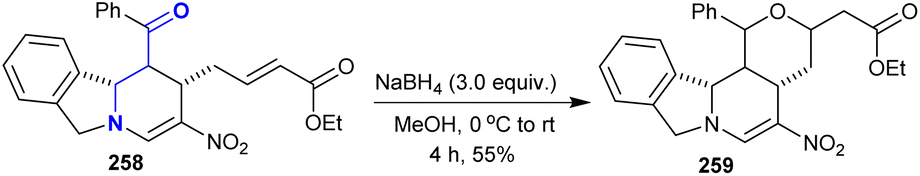DOI:
10.1039/D2RA03864A
(Review Article)
RSC Adv., 2022,
12, 24681-24712
Recent progress in the chemistry of β-aminoketones
Received
23rd June 2022
, Accepted 17th August 2022
First published on 31st August 2022
Abstract
The β-aminoketone moiety has been found to be the basic skeleton of several drugs such as the amine salts “tolperisone (vasodilation)” and “oxyfedrine (therapeutic coronary disease)”, and fluoroaryl derivatives such as “sitagliptin (antidiabetic)”. The objective of this review is to summarize and highlight the chemistry of compounds reported with a β-aminoketone core in the last five years regarding their synthetic strategies, chemical reactivity, and mechanistic synthetic pathways. In the different sections, we categorize the synthesis of β-aminoketones by Mannich reactions via catalytic, non-catalytic, and one-pot procedures. Also, the synthesis of the investigated compounds is accomplished by condensation reactions, from propargylic alcohols, reductive hydroamination, alkylation, carbonylative coupling, and acid hydrolysis of metal complexes. The aim of this review is to provide details for the synthesis of piperidines, morpholinones, piperazinones, dihydroxy-2-oxopyrroles, spirocyclic systems, imidazolines, indolizines, pyrido-isoindoles, aminoalcohols, metal complexes, fluoxetine, sotolon, (S)-ketamine, indolines, and benzoazepinones.
 Mohamed M. Hammouda | Dr. Mohamed M. Hammouda was born in Mansoura, Egypt, 1983. He received his BSc in 2004 from the Faculty of Science, Mansoura University, Egypt, and his MSc in 2008 from the Faculty of Science, Mansoura University, Mansoura, Egypt. He obtained his PhD in Organic Chemistry in 2013 from the Faculty of Science, Mansoura University, Egypt (PhD thesis Synthesis and Reactions of some New Isatin Mannich Bases and Related Compounds of Expected Biological Activity). In 2013 he joined the Erasmus Mundus Postdoctoral Fellowship, Laboratory of Organic and Bio-Organic Synthesis, Gent University, Belgium and conducted post-doctoral research on the development of new types of chiral catalysts for a wide variety of enantioselective reactions. In 2017 he joined the Chemistry Department, Faculty of Science, Mansoura University as a Lecturer of Organic Chemistry, “Synthesis of nitrogen-containing compounds for antioxidant activity”. In 2021 he joined the Department of Chemistry, College of Science and Humanities in Al-Kharj, Prince Sattam Bin Abdulaziz University, Saudi Arabia as an Assistant Professor of Organic Chemistry. |
 Khaled M. Elattar | Dr. Khaled M. Elattar was born in Menyet Sammanoud, Aga, El-dakahlia, Egypt (1979). He received his BSc in 2002 from the Faculty of Science, Mansoura University, Egypt, and MSc in 2006 from the Faculty of Science, Benha University, Benha, Egypt. He obtained his PhD in Organic Chemistry in 2011 from the Faculty of Science, Mansoura University, Egypt (PhD supervisors: Prof. A. A. Fadda and Prof. A. S. Fouda). He was a Lecturer in Organic Chemistry, at the Faculty of Education of the Sert University, Sert, Libya from 2012 to 2015. He is a member of the Egyptian Chemical Society. He is a reviewer for some scientific international journals. His main research interests are in the field of organic synthesis, the synthesis of heterocyclic compounds of the pharmaceutical interest, and medicinal chemistry. He joined the editorial board of OA Journal – Pharmaceutics (2018). https://oa.enpress-publisher.com/index.php/Pha/about/editorialTeam. Currently, he is on the Editorial Board of the following journals: Journal of Applied Science, Asian Journal of Textile, Asian Journal of Applied Science, International Journal of Chemical Technology, and Current Research in Chemistry. Recently, he joined the Asian Council of Science Editors (ACSE). |
1. Introduction
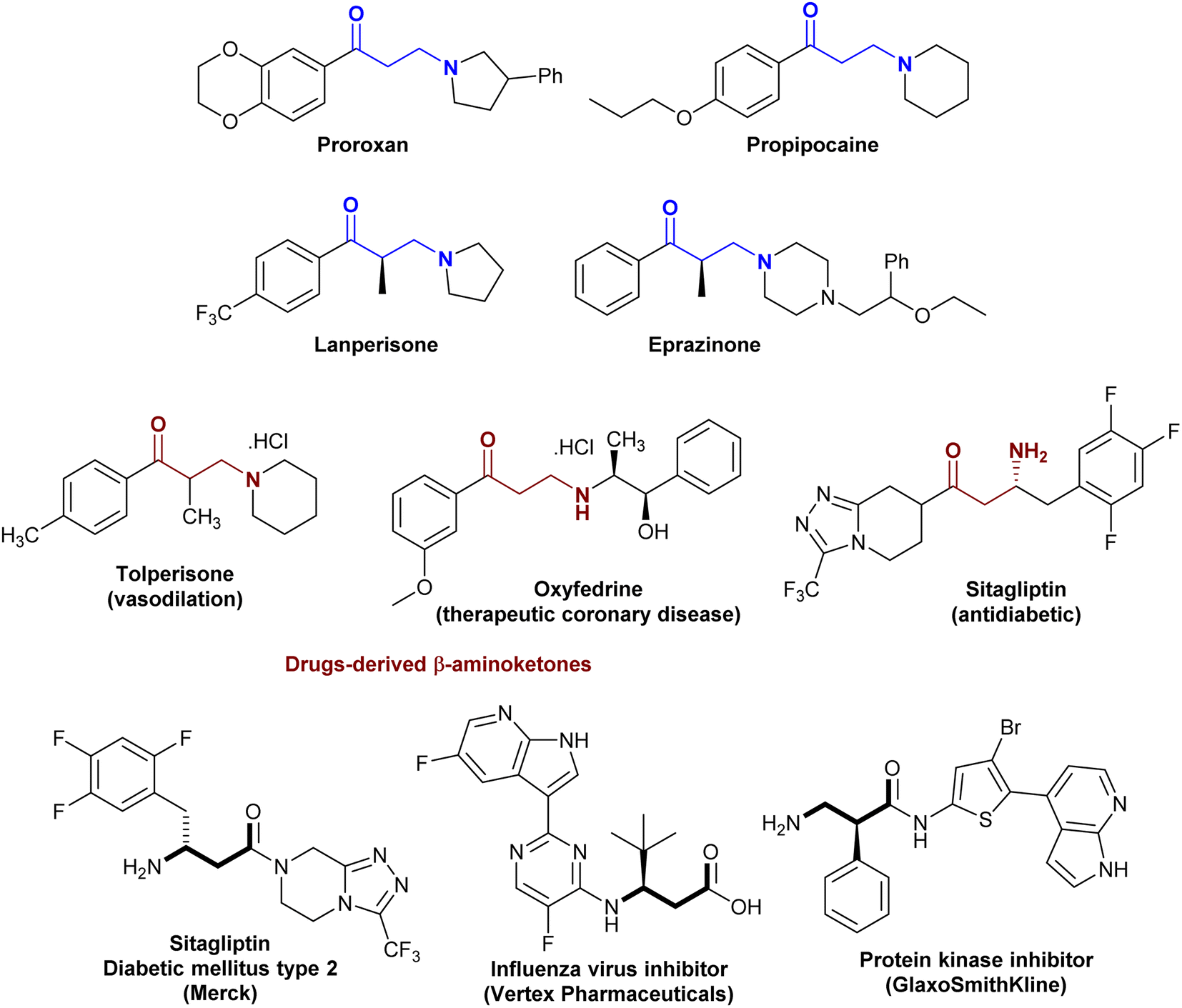
The class of β-aminoketones plays an important role in heterocyclic synthesis as one of the most vital skeletons employed in the preparation of various synthetic molecules and are extensively present in natural products, drugs, and bioactive molecules.1–4 As demonstrated in Fig. 1, proroxan acts as a nonselective α-adreno-blocker with peripheral and dominant action to delight and inhibit hypertonic predicaments and other types of sympathetic adrenal pathologies.5 Also, they are prevalent in native anesthetics such as propipocaine in pharmaceutical synthesis and medical treatment.6,7 In addition, the incorporation of β-aminoketones in drugs such as amine salts “tolperisone (vasodilation)” and “oxyfedrine (therapeutic coronary disease)”, and fluoroalkyl derivatives such as “sitagliptin (antidiabetic)”. Drugs derived from the β-aminoketone moiety are known to exhibit remarkable biological potency.8–10 Accordingly, many methods have been applied for the synthesis of β-aminoketones by amination at the β-position of fluoroalkyl.11,12 Furthermore, β-amino acids are a sub-class of β-aminoketones that have attracted considerable attention for the synthesis of bioactive peptides.13–16 The integral compounds with a β-amino acid skeleton display pharmaceutical active characters such as sitagliptin for the treatment of “diabetes mellitus type 2”, as influenza virus inhibitors and protein kinase inhibitors (Fig. 1).2–4,17
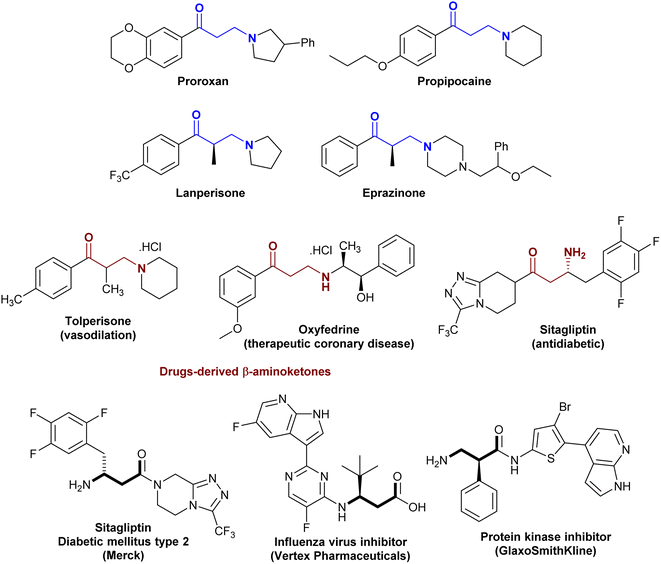 |
| | Fig. 1 Drug molecules consisting of β-aminoketone fragments. | |
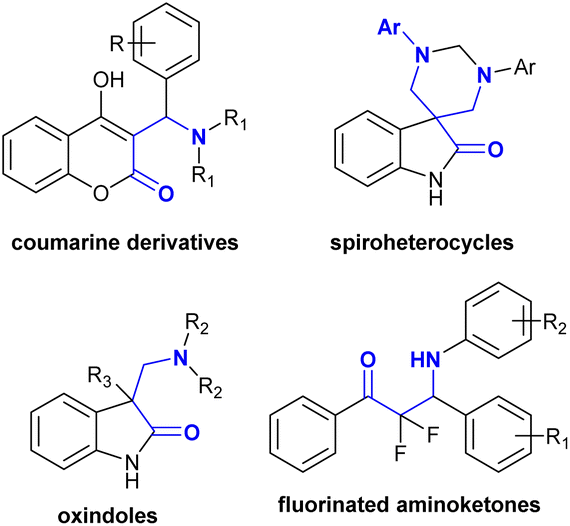
Many recent studies have focused on the development of different ways to prepare β-aminoketones with new technologies to obtain products with higher yields, improved reaction rates, and lower cost. Consequently, these strategies have been adopted for the synthesis of compounds with unique β-aminoketone skeletons. Compounds with β-aminoketone moieties were synthesized through the Mannich reaction18–20 from accessible enolates and imines with the formation of a C–C bond. Nevertheless, this reaction involved the use of a base with equimolar amounts for the synthesis of enolates, and frequently employed harsh reaction conditions with long reaction times.21 In another route, β-aminoketones were prepared via an aza-Michael reaction involving the addition of amines to α,β-unsaturated ketones,22–24 given that these reactions have a greater economic advantage than Mannich-type reactions. However, Michael reactions commonly require basic conditions,25,26 the utility of either stoichiometric or catalytic amounts of Lewis acids (e.g., Bi(NO3)3,27 CeCl3·7H2O,28 ad SmI2(THF)2 ref. 29), and frequently (toxic) organic solvents, and thus subsequent purification steps are necessary. Another methodology identified from previous research involved the utility of micellar solutions.30 Also, β-aminoketones can be prepared via the reduction of enaminones.31,32 The alternative synthetic routes also involved the direct reductive hydroamination of carbonyl alkynes through the chemo- and regioselective synthesis of enamines with the hydroamination of terminal alkynes under catalytic conditions.33–38 β-Aminoketones were efficiently prepared under mild, green, and catalyzed conditions.39–41 Specifically, Mannich reactions were further applied in the synthesis of a variety of adaptable compounds incorporating a β-aminoketone motif in water following green approaches, comprising coumarins,42 Betti bases,43 spiro heterocycles,44 oxindoles,45 and fluorinated aminoketones46 (Fig. 2).
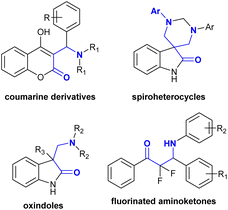 |
| | Fig. 2 Variety of versatile compounds with a β-aminoketone core. | |
This study is in continuation of our preceding plans and reports on emphasizing the chemistry of heterocycles with active moieties in the synthesis of heterocyclic compounds with varying biological activities and their impact in the fields of pharmaceutical and medicinal chemistry.47–61 The literature survey deals with the scope of synthetic approaches applied for the synthesis of β-aminoketones and ascertaining their reactivity against reactions with diverse reagents.
2. Literature patents
The literature reports indicate that many patents focused on the synthesis and reactivity of β-aminoketones, for instance, asymmetric hydrogenation,62 synthesis of amides,63 and chelation of metals.64 The synthesis of β-aminoketones has attracted attention from researchers in the field of organic synthesis, in which many reports highlighted the synthesis of β-amino alcohols,65 N-mono-substituted amino alcohols,66 β-aminoketones, and β-amino alcohols with optically active characters,67–70 and catalytic synthesis of β-aminoketones.71,72 Subsequently, the work on this moiety was extended to include the assessment of the biological features of β-aminoketones as anti-diabetic agents.73
3. Synthetic approaches
3.1. Multicomponent synthesis
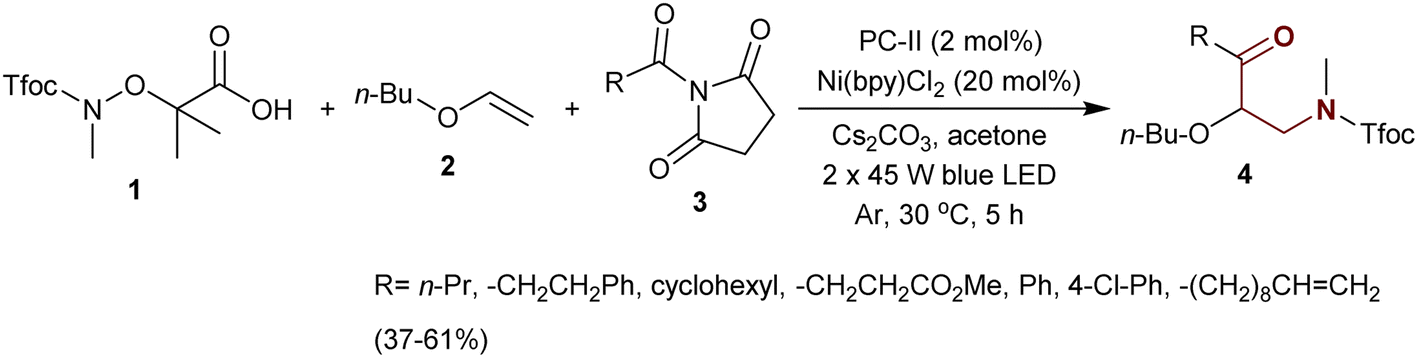
Jiang et al.74 designated three-component-type coupling reactions to synthesize a series of β-aminoketones 4. Specifically, the reaction of N-protected acid 1 with vinyl ether 2 and acyl succinimides 3 in acetone containing cesium carbonate under catalytic conditions gave the desired β-aminoketones 4 in moderate to low yields (37–61%) (Scheme 1). In these reactions, succinimides 3 act as electrophiles with N–C bond cleavage, and compound 1 acts as a radical source after N–O bond cleavage. The reactions were deliberated as the aminoacylation of the alkene-ether type as a route for the synthesis of β-aminoketones.
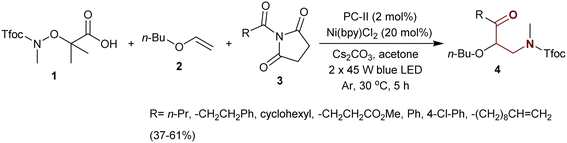 |
| | Scheme 1 Aminoacylation of 1-(vinyloxy)butane. | |
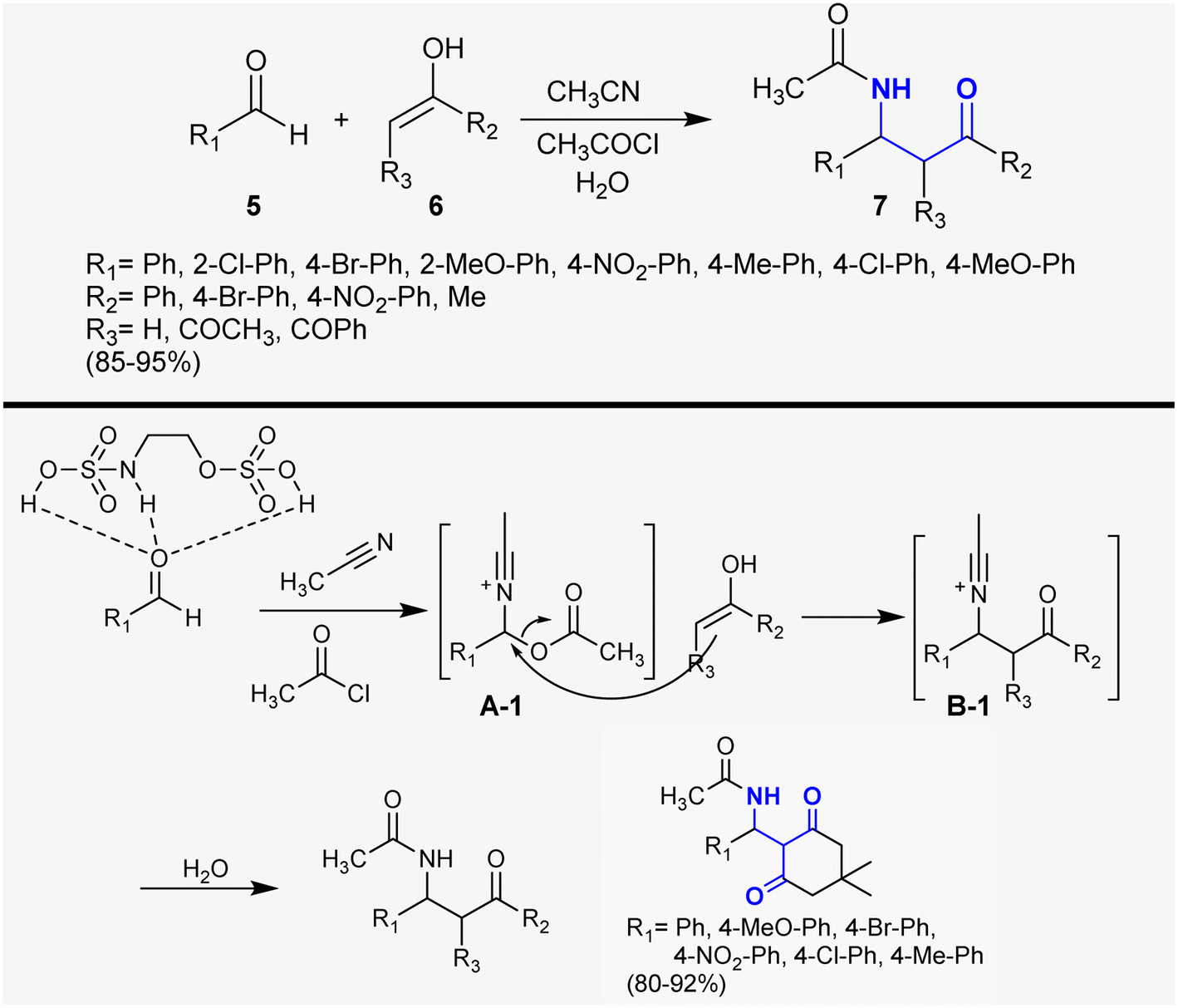
Hadizadeh et al.75 developed the synthesis of β-aminoketones 7 under 2-(sulfamoyloxy)ethyl hydrogen sulfate catalytic conditions. Therefore, the reactions of aldehydes 5 with ketones 6 in the enol form and acetonitrile in the presence of acetyl chloride afforded a series of β-aminoketones 7 in 85–95% yield (Scheme 2). This protocol is proficient to prepare these compounds without the formation of by-products and the yield of the products depends, in general, on the type of substituents linked to the aldehyde moiety using a recyclable catalyst. The catalyst activated the aldehyde carbonyl group for interaction with acetonitrile and acetyl chloride to generate intermediates A-1. Next, the interactions of the generated intermediates A-1 with the corresponding enolized ketones generated intermediates B-1, as described in the proposed reaction mechanism sequence.
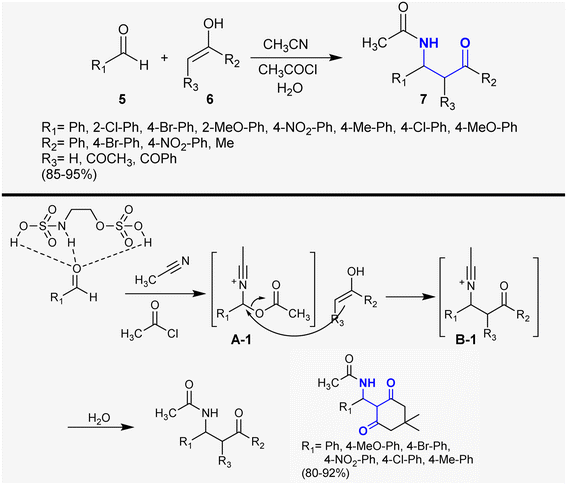 |
| | Scheme 2 Synthesis of β-aminoketones through multicomponent reactions. | |
3.2. Catalyzed Mannich reactions
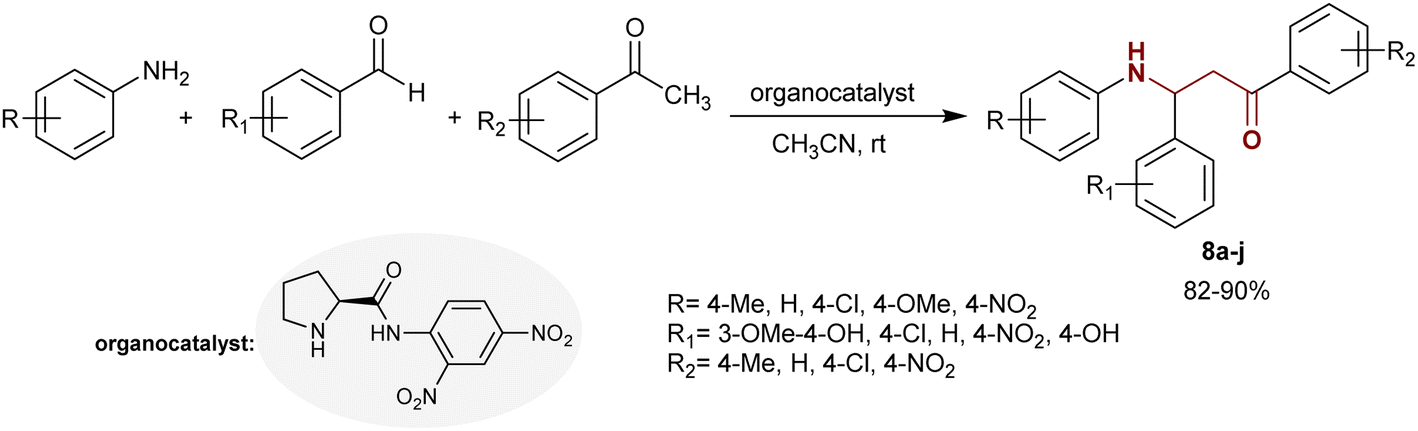
The Mannich reaction was adopted to prepare the asymmetric β-aminoketone series 8a–j in the presence of an organocatalyst, e.g., aryl pyrrolidine-carboxamide. Predominantly, the Mannich reaction of arylamines with aryl aldehydes and acetophenones in acetonitrile containing a catalyst gave the desired asymmetric β-aminoketone series 8a–j. The products obtained by this method possessed excellent properties (82–90%) depending on the nature of their substituents, which was preferable to have electron-withdrawing characters to achieve enantioselective β-aminoketones with the best yields under mild reaction conditions (Scheme 3).76
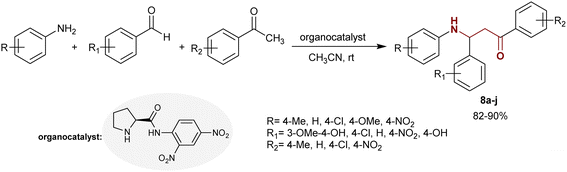 |
| | Scheme 3 Synthesis of β-aminoketones through Mannich reaction. | |
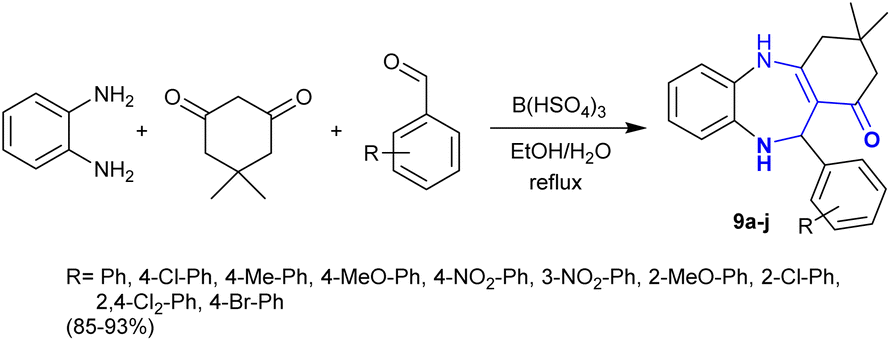
Heterocycles with a benzodiazepine skeleton are a privileged class in the fields of medicinal and applied chemistry. Benzodiazepines are safe and effective drugs for the treatment of anxiety, insomnia, agitation, seizures, muscle spasms, etc.77–80 Thus, Karimi-Jaberi et al.81 developed the synthesis of hexahydro-1H-dibenzo1,4 diazepinones 9a–j through one-pot multicomponent reactions utilizing a green approach. In this sequence, o-phenylenediamine reacted with dimedone and aryl aldehydes in a one-pot three-component procedure under reflux and catalytic conditions to afford the desired benzodiazepines 9 (Scheme 4). The use of boron hydrogen sulfate as an efficient catalyst in an ethanol/water mixture improved the yields and the rate of the reactions.
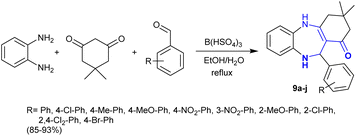 |
| | Scheme 4 Synthesis of benzo1,4 diazepinones. | |
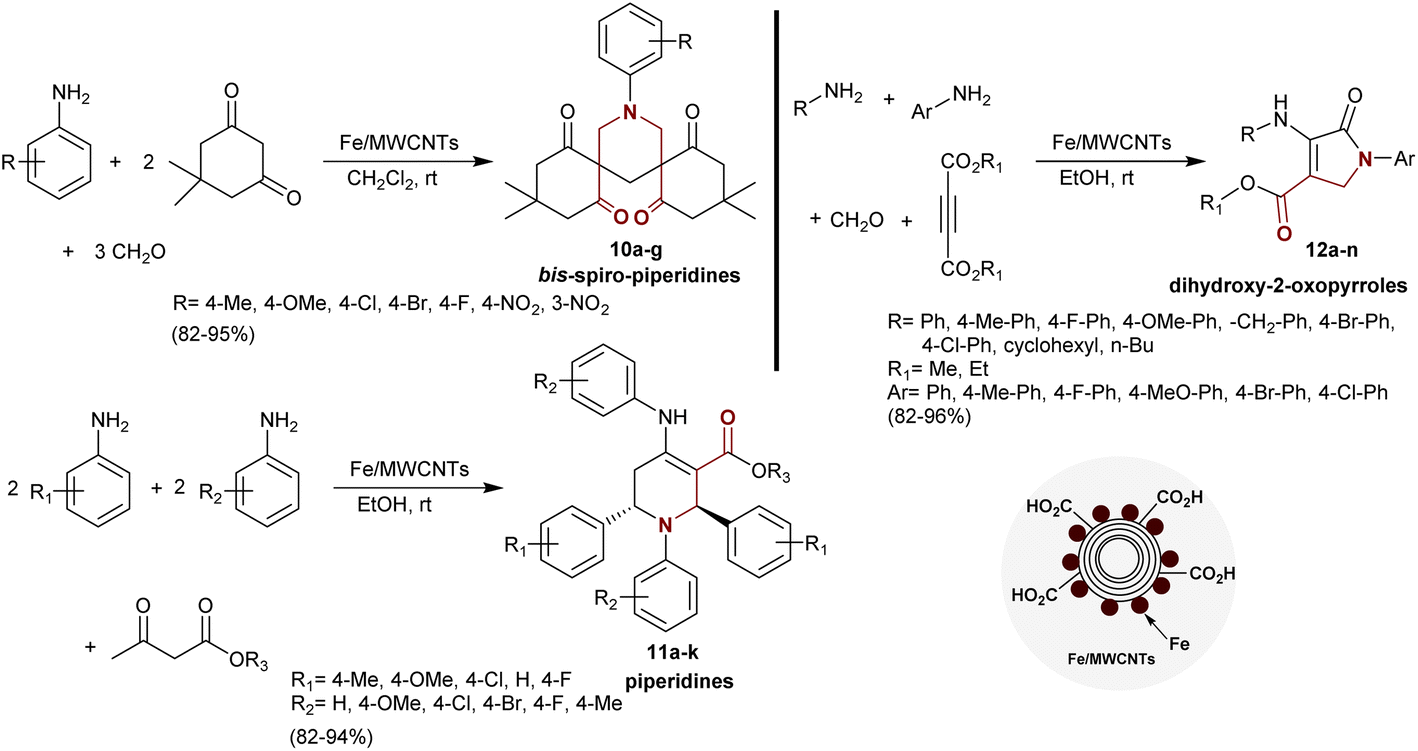
Sharghi et al.82 developed a method for the synthesis of bis-spiro-piperidines 10a–g, piperidines 11a–k, and dihydroxy-2-oxopyrroles 12a–n under nano catalytic conditions. Thus, multicomponent reactions of dimedone with aryl amines, and formaldehyde using Fe/MWCNTs as an effective and recyclable nano-catalyst gave the respective bis-spiro-piperidines 10a–g in excellent yields (82–95%). Also, multicomponent one-pot reactions of aryl amines (2 equiv.) with other aryl amines (2 equiv.) and β-ketoesters afforded the desired piperidines 11a–k. The previous reactions could be also applied using the same aryl amine with four equivalents in the reactions with β-ketoesters. Four-component reactions of alkyl-, aryl amines with formaldehyde, and alkyne diesters under the same reaction conditions gave the corresponding dihydroxy-2-oxopyrroles 12a–n. The yields of the products prepared from this protocol were excellent and the produced compounds with β-aminoketone moieties were simply separated (Scheme 5).
