
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelectivity profile comparison for certain γ-butyrolactone and oxazolidinone-based ligands on a sigma 2 receptor over sigma 1: a molecular docking approach†
Richie R. Bhandare *ab,
Dilep Kumar Sigalapalli*c,
Afzal B. Shaik*c,
Daniel J. Canneyd and
Benjamin E. Blass*d
*ab,
Dilep Kumar Sigalapalli*c,
Afzal B. Shaik*c,
Daniel J. Canneyd and
Benjamin E. Blass*d
aCollege of Pharmacy & Health Sciences, Ajman University, PO Box 340, Ajman, United Arab Emirates. E-mail: r.bhandareh@ajman.ac.ae
bCenter of Medical and Bio-allied Health Sciences Research, Ajman University, Ajman, United Arab Emirates
cDepartment of Pharmaceutical Chemistry, Vignan Pharmacy College, Jawaharlal Nehru Technological University, Vadlamudi, 522213, Andhra Pradesh, India. E-mail: dileepsigalapalli@gmail.com; bashafoye@gmail.com
dTemple University School of Pharmacy, 3307 North Broad Street, Philadelphia, PA 194140, USA. E-mail: benjamin.blass@temple.edu
First published on 11th July 2022
Abstract
Sigma receptors (σ1 R and σ2 R) are pharmacologically characterized membrane-bound receptors that bind a wide range of chemical compounds. Alzheimer's disease, traumatic brain injury, schizophrenia, and neuropathic pain have all been associated with abnormal σ2 activity. The σ2 receptor has recently been identified as a potential therapeutic target for inhibiting the formation of amyloid plaques. Numerous laboratories are now investigating the potential of σ2 ligands. Small molecule discovery is the focus of current research, with the goal of using target-based action to treat a variety of illnesses and ailments. Functionalized γ-butyrolactone and oxazolidinone-based ligands, in particular, are pharmacologically important scaffolds in drug discovery research and have been thoroughly examined for σ2 receptor efficacy. The purpose of this study was to evaluate the pharmacophoric features of different σ2 receptor ligands using in silico techniques. This study used a library of 58 compounds having a γ-butyrolactone and oxazolidinone core. To investigate the binding characteristics of the ligands with the σ2 receptor, a 3D homology model was developed. To understand the binding pattern of the γ-butyrolactone and oxazolidinone based ligands, molecular docking studies were performed on both σ1 and σ2 receptors. Furthermore, MM/GBSA binding energy calculations were used to confirm the binding of ligands on the σ2 over σ1 receptor. These in silico findings will aid in the discovery of selective σ2 ligands with good pharmacophoric properties and potency in the future.
1. Introduction
In 1976, W. R. Martin et al. published the results of a study on the impact of morphine, ketocyclazocine, and (±)-SKF-100047 in dogs with chronic spinal disease. They found that these three compounds elicited distinctly different responses and hypothesized that each was interacting with a different pharmacological target. They labeled these previously unidentified targets as the μ-opioid receptor (morphine type, MOR), the κ-opioid receptor (ketocyclazocine type, KOR), and the σ-opioid receptor (SKF-100047 like).1 Follow-up studies with the individual enantiomers of SKF-100047 revealed that each isomer interacted with a different biochemical target. (−)-SKF-100047 interacts with MOR and KOR to produce an opioid type response, but (+)-SKF-100047 produces a non-opioid response through the sigma receptor (σR).2 Eventually, it was determined that there are two subtypes of this receptor, which have been designated as sigma-1 (σ1) and sigma-2 (σ2).3 An X-ray structure of human σ1 was reported in 2016,4 but to date there are no known ligands for this receptor.The true nature of σ2, however, remained a mystery for nearly 40 years. In 2017, A. C. Kruse et al. demonstrated that this receptor is identical to the Transmembrane Protein 97 (TMEM97, also known as MAC30 (Meningioma-associated protein)),5 and an X-ray structure was published in 2021.6 Similar to σ1, there are no known natural functional σ2 ligands. It has been demonstrated that this protein is present in the endoplasmic reticulum (ER) and lysosomes where it binds to cholesterol, but the pharmacological role of σ2 has not been determined.7 Numerous disease states such as schizophrenia,8 Alzheimer's disease,9 neuropathic pain,10 traumatic brain injury,11 Niemann-Pick disease,12 and cancer13 have been linked to σ2 and this has prompted many research teams to investigate the potential therapeutic utility of σ2 ligands. Numerous in vivo active σ2 ligands have been identified and some have reached human clinical trials. UKH-1114 (1, σ2 Ki = 46 nM) is efficacious in animal models of pain,14 while Siramesine (2, σ2 Ki = 0.12 nM) is efficacious in animal models of depression and anxiety.15 The radioligand [18F]ISO-1 (3, σ2 Ki = 6.9 nM) has been studied as a possible PET ligand in the treatment of breast cancer,16 while CT1812 (4, σ2 Ki = 8.5 nM) has been the subject of clinical trials as a potential Alzheimer's disease therapy (Fig. 1).17
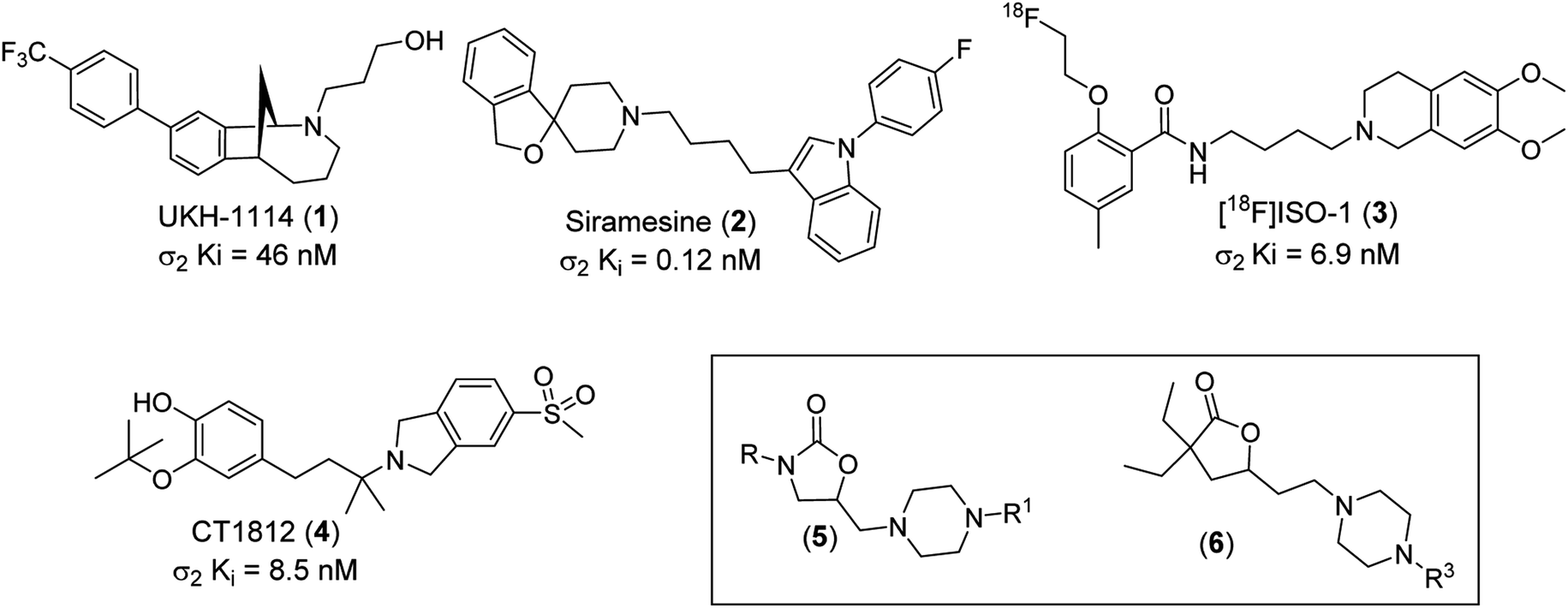 | ||
| Fig. 1 Structures of UKH-1114 (1), Siramesine (2), [18F]ISO-1 (3), CT1812 (4), oxazolidinone lead class (5) and γ-butyrolactones lead series (6). | ||
Recently, we reported our discovery of two novel classes of σ2 ligands, oxazolidin-2-ones (5) and functionalized γ-butyrolactones (6).18–20 The synthetic protocol for the synthesis of these two classes of compounds is outlined in the ESI† (Schemes 1 and 2). These compounds demonstrated a range of selectivity for σ2 over σ1. As part of an effort to develop a better understanding of the structural features that drive selectivity, we have studied generated in silico models of both receptors. In addition, we have conducted docking studies and calculated binding energies of the exemplary compounds from our previously disclosed σ2 ligands.
2. Materials and methods
2.1 Homology modeling
Homology modeling is a technique for creating and predicting an atomic resolution model of a target protein based on the experimentally established 3D structure of a homologous protein called the template protein. The four fundamental stages in homology modeling are (1) locating the template structure sequence, (2) matching the query sequence with the template structure sequence, (3) creating the query's model structure based on the template structure information, and (4) assessing the projected model. As a result, homology modeling may be used to predict the structure of unknown proteins, such as the human sigma 2 receptor.The model architectures of the human sigma 2 receptor were predicted using a homology modeling approach. Maestro was used for all computational and molecular modeling of the human sigma 2 receptor (Schrödinger, LLC, 2019-1).21 The human sigma 2 amino acid sequence, which consists of 176 residues, was retrieved from the Uniprot database (UniProt ID: Q5BJF2).22 PSI-BLAST was used to scan the nonredundant PDB database for template identification. The X-ray crystalline structure of the Bos taurus sigma-2 receptor bound to Z1241145220 (PDB ID: 7M95)23 exhibited measurable similarities to the query sequence and was therefore utilized to generate the model. Protein Data Bank was used to acquire the coordinates. The alignment of the template and the target was the initial stage. To choose the best alignment, the alignments were ordered by identities, score, positives, expectation value, and gaps, and statistically analyzed.
