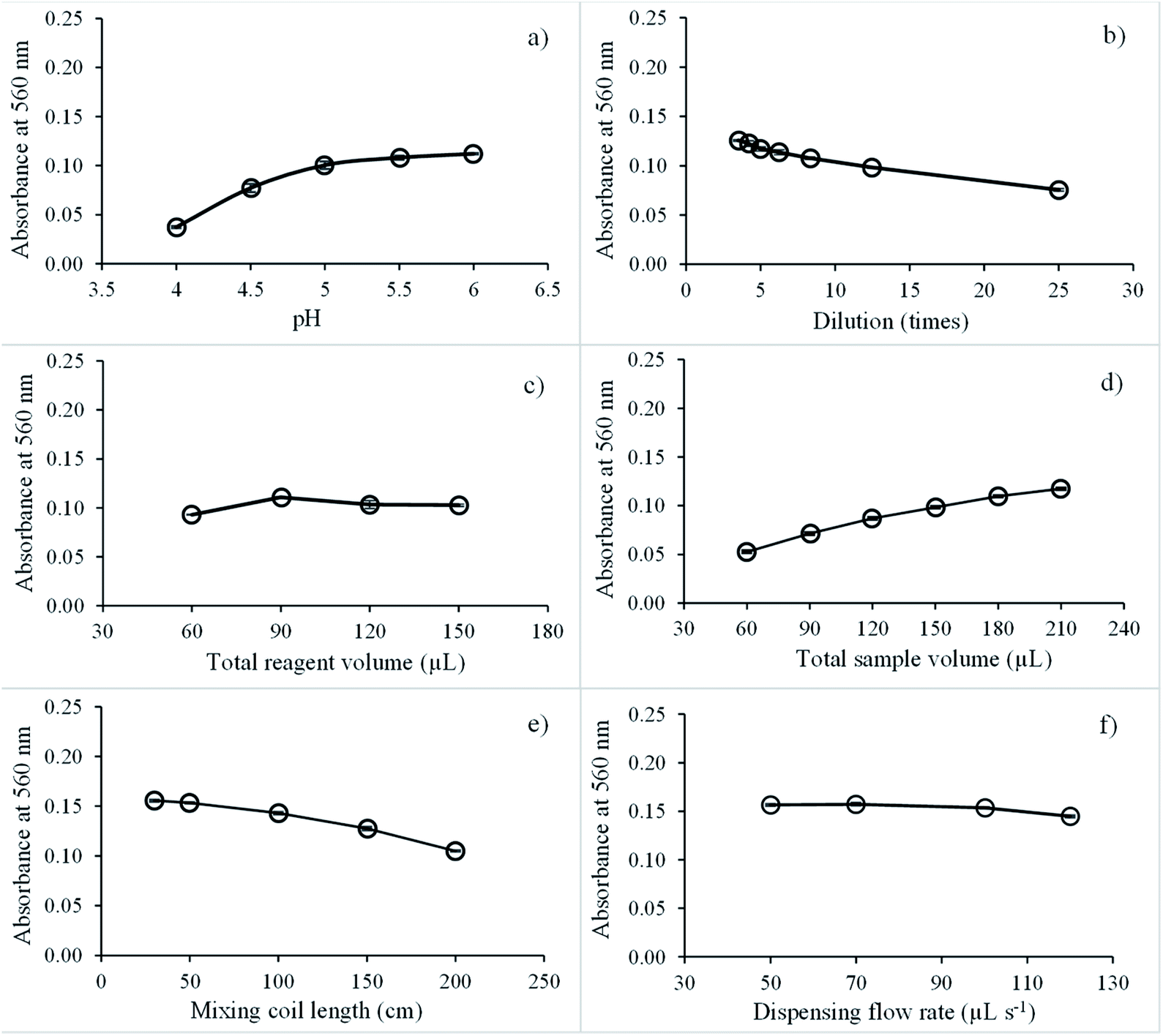 | ||
| Fig. 6 Effect of various parameters on SI for iron determination: (a) pH; (b) natural reagent dilution; (c) total aspirated reagent volume; (d) total aspirated sample volume; (e) mixing coil length; and (f) dispensing flow rate. | ||
Next, the effect of the natural reagent extract dilution on sensitivity was studied by diluting reagent extracts at 3.5 to 25 times with pH 5.5 buffer. Fig. 6b shows that the analytical signal decreased by 3.5 to 25 times. Dilution of natural reagent extracts was set at 5 times.