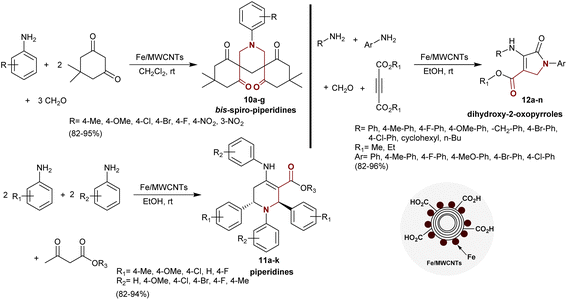 |
| | Scheme 5 Synthesis of β-aminoketones under nano-catalytic conditions. | |
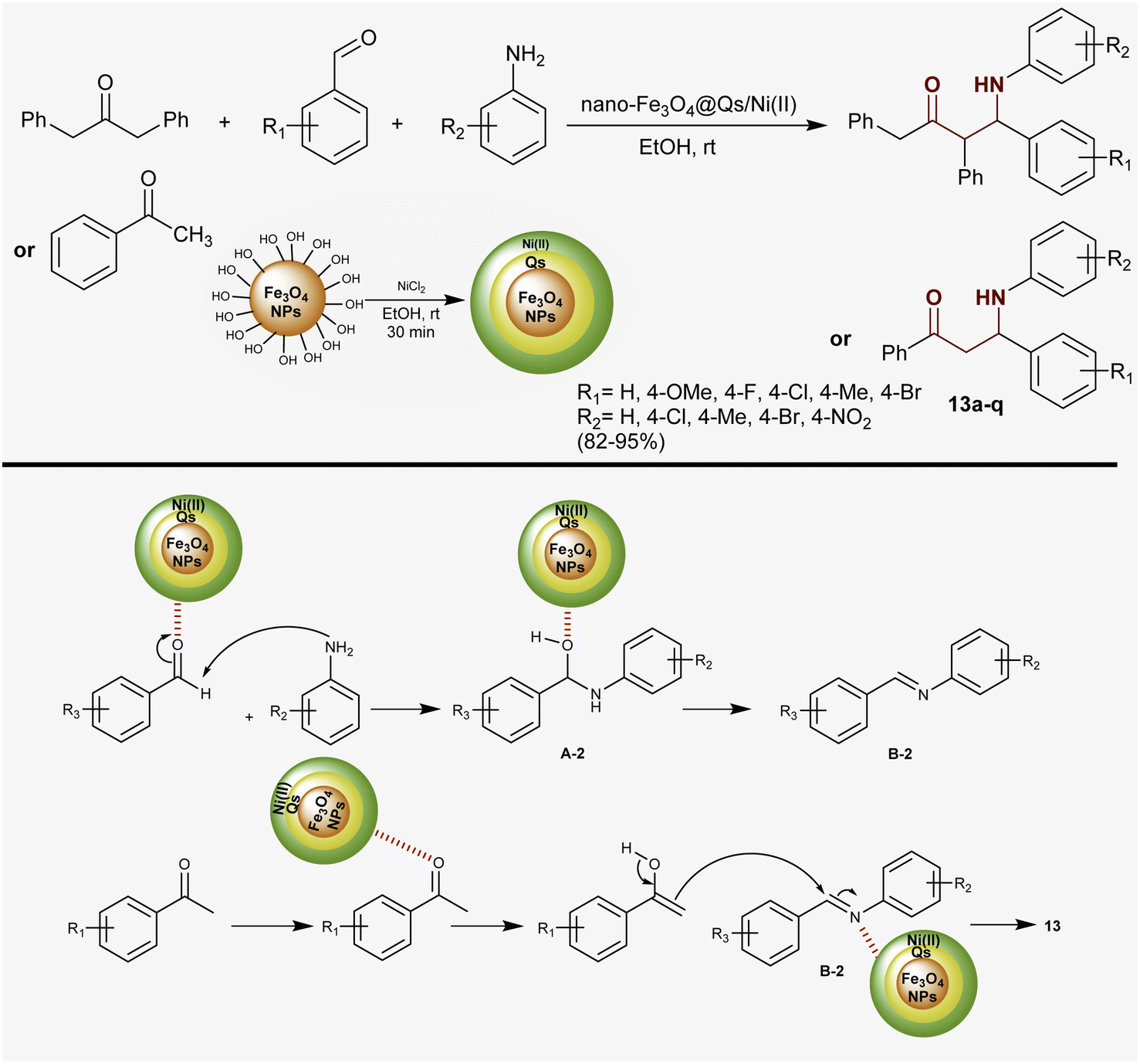
Heidarpour et al.83 developed a route for the synthesis of two series of β-aminoketones in remarkable yields through Mannich reactions under catalytic conditions. Therefore, multicomponent one-pot reactions of aryl aldehydes and amines with aryl/alkyl ketones such as acetophenone and 1,3-diphenylpropan-2-one catalyzed by nanomagnetic Fe3O4@Qs/Ni(II) in ethanol at room temperature yielded the anticipated β-aminoketones 13a–q (82–95%). In this method, the nano-catalyst was prepared and utilized with ease of separation and recyclability with good efficiency. The nano-catalyst activated the nucleophilic attack of amino groups at the aldehydic carbonyl group of aryl aldehyde to generate intermediates A-2. Condensation of A-2 formed intermediates B-2, which reacted with ketones after enolization by the action of the nano-catalyst to give the target products (Scheme 6).
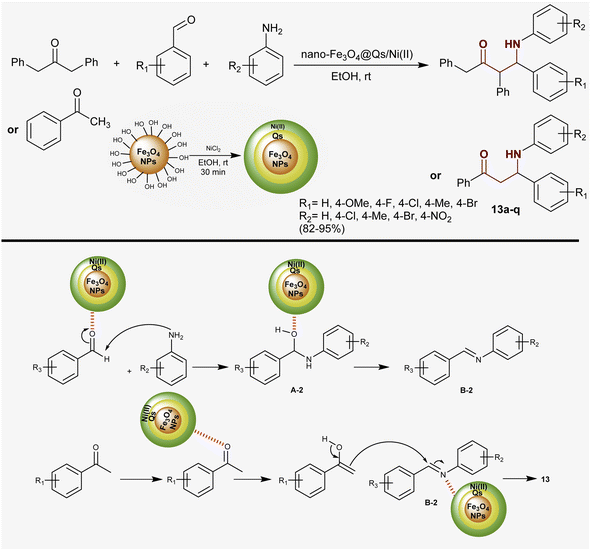 |
| | Scheme 6 Synthetic route and proposed mechanism for the formation of β-aminoketones. | |
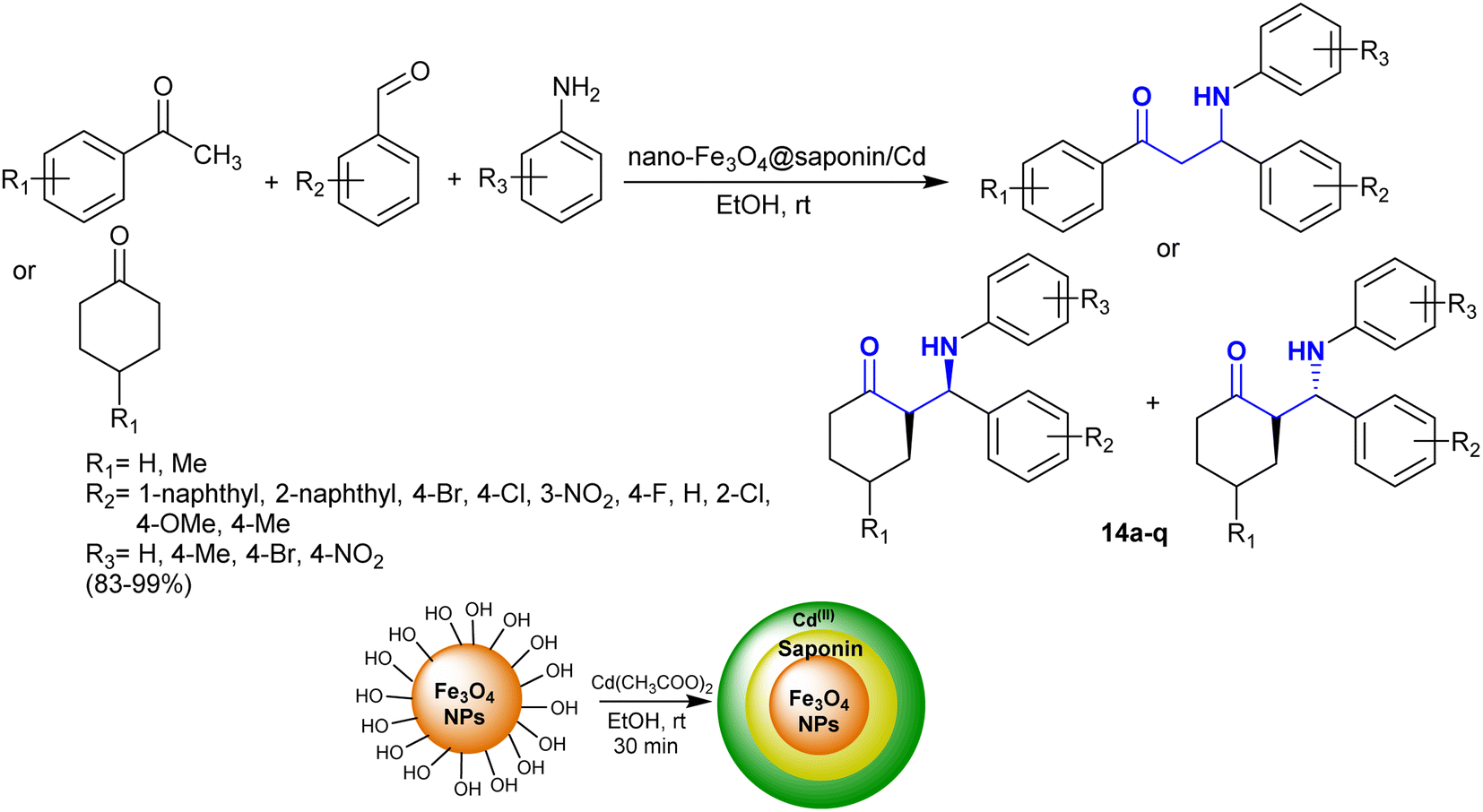
Kiani et al.84 developed the synthesis of Fe3O4@saponin/Cd as an efficient magnetic nanocatalyst for the synthesis of β-aminoketones with exceptional yields (83–99%) and reduced reaction time. In this route, the nanocatalyst is recyclable and prepared by the treatment of nano-Fe3O4@saponin with cadmium acetate. Thus, one-pot three-component reactions of aryl or cycloalkyl ketones with aryl aldehydes and aryl amines in ethanol at room temperature under catalytic conditions yielded the respective β-aminoketones 14a–q (Scheme 7).85–92 The sequence of these reactions is related to Mannich-type reactions in a green protocol. The nanocatalyst activated the aldehyde ketone to nucleophilic attack by the amine group, and consequently enabled the enolization of the aryl or cycloalkyl ketones in the condensation.
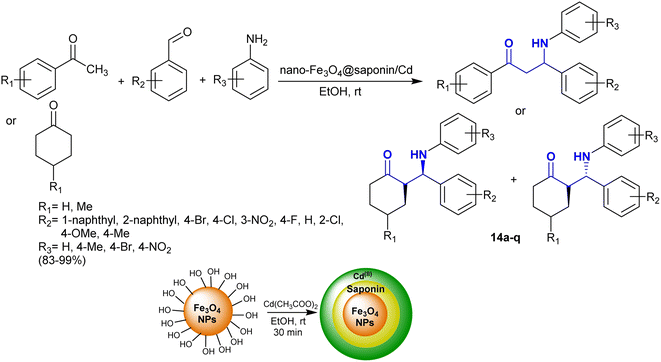 |
| | Scheme 7 Synthesis of β-aminoketones under catalytic conditions. | |
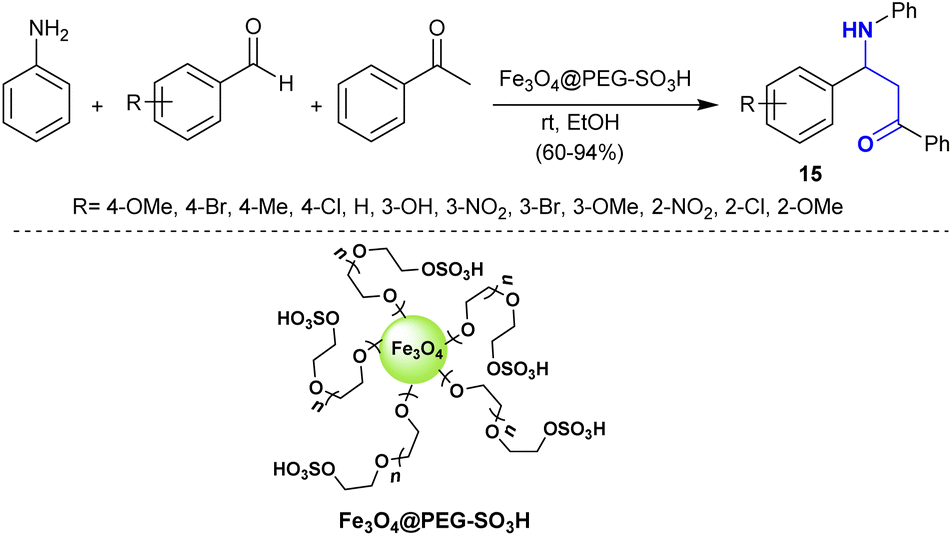
Maleki et al.93 developed the synthesis of β-aminoketones through Mannich reaction under nano catalytic conditions. Consequently, three-component one-pot reactions of aryl amines with aryl aldehydes, and acetophenone catalyzed by Fe3O4@PEG-SO3H yielded the target β-aminoketone compounds 15 (Scheme 8). This method involved the co-precipitation technique and characterization of the nanocatalyst for amended reaction yields and increased rates. This is a green technique given that the reactions were performed in ethanol at room temperature and the nanocatalyst is recyclable with high efficiency and easy preparation.
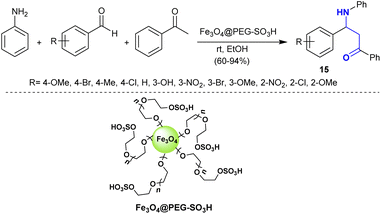 |
| | Scheme 8 Nanocatalytic synthesis of β-aminoketones. | |
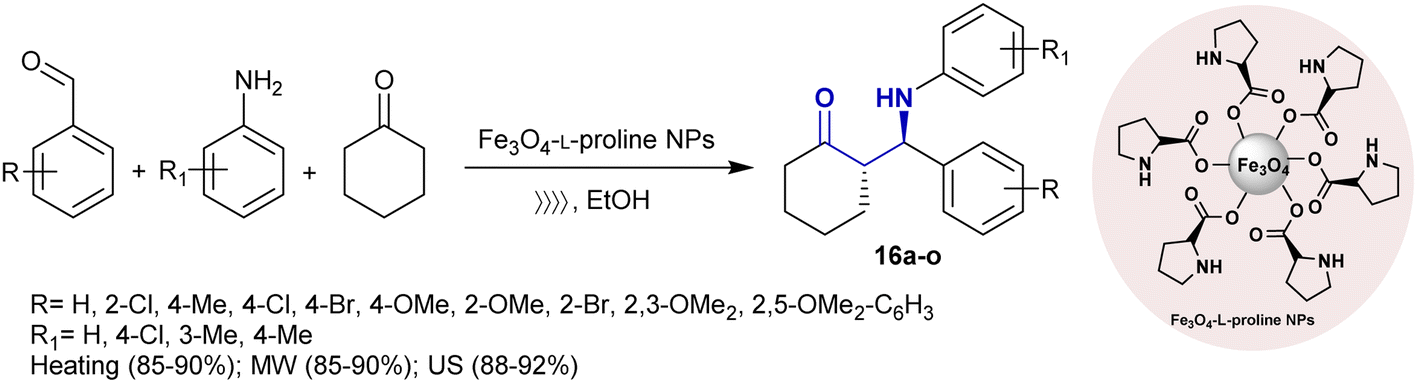
Safaei-Ghomi et al.94 developed the asymmetric synthesis of β-aminoketones 16a–o under magnetic nanoparticles, conventional, ultrasonic, and microwave irradiation conditions. Subsequently, the Fe3O4-L-proline nanoparticles catalyzed the Mannich reactions of aryl aldehydes with aryl amines and cyclohexanone to produce the preferred β-aminoketones with improved reaction yields under ultrasonic conditions than other conventional or microwave irradiation conditions (Scheme 9). The probable mechanism for the asymmetric Mannich reaction was previously reported using the same nanocatalyst under altered reaction conditions.95
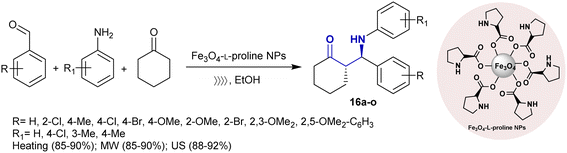 |
| | Scheme 9 Mannich reaction for the catalyzed synthesis of β-aminoketones. | |
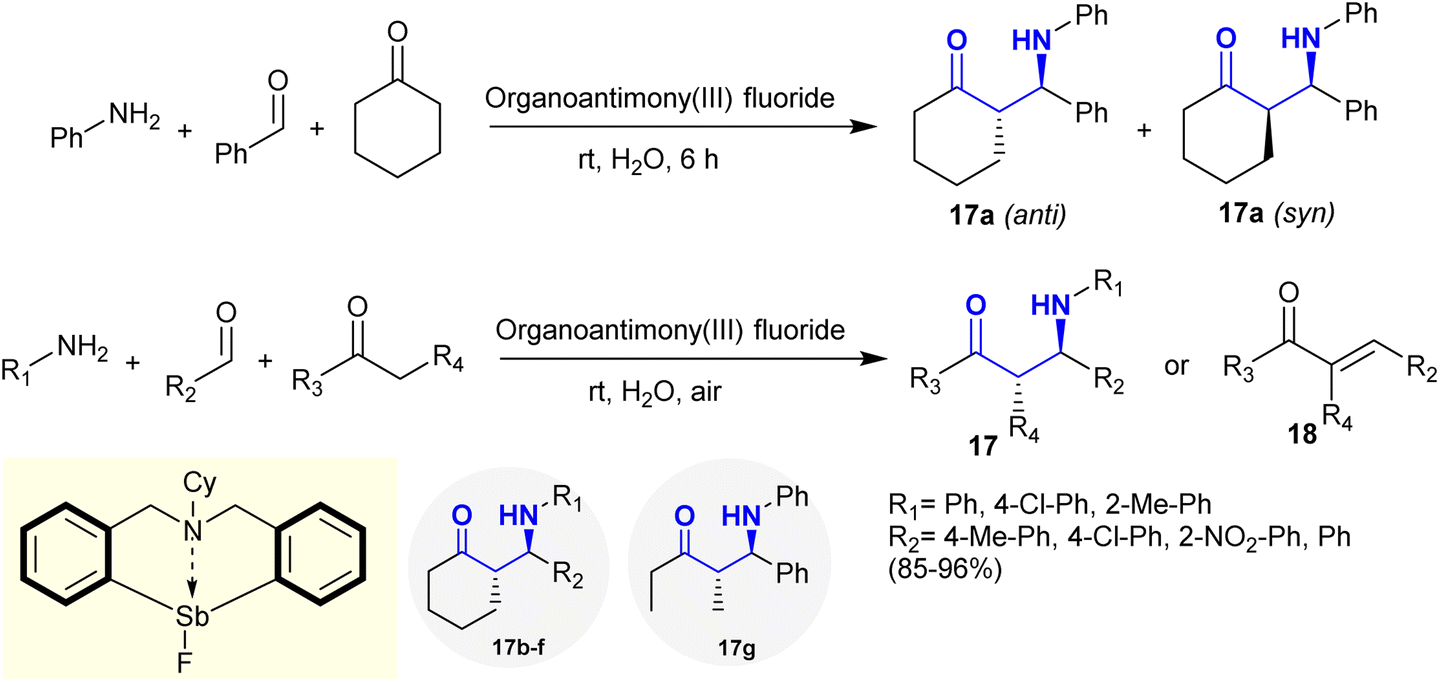
Mannich reactions of active methylene, i.e., cyclohexanone with primary amines, i.e., aniline and aldehydes, in water as an aqueous medium at room temperature yielded the desired β-aminoketones 17. The reaction is stereoselective under catalytic conditions using organoantimony(III) halides as a Lewis acid catalyst tolerant to water with the possible formation of anti and syn products. The best yield in the preparation of compound 17 was accomplished using organoantimony(III) fluoride with 98% yield (anti/syn: 98/2). The Sb–F moiety acts as a hydrogen bond acceptor in this reaction through the proposed catalytic pathway with the generation of a six-membered cyclic transition state. A series of β-aminoketones 17 was synthesized under the optimized conditions by Mannich-type reactions, which involved the reactions of amines with aldehydes and active methylene components. Cross condensation reactions may take place in the case of aliphatic amines with the formation of condensation products 18 in moderate to good yields (Scheme 10).96
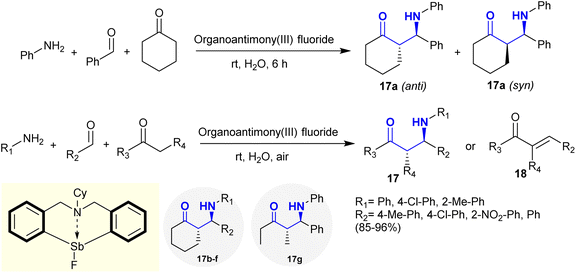 |
| | Scheme 10 Mannich reactions under Lewis acid catalytic conditions. | |
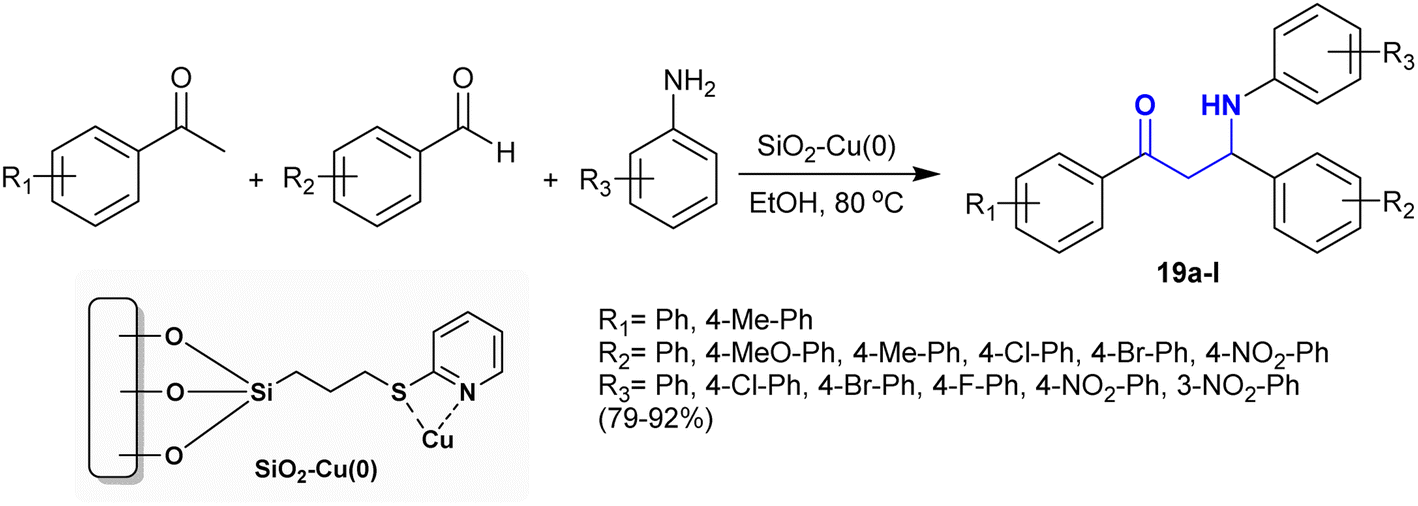
The synthesis of β-aminoketones was investigated by Gupta et al.97 applying a Mannich reaction under catalytic conditions. In this route, the Mannich reactions were catalyzed efficiently by catalytic silica-functionalized copper(0) nanoparticles. This procedure involved reactions of acetophenones with aryl aldehydes and aryl amines under the optimized conditions to afford the desired products of β-aminoketones 19 (Scheme 11). Heating at 80 °C was necessary for improved the reaction rate. The generation of product 19 was accomplished through C–C and C–N bond formation. The catalyst was recyclable for six runs of reusability and proficiently produced the products with excellent yields.