The Prime Module was used to create the homology models. In the model, the co-crystallized ligand Z1241145220 was retained. Using Prime functionality, loop refinement and numerous loop conformations were produced. Side-chain predictions and all-atom minimizations were used to score these conformations. The model was further optimized and minimized when the model construction calculations were completed.
2.2 Molecular docking studies
Schrödinger's Glide docking tool (Schrödinger, LLC, New York, 2019) was utilized to investigate ligand–receptor interactions. Glide is a novel method for finding favourable interactions between proteins and many ligands. As a result, Glide docking allows you to compare the binding mechanism and affinity of different ligands to the protein. In flexible docking, ligand posture refers to the location and orientation of a ligand with regard to the protein, as well as its shape. A number of hierarchical filters are used to analyze the ligand postures obtained by Glide docking. The ChemScore empirical scoring function is used by Glide. This algorithm detects the protein's and ligand's favourable hydrophobic and hydrogen-bonding interactions. Through many evaluation trials, Glide has been regarded as one of the best docking tools presently available.Customizing the protein, metal ions, and cofactors is simple using the Protein Preparation Wizard. The wizard fills in any missing residues around the protein's active site. The formal charges and ligand bond ordering were changed. Following these stages, the Impact Refinement module was used to perform restrained minimization (Impref, 2019). The revised ligand/protein/water structures were examined to guarantee the right formal charges, bond ordering, protonation states, and final changes were made to the protein structures. The receptor grid files were created using the prepared protein structures.
2.3 Prime MM/GBSA binding energy calculations
One of the most often used computational approaches for estimating relative binding affinities of target protein–ligand complexes is molecular mechanics with generalised born surface area (MM/GBSA). Through employing the MM/GBSA technology tools available in the Prime module of Schrodinger 2019-1, ligand-binding energies were calculated based on docking complex. The MM/GBSA calculations were performed using the protein–ligand complexes produced from the docking investigations. ΔGbind, the relative binding free energy, was calculated using the following equation:| ΔGbind = Ecomplex (minimized) − [Eligand(unbound, minimized) + Ereceptor(unbound, minimized)] |
3. Results and discussion
3.1 Homology modeling and binding site analysis
To the best of our knowledge, the crystal structure for sigma-2 receptor of Homo sapiens has not been reported yet. As a result, we used a comparative modeling approach to create a three-dimensional (3D) model for the sigma-2 receptor of H. sapiens. The homology model of the H. sapiens sigma-2 receptor was created in Schrödinger Suite 2019-1 (Schrödinger, LLC, New York, NY) using Meastro, Prime. The H. sapiens sigma intracellular receptor 2 query sequence was obtained from the universal protein resource (Uniprot, Entry Id: Q5BJF2). The template was chosen from the crystallographic structure of the Bos taurus sigma-2 receptor (PDB ID: 7M95, 2.41 resolution) with 78.4 percent sequence identity with the target. Prime homology modeling methods were used to create a 3D model of the human sigma-2 receptor. The Ramchandran plot and protein reports were used to analyze the predicted 3D structure. The sequence alignment of the sigma-2 receptor of H. sapiens (UniProt ID:Q5BJF2) and Bos Taurus (PDB ID: 7M95) is shown in Fig. 2. The homology-modeled structure of the human sigma-2 receptor and its Ramchandran plot are shown in Fig. 3 and 4. The Ramchandran plot of the human sigma-2 receptor structure revealed that >95 percent of residues are favored and allowed φ, ψ backbone conformational regions. | ||
| Fig. 2 Sequence alignment of sigma intracellular receptor 2 (σ2) from Homo sapiens (UniProt ID:Q5BJF2) and bovine sigma-2 receptor from Bos taurus (PDB ID: 7M95). | ||
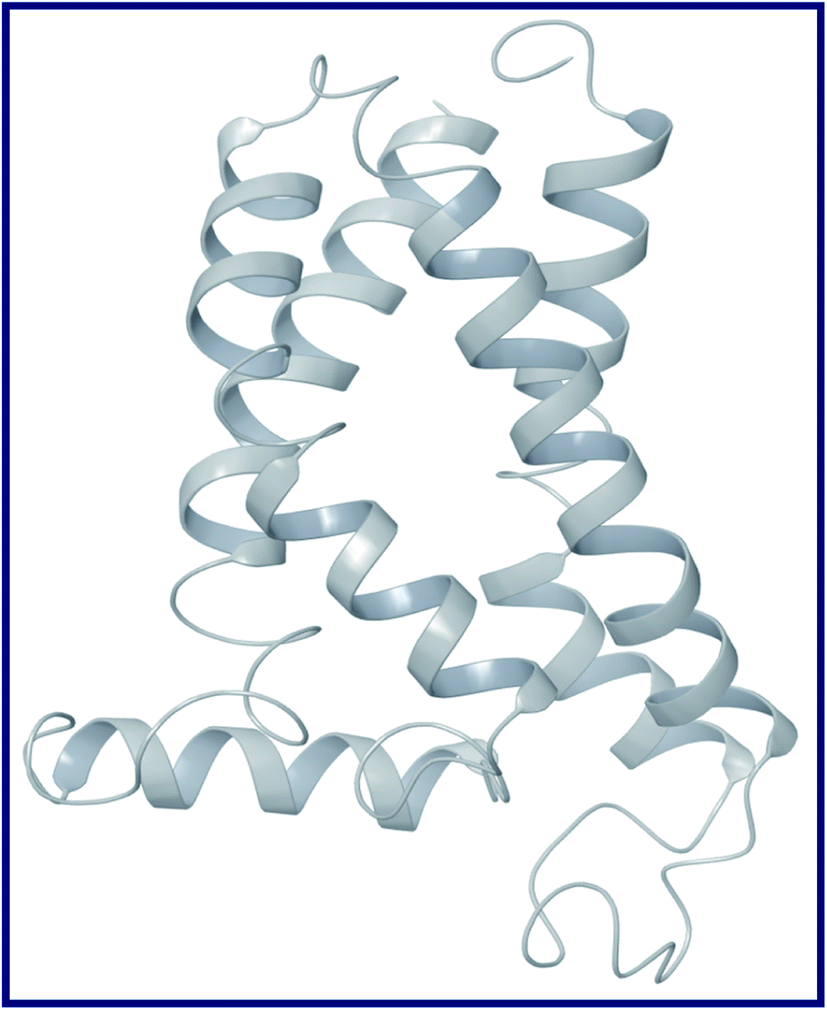 | ||
| Fig. 3 Homology modelled 3D structure of human sigma intracellular receptor 2 (σ2). | ||
 | ||
| Fig. 4 Ramachandran plot for the modeled sigma intracellular receptor 2 (σ2) of Homo sapiens. The plot is organized as follows: glycine, proline and all other residues are plotted as triangles, squares, and circles respectively. The red, yellow and white regions represent the favoured, allowed and the disallowed regions respectively. | ||
3.2 Molecular docking
All of the compounds were subjected to molecular docking studies in order to discover significant binding mechanisms responsible for their action on the sigma 1 and 2 receptors. Molecular docking was used to confirm the binding pose and conformation of these analogues. All of the compounds were docked into the active site of a homology-modeled sigma 2 receptor as well as the crystal structure of the sigma 1 receptor. The results of molecular docking, hydrogen bonding, and arene–arene interactions of compounds with sigma 1 and 2 receptors are shown in Tables 1 and S1.†| S. no | Ligand ID | Receptor name | Ki (nM) | Docking score | Interactions | ||
|---|---|---|---|---|---|---|---|
| H- bonds | Pi–Pi stacking | Hydrophobic | |||||
| a ND# not docked at the active site. | |||||||
| 1 | LACT11 | σ1 | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
−5.705 | — | Tyr103 | Val84, Trp89, Met93, Leu95, Ala98, Tyr103, Leu105, Phe107, Tyr120, Ile124, Phe133, Val162, Trp164, Met170, Ile178, Leu182, Ala185, Tyr206 |
| σ2 | 14 | −6.850 | — | Tyr50 | Ile24, Met28, Trp49, Tyr50, Leu59, Phe66, Phe69, Leu70, Cys72, Leu111, Ile114, Val146, Tyr147, Tyr150 | ||
| 2 | LACT21 | σ1 | 1168 | −5.681 | — | — | Val84, Ala86, Trp89, Met93, Leu95, Tyr103, Leu105, Phe107, Tyr120, Ile124, Phe133, Val162, Trp164, Met170, Ile178, Leu182, Phe184, Ala185, Tyr206 |
| σ2 | 44 | −7.550 | Asp29 | — | Ile24, Met28, Trp49, Tyr50, Leu59, Phe66, Phe69, Leu70, Leu111, Ile114, Val146, Tyr147, Tyr150 | ||
| 3 | LACT22 | σ1 | 195 | −4.693 | — | — | Val84, Trp89, Met93, Leu95, Ala98, Tyr103, Leu105, Phe107, Tyr120, Ile124, Phe133, Val152, Val162, Trp164, Met170, Ile178, Leu182, Phe184, Ala185, Leu186, Tyr206 |
| σ2 | 5.9 | −7.891 | Asp29 | — | Ile24, Met28, Trp49, Tyr50, Phe54, Leu59, Phe66, Leu70, Leu111, Ile114, Val146, Tyr147, Pro149, Tyr150 | ||
| 4 | LACT26 | σ1 | 10000 |
−4.983 | Glu172 | — | Val84, Trp89, Met93, Leu95, Ala98, Tyr103, Leu105, Tyr120, Ile124, Phe133, Val162, Trp164, Ile178, Leu182, Phe184, Ala185, Tyr206 |
| σ2 | 142 | −7.914 | Asp29, Glu73 | — | Ile24, Met28, Trp49, Tyr50, Phe54, Leu59, Phe66, Phe69, Leu70, Leu111, Ile114, Val146, Tyr147, Tyr150 | ||
| 5 | LACT29 | σ1 | 2167 | −6.486 | Glu172 | Tyr103, Tyr206 | Val84, Trp89, Met93, Leu95, Ala98, Tyr103, Leu105, Tyr120, Ile124, Phe133, Val152, Val162, Trp164, Ile178, Leu182, Phe184, Ala185, Tyr206 |