The effect of the total aspirated natural reagent extract volume was explored in the range of 60 to 150 μL, and the highest net signal was recorded at 90 μL, as presented in Fig. 6c. Above this volume, the net signal was almost constant because the absorbance of blank increased with increasing the total reagent aspirate volume. Therefore, the total aspirated volume of natural reagent was optimized at 90 μL.
Next, the effect of the total aspirated sample volume was examined in the range 60 to 210 μL. Absorbance continually increased from 60 to 210 μL, as illustrated in Fig. 6d. With increasing the total sample volume, the amount of iron analyst also increased, resulting in a high absorption value. However, absorption of the blank signal decreased slightly due to the dilution effect. Thus, 180 μL of the total aspirate sample volume was adopted for the next experiment since this provided suitable sensitivity.
The effect of mixing coil length on iron detection sensitivity was tested at 30, 50, 100, 150, and 200 cm. A 50 cm mixing coil length was found to be suitable because the reagent extract and iron(III) reacted immediately, as presented in Fig. 6e. Therefore, 50 cm of mixing coil length was selected for rapid analysis.
Finally, dispensing flow rates on iron sensitivity detection were studied at 50, 70, 100, and 120 μL s−1. The dispensing flow rate did not significantly affect sensitivity, as presented in Fig. 6f. Thus, 100 μL s−1 dispensing flow rate was selected for rapid analysis.
| Sample | Nitrite (mg L−1 ± SD) | Recovery nitrite% (RSD) | Iron (mg L−1 ± SD) | Recovery iron% (RSD) | ||||
|---|---|---|---|---|---|---|---|---|
| This method | NED | This method | NED | This method | FAAS | This method | FAAS | |
| 1 | nd | nd | 94(0.33) | 95(0.5) | nd | nd | 100(0.70) | 99(0.45) |
| 2 | 1.46 ± 0.06 | 1.50 ± 0.00 | 99(2.87) | 95(0.00) | 1.50 ± 0.03 | 1.40 ± 0.05 | 100(0.57) | 99(0.75) |
| 3 | nd | nd | 99(3.11) | 96(0.00) | nd | nd | 103(0.30) | 99(0.25) |
| 4 | 1.37 ± 0.05 | 1.48 ± 0.00 | 97(1.77) | 96(0.00) | nd | nd | 97(3.39) | 100(0.48) |
| 5 | 0.81 ± 0.01 | 0.79 ± 0.01 | 96(0.15) | 95(0.1) | 1.26 ± 0.05 | 1.28 ± 0.02 | 101(1.69) | 99(0.48) |
| 6 | nd | nd | 95(1.26) | 98(0.43) | nd | nd | 106(0.93) | 97(0.56) |
| 7 | nd | nd | 93(0.36) | 94(0.19) | 2.21 ± 0.01 | 2.13 ± 0.02 | 109(1.52) | 96(0.34) |