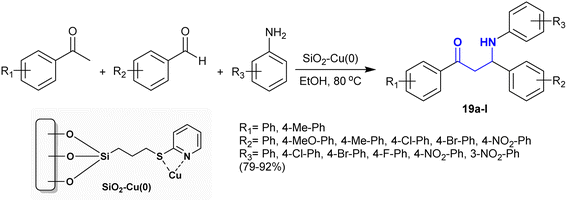 |
| | Scheme 11 Mannich reactions catalyzed by catalytic copper nanocluster. | |
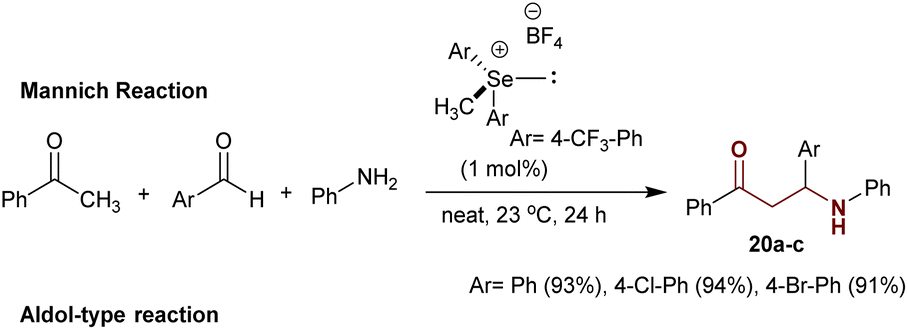
Precisely, the solvent-free reaction of acetophenone with aryl aldehydes and aniline under efficient catalytic conditions of chalcogenide (1 mol%), which was “prepared from diaryl selenium reaction with methyl iodide in the presence of AgBF4”, gave the anticipated β-aminoketones 20a–c in exceptional yields of 91–94%.98 In this case, the multicomponent reactions proceeded through a Mannich-type reaction (Scheme 12).99
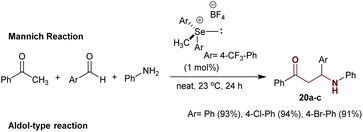 |
| | Scheme 12 Mannich reaction for the synthesis of β-aminoketones. | |
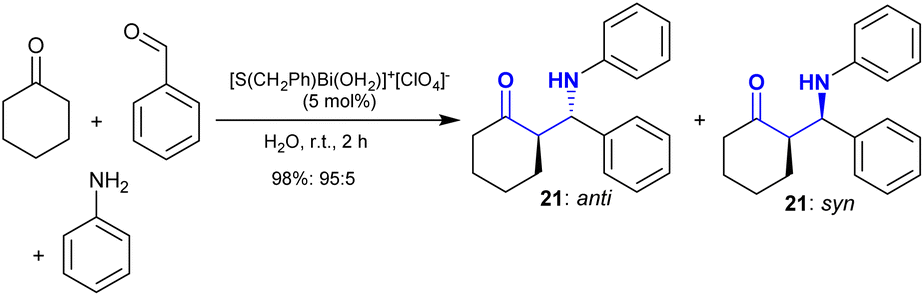
Iwanejko et al.100 reviewed the green approaches for the synthesis of β-aminoketones through the Mannich reaction. Particularly, Qui et al.101 recently developed the synthesis of β-aminoketones 21 through a green protocol under catalytic conditions. Therefore, the three-component one-pot reaction of benzaldehyde with cyclohexanone and aniline in water gave the desired Mannich adducts. The reaction is stereoselective with the formation of anti: syn adducts with (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) in a brilliant yield of 98% (Scheme 13). The Brønsted acid-surfactant enables the dispersion of the organic reactants in water to improve the product yield and reaction rate with high efficiency. The literature reports are rich with the utility of Brønsted acid-surfactants with free ionic liquids as catalysts, e.g., (BASCILs), ([TMBSA]HSO4), and ([DDPA]HSO4), in addition to quaternary ammonium salt surfactants, and BASCILs with Tos− and CH3SO3− organic ions.90,92,102,103
:5) in a brilliant yield of 98% (Scheme 13). The Brønsted acid-surfactant enables the dispersion of the organic reactants in water to improve the product yield and reaction rate with high efficiency. The literature reports are rich with the utility of Brønsted acid-surfactants with free ionic liquids as catalysts, e.g., (BASCILs), ([TMBSA]HSO4), and ([DDPA]HSO4), in addition to quaternary ammonium salt surfactants, and BASCILs with Tos− and CH3SO3− organic ions.90,92,102,103
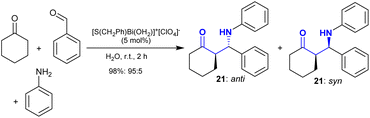 |
| | Scheme 13 Stereoselective Mannich-type reaction. | |
3.3. Non-catalyzed Mannich reactions
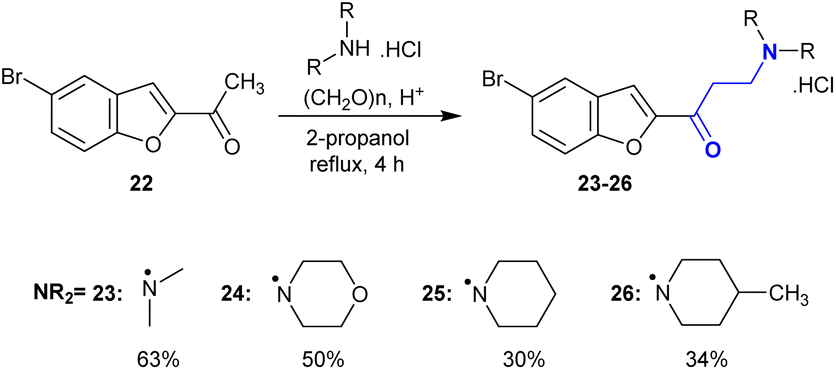
In another route, 1-(5-bromobenzofuran-2-yl)ethan-1-one (22) was subjected to Mannich reactions with hydrochloride salts of secondary amines and formaldehyde to give Mannich bases 23–26 (Scheme 14). The reactions were processed in iso-propanol under reflux conditions. The amine salts were prepared in situ by the addition of a catalytic amount of hydrochloric acid. Mannich base 23 produced a better yield (63%) through the aminomethylation step of compound 22 by reaction with dimethyl amine. This procedure efficiently produced the products after a short time. The products tended to decompose in solutions of water and DMSO with the loss of the amine group, which was the opposite for their solid state.104
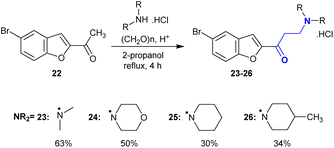 |
| | Scheme 14 Synthesis of Mannich bases. | |
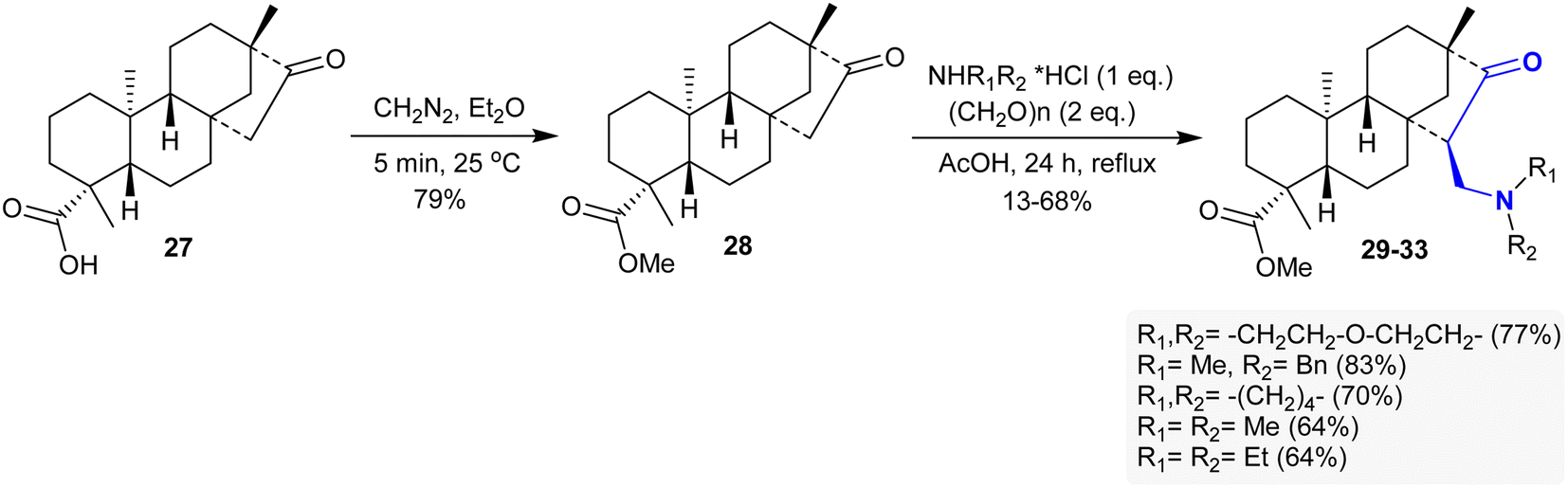
The methyl ester of isosteviol 28 was efficiently synthesized by treatment with diazomethane in diethyl ether, as reported by Coates et al.105 Particularly, the Mannich reaction of compound 28 with diverse secondary amine hydrochloride salts and paraformaldehyde in glacial AcOH yielded a series of β-aminoketones 29–33 in 64–83% yield (Scheme 15) following the method of Szakonyi et al.106 The products were stereo-selectively prepared with the formation of a diastereoisomer that has a structured 7R-configuration at the generated C15 stereo-center. The stereoselectivity in these reactions depended on the nature of the reacted amine salts. The cytotoxic results of β-aminoketones 29–33 demonstrated moderate to weak cytotoxic behaviors on A2780, SiHa, HeLa, MCF-7, and MDA-MB-231 human tumor cell lines (nearly IC50= >20 μM).107
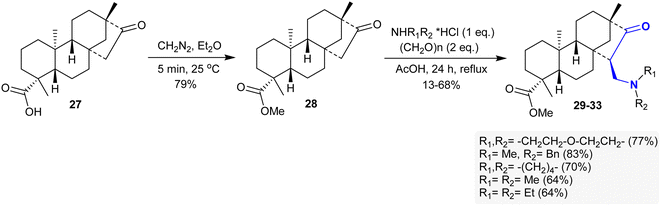 |
| | Scheme 15 Synthesis of β-aminoketones 29–33 via Mannich condensation. | |
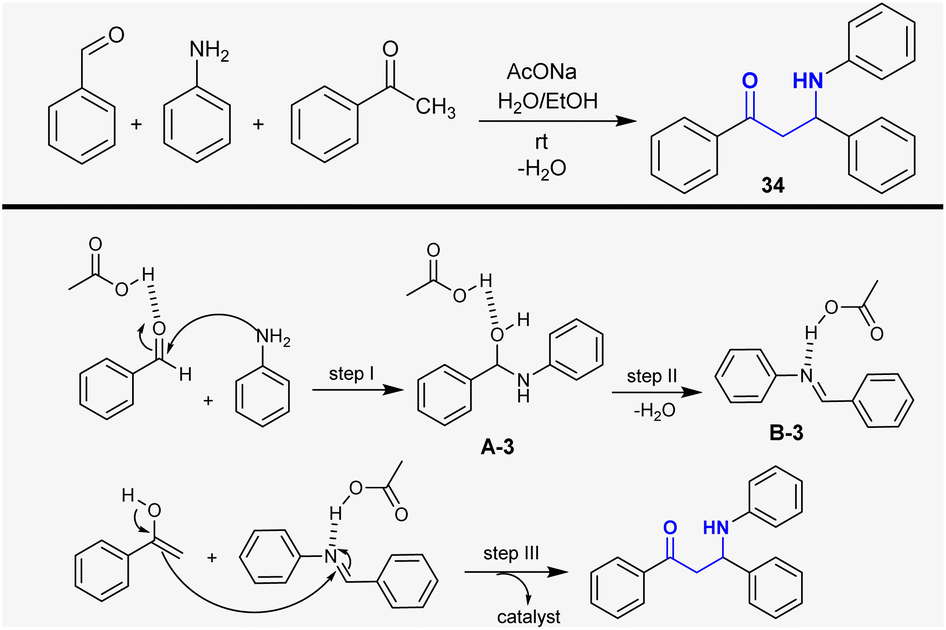
Habibi-Khorassani et al.108 developed the synthesis of β-aminoketone 34 through the Mannich reaction of benzaldehyde with acetophenone and aniline in a mixture of water and ethanol-containing sodium acetate. The kinetic parameters were studied for this reaction under catalytic conditions of sodium acetate. The reaction is second order based on its rate constant. The activation of the carbonyl group of the aldehyde proceeded by the action of the catalyst to initiate the reaction by amino group attack at the carbonyl group in the first step. Next, the condensation step was accomplished to give the Schiff base, which reacted with acetophenone to generate the β-aminoketone (Scheme 16).
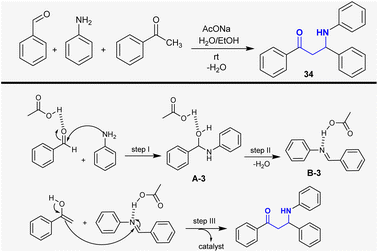 |
| | Scheme 16 Proposed mechanism for the synthesis of β-aminoketone through the Mannich reaction. | |
3.4. One-pot Mannich reaction
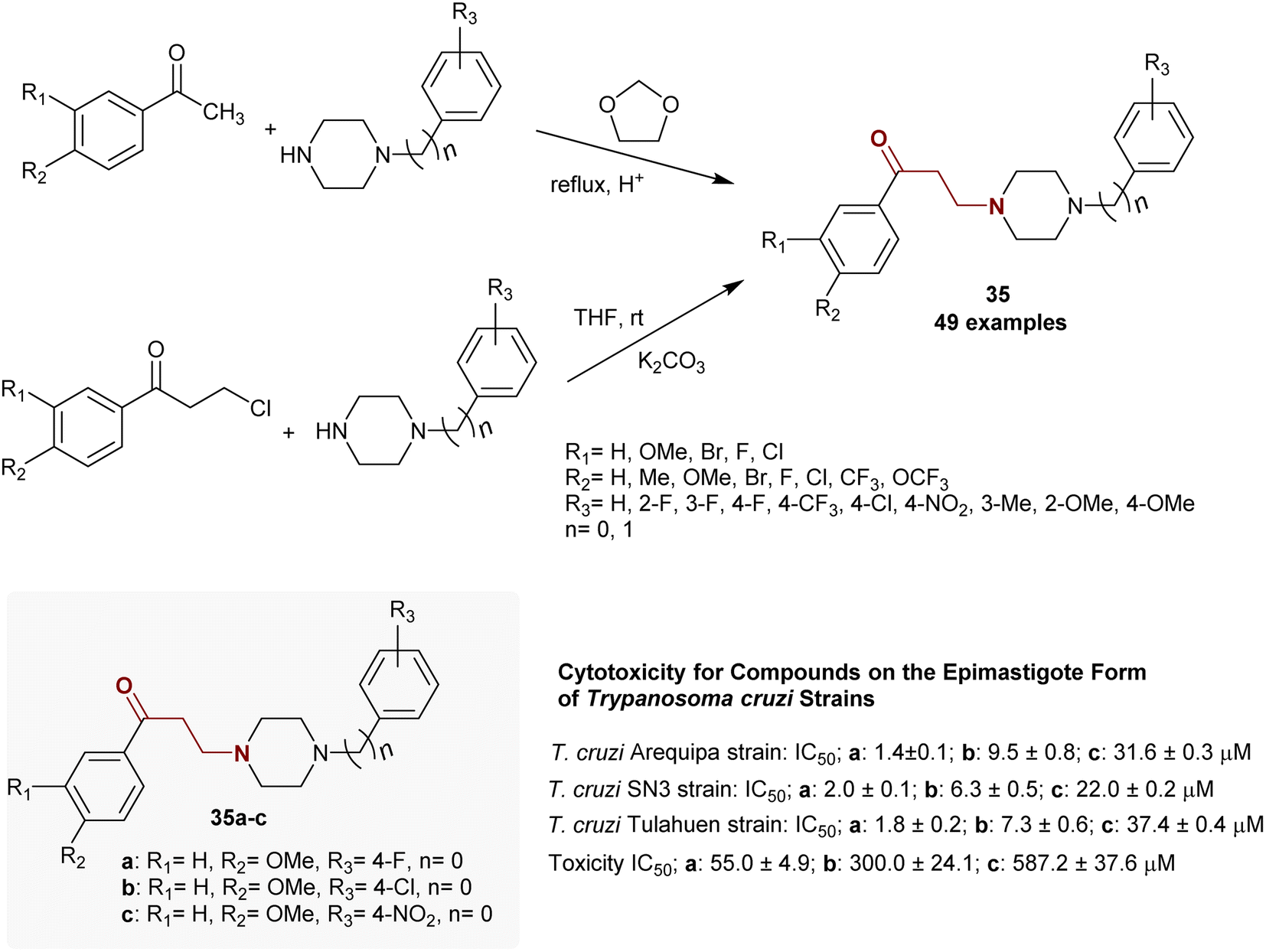
Martin-Escolano et al.109 reported an efficient, inexpensive, and fast technique for the preparation of a series of β-aminoketones. Thus, the synthesis of β-aminoketones 35 was accomplished by Mannich reactions of acetophenones with piperazines in the presence of 1,3-dioxolane as a source of formaldehyde under reflux conditions or by condensation of piperazines with β-halo ketones in THF containing potassium carbonate at room temperature (Scheme 17). The mechanism in the second route proceeded by aliphatic nucleophilic substitution for the investigated β-halo ketones by the piperazines. Most of these β-aminoketones 35 were prepared by Mannich reaction110 or by the method reported by Martinez-Esparza et al.111 to prepare the analogous product of β-aminoketone (R1 = R2 = H, R3 = 4-F, n = 0). Compounds 35a, 35b, and 35c revealed potent cytotoxic activities against epimastigote-type Trypanosoma cruzi strains with selective potency, as shown in Scheme 1.
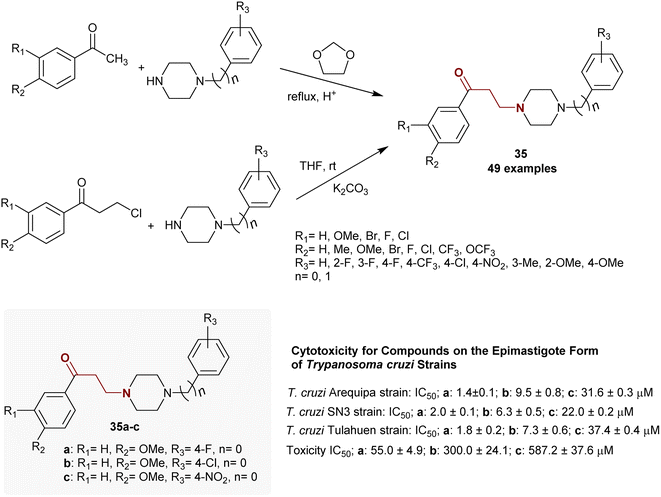 |
| | Scheme 17 Routes for the synthesis of β-aminoketones through one-step reaction. | |
3.5. The addition to imines and unsaturated compounds
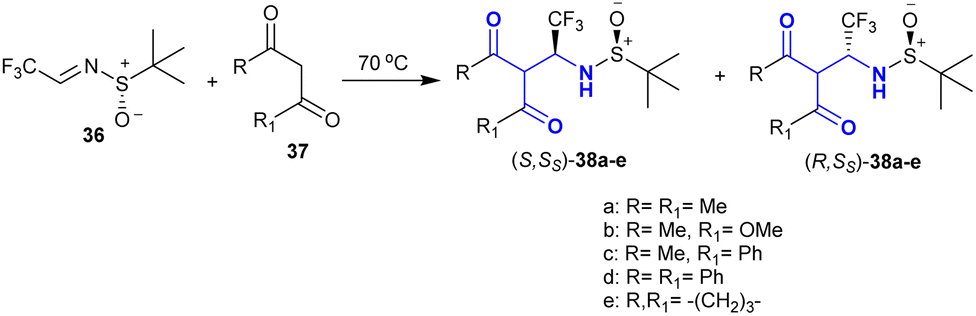
Mazzeo et al.112 utilized the enolizable feature of 1,3-dicarbonyl analogs 37 in an effortless reaction sequence with trifluoroacetaldimines 36 as effective electrophilic types. The different selectivity of the Mannich-type adducts 38a–e prepared by reactions of (S)–N-tert-butane-sulfinyl-trifluoro-acetaldimine with 1,3-dicarbonyl analogs depended on the reaction conditions and the structural nature of the reacted 1,3-dicarbonyl analogs. The reactions progressed under solvent-free conditions with tremendous yields and stereoselectivity. These reactions also have high synthetic potential for the preparation of chiral β-aminoketones. The same reactions may also proceed under DBU basic catalytic conditions in dichloromethane at a lower temperature but the yields of the products may be reduced, and also the stereoselectivity may be opposite to the free-solvent conditions for the same analog. The steric factor of the substituents completely affected the stereochemical of the obtained product (Scheme 18).
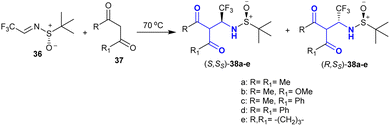 |
| | Scheme 18 Stereoselective synthesis of β-aminoketones. | |
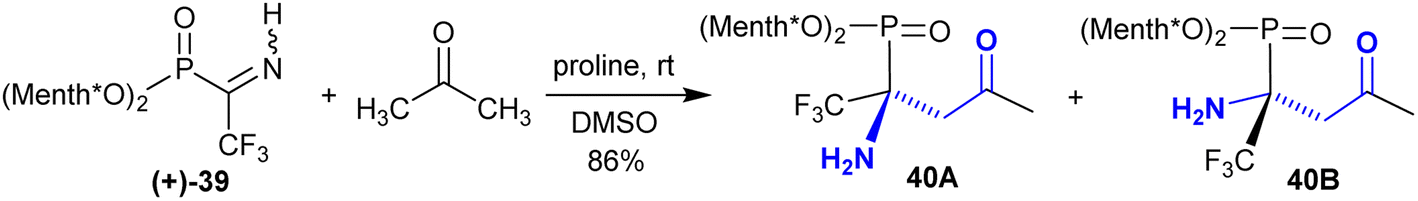
Rassukana et al.113 developed the synthesis of optically active O,O-dimethyl α-iminotrifluoro-ethyl-phosphonates 40. Thus, the reaction of NH-iminophosphonate (+)-39 with acetone under proline catalytic conditions enabled the synthesis of optically active β-aminoketones with a phosphonate moiety and the anticipated phosphonic acids. The reaction is diastereoselective reduction, leading to the preparation of chiral synthons under proline-catalyzed conditions (Scheme 19).
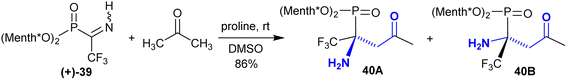 |
| | Scheme 19 Reaction of amino phosphonate (+)-1 with acetone. | |
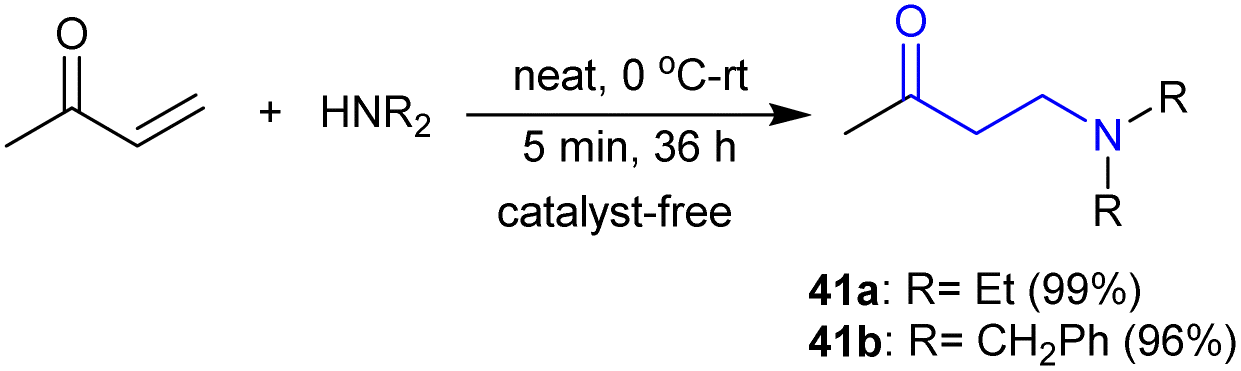
Fischer et al.114 reported an efficient method for the synthesis of β-aminoketones 41a and b in exceptional yields through the addition of secondary amines to α,β-unsaturated carbonyl compounds. This process involved the addition of an alkene double bond with C–N bond formation under catalyst-free conditions (Scheme 20).
 |
| | Scheme 20 Synthesis of β-aminoketones by addition to unsaturated ketones. | |
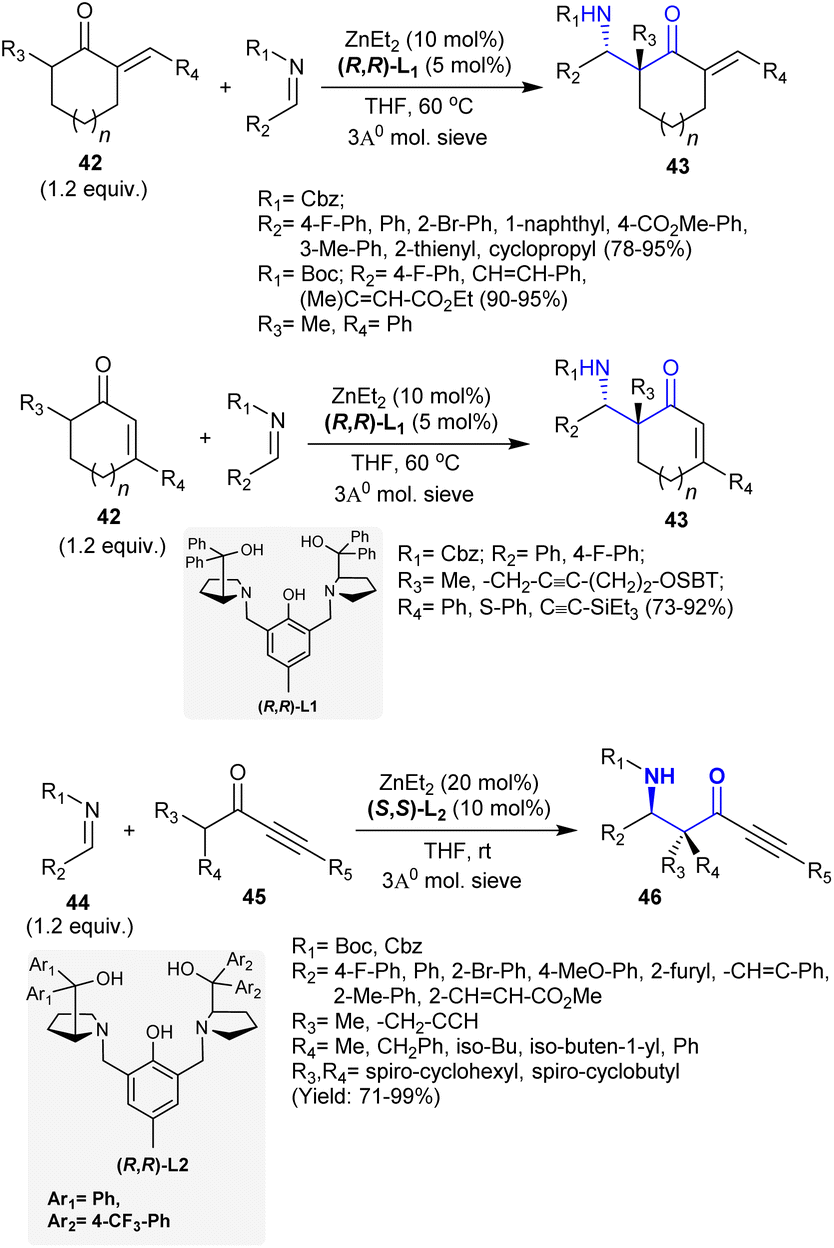
Owing to the biologically privileged impact of the β-aminoketone core, which is located in the basic skeleton of common biologically active alkaloids, Trost et al.115 developed the enantioselective synthesis of β-aminoketones 43 and 46 through Zn-ProPhenol-supported Mannich reactions. Thus, the reactions of N-carbamoyl imines with unsaturated ketones gave either cyclic or acyclic β-aminoketones 43 with quaternary stereocenters (Scheme 21). Significantly, the unsaturation presented through the nucleophile provided a convenient platform for structural variations. This method is a chemoselective and diastereoselective synthetic route for the synthesis of the target products with the aid of sustainable substrates and low catalyst packing, which does not affect its competence. Concisely, Mannich adducts including quaternary stereogenic centers were proficiently synthesized under Zn-ProPhenol catalytic conditions from reactions of exocyclic enones 42 as potential pronucleophiles with imines. Correspondingly, the reactions of imines 44 with alkynyl ketones 45 under catalytic conditions yielded the acyclic β-aminoketones.
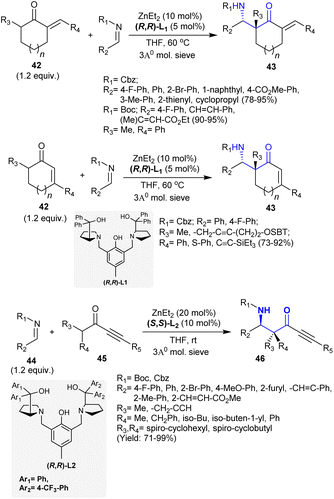 |
| | Scheme 21 Catalytic synthesis of cyclic and acyclic β-aminoketones. | |
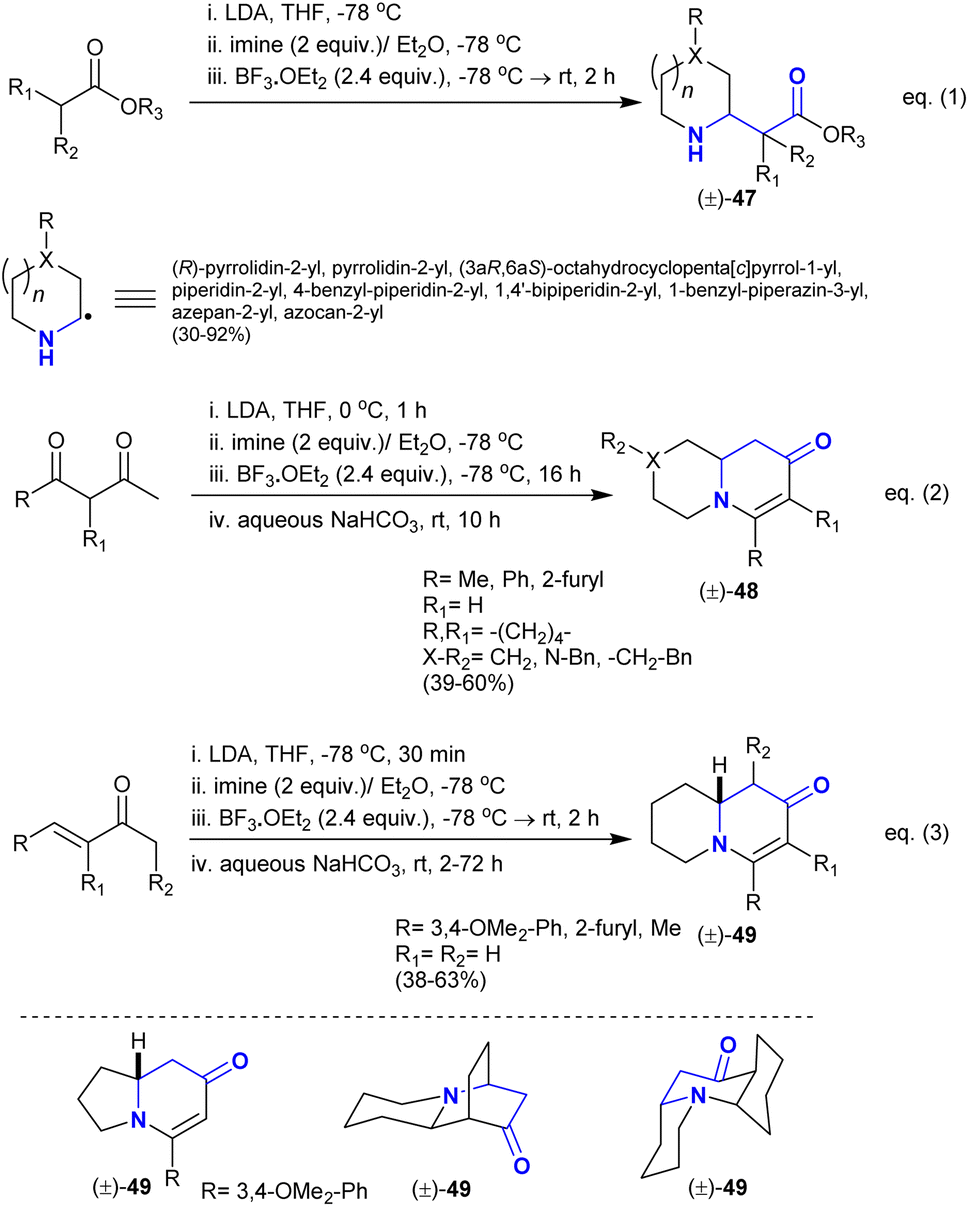
Kim et al.116 reported the α-alkylation of alicyclic imines with the nucleophilic enolate esters to synthesize a series of mono- and poly-cyclic β-aminoketones 47 with high diastereoselectivity (eqn. (1)). This method provided the modification of simple amines through C–H bond functionalization (Scheme 22). It also assisted the synthesis of natural target products such as (±)-myrtine. The esters were firstly treated with lithium diisopropylamide as a strong base before the addition of imines. The catalytic boron trifluoride etherate was the source of boron trifluoride, which acted as a Lewis acid catalyst to increase the nucleophilicity of esters. Alternatively, the use of 1,3-dicarbonyl compounds “dianions” as substrates instead of enolate esters tended to result in the preparation of a series of enaminones “pyridopyrazinones and quinolizinones” 48 (eqn. (2)). In this route, the base treatment supported the intramolecular condensation after the addition step with the annulation of the products. Also, the reactions of α,β-unsaturated ketones with in situ prepared imines under the same conditions yielded the bicyclic quinolizinones 49 (eqn. (3)). The mechanism of these reactions was proposed as the basic catalyst supported the nucleophilic addition and intramolecular heteroconjugation addition.
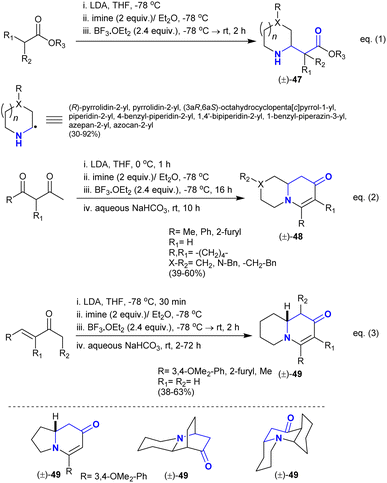 |
| | Scheme 22 Catalytic synthesis of piperidines, pyridopyrazinones, and quinolizinones. | |
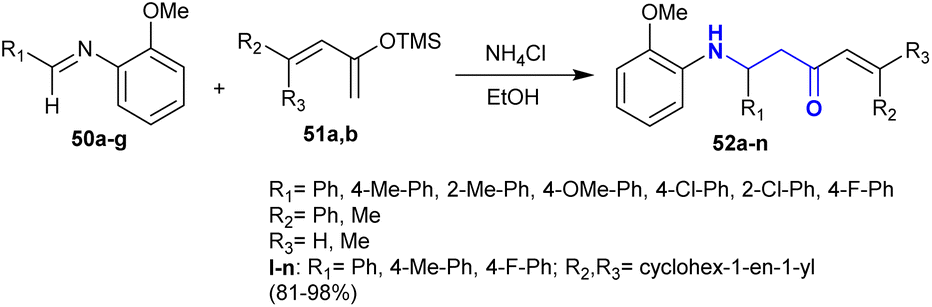
Fukumoto et al.117 reported a green protocol for the synthesis of Mannich-type products. Thus, the reactions of N-(2-methoxyphenyl)-1-arylmethanimine 50 with ((butadienyl)oxy)trimethylsilane 51 in the presence of ammonium chloride gave the respective β-aminoketones 52 (Scheme 23). The reactions are stereoselective for the formation of products 52 in excellent yield (81–98%). The mechanism of these reactions is projected as a Mannich-type reaction that involved the formation of β-aminoketones instead of a cyclization step followed by ring cleavage processes.