| σ2 | 32 | −7.915 | Asp29 | — | Ile24, Met28, Trp49, Tyr50, Leu59, Phe66, Phe69, Leu70, Leu111, Ile114, Val146, Tyr147, Tyr150 | ||
| 6 | LACT35 | σ1 | 125 | −7.416 | — | His154 | Val84, Trp89, Met93, Leu95, Tyr103, Leu105, Phe107, Tyr120, Ile124, Phe133, Val162, Trp164, Ile178, Leu182, Phe184, Ala185, Leu186 |
| σ2 | 6.1 | −10.62 | Asp29 | — | Ile24, Met28, Tyr50, Leu59, Phe66, Phe69, Leu70, Leu111, Ile114, Val146, Tyr147, Tyr150 | ||
| 7 | LACT37 | σ1 | 59 | −7.642 | — | Tyr103 | Val84, Trp89, Met93, Leu95, Ala98, Tyr103, Leu105, Phe107, Tyr120, Ile124, Phe133, Val162, Trp164, Met170, Ile178, Leu182, Phe184, Ala185, Tyr206 |
| σ2 | 2.8 | −9.880 | Asp29 | — | Ile24, Met28, Trp49, Tyr50, Leu59, Phe66, Leu70, Leu111, Ile114, Val146, Tyr147, Tyr150 | ||
| 8 | LACT41 | σ1 | 10000 |
−7.582 | Glu172 | — | Val84, Trp89, Met93, Leu95, Ala98, Tyr103, Leu105, Phe107, Tyr120, Ile124, Phe133, Val162, Trp164, Ile178, Leu182, Phe184, Ala185, Tyr206 |
| σ2 | 277 | −9.292 | Asp29 | — | Ile24, Met28, Tyr50, Leu59, Phe66, Phe69, Leu70, Leu111, Ile114, Val146, Tyr147, Tyr150 | ||
| 9 | OXAZ2 | σ1 | 10000 |
−5.445 | — | Tyr103 | Val84, Trp89, Met93, Leu95, Ala98, Tyr103, Leu105, Tyr120, Ile124, Phe133, Val152, Val162, Trp164, Ile178, Leu182, Phe184, Ala185, Leu186, Tyr206 |
| σ2 | 119 | −7.113 | Asp29 | Tyr50 | Ile24, Met28, Trp49, Tyr50, Leu59, Phe66, Leu70, Leu111, Ile114, Val146, Tyr147, Pro149, Tyr150 | ||
| 10 | OXAZ3 | σ1 | 10000 |
ND | — | — | — |
| σ2 | 465 | −7.708 | Asp29, Glu73 | Phe54 | Ile24, Met28, Trp49, Tyr50, Phe54, Leu59, Phe66, Phe69, Leu70, Leu111, Ile114, Val146, Tyr147, Pro149, Tyr150 | ||
| 11 | OXAZ4 | σ1 | 10000 |
ND | — | — | — |
| σ2 | 206 | −7.627 | Asp29 | Phe54 | Ile24, Met28, Trp49, Tyr50, Phe54, Leu59, Phe66, Leu70, Phe81, Tyr103, Leu111, Ile114, Val146, Tyr147, Pro149, Tyr150 | ||
| 12 | OXAZ5 | σ1 | 10000 |
−4.569 | — | Tyr103, Tyr206 | Val84, Trp89, Met93, Leu95, Ala98, Tyr103, Leu105, Tyr120, Ile124, Phe133, Val152, Val162, Trp164, Ile178, Leu182, Phe184, Ala185, Leu186, Tyr206 |
| σ2 | 530 | −6.371 | — | — | Ile24, Met28, Leu46, Trp49, Tyr50, Phe54, Leu59, Phe66, Phe69, Leu70, Phe81, Tyr103, Leu111, Ile114, Val146, Tyr147, Tyr150 | ||
| 13 | OXAZ6 | σ1 | 10000 |
ND | — | — | — |
| σ2 | 192 | −7.609 | Asp29 | Ile24, Met28, Trp49, Tyr50, Phe54, Leu59, Phe66, Phe69, Leu70, Leu111, Ile114, Vall46, Tyr147, Pro149, Tyr150 | |||
| 14 | OXAZ7 | σ1 | 10000 |
ND | — | — | — |
| σ2 | 91 | −8.164 | Asp29 | — | Ile24, Met28, Leu46, Trp49, Tyr50, Phe54, Leu59, Phe66, Phe69, Leu70, Leu111, Ile114, Vall46, Tyr147, Tyr150 | ||
| 15 | OXAZ8 | σ1 | 2847 | −1.104 | — | Trp121 | Tyr120, Trp121, Ala183, Phe184, Ala187, Phe191 |
| σ2 | 36 | −10.15 | — | — | Ile24, Met28, Leu46, Trp49, Tyr50, Phe54, Leu59, Phe66, Phe69, Leu70, Leu111, Ile114, Vall46, Tyr147, Tyr150 | ||
| 16 | OXAZ13 | σ1 | 10000 |
ND | — | — | — |
| σ2 | 49 | −8.832 | Asp29 | Tyr150 | Ile24, Met28, Trp49, Tyr50, Phe54, Leu59, Phe66, Phe69, Leu70, Leu111, Ile114, Vall46, Tyr147, Pro149, Tyr150 | ||
The initial set of compounds (LACT1–LACT41) comprised of a series of γ-butyrolactone analogues with a heterocyclic primary core and varied electron donating, electron withdrawing, hydrophobic, H-bond acceptor, and donor groups. When compared to the 1-diethyl lactone derivative (LACT1) of the γ-butyrolactone family, the 1,1-dimethyl lactone derivative (LACT2) demonstrates a restricted number of interactions with the active site of sigma 2 receptor. The active site residues (Asp29, Glu73) of σ2 R formed two hydrogen bonds with LACT1. The protonated N-atom of phenyl piperazine serves as an H-bond donor, forming an H-bond with Asp29 (d = 2.57 Å). Butyrolactone's carbonyl oxygen atom functions as an H-bond acceptor, forming an H-bond with Glu73 (d = 2.49 Å). Furthermore, piperazine's N-atom forms a Pi–cation interaction with Tyr147. LACT1 has also exhibited a variety of hydrophobic interactions with active site residues. The phenyl ring of LACT1 forming a Pi–Pi (arene–arene) contact with Tyr50. Furthermore, LACT1 formed only one Pi–Pi interaction with the σ1 R residue Tyr103 and made no H-bond interactions with it. There are fewer interactions with both σ1 and σ2 R for the 1,1-dimethyl lactone derivative (LACT2).
LACT3 with carbonyl group on its ethyl linker chain has lower docking scores towards both σ1 & σ2 R. LACT4 with piperazin-2-one ring has average docking scores with fewer interactions on σ1 & σ2 R. LACT5 with piperadine ring has shown good docking scores with σ1 & σ2 R. The N-atom of piperadine scaffold has made two strong Pi–cation interactions with the active site residues Tyr147 (d = 5.59 Å) and Tyr150 (d = 4.55 Å) of σ2 R. Interestingly, the N-atom of piperadine also made a salt bridge with Asp29 (d = 3.07 Å) of σ2 R. Further, the phenyl ring of LACT5 established two Pi–Pi interactions with the active site residues Tyr103, Tyr206 of σ1 R. In case of LACT6, the protonated nitrogen atom was observed at cyclohexyl connected N-atom of the piperazine ring and this N-atom interact with Asp29 via salt-bridge. In addition, the protonated N-atom of the piperazine ring has made a Pi–cation interaction Tyr150 of σ2 R. Further, the protonated N-atom of the piperazine ring of LACT6 has established an H-bond and Pi–cation interaction with Glu172 and Phe107 of σ1 R, respectively. Compounds with electron withdrawing substituent's like CN (LACT7), CF3 (LACT8), and Cl (LACT9) in the 4-position of the phenyl ring has shown good docking scores with both the σ1 & σ2 R. The protonated N-atom of the piperazine ring of LACT7, LACT8 and LACT9 has shown a salt-bridge and Pi–cation interaction with Asp29 and Tyr147 of σ2 R, respectively. Additionally, an H-bond interaction was observed in between LACT9 and Val146 of σ2 R. Further, a salt-bridge and a Pi–cation interaction were observed for both LACT7, LACT8 with Glu172 and Phe107 of σ1 R, respectively. A Pi–Pi interaction was observed for 4-trifluoromethyl substituted phenyl ring of LACT8, LACT9 with Tyr103 and Leu182 of σ1 R, respectively.
Compounds with electron donating substituent's like OMe (LACT10) and Me (LACT11) in the 4-position of the phenyl ring has shown good docking scores on σ2 R over σ1 R. LACT10 established two hydrogen bonds with the active site residues (Asp29, Glu73) of σ2 R. The protonated N-atom of phenyl piperazine has made an H-bond interaction with Asp29 and the carbonyl oxygen atom of butyrolactone shown H-bond interaction with Glu73. Further, the N-atom of piperazine is established a Pi–cation interaction with Tyr147. With respective to the σ1 R, compound LACT10 has formed an H-bond interaction with Glu172 and Pi–Pi interaction with His154. The detailed binding pose and protein–ligand interactions of LACT11 with σ2 R was depicted in Fig. 5. 4-Me substituted phenyl ring of LACT11 established a Pi–Pi interaction with Tyr50 and N-atom of phenyl piperazine has made a Pi–cation interaction Tyr147. Further, several hydrophobic interactions were observed for LACT11 and the active site residues of σ2 R, which stabilizes the lodging of LACT11 in the active pocket.