| 8 | 0.84 ± 0.05 | 0.71 ± 0.01 | 95(1.12) | 93(0.10) | nd | nd | 103(1.76) | 97(0.79) |
| 9 | nd | nd | 91(0.55) | 96(0.11) | 1.82 ± 0.05 | 1.63 ± 0.02 | 100(0.98) | 99(0.29) |
| 10 | nd | nd | 95(0.93) | 96(0.15) | 2.20 ± 0.04 | 2.14 ± 0.01 | 96(0.55) | 99(0.29) |
| 11 | 0.90 ± 0.02 | 1.11 ± 0.01 | 86(0.19) | 95(0.10) | 0.91 ± 0.04 | 0.98 ± 0.01 | 99(0.76) | 98(0.42) |
| 12 | 0.23 ± 0.03 | 0.21 ± 0.01 | 90(1.45) | 96(0.10) | 0.84 ± 0.05 | 0.97 ± 0.01 | 101(0.98) | 97(0.37) |
| 13 | 1.10 ± 0.01 | 1.01 ± 0.03 | 88(1.01) | 95(0.5) | 1.17 ± 0.02 | 1.10 ± 0.00 | 105(0.23) | 98(0.61) |
| 14 | 2.75 ± 0.01 | 2.52 ± 0.01 | 93(1.19) | 98(0.10) | 0.81 ± 0.01 | 0.97 ± 0.02 | 93(2.91) | 95(0.19) |
| 15 | 0.96 ± 0.02 | 0.98 ± 0.00 | 91(0.25) | 96(0.00) | nd | nd | 101(1.93) | 98(0.22) |
| 16 | 0.37 ± 0.03 | 0.32 ± 0.01 | 98(1.37) | 96(0.15) | 2.68 ± 0.02 | 2.89 ± 0.01 | 94(2.08) | 96(0.17) |
| 17 | nd | nd | 91(1.68) | 95(0.62) | 1.24 ± 0.04 | 1.21 ± 0.01 | 89(2.27) | 99(0.44) |
| 18 | 1.10 ± 0.01 | 1.12 ± 0.01 | 95(0.72) | 95(0.20) | nd | nd | 84(2.31) | 96(0.11) |
| 19 | 0.47 ± 0.02 | 0.42 ± 0.01 | 104(0.76) | 96(0.23) | nd | nd | 85(2.28) | 97(0.15) |
| 20 | nd | nd | 99(1.88) | 94(0.16) | nd | nd | 88(2.22) | 97(0.44) |
Likewise, tolerance limits of ions for iron were Na+ (1000 mg L−1), SO42− and Ca2+ (600 mg L−1), Mg2+ (500 mg L−1), K+ (200 mg L−1), Cr(VI) and NO2− (20 mg L−1), Cu2+ (15 mg L−1), CO32−, Al3+, and Zn2+ (10 mg L−1), and Hg2+, Mn2+, Pb2+, Ni2+, and Cd2+ (5 mg L−1). Results indicated that most ions did not interfere with nitrite and total iron determination, while the most serious interferences were Hg2+, Cr(VI), Cd2+, Cu2+, and Mn2+, Pb2+, Ni2+, and Cd2+ on nitrite and total iron determination, respectively. These ions were present at low concentrations in surface water samples,52–54 and our proposed system proved suitable for the dual determination of nitrite and total iron.
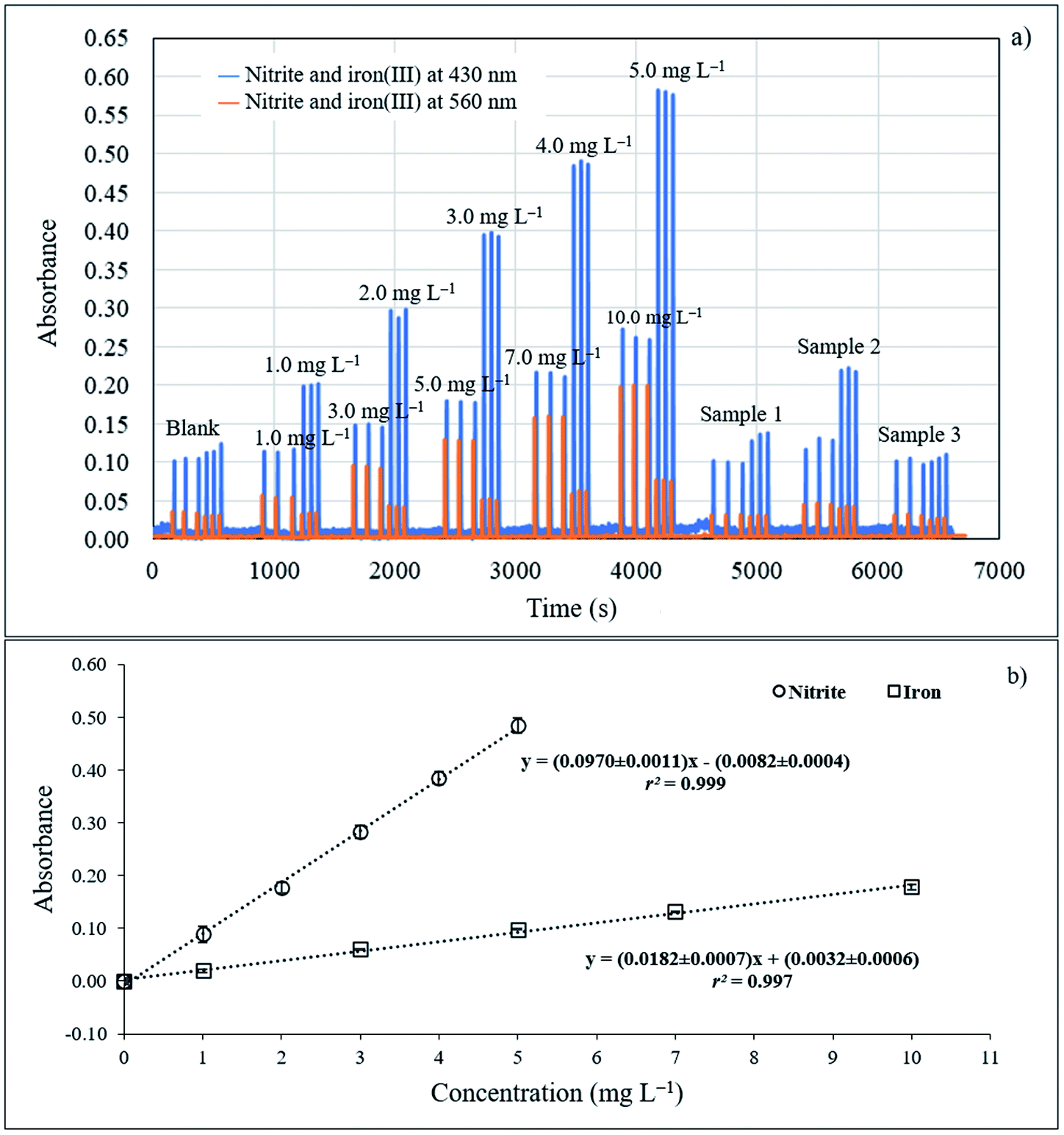 | ||
| Fig. 7 (a) SI graph of nitrite and iron at −430 nm and −560 nm and (b) calibration graph for nitrite and iron. | ||
Conclusions