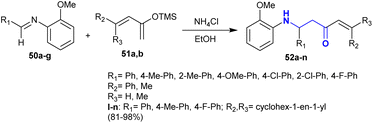 |
| | Scheme 23 Synthesis of β-aminoketones via Mannich-type reactions. | |
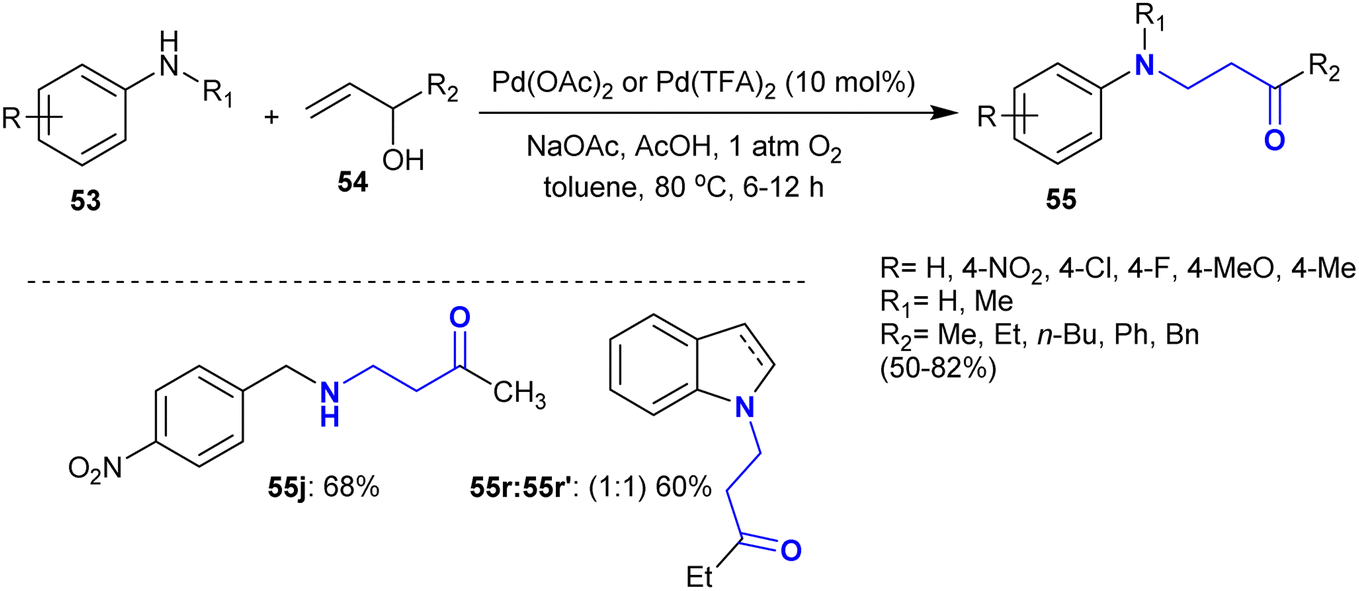
Kumar et al.118 developed the synthesis of cyclic β-aminoketones 55 through an exceptional and convenient procedure. Therefore, the oxidative coupling supported by catalytic palladium acetate of allyl alcohol derivatives 54 with aryl amines 53 gave moderate to worthy yields of the desired β-aminoketones 55 (Scheme 24). In these reactions, the N-alkylation is more favorite than the N-allylation process, in which the conjugate addition is preferable to the allylic amination as the suggested reaction mechanism.
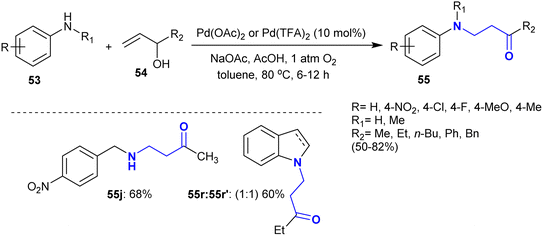 |
| | Scheme 24 Conjugate addition of the aryl amines to allyl alcohols. | |
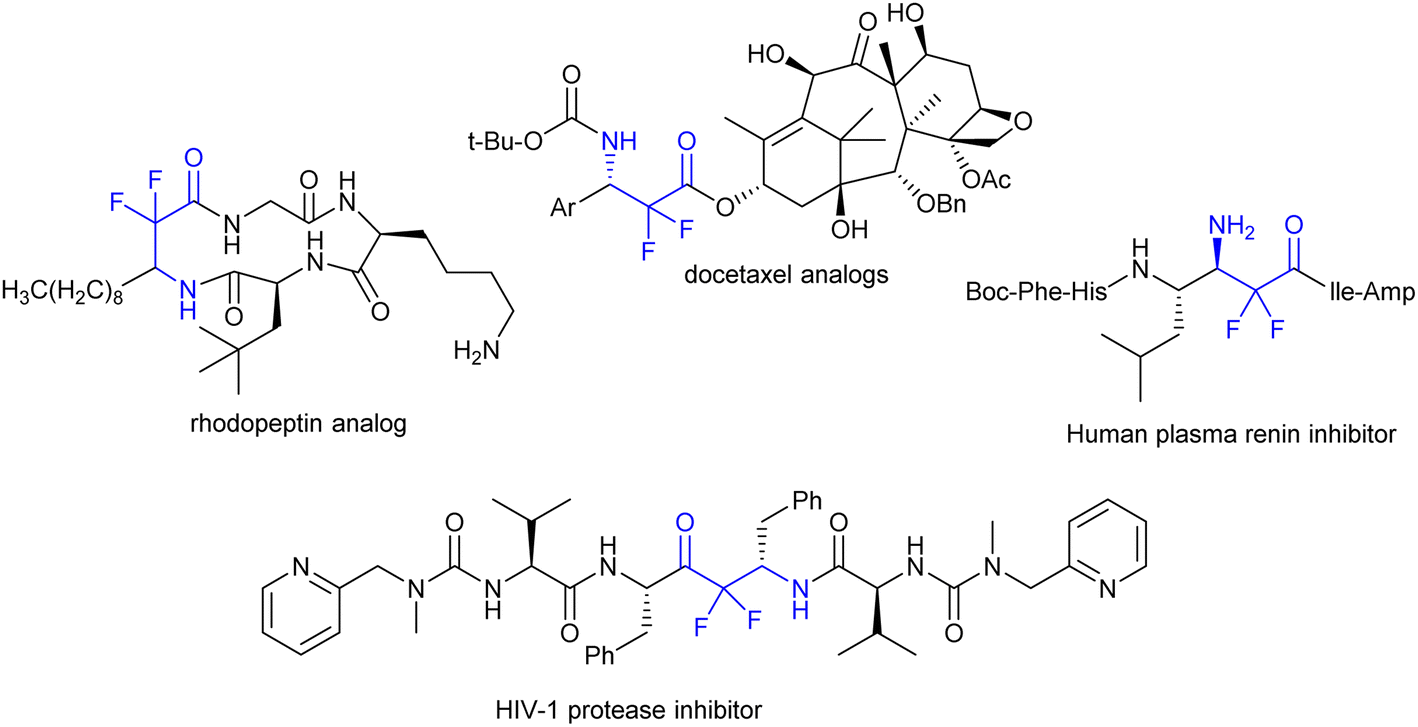
Many studies have focused on the preparation of fluorocarbon compounds, especially those containing trifluoromethyl, mono fluoro, and difluoromethyl groups. The advantage of the difluoromethylene group is that it is not a terminal group, and therefore, it is difficult to apply modern strategies from the last stage of fluoridation easily. Stunningly important compounds that contain a difluoromethylene group are difluoro-β-amino acid and α,α-difluoro-β-amino carbonyl groups.119 These basic skeletons were found to constitute the chemical composition of some vital compounds applied as drugs120,121 or inhibitors of enzyme activity such as rhodopeptin, docetaxel analogs, human plasma renin, and HIV-1 protease inhibitors (Fig. 3).122–124
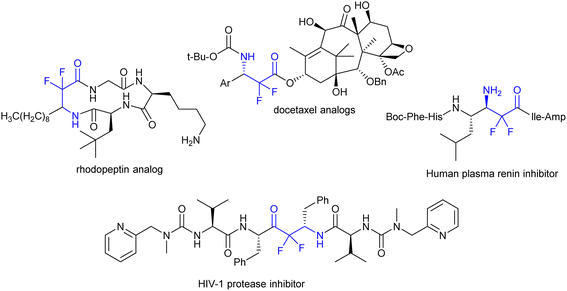 |
| | Fig. 3 Examples of privileged bioactive structures of compounds incorporating a difluoromethylene group. | |
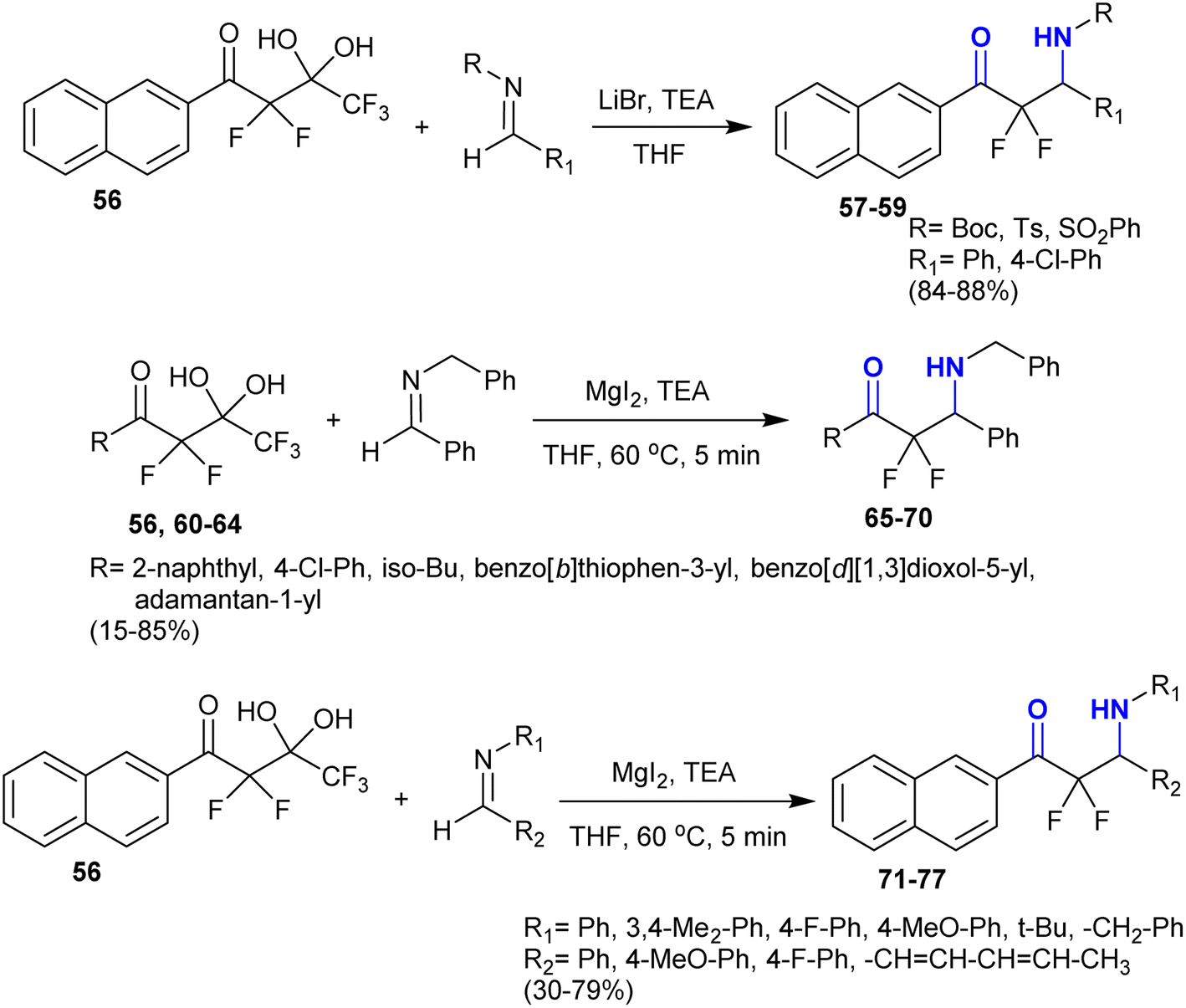
Accordingly, Nguyen et al.119 developed the synthesis of β-aminoketones incorporating a difluoromethylene group using magnesium iodide and organic bases. The addition reactions of fluorinated gem-diol 56 “difluoroenolates” as extremely α-fluorinated gem-diols with “unactivated imines” in THF containing triethylamine and lithium bromide yielded the desired α,α-difluoro-β-aminoketones 57–59. The reactions of fluorinated gem-diol 56, and 60–64 with imines “N-benzylidenebenzylamine” in THF containing magnesium iodide yielded the α,α-difluoro-β-aminoketones 65, and 66–70. The reaction of diol 56 with imine in the presence of lithium bromide or lithium chloride failed to provide product 65. Also, the addition of fluorinated gem-diol 56 to imines in the presence of magnesium iodide gave the desired α,α-difluoro-β-aminoketones 71–77 (Scheme 25) under mild conditions through the iminoaldol reaction model. This protocol provided the challenging preparation of the products without the need for the protection of groups or activation of the imines.
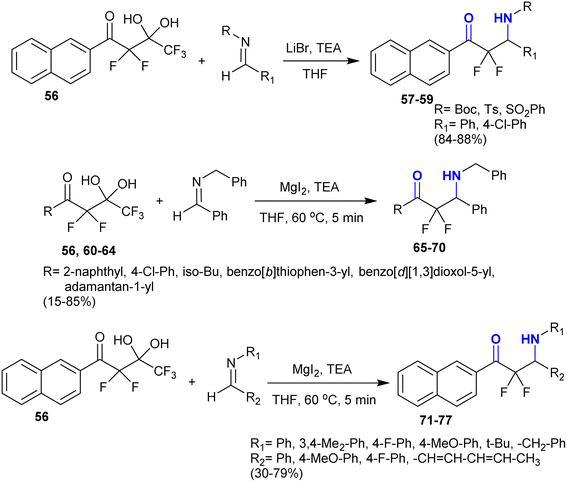 |
| | Scheme 25 Synthesis of α,α-difluoro-β-aminoketones. | |
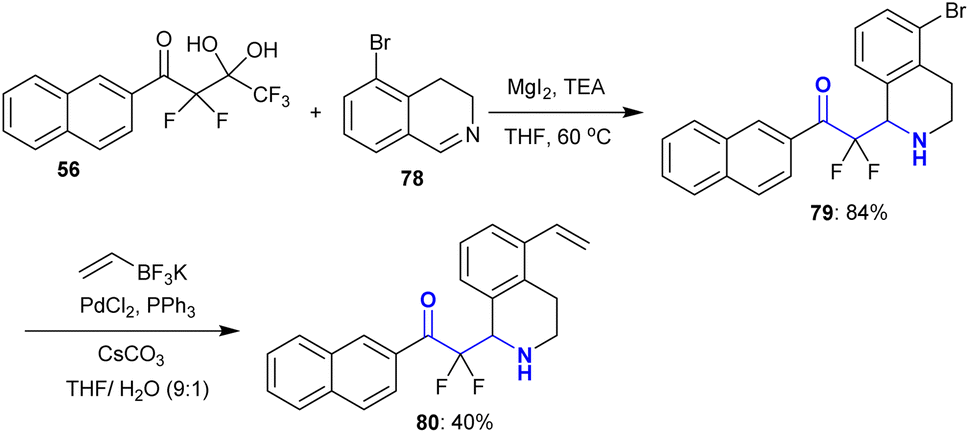
This strategy was extended to the addition reactions of fluorinated gem-diol 56 “difluoroenolates” to 5-bromo-3,4-dihydroisoquinoline 78 under the same previous conditions using magnesium iodide to prepare 2,2-difluoro-1-(naphthalenyl)ethanone 79 with excellent yield. Alkylation of 79 with vinyl trifluoroborate125 through Suzuki–Miyaura cross-coupling reaction in the presence of palladium(II) chloride, triphenyl phosphine, and cesium carbonate afforded the corresponding tetrahydroisoquinoline 80 in moderate yield (40%) (Scheme 26). This protocol verified that the structural divergence of tetrahydroquinolines can be rapidly constructed.119
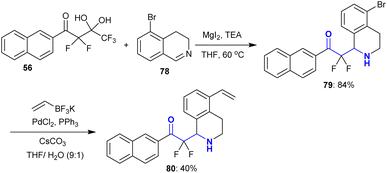 |
| | Scheme 26 Synthesis and alkylation of α,α-difluoro-β-aminoketones. | |
3.6. Condensation reactions
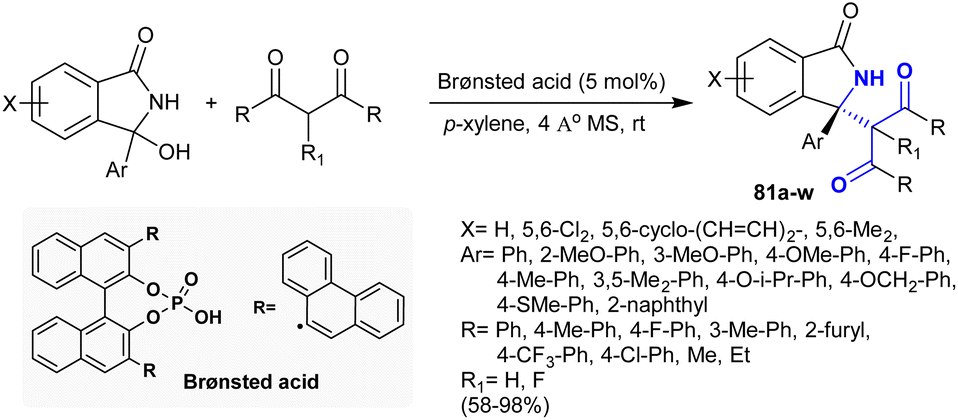
Treatment of 1,3-dicarbonyl compounds with cyclic N-acyl ketimines under Brønsted acid-catalyzed conditions in p-xylene afforded the anticipated 3-oxoisoindolines 81 in 58–98% yield (Scheme 27). The reactions progressed to yield the products by asymmetric Mannich-type addition type. Also, the products bearing stereocenter chiral carbon were obtained with exceptional yield (up to 98%) and reasonable to excessive enantioselectivity (up to 95% ee). The suggested mechanism of this reaction type involved dehydration of the desired 3-hydroxyisoindolinone to in situ generate N-acyl ketimines. Subsequent dual activations were attained for N-acyl ketimine ions in the transition state together with 1,3-dicarbonyl compounds by the influence of the bifunctional catalytic phosphoric acid. Lastly, nucleophilic attacks took place on the re-face of the planar ketimine through the construction of ten-membered transition-states to furnish oxoisoindolines with the (S) configuration.126
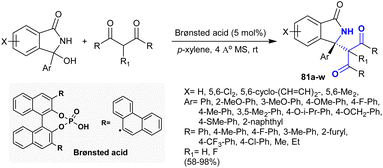 |
| | Scheme 27 Synthesis of β-aminoketones through Mannich-type addition. | |
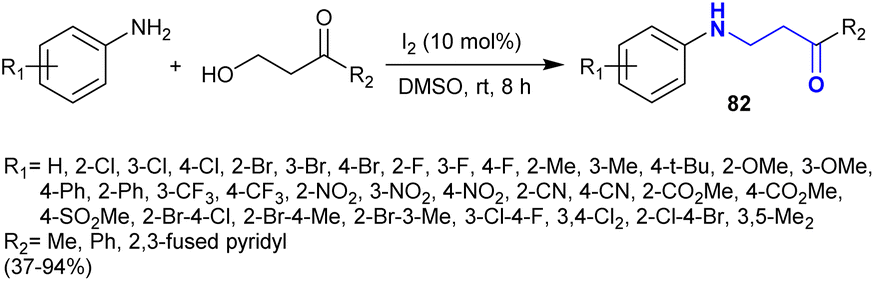
A series of β-aminoketones 82 was efficiently synthesized by Miao et al.127 through a catalytic coupling of aryl amines with β-hydroxyketones (Scheme 28). Hence, the reactions were catalyzed by iodine at room temperature, providing a facile procedure for the synthesis of the target compounds with relatively good yields based on the structural nature of the substituents. The iodine acts as an oxidant and metallic catalyst, enabling the oxidation of the hydroxyl group to the respective aldehyde with the release of HI molecules through the condensation with the amino group. This reaction is selective without the involvement of aza-Michael addition. The final step is the formation of the products through the addition of the H2 molecule “generated by the decomposition of HI to H2 and I2 owing to the intensive reducing impact of HI” to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond of the latter-formed intermediate.
N bond of the latter-formed intermediate.
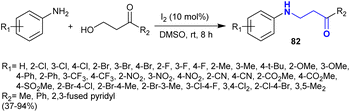 |
| | Scheme 28 Coupling of amines with β-hydroxyketones. | |
3.7. Synthesis of piperidines
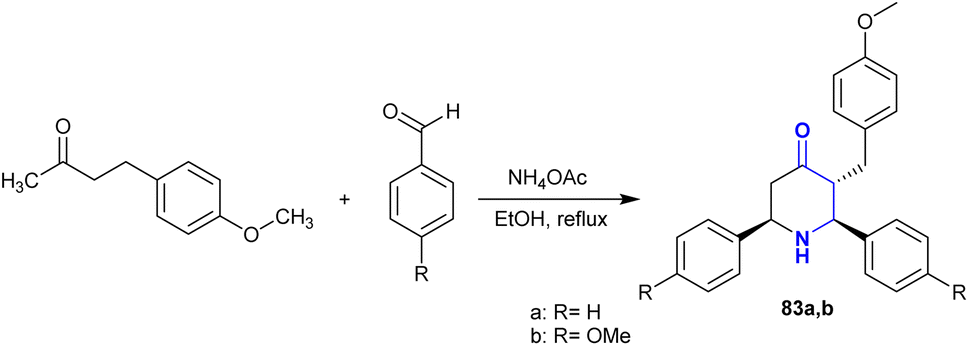
A successful piperidine ring cyclization was achieved by a Mannich condensation reaction from raspberry ketone methyl ether, as demonstrated by Shanthi et al.128 Thus, piperidinone with cyclic β-aminoketone moieties 83 was synthesized by the multicomponent Mannich reaction of methyl ether of raspberry ketone with aryl aldehydes, and ammonium acetate under reflux and catalyst-free conditions (Scheme 29). The stereochemistry of the constructed piperidinones was verified by the perceived coupling constants, in which the chair configuration with the substituents preferred the equatorial orientation, as estimated by 2D-NMR.
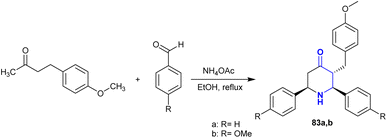 |
| | Scheme 29 Mannich reaction for the synthesis of piperidinone compounds. | |
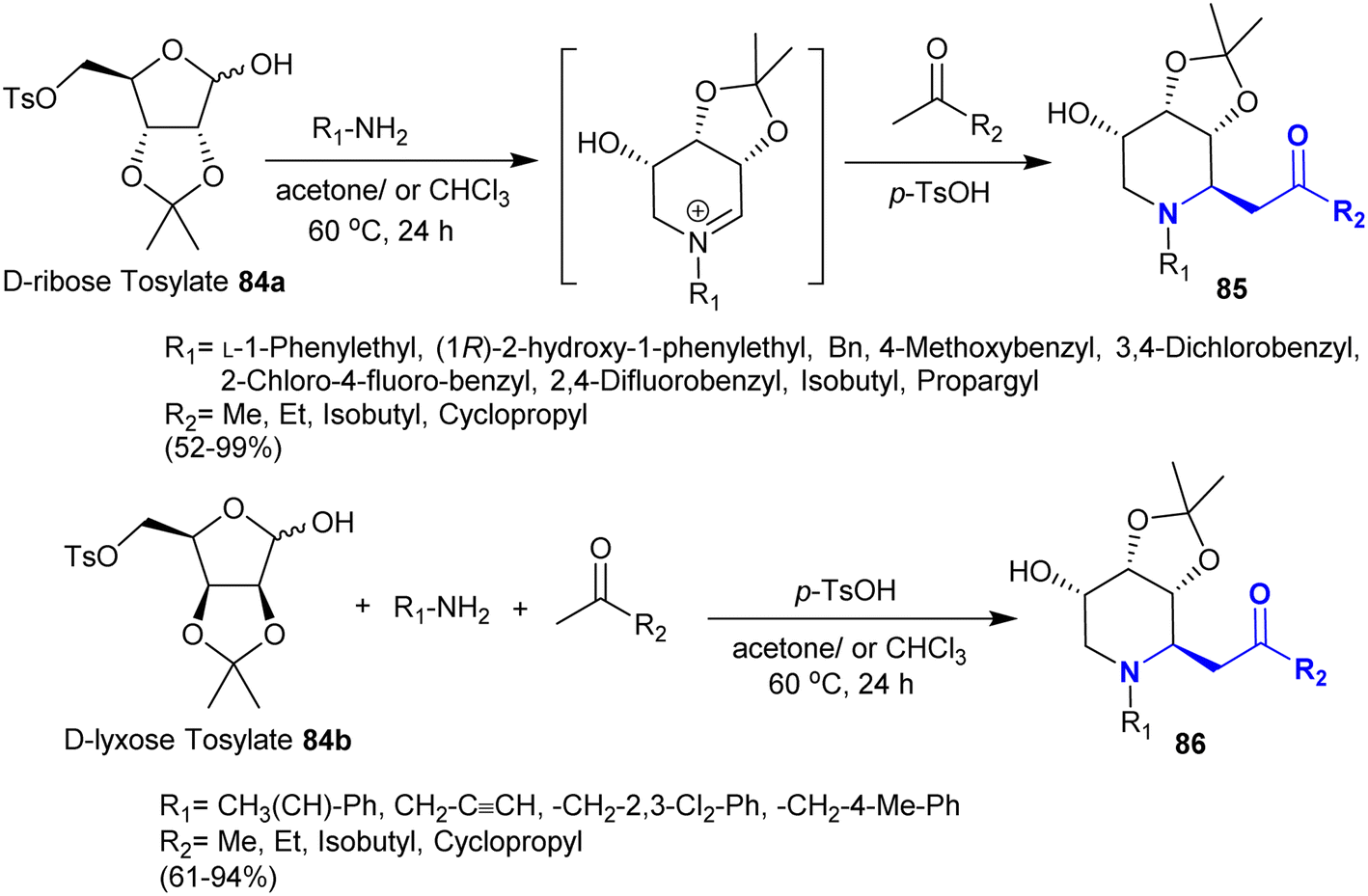
The synthesis of stereoselective bicyclic aza-sugar analogs was recently explored by Yuan et al.129 Subsequently, they130 interestingly reported a stereoselective synthetic route for the preparation of hydroxy-piperidines and imino-sugar C-glycoside analogs. The products in this route are potent glycosidase inhibitors prepared in exceptional yields. Consequently, Mannich reaction of D-ribose- and D-lyxose tosylates 84a and 84b, each with substituted amines and ketones in a one-pot procedure yielded the target compounds 85 and 86, respectively (Scheme 30). The reactions of this type proceeded under p-toluenesulfonic acid as a convenient catalyst in acetone for improved yields and reinforced reaction rate.