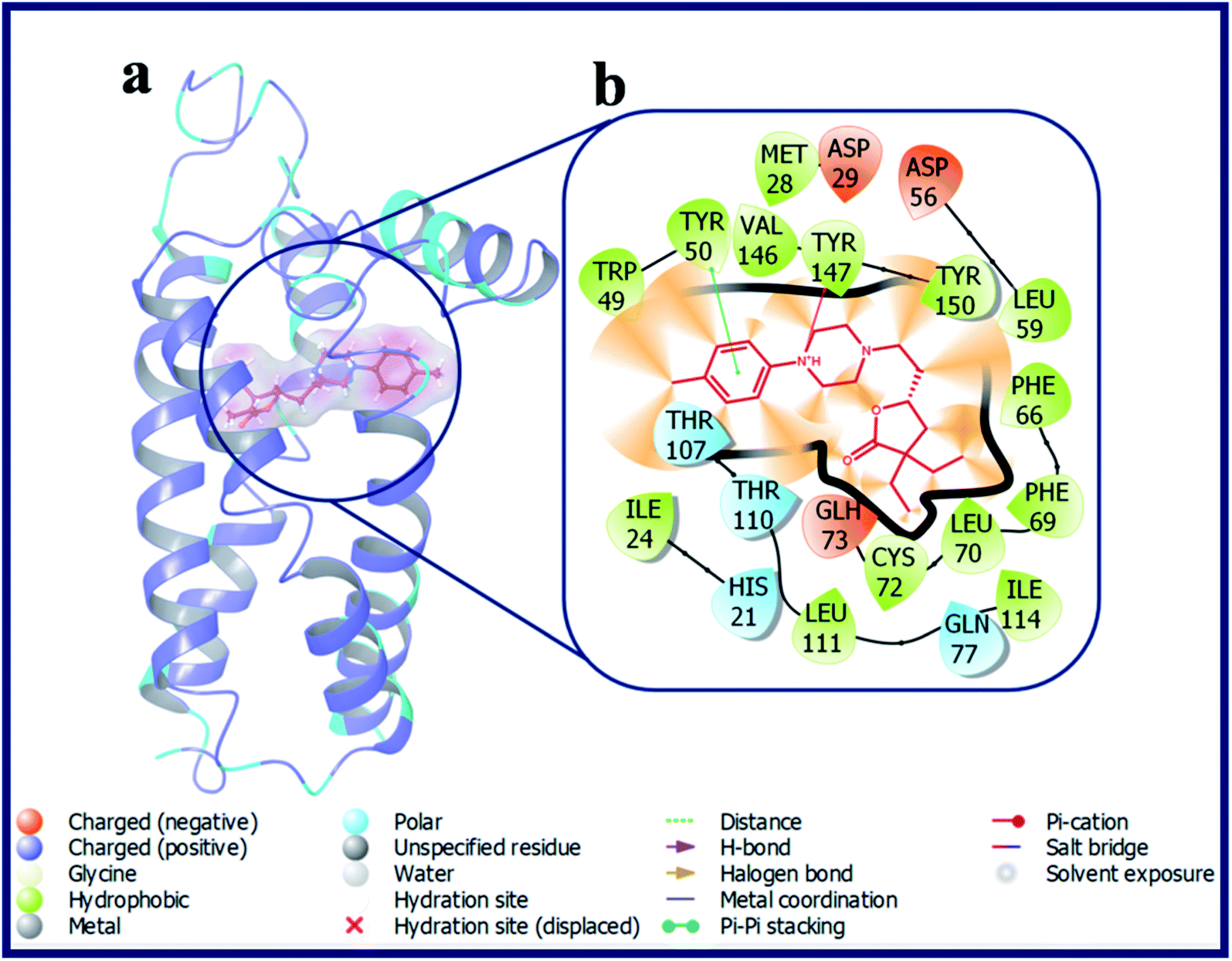 | ||
| Fig. 5 (a) Docking pose of compound LACT11 (purple colour stick) and (b) its ligand–protein interactions in the active site of modeled sigma intracellular receptor 2. | ||
Compounds LACT12–LACT14 include electron withdrawing substituent's at 3-position of the phenyl ring based derivatives endowed with an improved docking scores with σ2 R if compared with the σ1 R. The carbonyl oxygen atom of butyrolactone of LACT12 has shown an H-bond interaction with Glu73. Further, the protonated N-atom of phenyl piperazine of both LACT12 and LACT13 has made a salt-bridge and a Pi–cation interaction with Asp29 and Tyr147 of σ2 R, respectively. LACT14 established two hydrogen bonds with the active site residues Asp29 (d = 2.29 Å), Glu73 (d = 2.3 Å) and a Pi–cation interaction with Tyr147of σ2 R. Further, compound LACT15 and LACT16 with electron donating substituent's at 3-position of the phenyl ring showed almost similar docking scores on both σ1 & σ2 R.
Interestingly, compound LACT17–LACT19 with electron withdrawing substituent's at 2-position of the phenyl ring showed good docking scores on σ2 R if compared with the σ1 R. LACT17, LACT18 and LACT19 has shared a common H-bond interaction, as well as Pi–cation interaction with the active site residues Asp29 and Tyr147 of σ2 R, respectively. Additionally, compounds with electron donating substituent's at 2-position of the phenyl ring (LACT20 and LACT21) has also shown good docking scores on σ2 R over σ1 R. Mounting the steric bulk in the 2-position of the phenyl ring by placing an isopropyl group (LACT22) improved σ2 R selectivity. Fig. 6 displays the docking pose of LACT22 and its ligand–protein interactions in the active site of σ2 R. LACT23 with 2,4-di-Me substitution on phenyl ring displayed high docking score for both σ1 & σ2 R. LACT24 with 2-pyridine ring showed poor docking score at σ2 R. In this analogue, the 2-pyridine ring was involved in the Pi–Pi stacking interaction with Tyr50 of σ2 R. With respective to the σ1 R, compound LACT24 has formed an H-bond interaction with Glu172 (d = 2.55 Å). In case of 3-pyridine ring containing analogue (LACT25) and 4-pyridine ring containing analogue (LACT26), we observed a common H-bond interactions (Asp29, Glu73) and Pi–cation interaction (Tyr147) with σ2 R. In addition, the 3-pyridine ring of LACT25 also involved in the Pi–Pi stacking interaction with Tyr50 of σ2 R. Further, LACT25 did not show any H-bond, Pi–Pi, salt-bridge and Pi–cation interactions with σ1 R. LACT26 exhibited an H-bond between protonated N-atom of phenyl piperazine and Glu172, and Pi–cation interaction between N-atom of 4-pyridine ring and His154 of σ1 R.
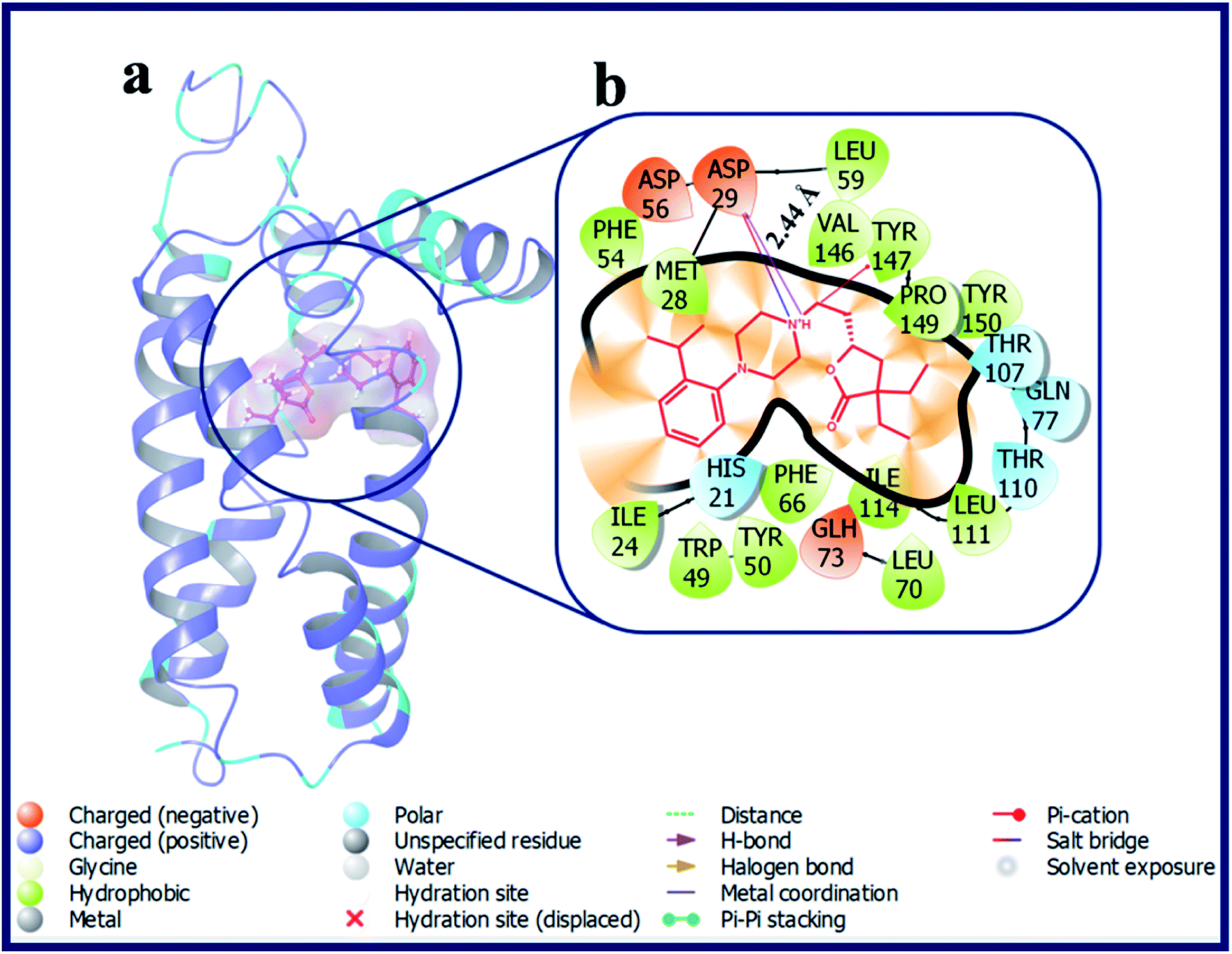 | ||
| Fig. 6 (a) Docking pose of compound LACT22 (purple colour stick) and (b) its ligand–protein interactions in the active site of modeled sigma intracellular receptor 2. | ||
LACT27 with linker chain length of “3C” displayed good docking score on σ2 R over σ1 R. The protonated N-atom of phenyl piperazine ring of LACT27 established an H-bond, salt-bridge, Pi–cation interactions with Val146 (d = 2.56 Å), Asp29 (d = 4.68 Å), and Tyr147 (d = 4.51 Å) of σ2 R, respectively. Compound LACT28 include linker chain length of “4C” also shown greater docking score on σ2 R over σ1 R. LACT28 established an H-bond with the active site residue Asp29 (d = 2.34 Å) and a Pi–cation interaction with Tyr147 (d = 5.06 Å) of σ2 R. 1-Naphthyl piperazine containing γ-butyrolactone analogue (LACT29) displayed good docking score on σ2 R. LACT29 made an H-bond with the active site residue Asp29 (d = 2.13 Å) and a Pi–cation interaction with Tyr147 (d = 4.98 Å) of σ2 R. With respective to the σ1 R, the protonated N-atom of piperazine ring of LACT29 has formed an H-bond interaction with Glu172 (d = 2.33 Å) and the 1-naphthyl part made two Pi–Pi interactions with the active site residues Tyr103 and Tyr206 of σ1 R. The 4-pyrimidine ring containing γ-butyrolactone analogue (LACT30) displayed poor docking scores on both σ1 and σ2 R.