A single automatic and greener sequential injection spectrophotometric system was developed for dual determination of nitrite and iron by employing the natural reagent of a simple single aqueous extract from Areca catechu Linn. For nitrite, tannic acid in the extract can replace the NED in the conventional Griess reaction, while tannic acid in the extract can also form a colored complex with iron. Colorimetric detection can be made for both. The SI system was applied to real surface water samples. The results agree with the reference methods. The developed procedure offers green chemistry analysis via the benefits of down scaling operation with the newly designed SI system and the use of simple single aqueous extract of natural reagent. This serves several goals of UN-SDGs.Author contributions
Kraingkrai Ponhong: conceptualization, resources, investigation, methodology, validation, data curation, formal analysis, writing – original draft, and writing – review & editing. Watsaka Siriangkhawut: writing – review & editing. Chang Young Lee: writing – review & editing. Norio Teshima: writing – review & editing. Kate Grudpan: writing – review & editing and supervision. Sam-ang Supharoek: investigation, methodology, validation, data curation, formal analysis, visualization, writing – original draft, and writing – review & editing. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Mahidol University Amnatcharoen Campus, Department of Chemistry, Faculty of Science, Mahasakham University for supporting all the instruments. The authors also gratefully acknowledge all funding support. This research project was financially supported by Thailand Science Research and Innovation (TSRI) 2021 (6406029/2564) and Mahidol University under the New Discovery and Frontier Research Grant NDFR 14/2563. The authors also thank Mahidol University Amnatcharoen Campus. The new research grant is gratefully acknowledged by S.-A. S. K. P. would like to thank the Center of Excellence for Innovation in Chemistry (PERCH-CIC) and the Ministry of Higher Education, Science, Research and Innovation for financial support. K. G. acknowledges Center of Excellence for Innovation in Analytical Science and Technology for Biodiversity-based Economic and Society, Chiang Mai University.References
- A. Menció, J. Mas-Pla, N. Otero, O. Regàs, M. Boy-Roura, R. Puig, J. Bach, C. Domènech, M. Zamorano, D. Brusi and A. Folch, Sci. Total Environ., 2016, 539, 241–251 CrossRef PubMed.