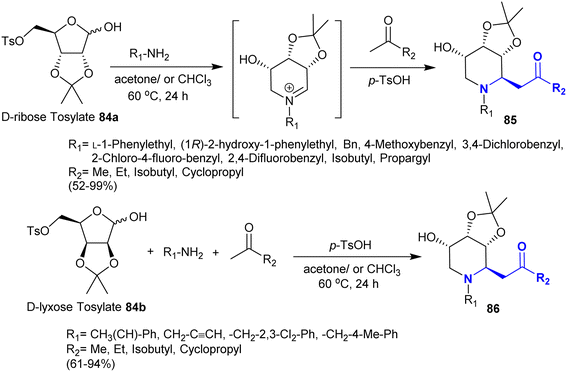 |
| | Scheme 30 Synthesis of β-aminoketones under acid-catalyzed conditions. | |
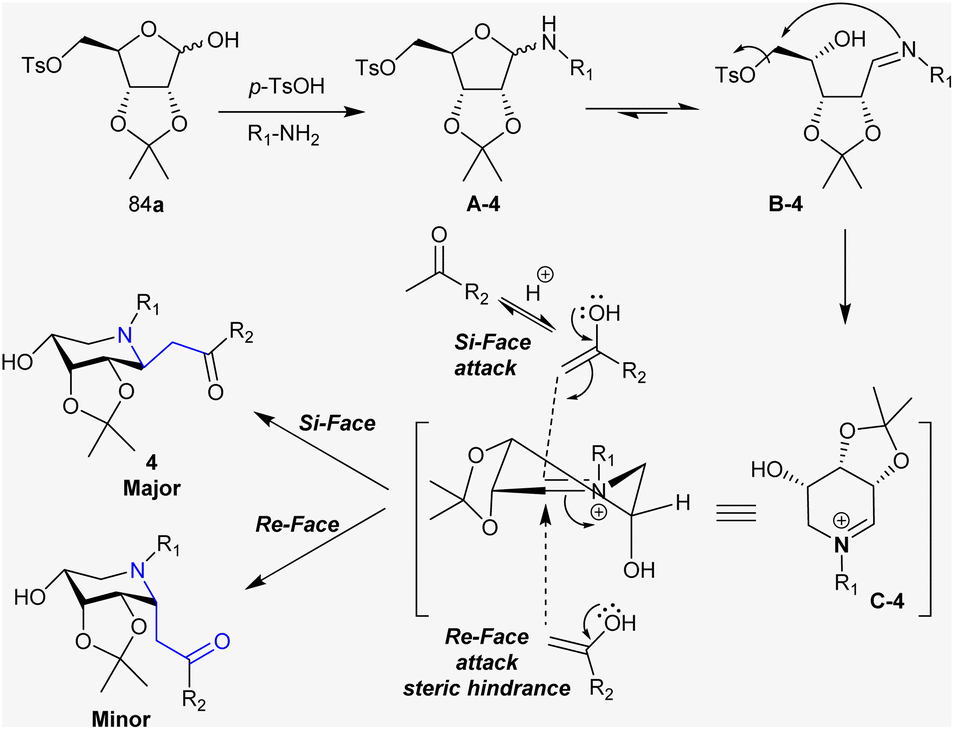
Under acid-catalyzed conditions, the condensation of amines with D-ribose- and D-lyxose tosylates generated the cyclic hemiaminal (A-4). The hemiaminal ring cleavage in intermediate A-4 generated the imine intermediate B-4. Thus, intramolecular nucleophilic substitution attack of the NH group at the C-OTs carbon with the good leaving group character of the OTs group generated the cyclic iminium ion in the form of intermediate C-4. The acid catalyst promoted the enolization of the ketone, and hence attack of the α-carbon of the iminium ion carbon (intermediate C-4). Accordingly, the two possible products are known as Si-face and Re-face with the majority of Si-face products owing to the steric hindrance factor of the iso-propylidene group (Scheme 31).130
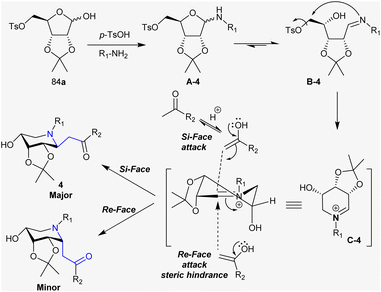 |
| | Scheme 31 Projected mechanism for the catalyzed synthesis of β-aminoketones. | |
3.8. Synthesis of morpholinones and piperazinones
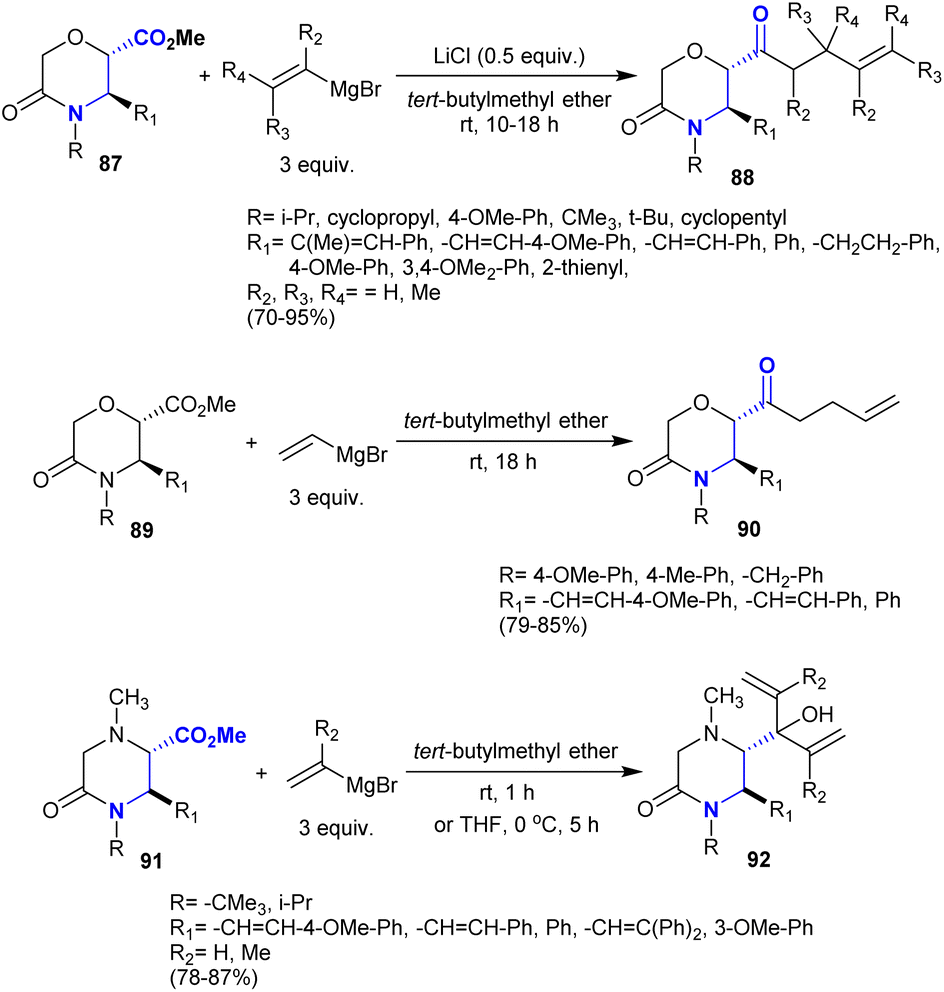
The direct synthesis of β-aminoketones from amides has been applied previously through sequential nucleophilic substitution at the carbonyl group by the reacted Grignard reagent such as vinyl magnesium bromide followed by Michael reaction.131 Recently, Farah et al.132 also reported the direct synthesis of β-aminoketones 88 from the addition of alkenyl Grignard reagents to methyl (2S,3R)-3,4-disubstituted-5-oxomorpholine-2-carboxylates “β-aminoketoesters” 87. The reactions progressed with the addition of lithium chloride in the absence of a transition metal catalyst to produce the desired lactam-bearing homo-allylic ketones. The products were achieved through a regioselective synthetic route with the generation of a new stereocenter carbon but it surprisingly maintained diastereoselectivity. A similar sequence was employed in the reactions of 5-oxomorpholine-2-carboxylates 89 and methyl (2S,3R)-1,3,4-trisubstituted-5-oxopiperazine-2-carboxylates 91 each with vinyl magnesium bromide reagents in tert-butyl methyl ether to give the β-aminoketones 90 and β-amino alcohol derivatives 92, respectively (Scheme 32).
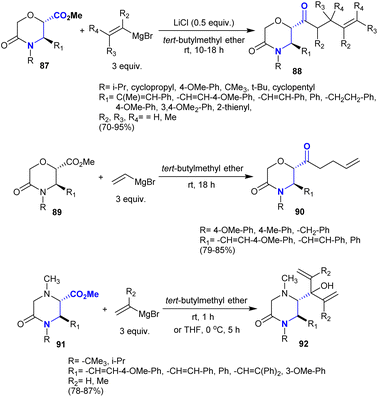 |
| | Scheme 32 Synthesis of substituted morpholinones and piperazinones. | |
3.9. Synthesis of spirocyclic β-aminoketones
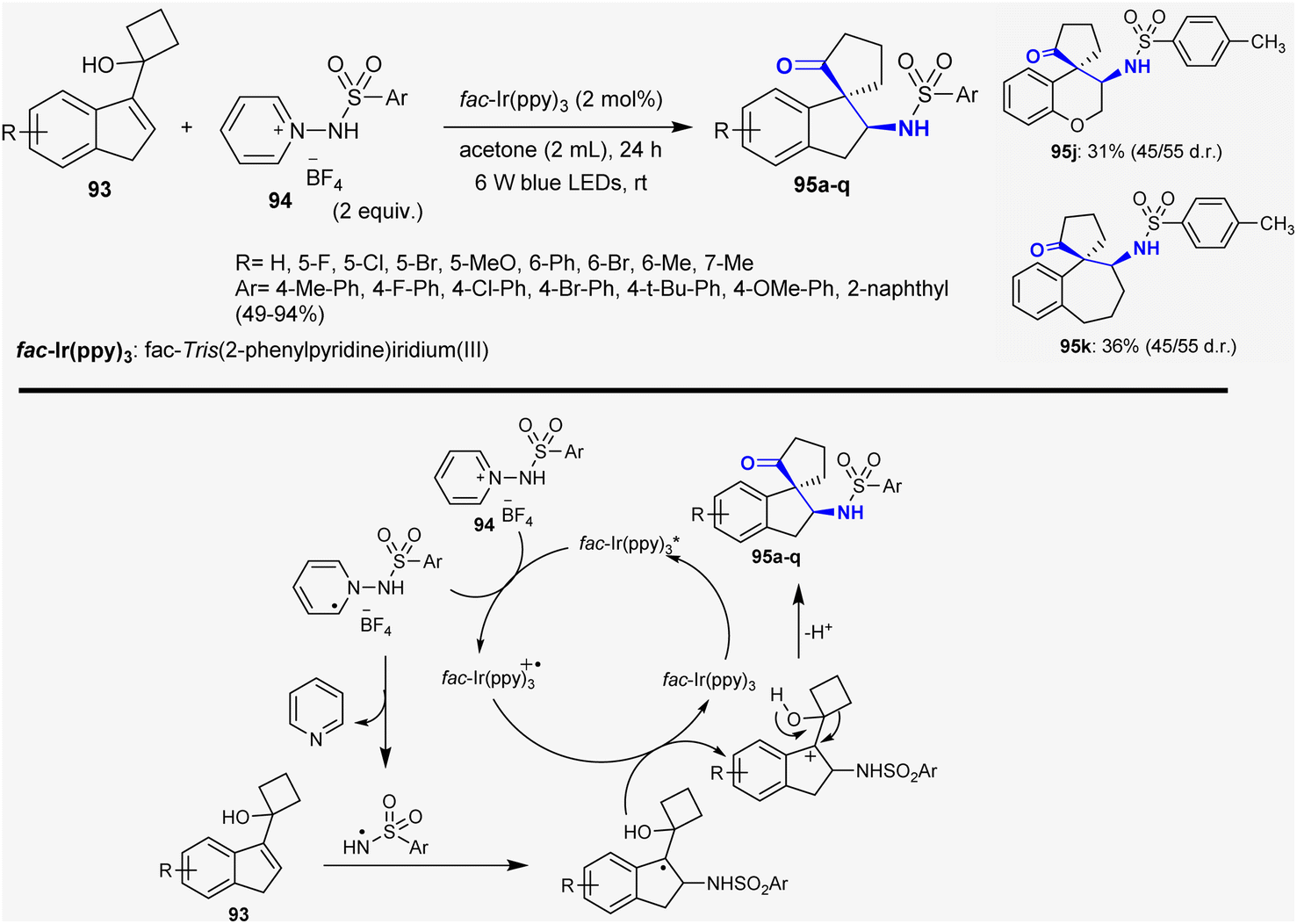
Ma et al.133 reported the synthesis of a series of N-(2-oxo-2′,3′-dihydrospiro[cyclopentane-1,1′-inden]-2′-yl)benzene-sulfonamides 95 through the reactions of cycloalkanol-1H-indenes with 1-(arylsulfonamido)pyridinium tetrafluoroborates. The reactions proceeded under catalytic and mild conditions using visible light photoredox to stimulate N-centered radical addition and semipinacol rearrangement of cycloalkanol with N-protected pyridinium salts. This method efficiently accessed a series of β-aminoketones of the spiro-cyclic type in acetone under fac-Ir(ppy)3 (2 mol%) catalytic conditions. The benefits of this performance are to enable extensive scope of substrates, respectable assembly tolerance, and simple synthetic route. The recommended mechanism for this type is a free radical mechanism involving N-centered radical addition and semipinacol rearrangement. Firstly, the reaction was activated by the visible-light-induced SET reduction of pyridinium salt 94 with the generation of radicals. The photo-excitation of the catalyst resulted in the generation of the radical intermediate with the generation of the catalyst in the oxidized form. Cleavage of the N–N bond by hemolytic fission-released pyridine is dependent on the efficiency of the reacted cycloalkanol-1H-indenes 93 to rapidly form the greatest stable alkyl radical. Next, oxidation of the previously formed intermediate led to the generation of a carbocation intermediate with the oxidation state of fac-Ir(ppy)3+˙, and the ground-state fac-Ir(ppy)3 was reinforced. Finally, semipinacol rearrangement took place for the intermediate to afford the target compounds 95 (Scheme 33).
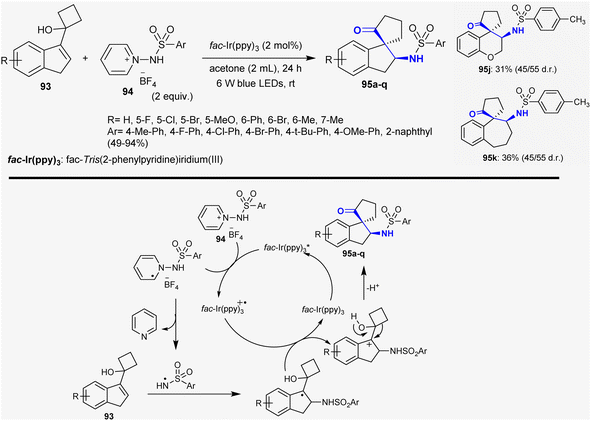 |
| | Scheme 33 Synthesis of β-aminoketones with the spiro-cyclic core. | |
3.10. Synthesis of imidazolines
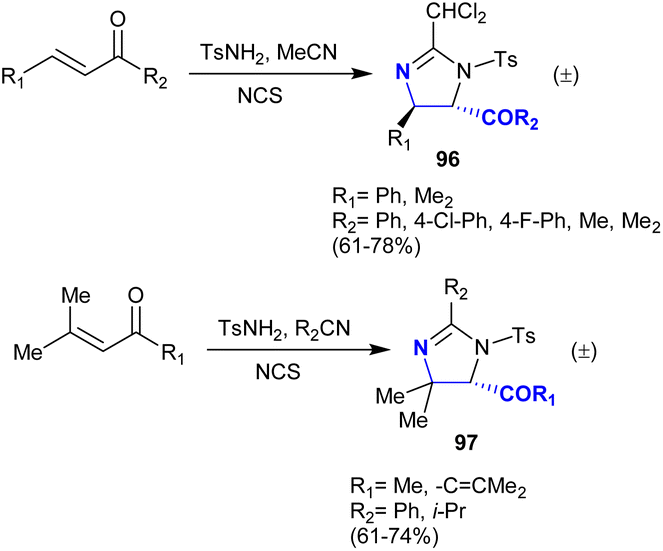
In another recently developed route, the imidazoline ring was constructed through the one-pot reaction of triflamide with silane in acetonitrile containing N-bromosuccinimide accompanied by the dehydrobromination of amidine with the aid of potassium carbonate.134 One of the most challenging topics in the field of organic synthesis is related to developing novel strategies of regio- and stereoselective approaches to obtain vicinal diamines.135–139 Timmons et al.140 developed an approach for the direct amination of enones, which is regio-, stereo-, and chemoselective deamination of enones employing electrophiles such as N-chlorosuccinimide and tosyl amine. The acetonitrile provided the nitrile group as a nucleophile for the nitrogen source. The projected mechanistic route involved the generation of an aziridinium intermediate produced from the reaction of tosyl amine hydrochloride with alkenes followed by [2 + 3] cycloaddition with aziridinium ring cleavage. In particular, α,β-unsaturated ketones reacted with tosyl amine, N-chlorosuccinimide, and acetonitrile to produce moderate to good yields of the deamination products 96 with high regioselectivity (>95). The stereoselectivity in the case of the reacted 1,3-di-aryl-prop-2-en-1-ones reached >95 of minor isomer, while no regio-isomers of the minor products were formed in the case of the reaction with 1,3-di-alkyl-prop-2-en-1-ones. Furthermore, α,β-unsaturated ketones reacted with tosyl amine, alkyl/aryl nitriles, and N-chlorosuccinimide to give the anticipated imidazolines 97 in 61–74% yield (Scheme 34).
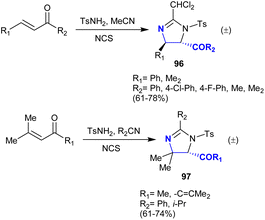 |
| | Scheme 34 Direct amination of α,β-unsaturated ketones. | |
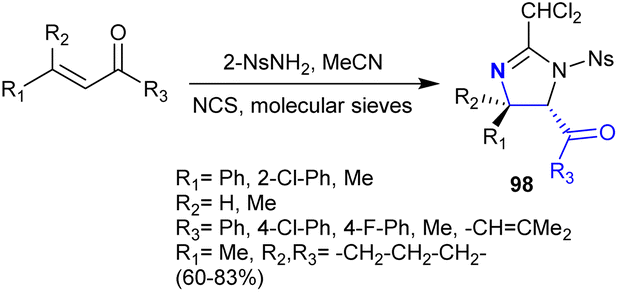
In 2004, Timmons et al.141 also extended their work on the regio- and stereoselective synthesis of imidazoline analogs 98 from the instantly accessible α,β-unsaturated ketones. In these reactions, N-chlorosuccinimide and 2-NsNH2 enable nitrogen electrophilic sources. The reactions chemoselectively yielded the products with the formation of side-products such as halo-amines (Scheme 35). The reactions of this type involved a mechanism of [2 + 3] cycloaddition, performed in the formation of the deamination products with remarkable regio- and stereoselectivity. The recorded stereoselectivity for this reaction type reached up to >95% in the case of the reactions with α,β-unsaturated ketones that have aryl substituents, while the alkyl substituents tended to have no selectivity. It is worth mentioning that the use of molecular sieves in the reaction of (E)-chalcone with 2-NsNCl2 and acetonitrile at room temperature resulted in the formation of the 2-(dichloromethyl)-4,5-dihydro-1H-imidazoline analog (CHCl2-imidazolines), while the reaction in the absence of molecular sieves at 50 °C led to the formation of 2-(trichloromethyl)-4,5-dihydro-1H-imidazoline (CCl3-imidazolines).
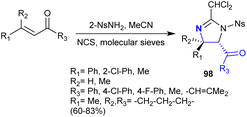 |
| | Scheme 35 Synthesis of imidazolidines. | |
3.11. Synthesis of indolizines
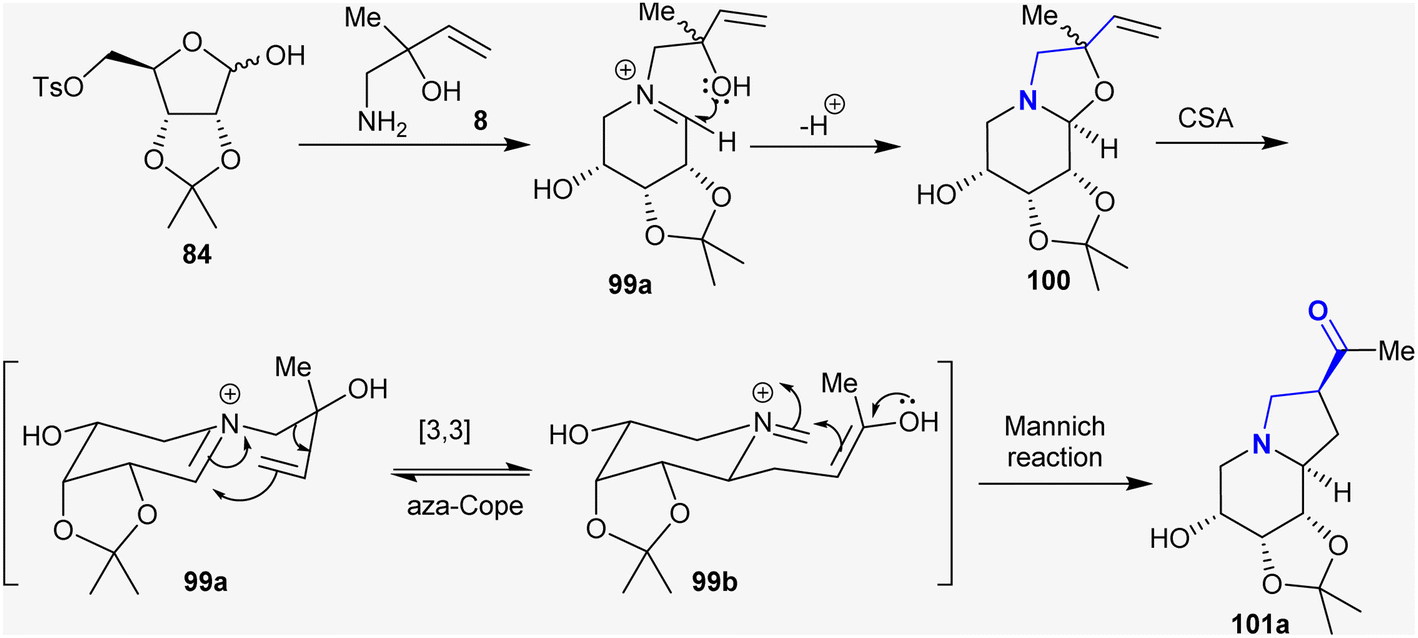
Previously, Chinthapally et al.142 developed a protocol for the stereoselective synthesis of polyhydroxy-2-acyl indolizidines through aza-Cope rearrangement–Mannich cyclization. This approach introduced the conceivable preparation of the prospective indolizidines and tetrahydroindolizidine-based imino-sugars in respectable yields. Camphor–sulfonic acid supported the reaction of substituted amino butanol with sugars such as ribose-tosylate to give the anticipated hydroxy-2-acyl indolizidines (Scheme 36).
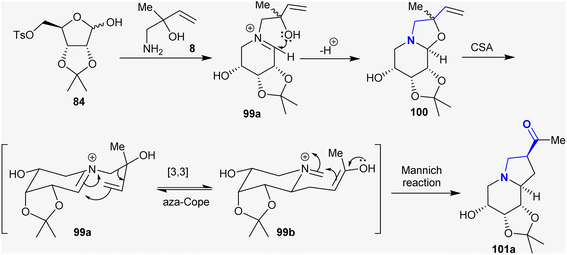 |
| | Scheme 36 Estimated mechanistic route for the synthesis of octahydro1,3 dioxolo-indolizines. | |
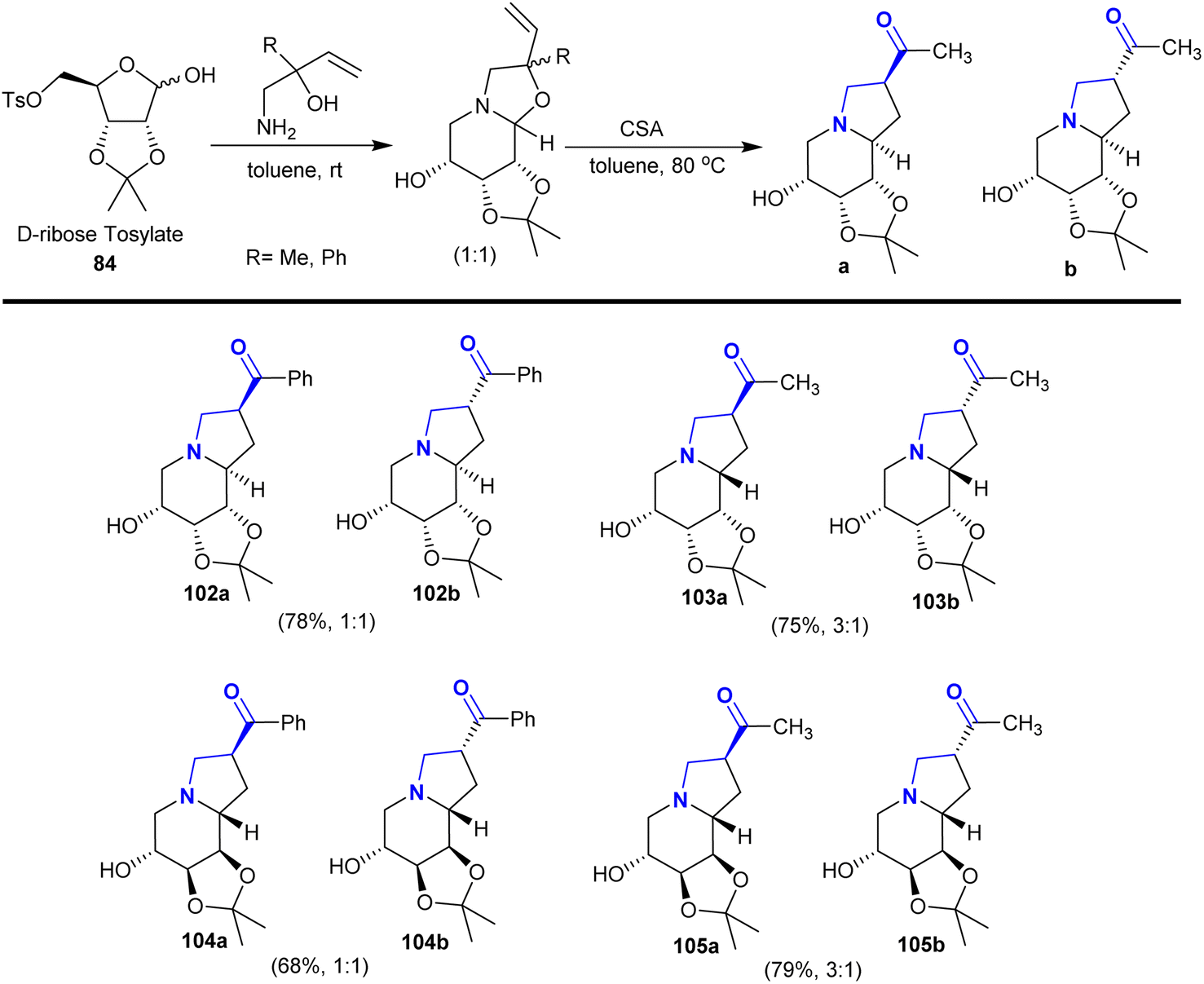
Consequently, the reactions of D-ribose tosylate 84 with 1-amino-2-substituted-but-3-en-2-ol derivatives in toluene at room temperature under catalytic conditions of camphor-sulfonic acid (CAS) yielded the anticipated octahydro1,3 dioxolo-indolizines142 (Scheme 37). Recently, researchers have focused on employing these reaction approaches to prepare a series of bicyclic aza-sugars 102–105 with glycosidic heteroatoms.129
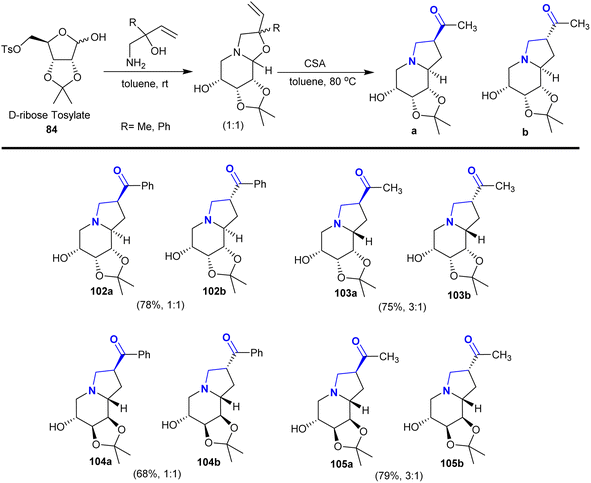 |
| | Scheme 37 Synthesis of polyhydroxy-2-acyl indolizidines. | |
3.12. Synthesis of pyrido-isoindoles
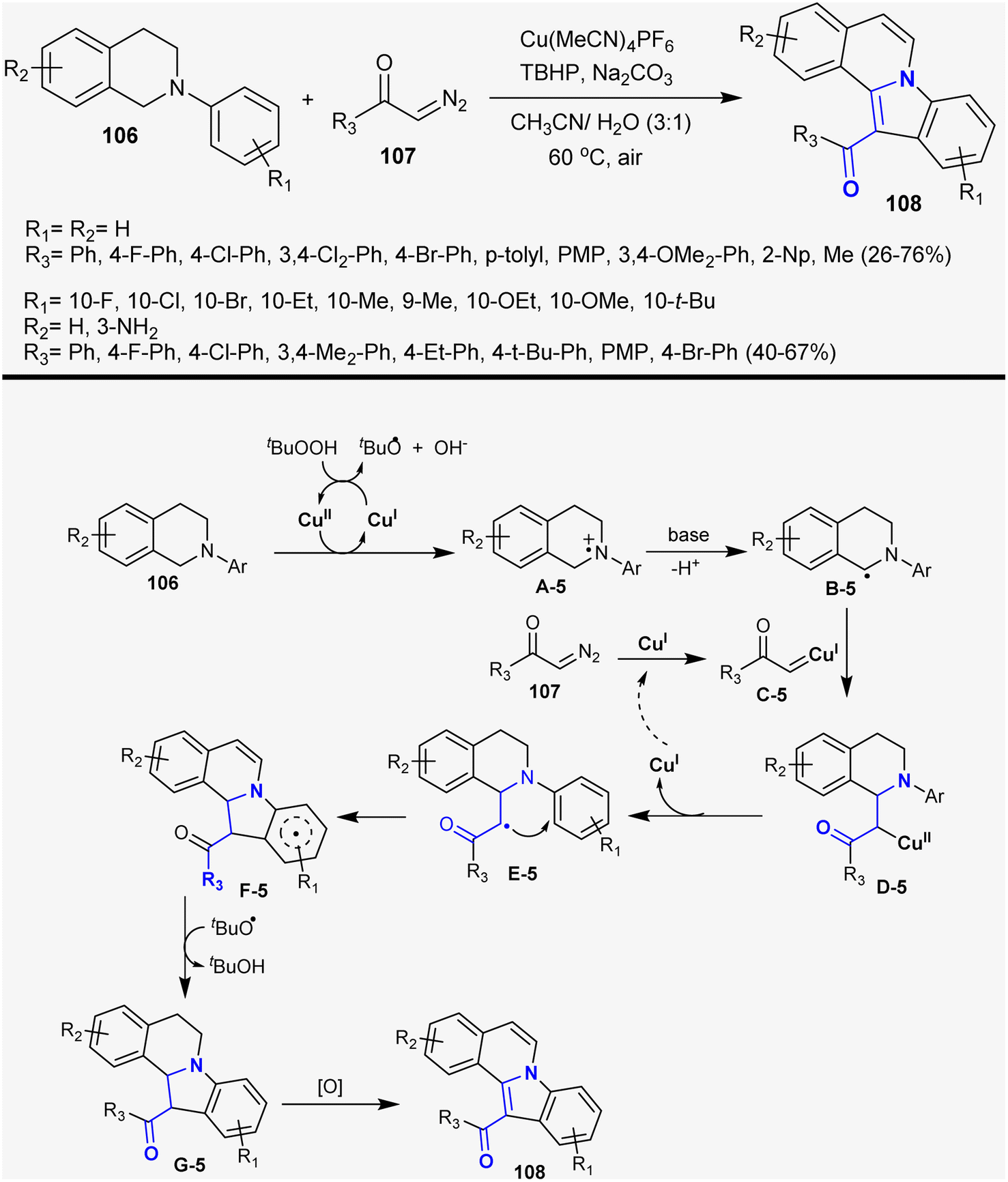
Zhang et al.143 reported the synthesis of indoloisoquinoline series 108 by the reaction of N-aryl-1,2,3,4-tetrahydroisoquinolines 106 with α-diazoketones 107. The reactions proceeded under Cu-catalyzed and oxidative conditions using tert-butyl hydroperoxide with the formation of the products in moderate to good yields (26–76%). The reaction sequence involved Cu-catalyzed [4 + 1] annulation, dediazotization of α-diazoketones, and oxidative dehydrogenation. Thus, the Cu(II) complex enabled electron transfer with the tert-amines generating amine radical cation intermediate A-5. Next, a proton was lost from the formed intermediate A-5 to generate the aminoalkyl radical B-5. Intermediate B-5 was trapped by carbene C-5, which was generated by the interaction of compound 107 with copper Cu(II)complex to generate intermediate D-5. Homolytic cleavage of intermediate D-5 generated intermediate E-5 with the release of the Cu(I) catalyst. The cyclization of the 5-endo-trig type intermediate E-5 followed by proton abstraction with the assistance of the t-BuO˙ base produced intermediate G-5. The final step is oxidative dehydrogenation to give the anticipated products 108 as cyclic β-aminoketones (Scheme 38).