Captivatingly, compounds with prospective piperazine bioisosteres like homopiperazine analogue (LACT31), 2,6-diazaspiro[3.3]heptane analogue (LACT32), and hexahydropyrrolo[3,4-c]pyrrole analogue (LACT33) shown excellent docking scores on σ2 R over σ1 R. LACT31, LACT32 and LACT33 has shared a common H-bond interaction, as well as Pi–cation interaction with the active site residues Asp29 and Tyr147 of σ2 R, respectively. Further, LACT32 and LACT33 have shown additional Pi–cation interaction with Tyr150 of σ2 R. Replacing of phenyl ring in LACT33 with 4-pyridine led to increase in number interactions with the active site residues of σ2 R. The N-atom of 4-pyridine of LACT34 established H-bond interaction with Gln77 (d = 2.61 Å), and also 4-pyridine formed a Pi–Pi interaction with His21 of σ2 R. Further, LACT34 has made H-bond interaction, as well as Pi–cation interaction with the active site residues Asp29 (d = 2.03 Å) and Tyr147 of σ2 R, respectively.
According to our calculations, fascinatingly, compound LACT35–LACT38 with tetrahydroisoquinoline scaffold have shown admirable docking scores with the σ2 R. Compound LACT35–LACT38 has shared a common H-bond interaction, as well as two Pi–cation interactions with the active site residues Asp29 and Tyr147 & Tyr150 of σ2 R, respectively. The protonated N-atom of tetrahydroisoquinoline involved in both H-bond and Pi–cation interactions. In addition, the carbonyl oxygen atom of butyrolactone of LACT35 is acting as H-bond acceptor and made an H-bond interaction with Gln77 (d = 3.38 Å). Fig. 7 illustrates the binding pose of compound LACT37 and its ligand–protein interactions in the active site of σ2 R. Similarly, compound LACT39–LACT41 with pyridine type nitrogen atom in the tetrahydroisoquinoline ring also exhibited the good docking scores and interactions against both σ1 and σ2 R, but their actual biological Ki values are not at all potent towards the both σ1 and σ2 R. The in silico docking scores for these compounds is probably due to the close structure resemblance with that of simple tetrahydroisoquinoline scaffold containing analogues (LACT35–LACT38).
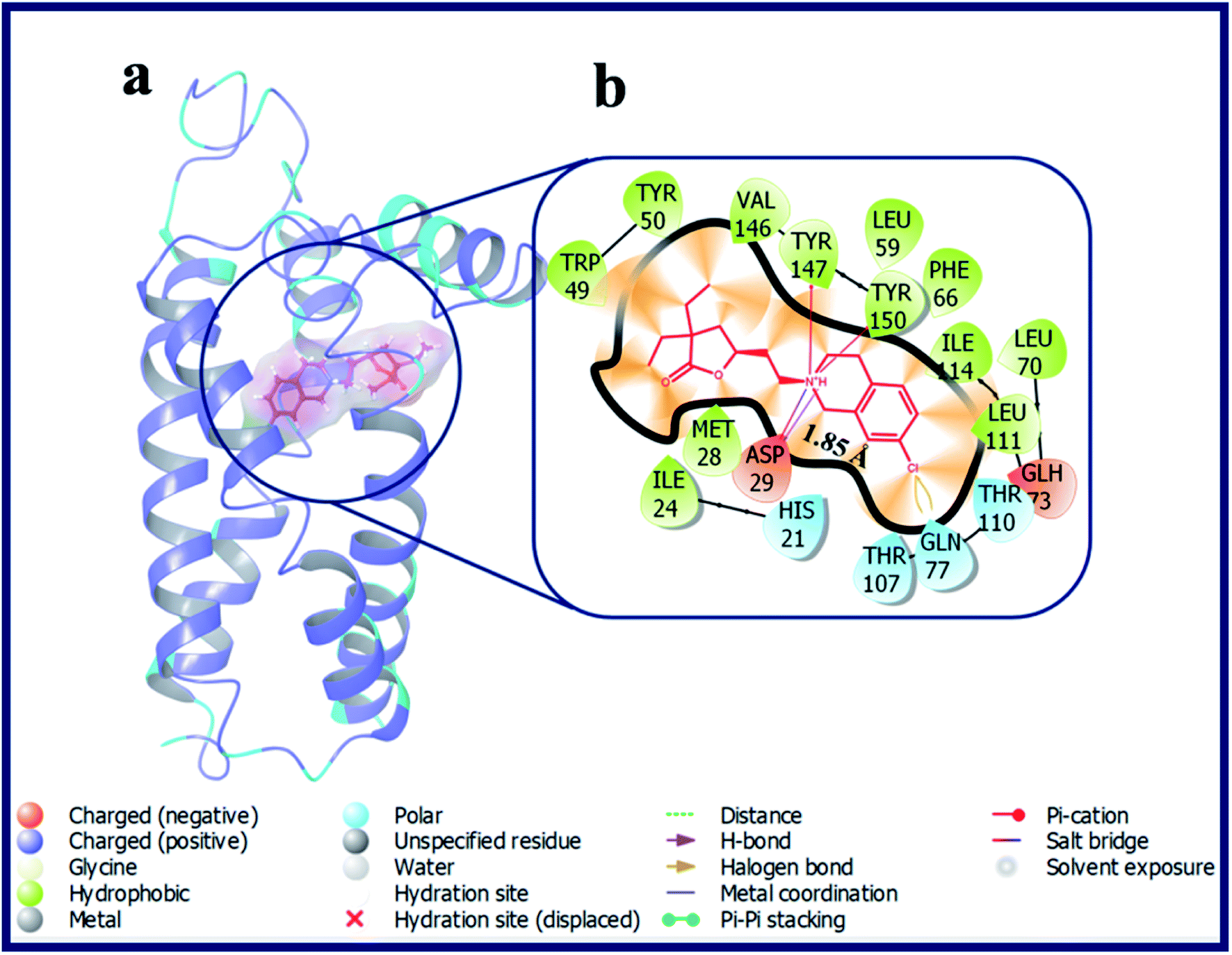 | ||
| Fig. 7 (a) Docking pose of compound LACT37 (purple colour stick) and (b) its ligand–protein interactions in the active site of modeled sigma intracellular receptor 2. | ||
The second set of compounds (OXAZ1–OXAZ17) consisted of a series of oxazolidinone-based derivatives with a heterocyclic primary core and various electron donating, electron withdrawing, hydrophobic, H-bond acceptor, and donor groups. Within this series, compounds have various substituent's capped at amide group of oxazolidin-2-one ring. The unsubstituted oxazolidin-2-one ring containing compound OXAZ1 displayed very poor docking scores towards both σ1 and σ2 R. Compounds with 3 to 6 membered cycloalkane group attachments, i.e. cyclopropyl (OXAZ2), cyclobutyl (OXAZ3), cyclopentyl (OXAZ4) and cyclohexyl (OXAZ5) have shown good to moderate docking scores against σ2 R. Compound OXAZ2 established one hydrogen bond with the active site residue (Asp29) of σ2 R. The protonated N-atom of phenyl piperazine is acting as H-bond donor and made an H-bond interaction with Asp29 (d = 1.80 Å). Further, the N-atom of piperazine is established two Pi–cation interactions with Tyr147 and Tyr150. Additionally, one of the phenyl groups of benzhydryl part established a Pi–Pi interaction with Tyr50. With respective to the σ1 R, the protonated N-atom of piperazine ring of OXAZ2 has formed a salt-bridge with Glu172 and one of the phenyl groups of benzhydryl part established a Pi–Pi interaction with Tyr103. OXAZ3 and OXAZ4 have shared a common H-bond interaction, as well as Pi–cation interaction and Pi–Pi interaction with the active site residues Asp29, Tyr147 and Phe54 of σ2 R, respectively. Further, OXAZ3 have shown additional H-bond interaction with Glu73 of σ2 R. On the contrary, compound OXAZ3 and OXAZ4 was not docked at the active site of σ1 R. Compound OXAZ5 exhibited a Pi–cation interaction with Tyr147 of σ2 R and two Pi–Pi stacking interactions with Tyr103 and Tyr206 of σ1 R.