- X. Liu and F. J. Millero, Mar. Chem., 2002, 77, 43–54 CrossRef CAS.
- F. B. Jensen, Comp. Biochem. Physiol., Part A: Mol. Integr. Physiol., 2003, 135, 9–24 CrossRef.
- H. Suzuki, K. Iijima, A. Moriya, K. McElroy, G. Scobie, V. Fyfe and K. E. McColl, Gut, 2003, 52, 1095–1101 CrossRef CAS.
- D. Khadija, A. Hicham, A. Rida, E. Hicham, N. Nordine and F. Najlaa, Environ. Challenges, 2021, 5, 100230 CrossRef CAS.
- N. Mounesh and K. R. Venugopala Reddy, Anal. Chim. Acta, 2020, 1108, 98–107 CrossRef PubMed.
- J. Yin, W. Gao, Z. Zhang, Y. Mai, A. Luan, H. Jin, J. Jian and Q. Jin, Electrochim. Acta, 2020, 335, 135660 CrossRef CAS.
- L. Fu, S. Yu, L. Thompson and A. Yu, RSC Adv., 2015, 5, 40111–40116 RSC.
- X. Liu, L. Guo, L. Cheng and H. Ju, Talanta, 2009, 78, 691–694 CrossRef CAS.
- X. Yin, Q. Chen, H. Song, M. Yang and H. Wang, Electrochem. Commun., 2013, 34, 81–85 CrossRef.
- D. He, Z. Zhang, Y. Huang and Y. Hu, Food Chem., 2007, 101, 667–672 CrossRef CAS.
- P. Mikuska, Z. Vecera and Z. Zdrahal, Anal. Chim. Acta, 1995, 316, 261–268 CrossRef CAS.
- J. Wu, X. Wang, Y. Lin, Y. Zheng and J.-M. Lin, Talanta, 2016, 154, 73–79 CrossRef CAS PubMed.
- S.-L. Lin, J.-W. Hsu and M.-R. Fuh, Talanta, 2019, 205, 120082 CrossRef CAS.
- L. He, K. Zhang, C. Wang, X. Luo and S. Zhang, J. Chromatogr. A, 2011, 1218, 3595–3600 CrossRef CAS PubMed.
- M. R. Khan, S. M. Wabaidur, Z. A. Alothman, R. Busquets and M. Naushad, Talanta, 2016, 152, 513–520 CrossRef CAS.
- F. Della Betta, L. Vitali, R. Fett and A. C. O. Costa, Talanta, 2014, 122, 23–29 CrossRef CAS.
- C. Merusi, C. Corradini, A. Cavazza, C. Borromei and P. Salvadeo, Food Chem., 2010, 120, 615–620 CrossRef CAS.
- N. Öztekin, M. S. Nutku and F. B. Erim, Food Chem., 2002, 76, 103–106 CrossRef.
- G. Daneshvar Tarigh and F. Shemirani, Talanta, 2014, 128, 354–359 CrossRef CAS.
- S. Biswas, B. Chowdhury and B. C. Ray, Talanta, 2004, 64, 308–312 CrossRef CAS.
- F. Gao, L. Zhang, L. Wang, S. She and C. Zhu, Anal. Chim. Acta, 2005, 533, 25–29 CrossRef CAS.
- P. Singh, M. K. Singh, Y. R. Beg and G. R. Nishad, Talanta, 2019, 191, 364–381 CrossRef CAS.
- S. Senra-Ferreiro, F. Pena-Pereira, I. Lavilla and C. Bendicho, Anal. Chim. Acta, 2010, 668, 195–200 CrossRef CAS.
- J. Zhao, Y. Lu, C. Fan, J. Wang and Y. Yang, Spectrochim. Acta, Part A, 2015, 136, 802–807 CrossRef CAS.
- M. N. Abbas and G. A. Mostafa, Anal. Chim. Acta, 2000, 410, 185–192 CrossRef CAS.
- N. Altunay and A. Elik, Food Chem., 2020, 332, 127395 CrossRef CAS.
- M. A. Feres and B. F. Reis, Talanta, 2005, 68, 422–428 CrossRef CAS PubMed.
- L. D. Nguyen, T. M. Huynh, T. S. V. Nguyen, D. N. Le, R. Baptist, T. C. D. Doan and C. M. Dang, J. Electroanal. Chem., 2020, 873, 114396 CrossRef CAS.