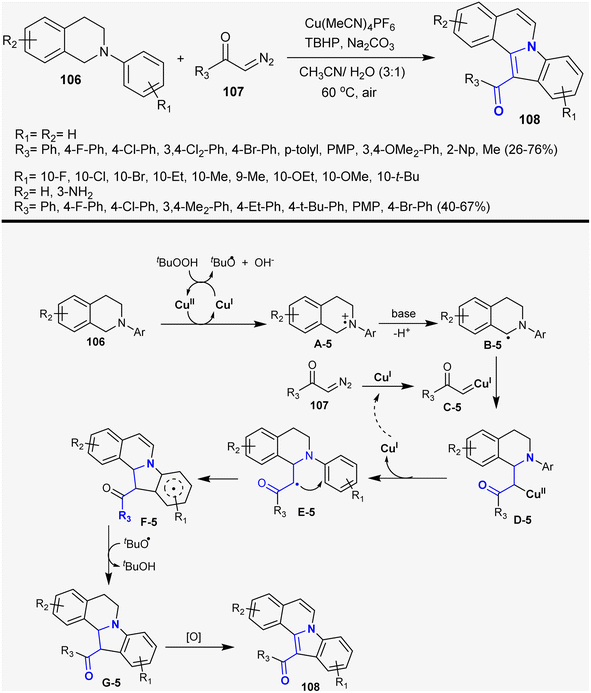 |
| | Scheme 38 Synthesis of aza-tetracyclic systems. | |
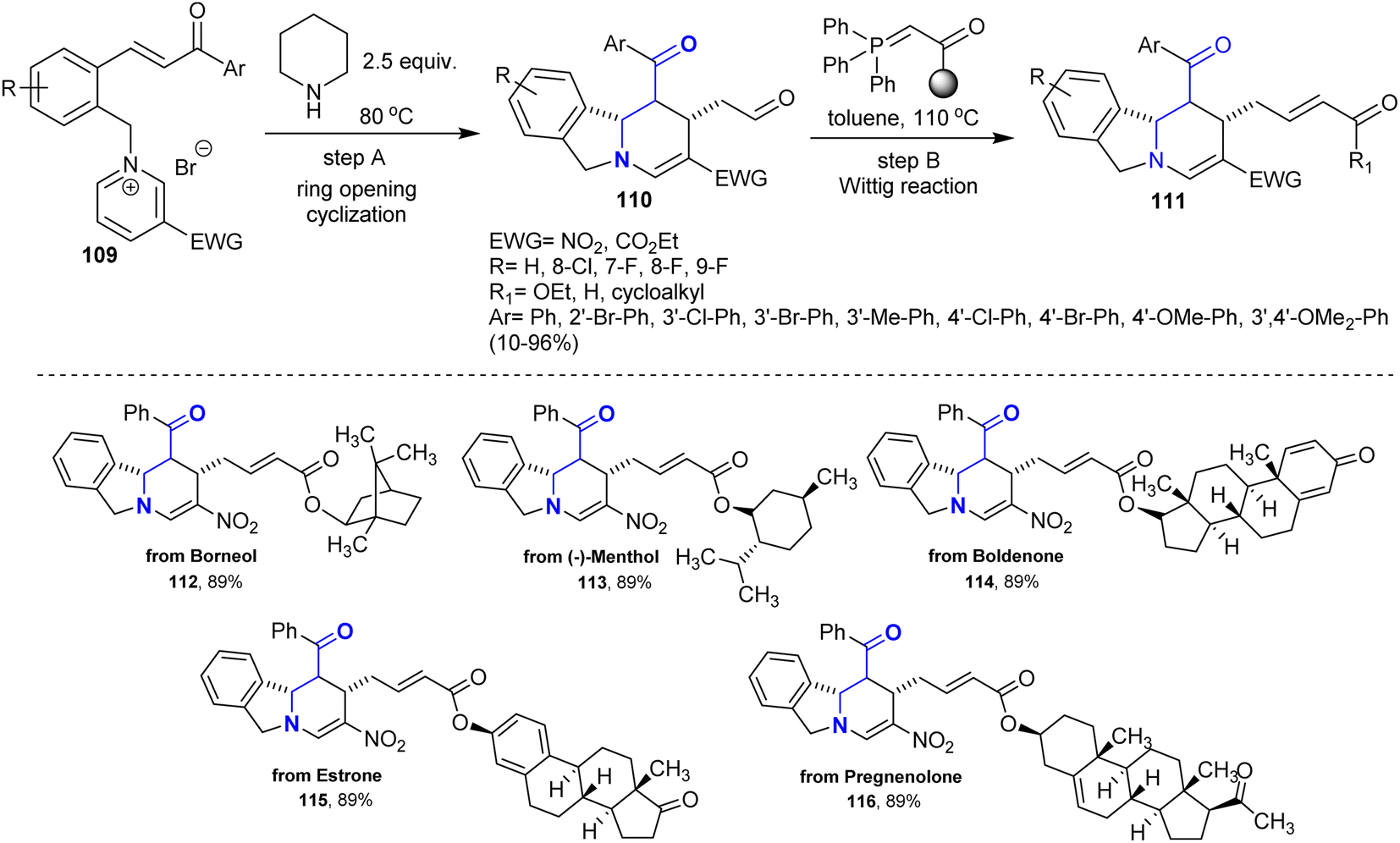
A sequence of reactions involving the synthesis of polycyclic isoindolines was efficiently developed by Wang et al.144 Thus, a series of tetrahydropyrido-isoindoles 110 was synthesized by treatment of chalcone-based pyridinium salts 109 with piperidine through ring-opening/ring-closure reactions. Two main factors that may affect the deconstruction of the pyridinium motif were defined as the exceptional binding of unstable in situ-formed cyclic β-aminoketones and the instability of the produced N,N-ketals. Alternatively, Wittig reactions of the anticipated isoindolines with several phosphorus ylides yielded isoindolines 111 with α,β-unsaturated ketone side chains (Scheme 39).
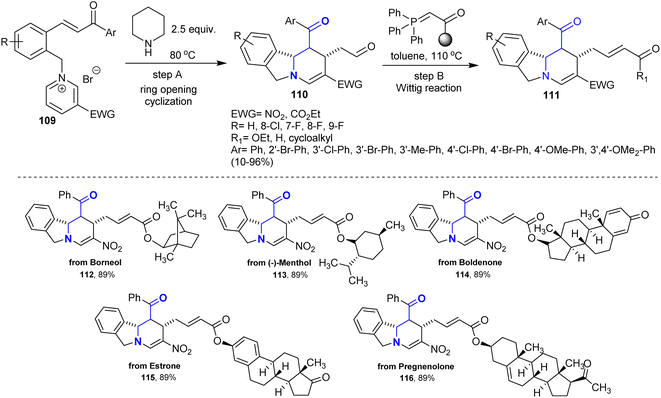 |
| | Scheme 39 Synthesis of tetrahydropyrido-isoindoles. | |
3.13. Synthesis from propargylic alcohols
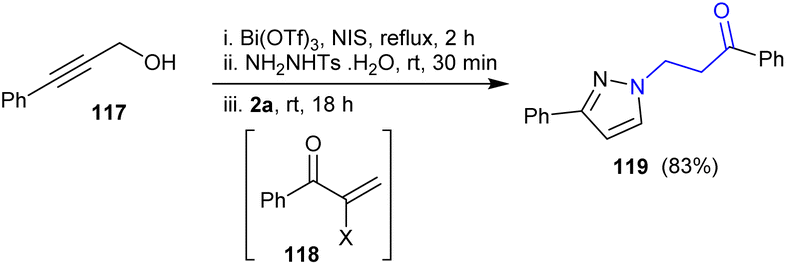
Guo et al.145 reported the synthesis of β-aminoketone from the reaction of 3-phenylprop-2-yn-1-ol 117 with N-Ts-substituted hydrazine. Therefore, 1-phenyl-3-(3-phenyl-1H-pyrazol-1-yl)propan-1-one 119 was prepared in 83% yield through the nucleophilic addition of the in situ-formed intermediate 118 and hydrazine derivative followed by cyclocondensation. The hydrazine derivative was applied in this reaction as a nucleophilic reagent in the one-pot synthesis for the construction of a pyrazole ring from the propargylic alcohol (Scheme 40).
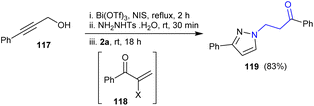 |
| | Scheme 40 Synthesis of 1-phenyl-3-(3-phenyl-1H-pyrazol-1-yl)propan-1-one. | |
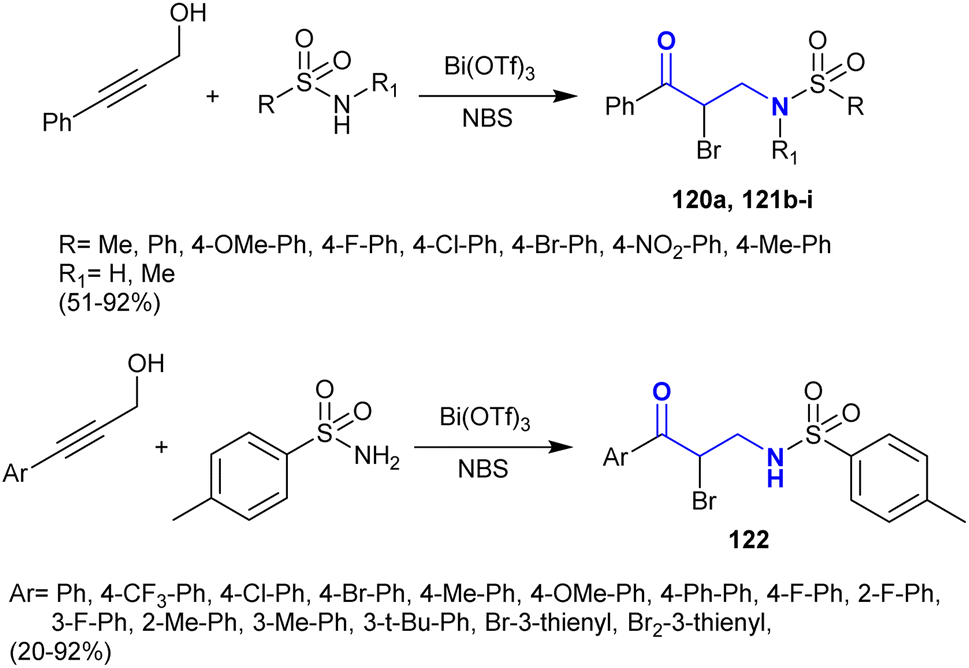
Zhang et al.146 developed a facile procedure for the synthesis of α-bromo-β-aminoketones 120–122 under catalytic conditions. Consequently, Bi(OTf)3-catalyzed reactions of 3-phenyl(aryl)prop-2-yn-1-ol with sulfonamides and N-bromosuccinamide as a source of the halogen yielded the desired series of α-bromo-β-aminoketones 120–122 (Scheme 41). This procedure was achieved in a one-pot reaction of the reacted materials. Tandem Meyer–Schuster-type rearrangements were proposed for these reactions followed by intermolecular Michael addition reactions of α,β-unsaturated ketones with sulfonamides. The substituents at the C4 position of the phenyl ring are preferred to achieve a shortened reaction time and good yields. Substituents with high electron density on the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond increased the reaction rate. Additionally, the steric hindrance factor of the substituents at the para position of the phenyl ring does not affect the product yield.
C bond increased the reaction rate. Additionally, the steric hindrance factor of the substituents at the para position of the phenyl ring does not affect the product yield.
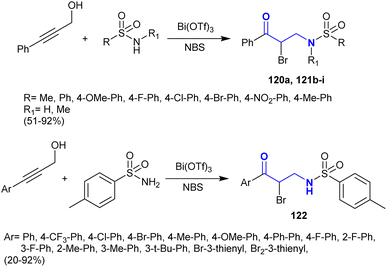 |
| | Scheme 41 Synthesis of α-bromo-β-aminoketones. | |
Laserna et al.147 developed gold-catalyzed reactions of hydroxy-alkynes with anilines to efficiently prepare a series of β-aminoketones 123 (Scheme 42). The mechanism of the reactions, in this case, is related to the efficiency of the catalyst to enable the Meyer–Schuster rearrangement148 of the propargylic alcohols in the first step, and then the conjugated addition of the primary amine to the generated enones. The solvent effect in this reaction type is related to the selectivity of the reaction, and thus a toluene and methanol mixture with a 98:2 molar ratio is preferrable. Even though the reactions are tolerant of both electron-deficient and electron-rich aniline substituents, primary and secondary alcohols are appropriate substrates. The nucleophile addition was related to the elimination of the adjacent hydroxyl group.149 Consistently, the imine reduction after hydroamination provided the analogous amino alcohol in 72% yield as a mixture of diastereomers (85:15).
 |
| | Scheme 42 Addition of amines to alkynols. | |
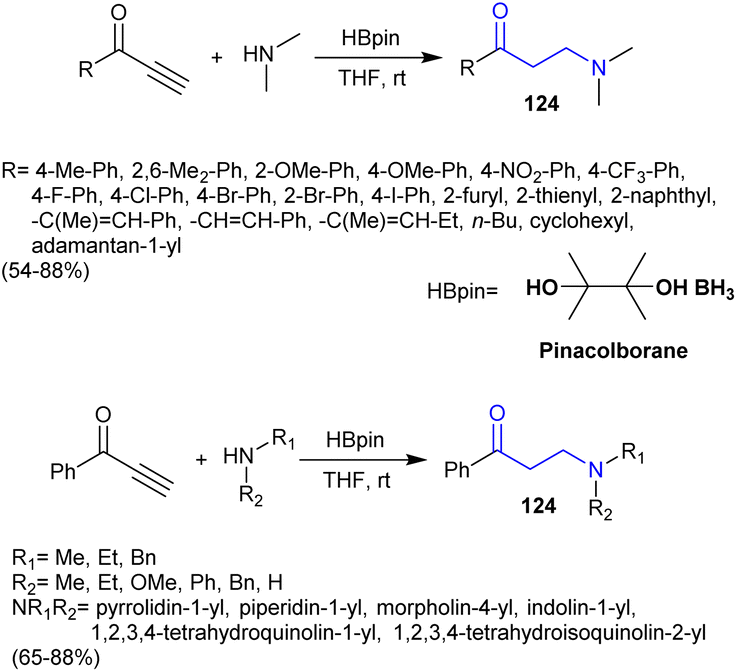
3.14. Reductive hydroamination of ynones
Fu et al.150 employed the reductive hydroamination of ynones for the proficient synthesis of β-aminoketones 124. In this route, the metal-free reductive hydroamination of alkynones with secondary amines under mild reaction conditions yielded the anticipated β-aminoketones in moderate to good yields. The reactions involved the hydroamination of CC bonds through the addition of the amines with the aid of pinacolborane catalyst (Scheme 43). The reaction mechanism may follow the interaction of the amine with the pinacolborane catalyst-generated intermediates that interacted with ynones. Afterward, coordination of the aminoborane with ynones tended to the formation of the reductive complex. The reactions could also have followed Michael addition to form the hydroamination product, which was reduced by hydride, enolized, and protonolysis of a carbon–boron bond with the amines to afford the respective ketamine.
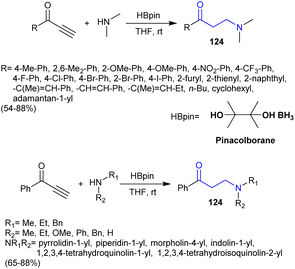 |
| | Scheme 43 Synthesis β-aminoketones from ynones. | |
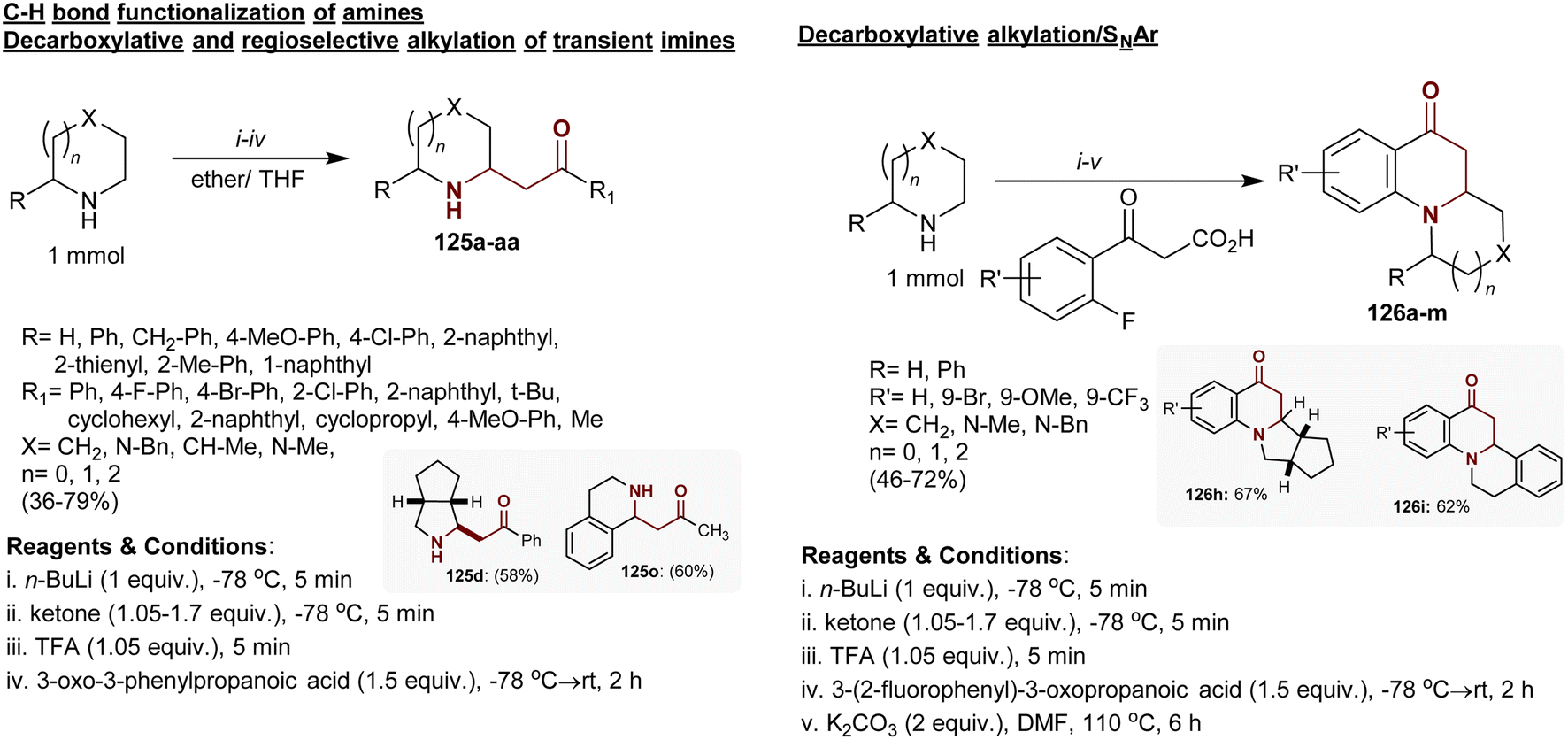
3.15. Alkylation of alicyclic imines with β-ketoacids
Regioselective alkylation and decarboxylation of alicyclic imines with β-ketoacids yielded the respective β-aminoketones 125 in poor to excellent yields (36–79%). The reactions tended to C–H bond cleavage at the α-position of imines accompanied by C–C bond formation. The reactions enabled the alkylation of imines at the α-position through regioselective substitutions. Ten analogs were obtained as trans-diastereomers although the cis-isomers were thermodynamically stable with probable interconversion of the products. Also, decarboxylative alkylation of imines with o-fluoroaryl-β-ketoacids followed by a nucleophilic substitution step yielded the polycyclic quinolinone products 126a–m with a β-aminoketone moiety without isolation of the intermediates (Scheme 44).151
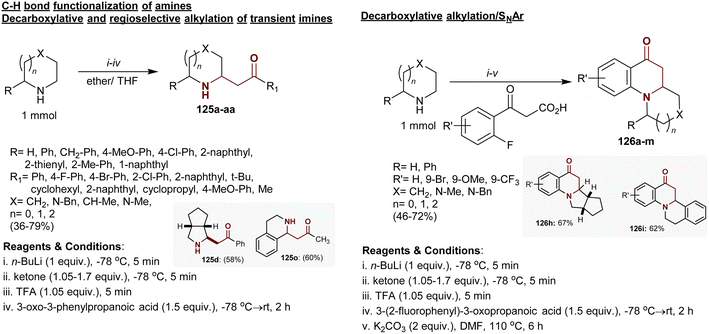 |
| | Scheme 44 Regioselective alkylation of alicyclic imines. | |
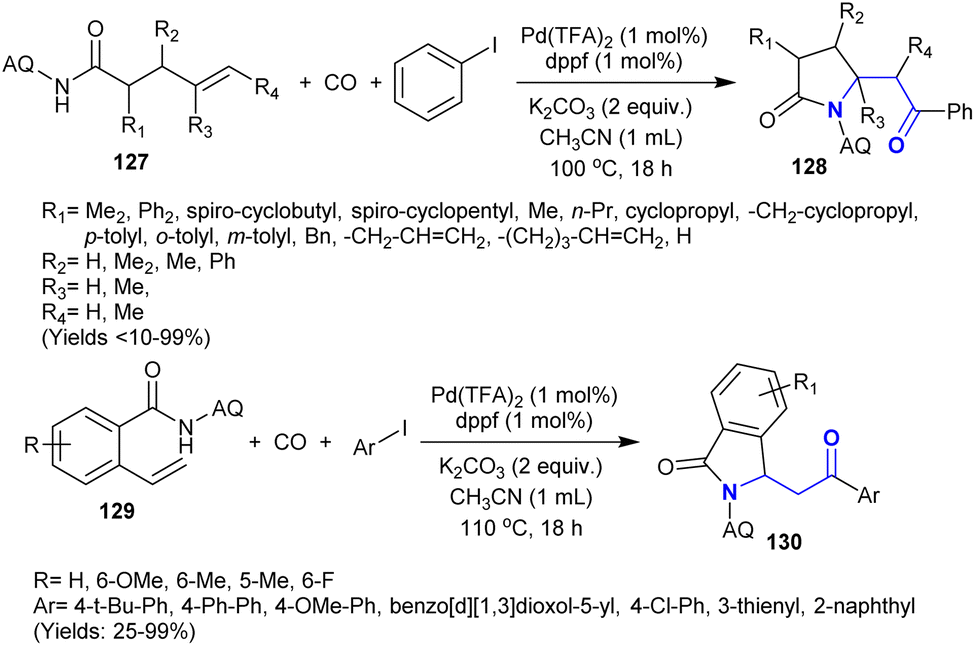
3.16. Carbonylative coupling
Owing to the difficulties of the reactions intended for the coupling of aryl halides with unactivated alkenes, Peng et al.152 developed a protocol involving these reactions under catalytic conditions. Thus, the carbonylative coupling and amination were developed under palladium-catalyzed conditions to synthesize a sequence of β-aminoketones 128 and 130. With the support of a leading group (8-aminoquinoline “AQ”), the identity of the olefin to acyl–palladium complex could be improved, thus stimulating the acyl-palladation crosswise of the CC double bonds. A broad range of β-aminoketones was synthesized in reasonable to exceptional yields with broad regioselectivity employing 4-pentenoic 127 and 2-vinyl benzoic amides 129 as the preliminary materials. This performance involved the generation of two C–C bonds in addition to one C–N bond and delivered a technique for the carbonylative coupling of the desired unactivated alkenes (Scheme 45).
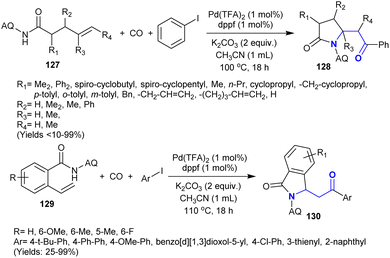 |
| | Scheme 45 Synthesis of β-aminoketones via carbonylative coupling of unactivated alkenes. | |
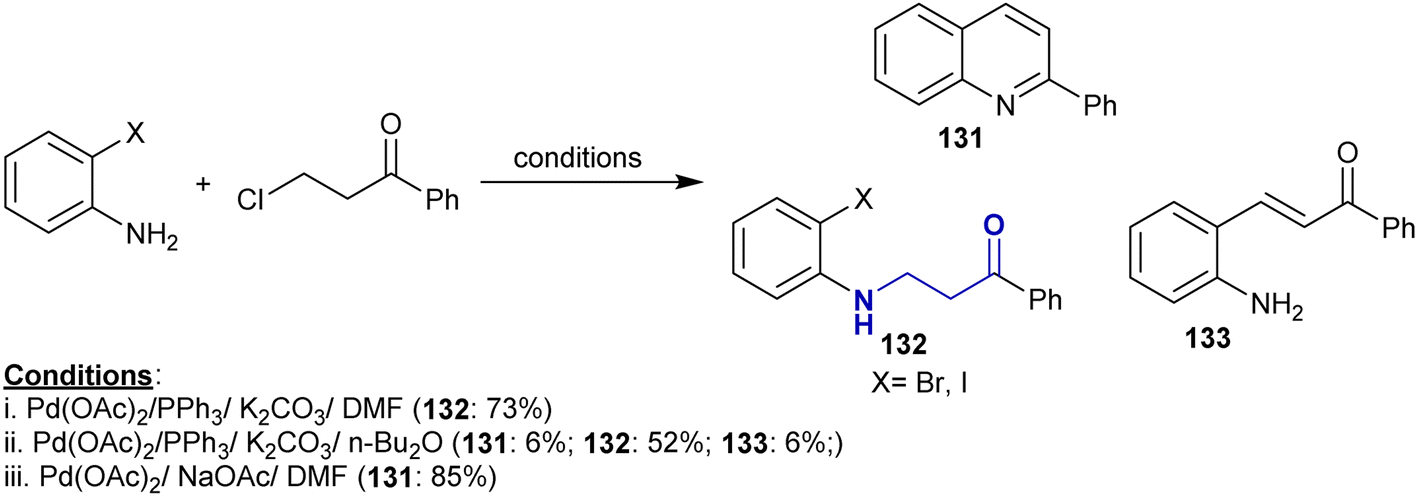
3.17. Coupling of halo ketones with amines
The palladium-catalyzed cascade reactions of o-halo-anilines with 3-chloro-1-phenylpropan-1-one were accomplished under different conditions to prepare a series of quinolones. The various conditions led to the formation of side products such as β-aminoketones 132 and arylidene 133, where the Michael sequence reaction generated β-aminoketones 132 and the Heck reaction generated the desired arylidene 133 via β-hydride elimination. The use of palladium acetate and triphenyl phosphine in DMF containing potassium carbonate yielded β-aminoketones 132 as the only product, while the use of n-Bu2O instead of DMF led to the highest yield of β-aminoketones 132, and the use of sodium acetate as a base in the absence of ligand only gave the aryl quinoline 131 (Scheme 46).153
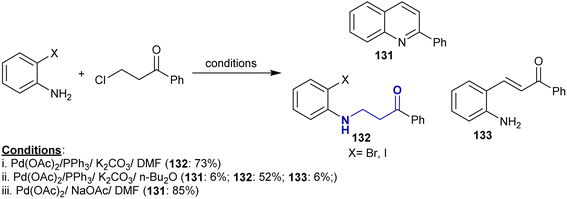 |
| | Scheme 46 Synthesis of β-aminoketones via cascade reactions under palladium-catalyzed conditions. | |
3.18. Acid hydrolysis of metal complexes
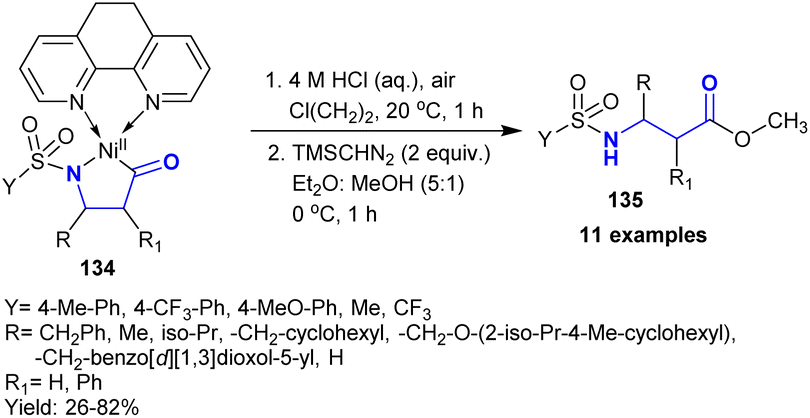
Ravn et al.1 developed the synthesis of β-aminoketones 135 in low to good yields (26–82%) through an appropriate and proficient synthetic route for the combination of a single carbon isotope. Thus, the acid hydrolysis of nickel complexes 134 under atmospheric conditions afforded β-aminoketones 135 (Scheme 47). This method enabled easy product purification. The products were formed through the cleavage of M–C and M–N bonds.