Compound with phenyl ring (OXAZ6) has showed good docking score against σ2 R but not docked to the active site of σ1 R. The protonated N-atom of piperazine ring of OXAZ6 has formed an H-bond with Asp29 (d = 1.85 Å) and a Pi–cation interaction with Tyr147 of σ2 R. Further, the phenyl group of benzhydryl part established a Pi–Pi interaction with Tyr50. OXAZ7 with benzyl substituent showed good docking score with σ2 R, but not docked at σ1 R active pocket. OXAZ8 with 4-F-benzyl substituent showed increased docking score towards σ2 R, and poor score with σ1 R. Better activity of OXAZ8 with σ2 R is probably due to the strong salt-bridge formation between N-atom of piperazine ring and Asp29 of σ2 R. OXAZ8 also exhibited a Pi–cation interaction with Tyr147 and Pi–Pi interaction with Phe54 of σ2 R. In contrast, the presence of electron donating groups, such as 4-OMe (OXAZ9) and 4-Me (OXAZ10) on benzyl part led to decrease in docking scores against σ2 R. Further, OXAZ9 and OXAZ10 showed very poor binding scores with σ1 R. Fig. 8 illustrates the docking pose of compound OXAZ8 and its ligand–protein interactions in the active site of σ2 R. Fig. 9 displays the ligand–protein interactions for the compounds LACT11, LACT22, LACT37 and OXAZ8 at the active site of σ1 R.
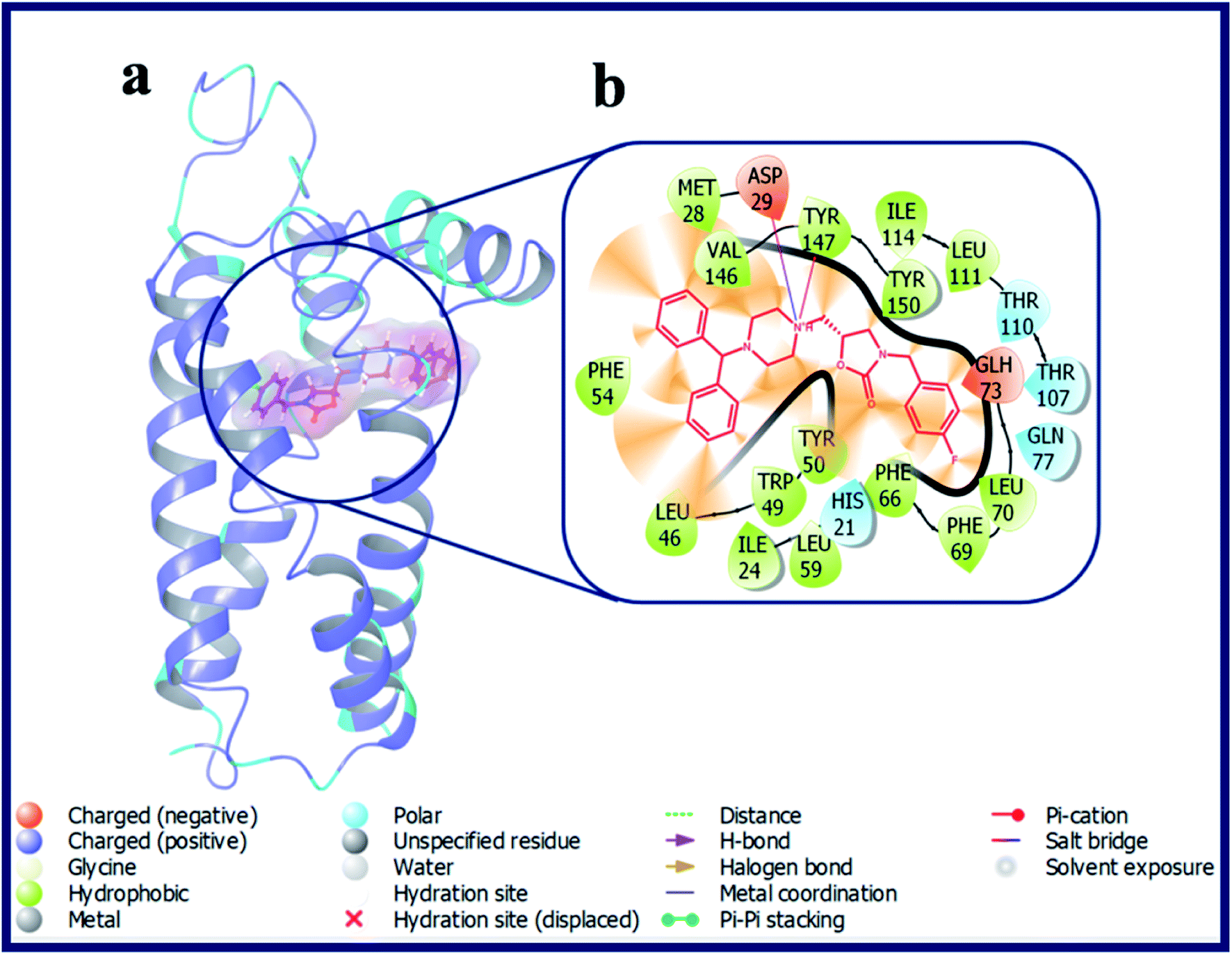 | ||
| Fig. 8 (a) Docking pose of compound OXAZ8 (purple colour stick) and (b) its ligand–protein interactions in the active site of modeled sigma intracellular receptor 2. | ||
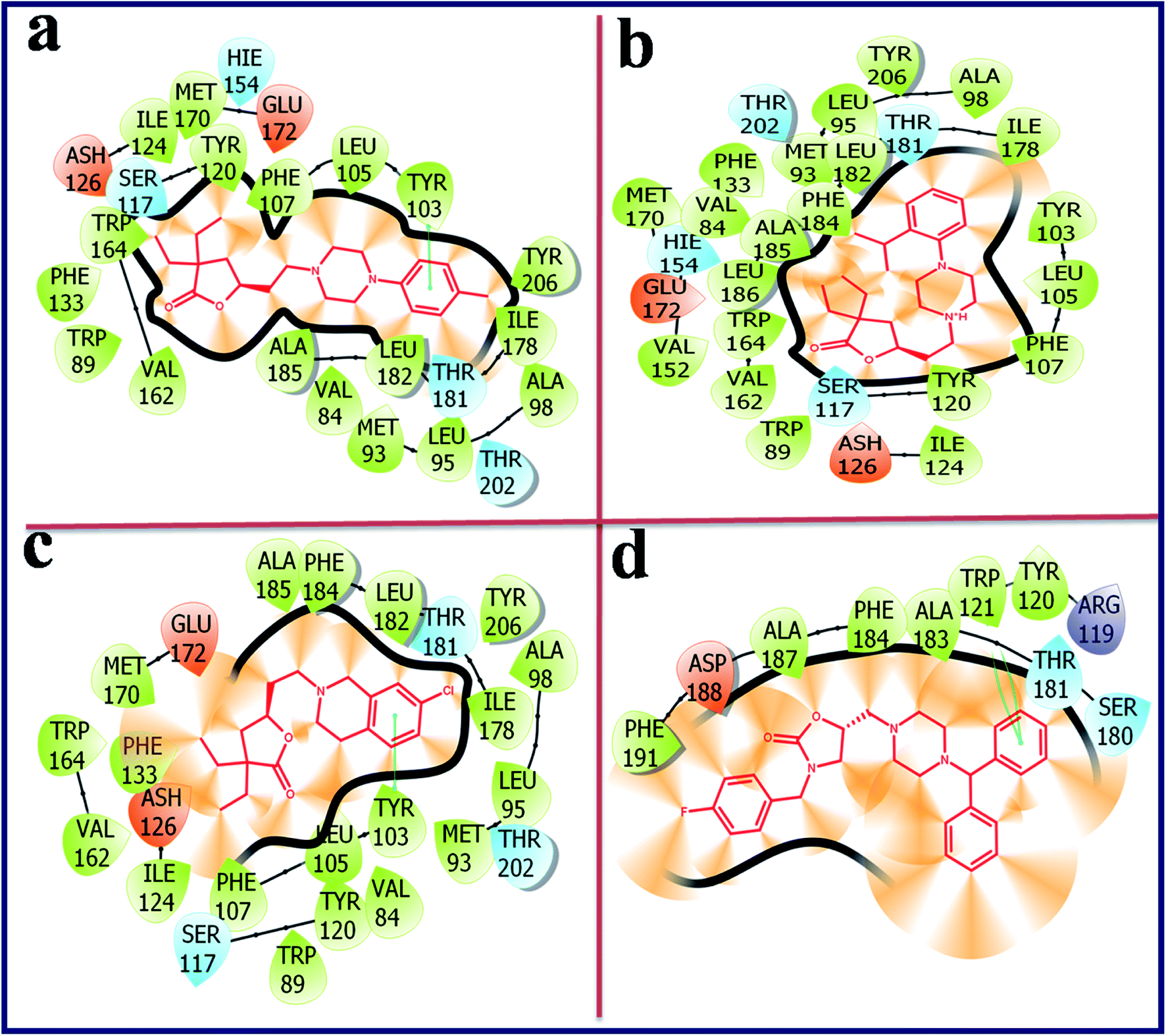 | ||
| Fig. 9 Ligand–protein interaction diagram for the compound LACT11 (a), LACT22 (b), LACT37 (c) and OXAZ8 (d) at the active site of human sigma intracellular receptor 1 (PDB ID: 5HK1). | ||
Compounds with a cyclohexane ring (OXAZ11) and tetrahydropyran ring (OXAZ12) also showed moderate binding with σ2 R and very poor binding with σ1 R. Compound OXAZ13 with phenethyl group displayed good docking score with σ2 R but not docked against σ1 R active pocket. The protonated N-atom of piperazine ring of OXAZ13 has formed an H-bond with Asp29 (d = 1.95 Å) and a Pi–cation interaction with Tyr147 of σ2 R. Further, one of the phenyl groups of benzhydryl part established a Pi–Pi interaction with Tyr150. Further, OXAZ14 with 4-F-phenethyl group showed poor binding with σ2 R and σ1 R. OXAZ15 with 4-OMe-phenethyl group showed an H-bond interaction with Asp29 (d = 2.04 Å) and a Pi–cation interaction with Tyr147 of σ2 R. OXAZ16 with 4-Me-phenethyl group showed poor docking with σ2 R and it exhibited two Pi–cation interactions with Tyr50 and Tyr147 of σ2 R. OXAZ17 with phenylpropyl also displayed an H-bond interaction with Asp29 (d = 2.34 Å) and a Pi–cation interaction with Tyr147 of σ2 R. Further, compound OXAZ15, OXAZ16 and OXAZ17 showed very poor binding score with the active site of σ1 R.