- B. Haghighi and A. Safavi, Anal. Chim. Acta, 1997, 354, 43–50 CrossRef CAS.
- J. Proch and P. Niedzielski, Talanta, 2021, 231, 122403 CrossRef CAS PubMed.
- S. Liu, M. Zhao and C. Deng, J. Chromatogr. A, 1992, 598, 298–302 CrossRef CAS.
- B. F. Reis, M. Knochen, G. Pignalosa, N. Cabrera and J. Giglio, Talanta, 2004, 64, 1220–1225 CrossRef CAS PubMed.
- B. M. Soares, R. F. Santos, R. C. Bolzan, E. I. Muller, E. G. Primel and F. A. Duarte, Talanta, 2016, 160, 454–460 CrossRef CAS PubMed.
- D. J. Leao, M. M. S. Junior, G. C. Brandao and S. L. C. Ferreira, Talanta, 2016, 153, 45–50 CrossRef CAS PubMed.
- M. H. Mashhadizadeh, M. S. Azimi, M. Pesteh, I. Sheikhshoaei, M. M. Ardakani and M. A. Karimi, Spectrochim. Acta, Part B, 2008, 63, 889–892 CrossRef.
- J. Bok-Badura, A. Jakóbik-Kolon, M. Turek, S. Boncel and K. Karoń, RSC Adv., 2015, 5, 101634–101640 RSC.
- L. Ganranoo, R. Chokchaisiri and K. Grudpan, Talanta, 2019, 191, 307–312 CrossRef CAS.
- K.-W. Cha and C.-I. Park, Talanta, 1996, 43, 1335–1340 CrossRef CAS.
- T. C. F. Ribas, R. B. R. Mesquita, T. Moniz, M. Rangel and A. O. S. S. Rangel, Talanta, 2020, 216, 120925 CrossRef CAS PubMed.
- S. Ohno, N. Teshima, T. Sakai, K. Grudpan and M. Polasek, Talanta, 2006, 68, 527–534 CrossRef CAS.
- N. Kotchabhakdi, C. Seanjum, K. Kiwfo and K. Grudpan, Microchem. J., 2021, 162, 105860 CrossRef CAS.
- W. Siriangkhawut, P. Didpinrum, Y. Khanhuathon, K. Ponhong and K. Grudpan, Anal. Lett., 2020, 53, 887–904 CrossRef CAS.
- N. Jaikrajang, S. Kruanetr, D. J. Harding and P. Rattanakit, Spectrochim. Acta, Part A, 2018, 204, 726–734 CrossRef CAS.
- B. Weerasuk, S.-a. Supharoek, K. Grudpan and K. Ponhong, J. Iran. Chem. Soc., 2022, 19, 741–751 CrossRef CAS.
- C.-K. Wang, W.-H. Lee and C.-H. Peng, J. Agric. Food Chem., 1997, 45, 1185–1188 CrossRef CAS.
- Z. Fu and R. Chen, J. Anal. Methods Chem., 2019, 2019, 3894571 Search PubMed.
- M. S. Masoud, A. E. Ali, S. S. Haggag and N. M. Nasr, Spectrochim. Acta, Part A, 2014, 120, 505–511 CrossRef CAS.
- P. Griess, Ber. Dtsch. Chem. Ges., 1879, 12, 426–428 CrossRef.
- I. A. Mohammed and A. Mustapha, Molecules, 2010, 15, 7498–7508 CrossRef CAS PubMed.
- J. Iglesias, E. García de Saldaña and J. A. Jaén, Hyperfine Interact., 2001, 134, 109–114 CrossRef CAS.
- M. Rajfur, A. Kłos and M. Wacławek, Bioelectrochem, 2010, 80, 81–86 CrossRef CAS PubMed.
- R. Mingkhwan and S. Worakhunpiset, Int. J. Environ. Res. Public Health, 2018, 15, 1890–1899 CrossRef.
- I. T. Somé, A. K. Sakira, D. Mertens, S. N. Ronkart and J.-M. Kauffmann, Talanta, 2016, 152, 335–340 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra03870f |
| This journal is © The Royal Society of Chemistry 2022 |