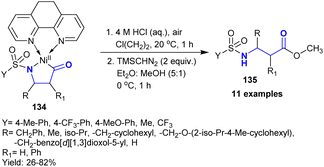 |
| | Scheme 47 Synthesis of β-aminoketones under acid hydrolysis conditions. | |
4. Synthetic applications
4.1. Aminoalcohol and oxime synthesis
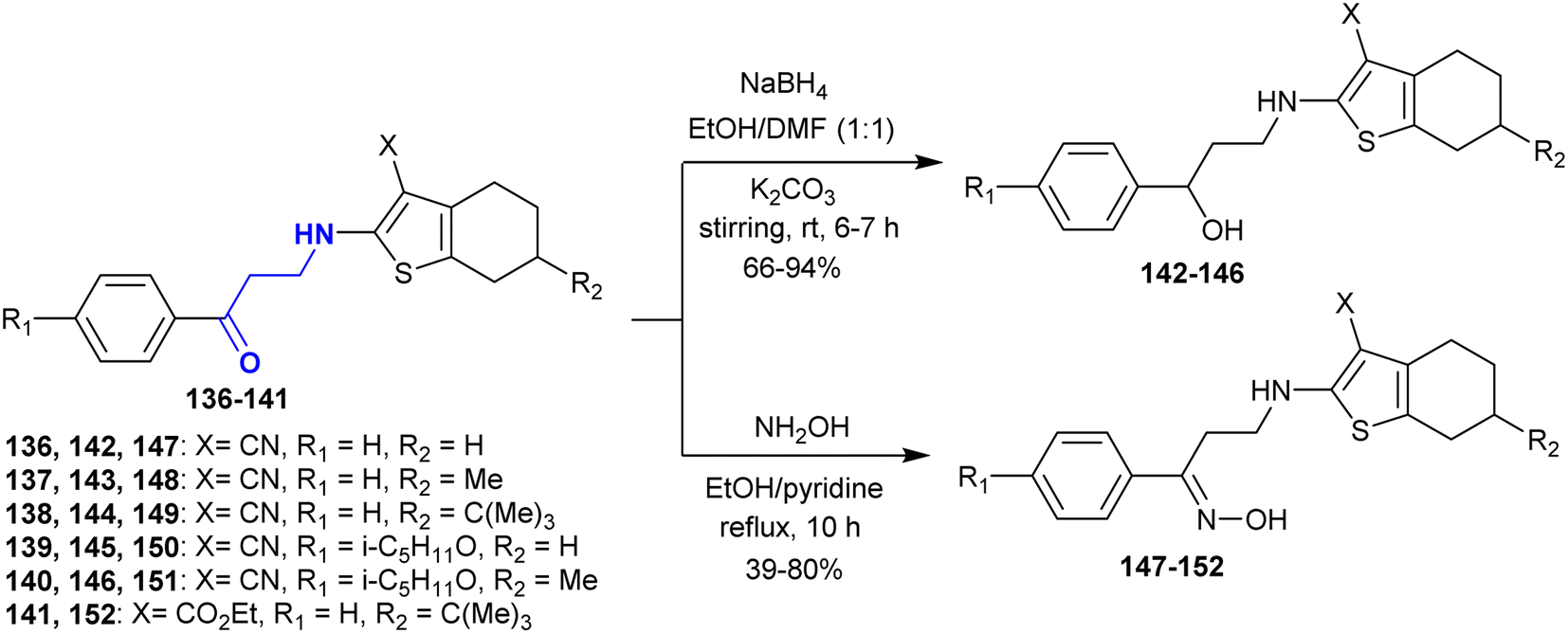
The selective reduction of β-aminoketones 136–141 with sodium borohydride in the presence of potassium carbonate yielded a series of amino alcohols 142–146. Alternatively, the reactions of β-aminoketones 136–141 with hydroxyl amine hydrochloride gave the corresponding oximes 147–152, respectively. These reactions proceeded under catalytic conditions of pyridine through nucleophilic attack of the amino group at the carbonyl group followed by condensation (Scheme 48). Compound 149 revealed the most potent antioxidant character given that it can inhibit the oxidation caused by free radicals through the generation of lipid peroxides by about 30%. In addition, compounds 142 and 144 displayed moderate activities, while compound 148 displayed reduced activity. Compounds 145, 146, and 151 are capable of acting on the free radical oxidation step reactions of lipids in the liver. Among them, compound 142 presented the greatest sensitivity against blood coagulation, while compounds 143, 144, and 147–151 had reduced activity compounds 145, 146, and 152 were inactive.154
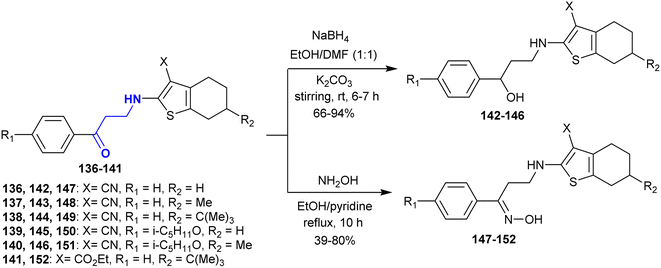 |
| | Scheme 48 Synthesis of aminoalcohols and oximes. | |
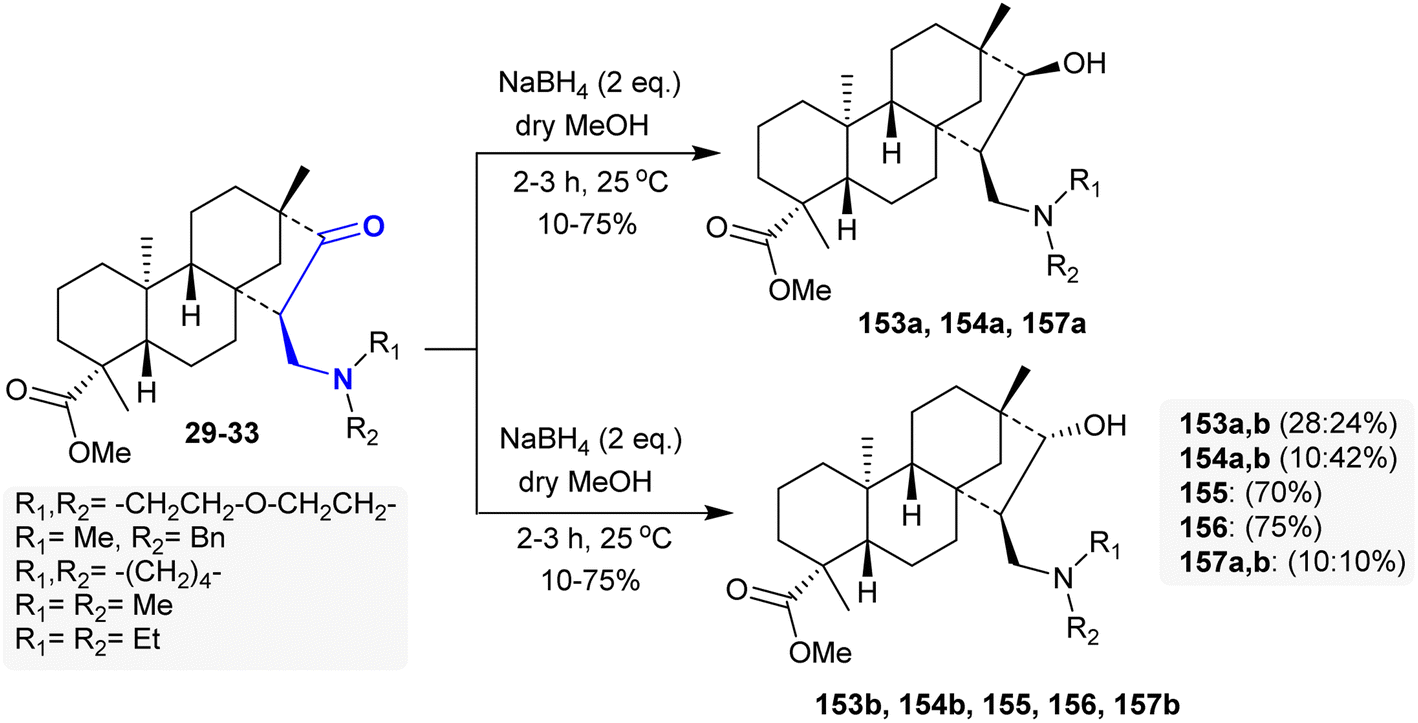
The respective β-aminoketone “methyl esters of isosteviol” 29–33 underwent selective reduction by treatment with sodium borohydride in dry methanol under mild reaction conditions to give 1,3-amino alcohols 153–157 through hydroxy-formylation processes. The 1,3-amino alcohol products are diastereomeric mixtures in some cases. Thus, the reduction of β-aminoketones 31 and 32 yielded products 155 and 156, respectively, with high stereoselectivity as single diastereoisomers (Scheme 49). Remarkable cytotoxic results were estimated for the 1,3-amino alcohol derivatives against A2780, SiHa, HeLa, MCF-7, and MDA-MB-231 human tumor cell lines. Compounds 154a and b are the most potent cytotoxic agents against HeLa (IC50 = 10.17 and 13.65 μM), SiHa (IC50 = 12.20 and 14.34 μM), MDA-MB-231 (IC50 = 9.20 and 9.26 μM), MCF-7 (IC50 = 17.29 and 17.24 μM), and A2780 (IC50 = 16.08 and 14.93 μM), respectively. The transformation of the β-aminoketones into 1,3-amino alcohols improved the cytotoxic effects of these compounds. In addition, the incorporation of N-benzyl substituents in the amino group is crucial for improved cytotoxic influence and privileged antiproliferative characters.107
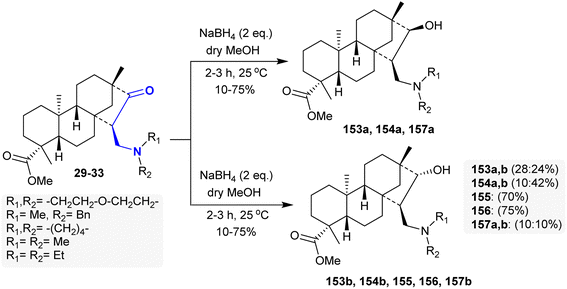 |
| | Scheme 49 Synthesis of amino alcohols. | |
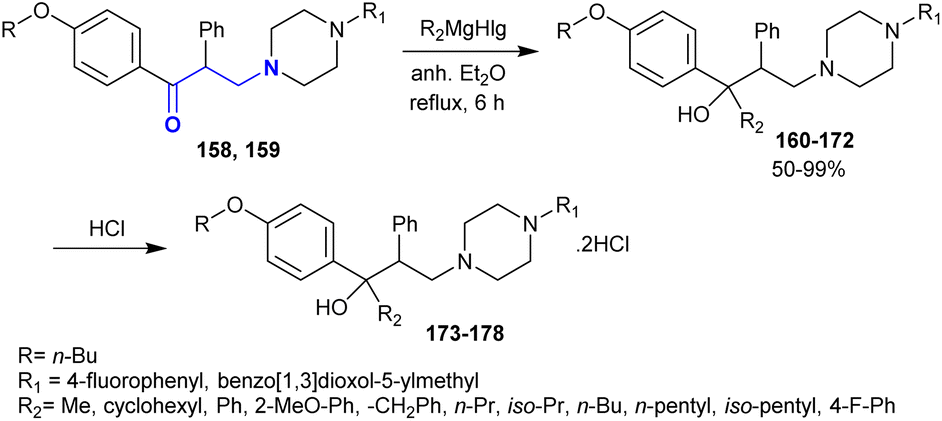
Owing to the privileged biological potency of tertiary amino alcohols,155,156 Gevorgyan et al.154 synthesized a series of tertiary piperazinyl amino alcohols 160–172 by the reaction of β-aminoketones 158 and 159 with a Grignard reagent “prepared from metallic magnesium with alkyl(aryl)halide in anhydrous diethyl ether”. The dihydrochloride salts of some of these compounds 173–178 were prepared by treatment with hydrochloric acid (Scheme 50). The impacts of the synthesized compounds 162, 163, 168, 170, 175, and 178 against C-180 mouse tumor DNA methylation were studied in vivo. The incubation step was accomplished for the drug sample with a concentration of 3 × 10−6 M with the tumor cell line at 37 °C for 24 h. The influence of the compounds on inhibiting DNA methylation was estimated. It was found that compound 162 “R = n-Bu, R1 = 4-fluorophenyl, R2 = phenyl” inhibited DNA methylation by 51.6%. The incorporation of the methoxy group at the o-position of the phenyl group reduced the activity to 37.5%.
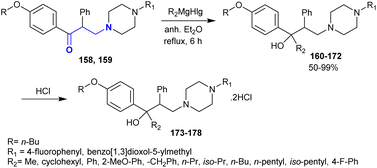 |
| | Scheme 50 Synthesis of amino alcohols and their dihydrochloride salts. | |
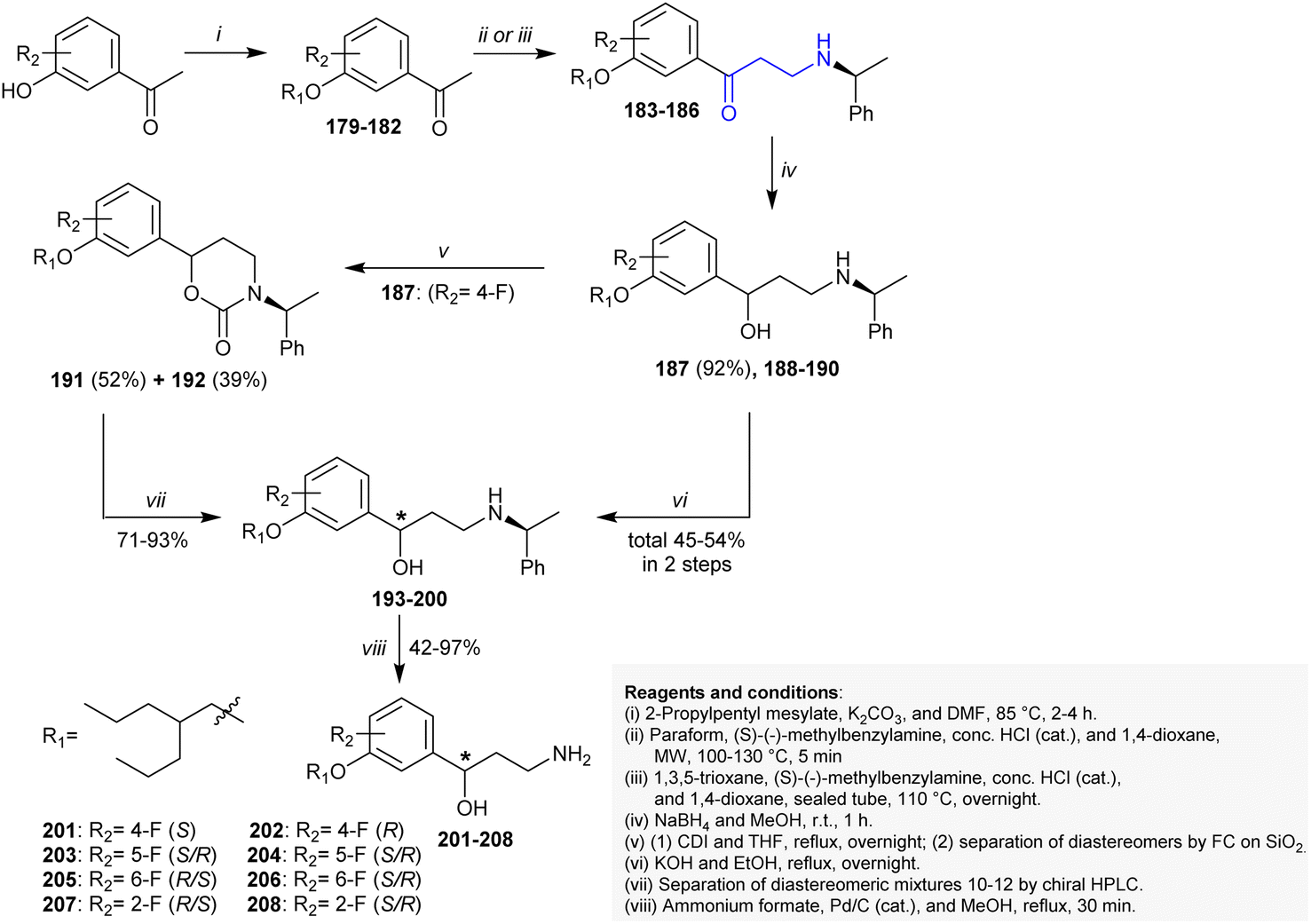
Blum et al.157 developed a synthetic route for the preparation of enantiopure γ-amino alcohols 201–208 from the reactive precursors β-aminoketones 183–186. Thus, the synthesis of this series is based on the preparation of the reactive precursor β-aminoketones 183–186. In this route, acetophenones reacted with 2-propylpentyl mesylate in DMF/K2CO3 under heating conditions followed by a Mannich three-component reaction with chiral benzylamines under microwave irradiation conditions to yield β-aminoketones 183–186. The reactions proceeded under HCl catalytic conditions with the preparation of compounds 183 and 185 in moderate yields (47%) together with insufficient yields of compounds 184 and 186 (<20%). Alternatively, a proficient method was applied for the preparation of compounds 184 (34%) and 186 (78%) by heating compounds 180 and 182 with 1,3,5-trioxane in a sealed pressure tube. The reduction of compounds 183–186 with sodium borohydride in methanol gave an analogous diastereomer mixture of N-protected γ-amino alcohols 187–190. The preparative chiral HPLC technique was applied to separate diastereomeric mixtures of compounds 187–190, respectively. Nitrogen deprotection of compounds 193–200 was accomplished by reductive hydrogenation with Pd/C in the presence of ammonium formate to yield the enantiomeric γ-hydroxyalkyl amine products 201–208, respectively (Scheme 51).
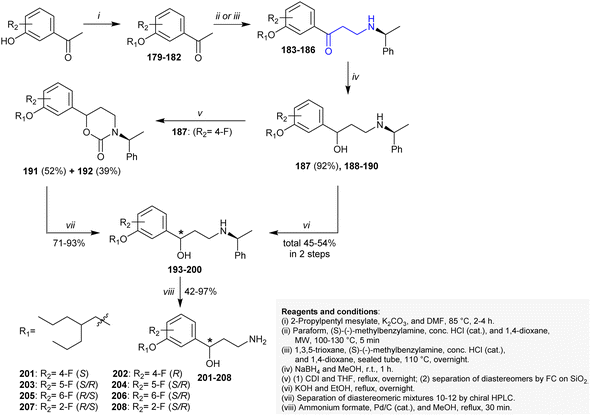 |
| | Scheme 51 Synthesis of amino alcohols. | |
4.2. Synthesis of metal complexes
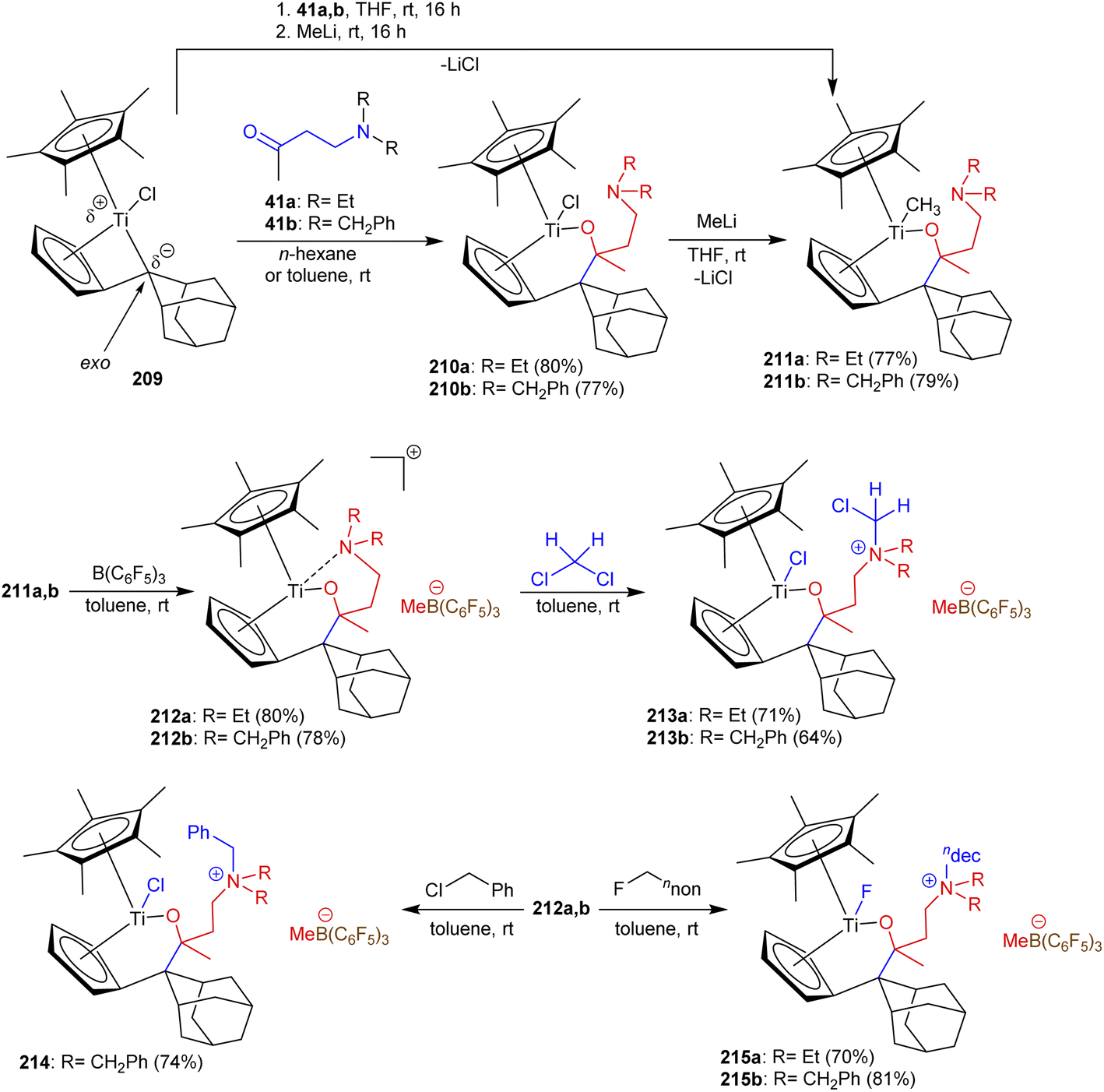
As reported by Fischer et al.,114 β-aminoketones 41a and b were involved in the exceedingly proficient synthesis of titanium complexes. Therefore, the monopentafulvene complex 1 reacted with β-aminoketones 41a and b as bidentate O,N-ligands and yielded the cationic titanium complexes 212a and b through multistep synthesis, respectively. The reactions involved treatment with ligands in n-hexane or toluene at room temperature followed by treatment with methyl lithium and the strong Lewis acid B(C6F5)3 for activation. Subsequently, the cationic complexes 213 were prepared by the treatment of 212 with dichloromethane with C–Cl bond cleavage. Besides, treatment of 212a with benzyl chloride in toluene afforded titanium complex 214 in good yield through the formation of a Ti–Cl bond. Predominantly, treatment of titanium complexes 212a and 212b with n-fluorodecane under the same previous conditions yielded complexes 215a and 215b, respectively, over the formation of Ti–F bonds (Scheme 52).
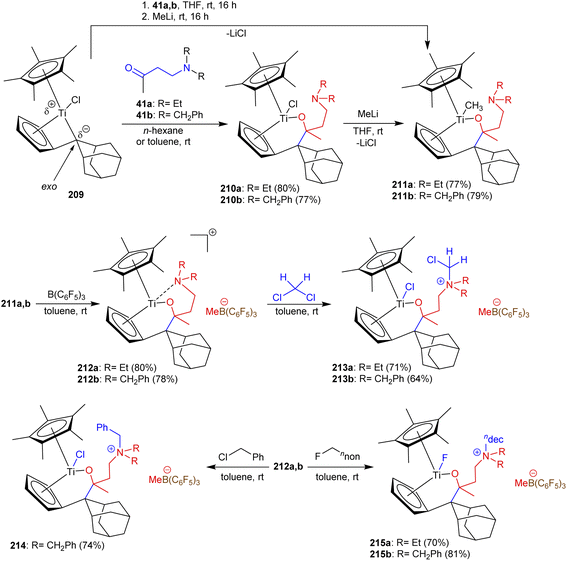 |
| | Scheme 52 Synthesis of cationic titanium complexes. | |
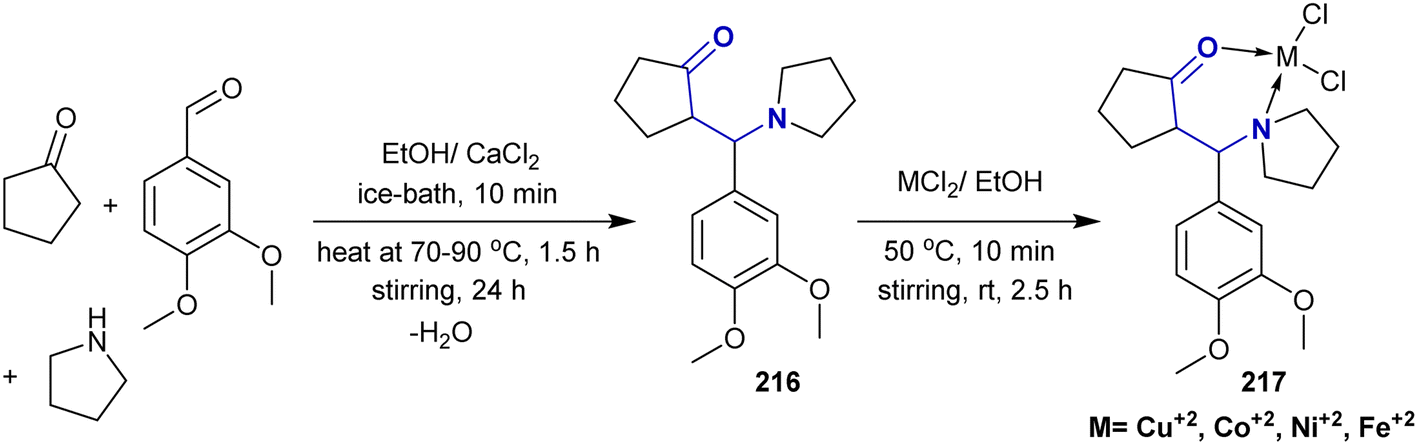
According to Liaqat et al.,158 Mannich base 216 was prepared as a ligand for the synthesis of metal complexes. Thus, the multicomponent reaction of cyclopentanone with 3,4-dimethoxybenzaldehyde and pyrrolidine in equimolar ratios in ethanol containing calcium chloride gave the corresponding Mannich base 216. Consequently, treatment of the Mannich base ligand with equimolar ratios of the anticipated metal chlorides yielded the respective metal complexes 217 (Scheme 53).
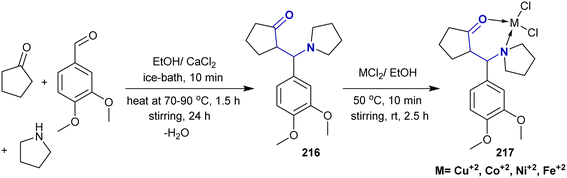 |
| | Scheme 53 Synthesis of β-aminoketones and complex formation. | |
4.3. Synthesis of fluoxetine derivatives
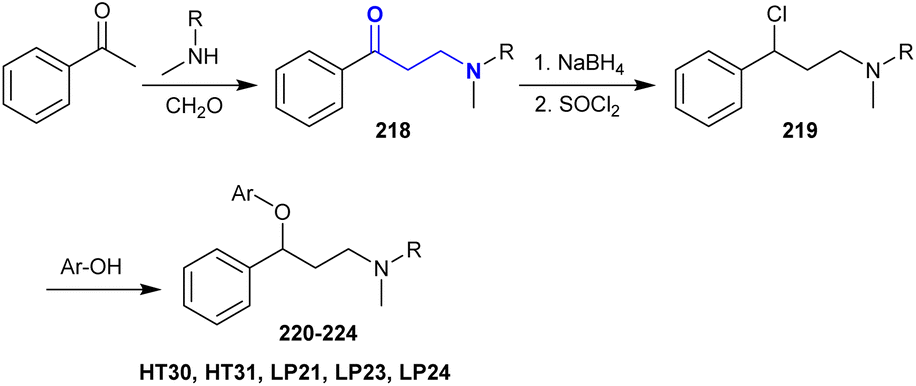
A series of β-aminoketones was synthesized by the Mannich reaction of acetophenone with methyl amines and formaldehyde in ethanol under reflux conditions. Subsequent reduction of β-aminoketones 218 followed by treatment with thionyl chloride in dichloromethane under heating conditions yielded the respective chloro-phenyl propane-amines 219. Fluoxetine derivatives 220–224 were efficiently synthesized through reactions of amines 219 with 3- or 4-trifluoromethyl phenol, 4-hydroxybenzaldehyde or 4-hydroxybenzonitrile, respectively, under gentle heating in DMF catalyzed by potassium carbonate (Scheme 54).159
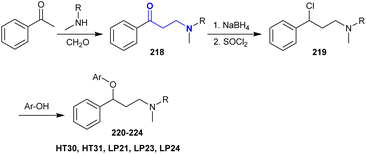 |
| | Scheme 54 Synthesis of fluoxetine analogs. | |
4.4. Synthesis of sotolon
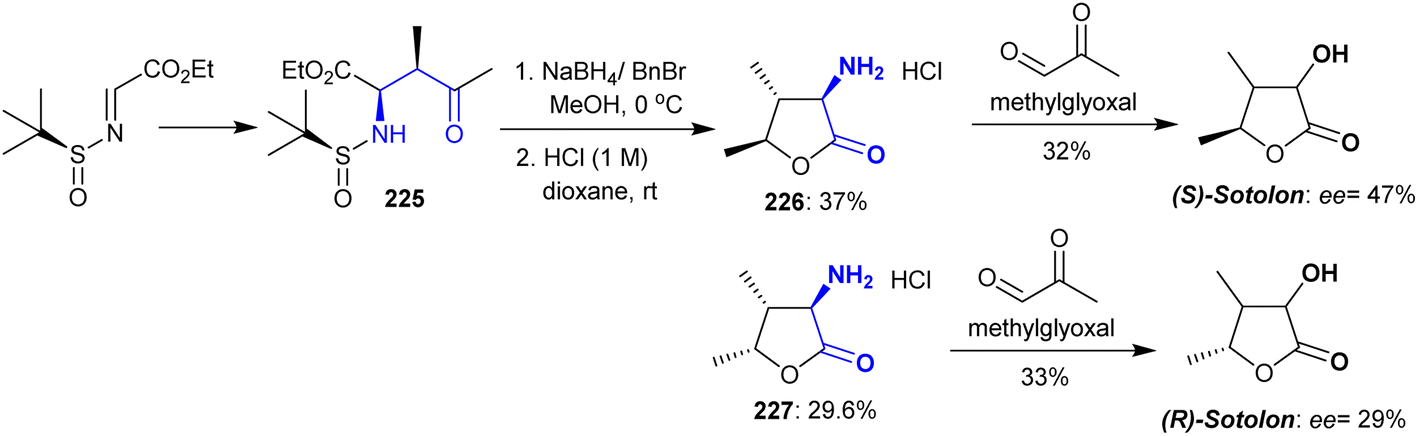
The synthesis of sotolon was developed by Trang et al.160 from cyclic β-aminoketones 226 and 227 through their reactions with methylglyoxal. An auxiliary chiral starting precursor such as (S)–N-tert-butane sulfinimine was incorporated in the synthesis of sotolon as an optically active compound. Consequently, the reduction of ethyl (2R,3R)-2-(((S)-tert-butylsulfinyl)amino)-3-methyl-4-oxopentanoate with sodium borohydride in the presence of benzyl bromide, followed by treatment with hydrochloric acid resulted in sulfinyl group cleavage and aminolactone ring closure with the generation of two optically pure products 226 and 227 as a racemic mixture (ee > 99%). The transformation of amine hydrochlorides into the optically active (S)- and (R)-sotolon was processed in the presence of a phosphate buffer solution (Scheme 55). Hence, the plausible mechanism of this reaction was tandem isomerization-aldolization and isomerization-Mannich reactions.