3.3 Prime MM/GBSA binding energy calculations
To further authenticate the binding affinity of docked γ-butyrolactone and oxazolidinone-based ligands at the active sites of the sigma 1 and sigma 2 receptors, binding free energy was calculated using the molecular mechanics generalised born surface area (MM-GBSA) approach available in the Prime module. Table 2 summarizes the binding energies of certain γ-butyrolactone and oxazolidinone-based ligands based on biological Ki values and the σ2/σ1 ratio.| S. no. | Ligand name | Receptor name | aΔGBind | bΔGBind Coulomb | cΔGBind covalent | dΔGBind H-bond | eΔGBind lipo | fΔGBind solv GB | gΔGBind packing | hΔGBind vdW |
|---|---|---|---|---|---|---|---|---|---|---|
| a Free energy of binding.b Coulomb energy.c Covalent energy (internal energy).d Hydrogen bonding correction.e Lipophilic energy.f Electrostatic solvation energy.g Pi–pi packing correction.h van der Waals energy.i PD144418 = 3-(4-methylphenyl)-5-(1-propyl-3,6-dihydro-2H-pyridin-5-yl)-1,2-oxazole.j Z1241145220 = 3-[1-(3-phenylpropyl)-1,2,3,6-tetrahydropyridin-4-yl]-1H-pyrrolo[2,3-b]pyridine. | ||||||||||
| 1 | LACT11 | σ1 | −72.692 | −36.141 | 5.125 | −0.110 | −64.419 | 51.599 | −0.516 | −28.230 |
| σ2 | −84.578 | −12.003 | 3.137 | −0.042 | −65.936 | 18.129 | −0.182 | −27.682 | ||
| 2 | LACT21 | σ1 | −70.828 | −42.736 | 15.959 | −0.490 | −64.396 | 51.490 | −0.803 | −29.853 |
| σ2 | −100.593 | −18.207 | 1.251 | −0.751 | −61.938 | 27.402 | −0.546 | −47.804 | ||
| 3 | LACT22 | σ1 | −82.630 | −50.328 | 12.790 | −0.742 | −75.122 | 59.170 | −0.875 | −27.523 |
| σ2 | −93.976 | −8.363 | 2.000 | −0.564 | −63.601 | 22.203 | −0.137 | −45.515 | ||
| 4 | LACT26 | σ1 | −62.239 | 2.854 | 7.838 | −0.012 | −57.765 | 22.889 | −0.996 | −37.048 |
| σ2 | −107.986 | 1.463 | 7.707 | −0.751 | −61.362 | −4.223 | −0.446 | −50.375 | ||
| 5 | LACT29 | σ1 | −60.303 | −1.503 | 16.626 | −0.042 | −70.189 | 34.138 | −0.958 | −38.376 |
| σ2 | −107.352 | −12.577 | 2.049 | −0.886 | −66.412 | 20.676 | −0.822 | −49.380 | ||
| 6 | LACT35 | σ1 | −67.810 | −2.586 | 6.614 | −0.012 | −57.729 | 26.364 | −0.485 | −39.977 |
| σ2 | −95.993 | −15.515 | 5.009 | −0.914 | −61.545 | 23.716 | −0.093 | −46.650 | ||
| 7 | LACT37 | σ1 | −84.106 | −4.010 | 6.139 | −0.011 | −67.887 | 24.824 | −0.371 | −42.789 |
| σ2 | −90.984 | −22.915 | 9.645 | −1.181 | −57.791 | 28.408 | −0.001 | −47.149 | ||
| 8 | LACT41 | σ1 | −65.730 | −0.511 | 5.299 | −0.039 | −54.189 | 26.266 | −1.028 | −41.529 |
| σ2 | −95.398 | −14.991 | 5.238 | −0.901 | −59.999 | 21.977 | −0.093 | −46.629 | ||
| 9 | OXAZ2 | σ1 | −2.382 | 2.861 | 27.555 | −0.037 | −72.857 | 29.786 | −3.059 | 13.370 |
| σ2 | −99.275 | −3.132 | 8.131 | −0.925 | −62.137 | 9.443 | −1.060 | −49.596 | ||
| 10 | OXAZ3 | σ1 | — | — | — | — | — | — | — | — |
| σ2 | −114.210 | −12.314 | 3.431 | −1.004 | −65.374 | 12.458 | −1.244 | −50.162 | ||
| 11 | OXAZ4 | σ1 | — | — | — | — | — | — | — | — |
| σ2 | −111.426 | −12.105 | 2.296 | −0.810 | −63.686 | 17.158 | −1.379 | −52.898 | ||
| 12 | OXAZ5 | σ1 | −2.018 | 4.574 | 32.115 | −0.004 | −81.300 | 32.921 | −2.689 | 12.365 |
| σ2 | −120.283 | −14.860 | 9.246 | −0.038 | −68.995 | 15.282 | −0.186 | −60.732 | ||
| 13 | OXAZ6 | σ1 | — | — | — | — | — | — | — | — |
| σ2 | −97.551 | −5.157 | 9.508 | −0.711 | −57.962 | 11.774 | −0.987 | −54.016 | ||
| 14 | OXAZ7 | σ1 | — | — | — | — | — | — | — | — |
| σ2 | −103.179 | −3.886 | 5.441 | −0.478 | −61.168 | 11.805 | −0.022 | −54.871 | ||
| 15 | OXAZ8 | σ1 | −45.625 | −4.248 | 3.120 | −0.224 | −26.243 | 16.691 | −0.444 | −34.277 |
| σ2 | −103.320 | −10.931 | 10.362 | −0.415 | −63.127 | 15.993 | −0.220 | −54.983 | ||
| 16 | OXAZ13 | σ1 | — | — | — | — | — | — | — | — |
| σ2 | −104.800 | −11.373 | 5.611 | −0.774 | −63.712 | 18.488 | −1.227 | −51.812 | ||
| 17 | iPD144418 | σ1 | −92.772 | −52.669 | 4.382 | −0.784 | −52.904 | 56.883 | −0.219 | −47.460 |
| 18 | jZ1241145220 | σ2 | −92.982 | −23.867 | 4.044 | −1.290 | −51.544 | 29.613 | −0.276 | −49.662 |
The estimated binding free energy of docked poses of molecules with σ1 receptor ranged from −2.018 to −84.106 kcal mol−1, whereas the BFG score for the σ2 receptor ranged from −84.578 to −114.210 kcal mol−1. The study shows that produced chemicals bind to the σ2 receptor at a higher rate than those that bind to the σ1 receptor. The docking complexes for the chemicals OXAZ3, OXAZ4, OXAZ6, OXAZ7, and OXAZ13 were not identified. So, their binding energies were assumed to be 0 kcal mol−1. The compound OXAZ3 with the σ2 receptor has the greatest BFG score of around −114.210 kcal mol−1. The van der Waal energy (GvdW = −50.162 kcal mol−1) and lipophilic energy (Glipo = −65.374 kcal mol−1) terms are the major contributors to OXAZ3 binding activity in the active pocket of the σ2 receptor, while coulombic energy (Gcou = −12.314 kcal mol−1) moderately favours ligand binding, according to this study. Furthermore, the ligand's binding activity inside the active site of the σ2 receptor is deemed unfavourable due to covalent energy and electrostatic solvation energy terms. Nonetheless, when compared to the σ1 receptor, the compounds had lower BFG, van der Waal energy, lipophilic energy, and coulombic energy. The results show that the produced compounds had a higher affinity for the σ2 receptor than the σ1 receptor.
4. Conclusion
In this work, essential components responsible for the binding process between the σ2 receptor and its ligands were identified utilizing molecular docking and MM/GBSA binding energy calculations. Molecular docking study provides a useful prediction of structural features for certain series of compounds belonging to γ-butyrolactone and oxazolidinones to bind at the active sites of σ1 and σ2 receptor. A 3D homology model for the σ2 receptor was built to perform the modeling studies. The Ramchandran plot examination was conducted to assess the correctness of modeled σ2 receptor. The putative binding site on the modeled σ2 receptor was identified using SiteMap analysis. In this present work, a total of 58 compounds have been used. The molecules were docked into both the active site of a homology-modeled σ2 receptor and the crystal structure of the σ1 receptor. The structural features required for ligand binding to σ1 and σ2 receptors were studied in 2D and 3D. With the σ2 receptor, the ligands showed higher BFG, van der Waal energy, lipophilic energy, and coulombic energy. The top-ranked molecules, according to the above study, had much more interactions and binding energy for the σ2 receptor than the σ1 receptor. Thus, the present study provides an understanding of the specific pharmacophoric features and interactions of ligands that are responsible for σ2 selectivity over σ1 receptor. Future research on these molecules might lead to the discovery of novel and potential selective σ2 ligands.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors (DKS, ABS) would like to acknowledge Vignan Pharmacy College, Vadlamudi, Andhra Pradesh, India. RRB would like to thank Deanship of Graduate Studies and Research, Ajman University, UAE for their support in providing partial assistance in article processing charges of this manuscript. DC and BB would like to extend their thanks to Dean's office at Temple School of Pharmacy for their support in the preparation of this manuscript.References
- W. R. Martin, C. E. Eades, J. A. Thompson and R. E. Huppler, The effects of morphine and nalorphine-like drugs in the non-dependent and morphine-dependent chronic spinal dog, J. Pharmacol. Exp. Ther., 1976, 197(3), 517–532 CAS.
- T. P. Su, Evidence for sigma opioid receptor: binding of [3H]SKF-10047 to etorphine inaccessible sites in guinea-pig brain, J. Pharmacol. Exp. Ther., 1982, 223(2), 284–290, DOI:10.1007/s00044-020-02574-9.
- W. D. Bowen, B. R. de Costa, S. B. Hellewell, J. M. Walker and K. C. Rice, [3H]-(+)-Pentazocine: a potent and highly selective benzomorphan-based probe for sigma-1 receptors, Mol. Neuropharmacol., 1993, 3, 117–126 CAS.
- H. R. Schmidt, S. Zheng, E. Guripinar, A. Koehl, A. Manglik and A. C. Kruse, Crystal structure of the human σ1 receptor, Nature, 2016, 532(7600), 527–530, DOI:10.1038/nature17391.
- A. Alon, H. R. Schmidt, M. D. Wood, J. J. Sahn, S. F. Martin and A. C. Krusea, Identification of the gene that codes for the σ2 receptor, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(27), 7160–7165, DOI:10.1073/pnas.1705154114.
- A. Alon, J. Lyu, J. M. Braz, T. A. Tummino, V. Craik, M. J. O'Meara, C. M. Webb, D. S. Radchenko, Y. S. Moroz, X. P. Huang, Y. Liu, B. L. Roth, J. J. Irwin, A. I. Basbaum, B. K. Shoichet and A. C. Kruse, Structures of the σ2 receptor enable docking for bioactive ligand discovery, Nature, 2021, 600(7890), 759–764, DOI:10.1038/s41586-021-04175-x.
- F. Bartz, L. Kern, D. Erz, M. Zhu, D. Gilbert, T. Meinhof, U. Wirkner, H. Erfle, M. Muckenthaler, R. Pepperkok and H. Runz, Identification of cholesterol-regulating genes by targeted RNAi screening, Cell Metab., 2009, 10(1), 63–75, DOI:10.1016/j.cmet.2009.05.009.
- L. Guo and X. Zhen, Sigma-2 receptor ligands: neurobiological effects, Curr. Med. Chem., 2015, 22(8), 989–1003, DOI:10.2174/0929867322666150114163607.
- (a) B. Yi, J. J. Sahn, P. M. Ardestani, A. K. Evans, L. L. Scott, J. Z. Chan, S. Iyer, A. Crisp, G. Zuniga, J. T. Pierce, S. F. Martin and M. Shamloo, Small molecule modulator of sigma 2 receptor is neuroprotective and reduces cognitive deficits and neuroinflammation in experimental models of Alzheimer's disease, J. Neurochem., 2017, 140(4), 561–575, DOI:10.1111/jnc.13917; (b) N. J. Izzo, A. Staniszewski, L. To, M. Fa, A. F. Teich, F. Saeed, H. Wostein, T. Walko, A. Vaswani, M. Wardius, Z. Syed, J. Ravenscroft, K. Mozzoni, C. Silky, C. Rehak, R. Yurko, P. Finn, G. Look, G. Rishton, H. Safferstein, M. Miller, C. Johanson, E. Stopa, M. Windisch, B. Hutter-Paier, M. Shamloo, O. Arancio, H. LeVine and S. M. Catalano, Alzheimer's therapeutics targeting amyloid beta 1−42 oligomers I: Abeta 42 oligomer binding to specific neuronal receptors is displaced by drug candidates that improve cognitive deficits, PLoS One, 2014, 9(11), e111898, DOI:10.1371/journal.pone.0111898; (c) N. J. Izzo, J. Xu, C. Zeng, M. J. Kirk, K. Mozzoni, C. Silky, C. Rehak, R. Yurko, G. Look, G. Rishton, H. Safferstein, C. Cruchaga, A. Goate, M. A. Cahill, O. Arancio, R. H. Mach, R. Craven, E. Head, H. LeVine, T. L. Spires-Jones and S. M. Catalano, Alzheimer's therapeutics targeting amyloid beta 1−42 oligomers II: Sigma-2/PGRMC1 receptors mediate Abeta 42 oligomer binding and synaptotoxicity, PLoS One, 2014, 9(11), e111899, DOI:10.1371/journal.pone.0111899.
- J. J. Sahn, G. L. Mejia, P. R. Ray, S. F. Martin and T. J. Price, Sigma 2 receptor/Tmem97 agonists produce long lasting antineuropathic pain effects in mice, ACS Chem. Neurosci., 2017, 8(8), 1801–1811, DOI:10.1021/acschemneuro.7b00200.
- E. Vazquez-Rosa, M. R. Watson, J. J. Sahn, T. R. Hodges, R. E. Schroeder, C. J. Cintron-Perez, M. K. Shin, T. C. Yin, J. L. Emery, S. F. Martin, D. J. Liebl and A. A. Pieper, Neuroprotective Efficacy of a Sigma 2 Receptor/TMEM97 Modulator (DKR-1677) after Traumatic Brain Injury, ACS Chem. Neurosci., 2019, 10(3), 1595–1602, DOI:10.1021/acschemneuro.8b00543.
- D. Ebrahimi-Fakhar, L. Wahlster, F. Bartz, J. Werenbeck-Ueding, M. Praggastis, J. Zhang, B. Joggerst-Thomalla, S. Theiss, D. Grimm, D. S. Ory and H. Runz, Reduction of TMEM97 increases NPC1 protein levels and restores cholesterol trafficking in Niemann-pick type C1 disease cells, Hum. Mol. Genet., 2016, 25(16), 3588–3599, DOI:10.1093/hmg/ddw204.
- (a) B. J. Vilner, C. S. John and W. D. Bowen, Sigma-1 and Sigma-2 receptors are expressed in a wide variety of human and rodent tumor cell lines, Cancer Res., 1995, 55(2), 408–413 CAS; (b) G. Asong, X. Y. Zhu, B. Bricker, T. Andey, F. Amissah, N. Lamango and S. Y. Ablordeppey, New analogs of SYA013 as sigma-2 ligands with anticancer activity, Bioorg. Med. Chem., 2019, 27(12), 2629–2636, DOI:10.1016/j.bmc.2019.04.012.
- (a) J. J. Sahn, G. L. Mejia, P. R. Ray, S. F. Martin and T. J. Price, Sigma 2 Receptor/Tmem97 Agonists Produce Long Lasting Antineuropathic Pain Effects in Mice, ACS Chem. Neurosci., 2017, 8(8), 1801–1811, DOI:10.1021/acschemneuro.7b00200; (b) W. D. Bowen, C. M. Bertha, B. J. Vilner and K. C. Rice, Antinociceptive CB-64D and CB-184: ligands with high σ2 receptor affinity and subtype selectivity, Eur. J. Pharmacol., 1995, 278(3), 257–260, DOI:10.1016/0014-2999(95)00176-L.
- J. Perregaard, E. K. Moltzen, E. Meier and C. Sánchez, Sigma ligands with subnanomolar affinity and preference for the sigma 2 binding site. 1. 3-(omega-aminoalkyl)-1H-indoles, J. Med. Chem., 1995, 38(11), 1998–2008, DOI:10.1021/jm00011a019.
- E. S. McDonald, R. K. Doot, A. J. Young, E. K. Schubert, J. Tchou, D. A. Pryma, M. D. Farwell, A. Nayak, A. Ziober, M. D. Feldman, A. DeMichele, A. S. Clark, P. D. Shah, H. Lee, S. D. Carlin, R. H. Mach and D. A. Mankoff, Breast Cancer 18F-ISO-1 Uptake as a Marker of Proliferation Status, J. Nucl. Med., 2020, 61(5), 665–670, DOI:10.2967/jnumed.119.232363.
- G. M. Rishton, G. C. Look, Z. J. Ni, J. Zhang, Y. Wang, Y. Huang, X. Wu, N. J. Izzo, K. M. LaBarbera, C. S. Limegrover, C. Rehak, R. Yurko and S. M. Catalano, Discovery of Investigational Drug CT1812, an Antagonist of the Sigma-2 Receptor Complex for Alzheimer's Disease, ACS Med. Chem. Lett., 2021, 12(9), 1389–1395, DOI:10.1021/acsmedchemlett.1c00048.
- B. E. Blass, R. Gao, K. M. Blattner, J. C. Gordon, D. A. Pippin and D. J. Canney, Synthesis and evaluation of novel, selective, functionalized γ-butyrolactones as sigma-2 ligands, Med. Chem. Res., 2022, 31(2), 337–349, DOI:10.1007/s00044-021-02831-5.
- B. E. Blass, R. R. Bhandare and D. J. Canney, Discovery of oxazolidinone-based heterocycles as subtype selective sigma-2 ligands, Med. Chem. Res., 2022, 31(3), 416–425, DOI:10.1007/s00044-022-02848-4.
- B. E. Blass, R. Gao, K. M. Blattner, J. C. Gordon, D. A. Pippin and D. J. Canney, Design, synthesis, and evaluation of novel, selective γ-butyrolactones sigma-2 ligands, Med. Chem. Res., 2021, 30(9), 1713–1727, DOI:10.1007/s00044-021-02771-0.
- Schrödinger Release 2019-1, Maestro, Schrödinger, LLC, New York, NY, 2019 Search PubMed.
- https://www.uniprot.org/uniprot/Q5BJF2.
- A. Alon, J. Lyu, J. M. Braz, T. A. Tummino, V. Craik, M. J. O'Meara, C. M. Webb, D. S. Radchenko, Y. S. Moroz, X. P. Huang, Y. Liu, B. L. Roth, J. J. Irwin, A. I. Basbaum, B. K. Shoichet and A. C. Kruse, Structures of the sigma 2 receptor enable docking for bioactive ligand discovery, Nature, 2021, 600, 759–764, DOI:10.1038/s41586-021-04175-x.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra03497b |
| This journal is © The Royal Society of Chemistry 2022 |