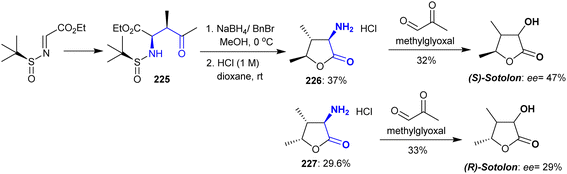 |
| | Scheme 55 Synthesis of optically active sotolon. | |
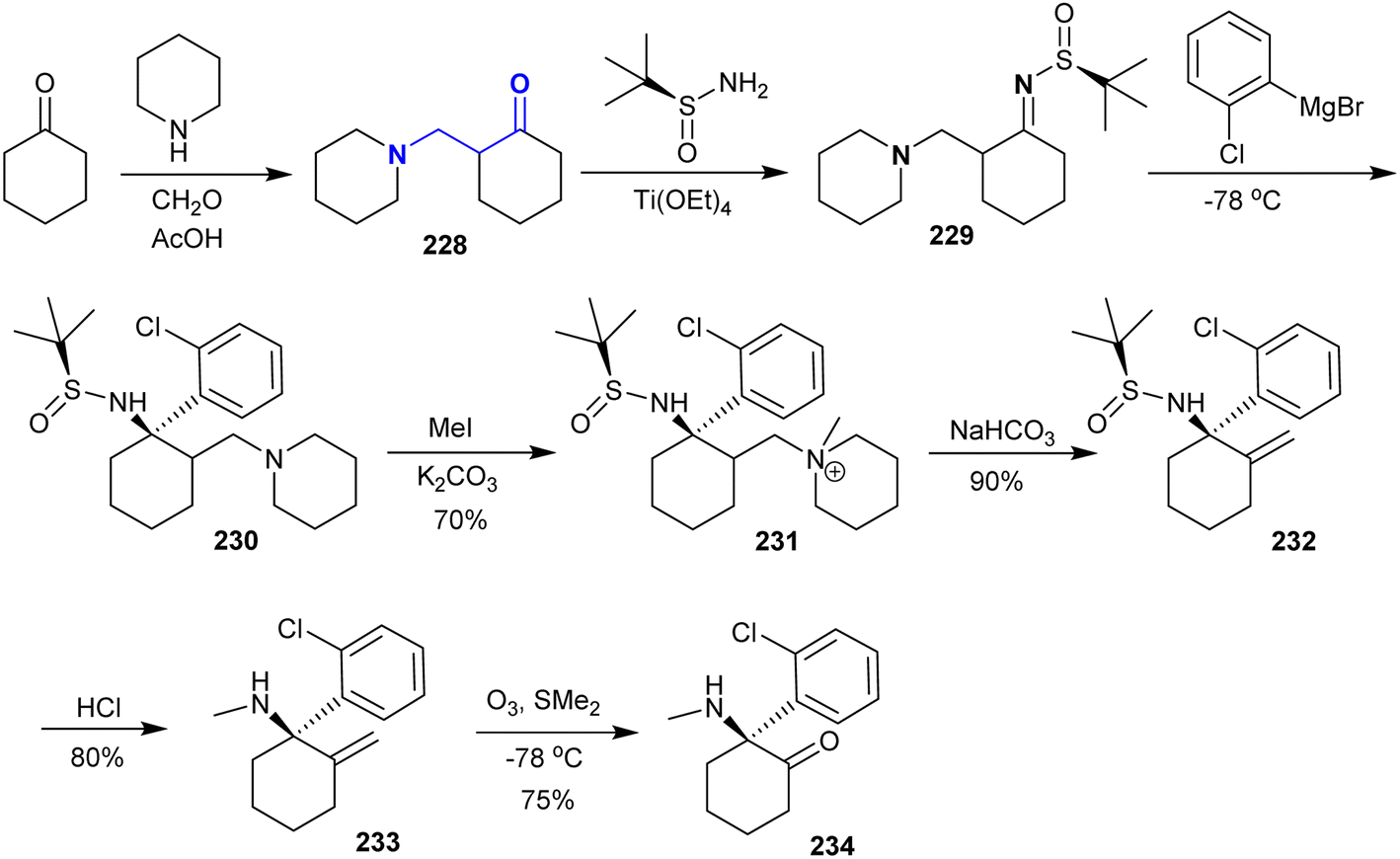
4.5. Asymmetric synthesis of (S)-ketamine
Gohari et al.161 reported the synthesis of (S)-ketamine from a multistep reaction sequence. Therefore, the Mannich reaction of cyclohexanone with piperidine and formaldehyde afforded the desired β-enaminone 228. Subsequently, β-enaminone 228 reacted with (S)-2-methylpropane-2-sulfinamide through the condensation step under tetraethoxytitanium catalytic conditions to give sulfiniylamin 229 in 85% yield. The reaction of 229 with Grignard reagent gave compound 230 in 80% yield, which was methylated with methyl iodide to produce the quaternary salt 231 (70% yield). Treatment of salt 231 with sodium bicarbonate afforded sulfinamide 232. Deprotection of the amino group by removal of the t-butyl sulfinyl group was achieved by treatment with hydrochloric acid to give (R)-1-(2-chlorophenyl)-N-methyl-2-methylenecyclohexan-1-amine (233). (S)-Ketamine 234 was enantioselectively synthesized in 75% (75% ee) yield in the final step through the ozonolysis step (Scheme 56).
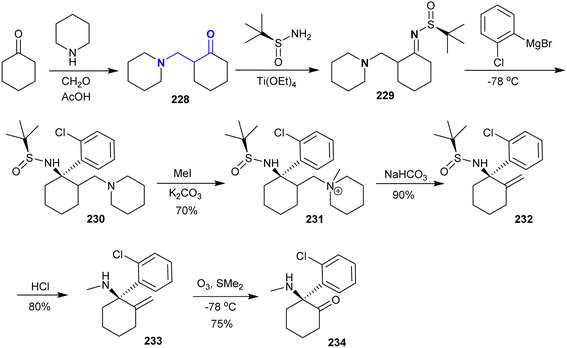 |
| | Scheme 56 Multistep synthesis of (S)-ketamine. | |
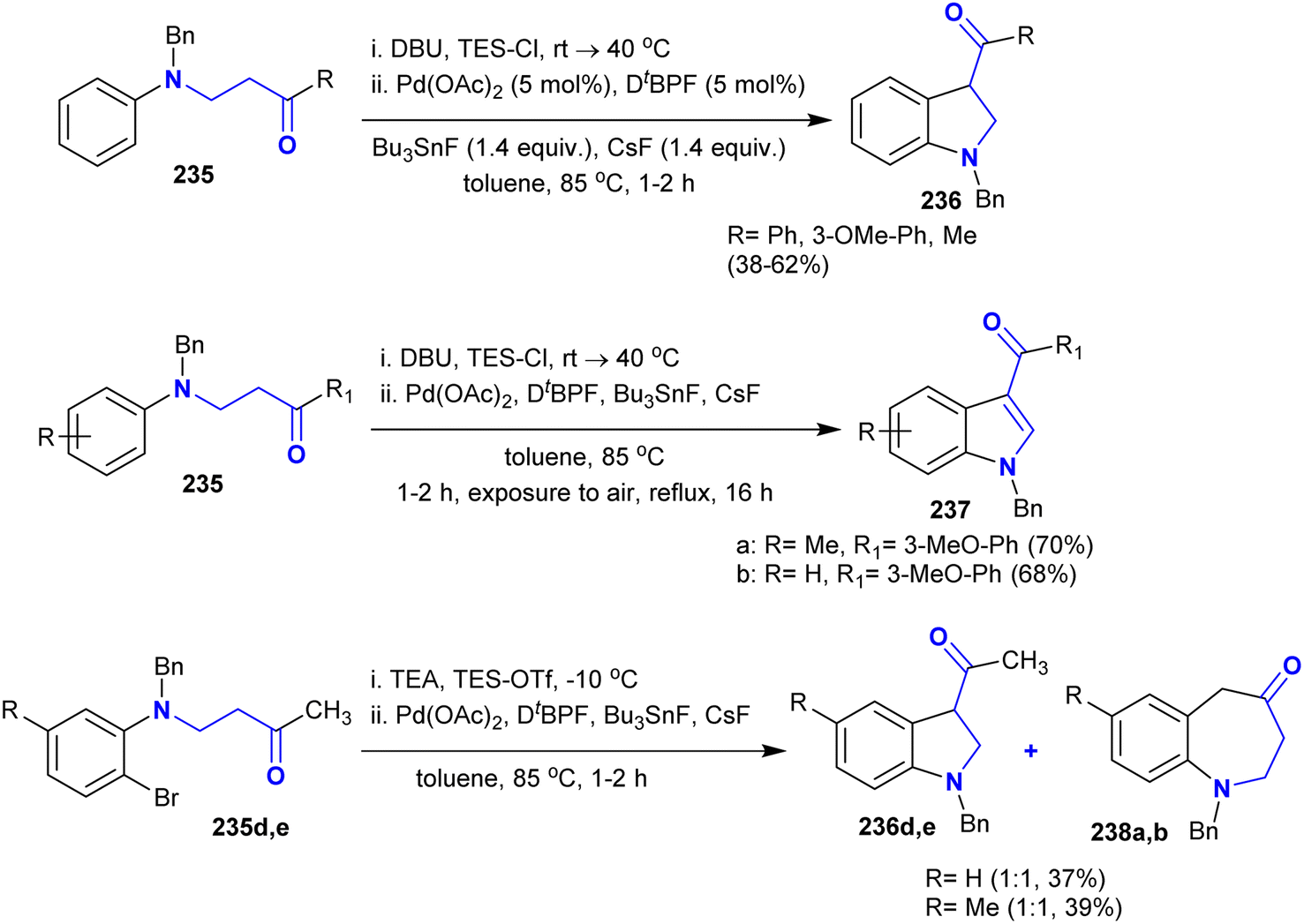
4.6. Synthesis of indolines and benzoazepinones
The procedure reported by Kumar et al.118 was extended to the synthesis of a series of indolines and benzoazepinones. Thus, the annulation of N-aryl-β-aminoketones under palladium-catalyzed conditions gave the target products 236–238 through intramolecular α-arylation with one-pot annulations (Scheme 57). The treatment of β-aminoketones with chlorotriethylsilane in the presence of DBU led to the in situ generation of the respective silylenol ethers, which were annulated by the Pd-catalyst with high selectivity. The reactions yielded azepinones and 3-acylindolines by the use of triethylamine and triethylsilyl trifluoro-methane-sulfonate for the enolization of β-aminoketones.
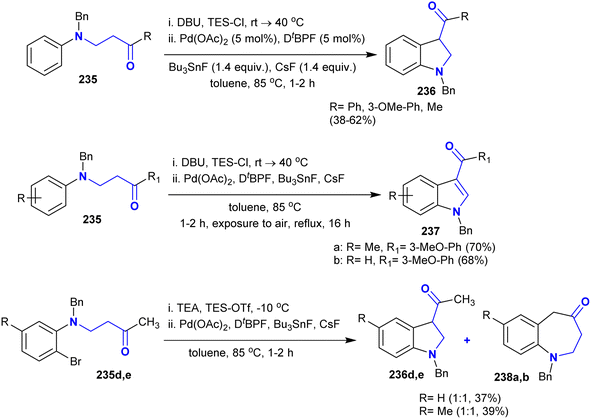 |
| | Scheme 57 Synthesis of indolines and benzoazepinones. | |
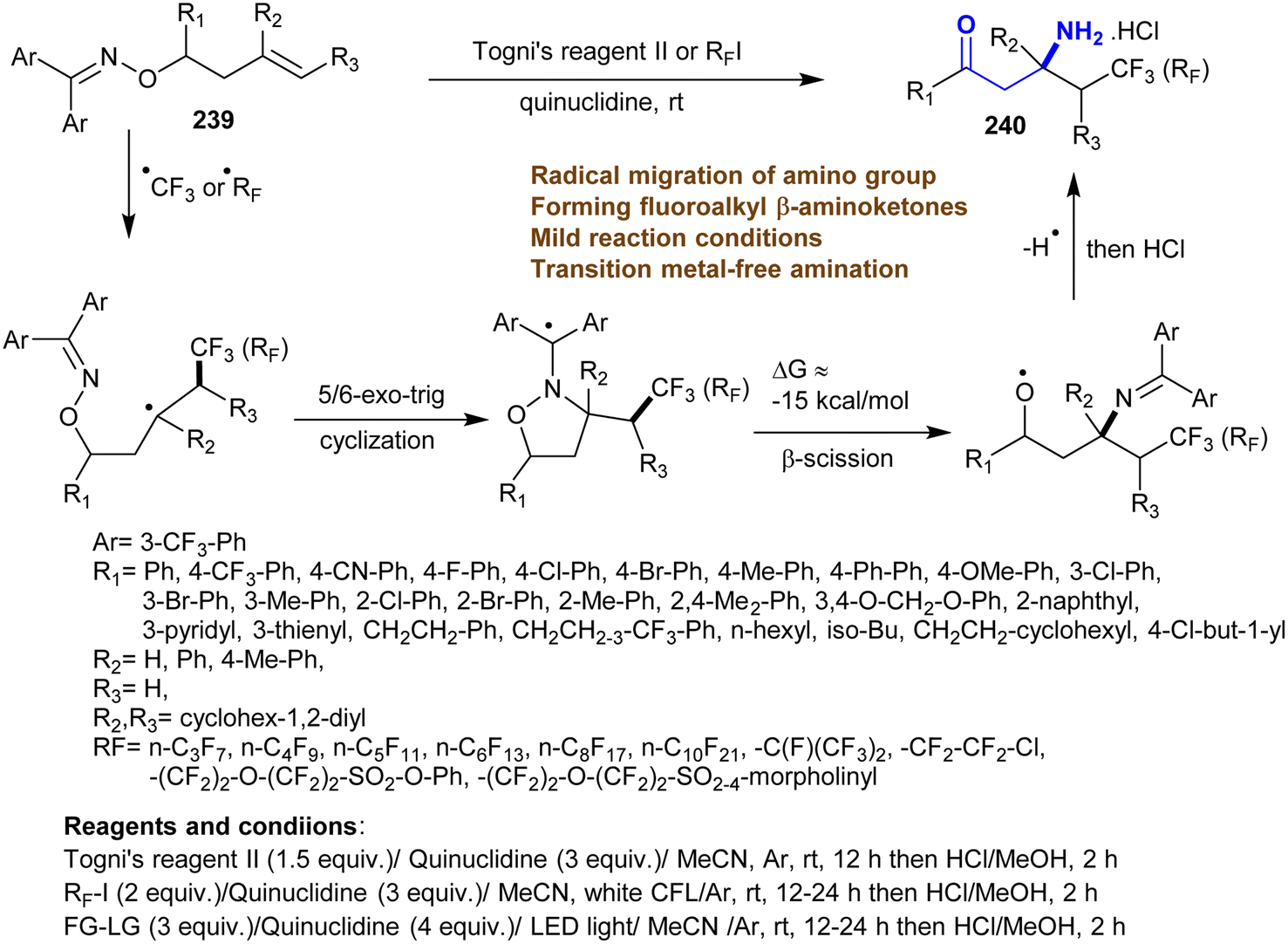
5. Miscellaneous approaches
The reactions of oximes 239 with quinuclidine as an electron-donor in the presence of Togni's reagent in acetonitrile under argon at 25 °C followed by hydrolysis via HCl gave the desired hydrochlorides of β-aminoketones 240 (Scheme 58). The amine was necessary for this conversion, where its absence led to no reaction. The effects of the substituents on the product yield were noticed clearly through the reactions using 4-MeO-Ph, Ph, and fluorenyl substituents instead of 3-CF3-Ph, which led to reduced yields of β-aminoketones 240. The mechanism of this type of reaction involved the addition of fluoroalkyl radicals on the alkene CC double bond with the radical formation in the C2 position. The fluoroalkyl radicals were produced from a complex of electron donor–acceptor of Togni's reagent II or fluoroalkyl iodides and quinuclidine. Subsequently, 5(6)-exo-trig cyclization of C2 radical on the nitrogen atom of ketoxime ethers followed by N–O bond cleavage with the formation of π bond between C–N. Other examples of radical sources can be applied using this protocol instead of Togni's reagent II and fluoroalkyl iodides.162
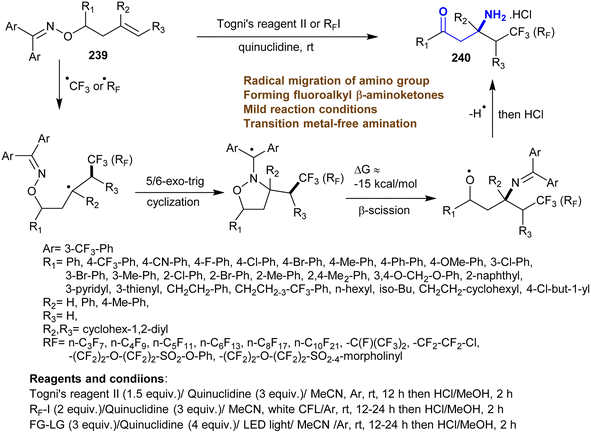 |
| | Scheme 58 Synthesis of β-aminoketones through amination and radical migration of amino group. | |
6. Reactivity
6.1. Amino group substitution
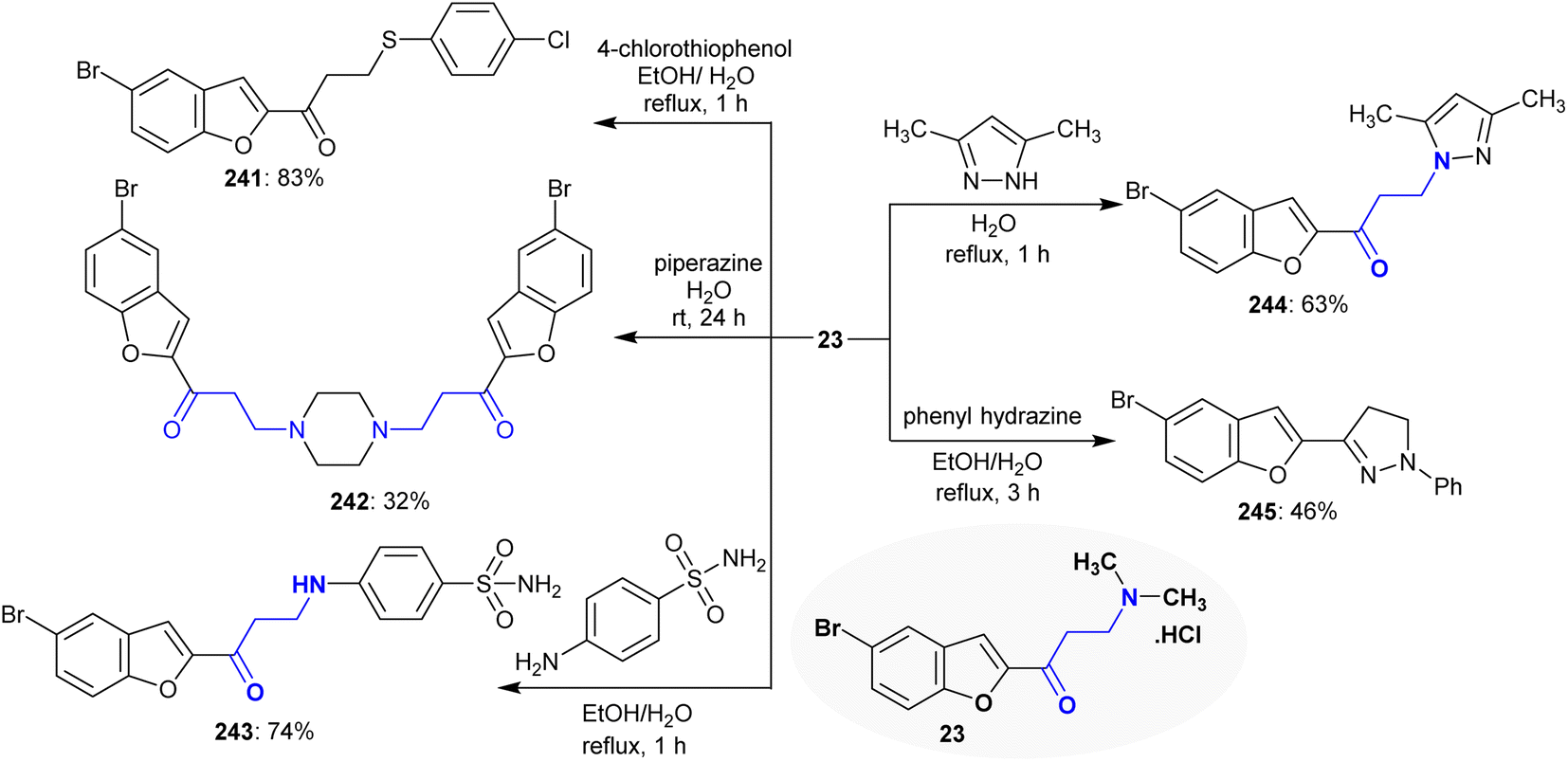
The reactivity of Mannich base 23 was investigated against a variety of nucleophiles, in which the product type depended on the reacted nucleophiles. Thus, Mannich base 23 reacted with 4-chlorothiophenol to give β-(arylmercapto)ketone 241, while its reaction with piperazine “aliphatic” in a 2:1 molar ratio yielded the symmetric bis(1-(5-bromobenzofuran-2-yl)propan-1-one) 7. Particularly, the reaction of Mannich base 23 with an acyclic aromatic amine such as 4-aminobenzenesulfonamide and cyclic 3,5-dimethyl-1H-pyrazole yielded the corresponding β-aminoketones 243 and 244, respectively. The reactions involved nucleophilic substitutions through the attack of the sulfur or amine nucleophiles at the β-carbon. Alternatively, the reaction of Mannich base 23 with phenyl hydrazine tended to the ring closure product 245 in moderate yield (Scheme 59). The first step was the nucleophilic substitution of the NH group of phenyl hydrazine followed by cyclocondensation.104
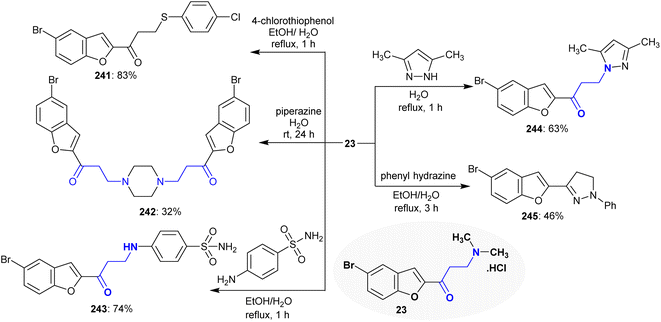 |
| | Scheme 59 Substitution reactions of β-amino group. | |
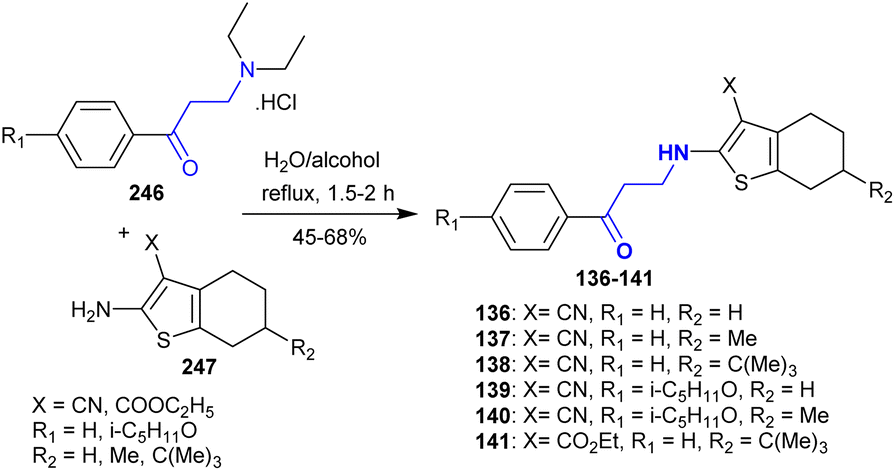
Gevorgyan et al.154 also developed the synthesis of a series of β-aminoketones 136–141 in a mixture of water and alcohol after a short time. Thus, 3-(diethylamino)-1-(aryl)propan-1-one hydrochlorides 246 reacted with tetrahydrobenzo[b]-thiophenes 247 under free-catalyst conditions to afford the desired β-aminoketones 136–141 (Scheme 60). The antioxidant evaluation assay demonstrated that compounds 136 and 139 displayed moderate activities for inhibiting the free radical reactions, while lower activities were recorded for compounds 137 and 140 and the active character for compound 141. Compounds 136, 137, and 139–141 presented the most sensitive shifts for the activation of the blood coagulation system.
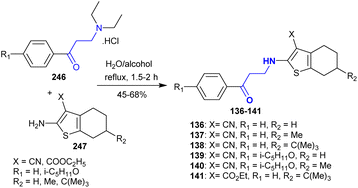 |
| | Scheme 60 Synthesis of β-aminoketones. | |
6.2. Reactivity of substituents
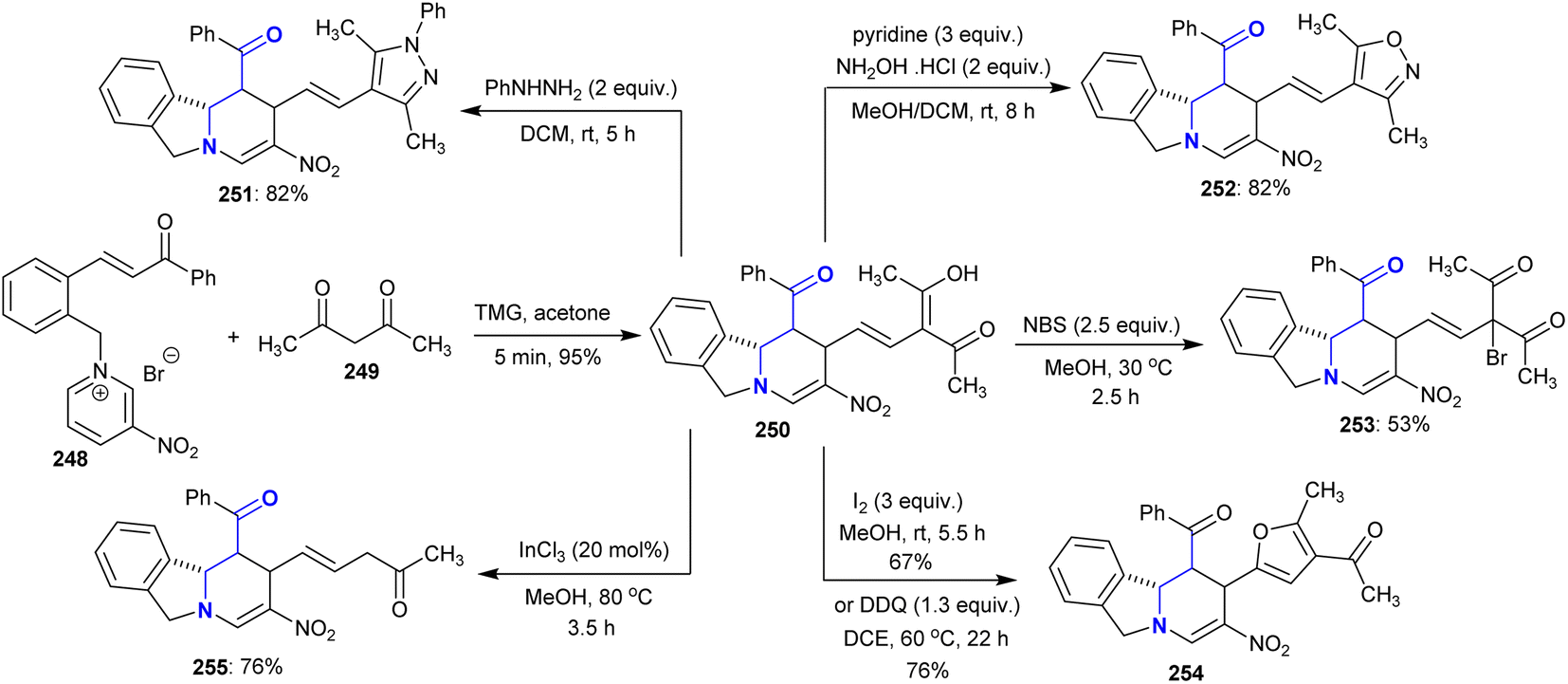
Under optimized conditions, tetrahydropyrido[2,1-a]isoindoline 250 was prepared by the reaction of acetyl acetone with chalcone-based pyridinium salt in acetone with excellent yield (95%). The reactivity of compound 250 was investigated through reactions with phenyl hydrazine, hydroxylamine hydrochloride, and N-bromosuccinimide to yield binary heterocycles and branched halides 251–253, respectively. Also, the oxidative cyclization in the presence of iodine gave the acetyl furan analog 254, and deacetylation of one acetyl group in the presence of InCl3 catalyst gave compound 255 in 76% yield (Scheme 61).144
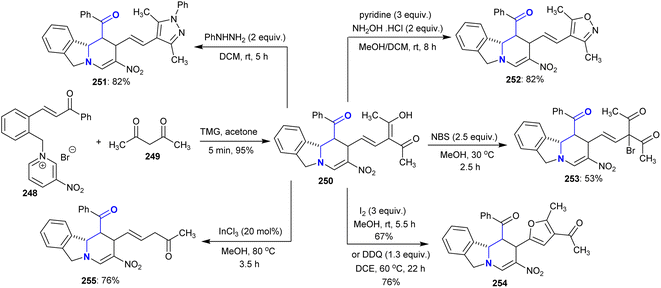 |
| | Scheme 61 Reactivity of tetrahydropyrido[2,1-a]isoindoline. | |
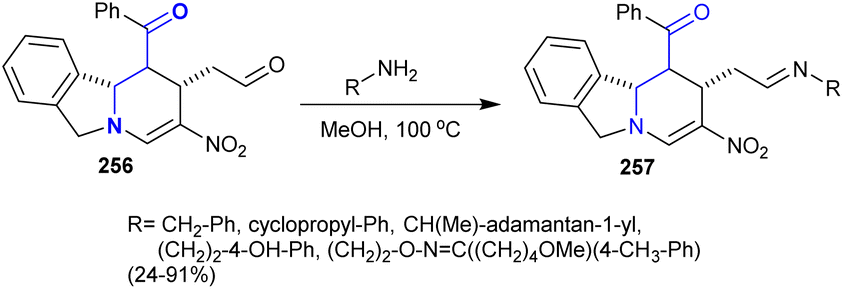
6.3. Synthesis of imines
In this case, tetrahydropyrido[2,1-a]isoindole 256 reacted with various amines in methanol under reflux conditions to give the desired imines 257 in 24–91% yield (Scheme 62). These imines are assumed to have potential biological and pharmaceutical interest.144
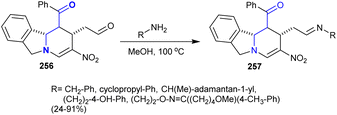 |
| | Scheme 62 Synthesis of imines. | |
6.4. Reductive cyclization
Treatment of tetrahydropyrido[2,1-a]isoindol-2-ylbut-2-enoate 258 with sodium boron hydride in methanol led to the formation of hexahydro-1H-pyranopyrido[2,1-a]isoindole 259 in a moderate yield (55%). Product 259 was obtained through the reduction process followed by Michael addition in a sequence of reductive cyclization (Scheme 63).144
 |
| | Scheme 63 Reductive cyclization. | |
7. Concluding remarks
Formerly, Gevorgyan et al.163 reviewed the same topic regarding the advances in the chemistry of β-aminoketones and published an update in 1985 regarding the chemical reactivity of this type of moiety. In 2008, Simplício et al.164 reviewed the amine prodrugs including the significance of β-aminoketones as the privileged motif of many drugs. Alternatively, Verkade et al.165 reviewed the utility of organocatalysts in asymmetric Mannich reactions, which was accompanied by the study of Allochio Filho et al.166 in 2017, where they reported multicomponent Mannich reactions including their various applications. The present study highlighted the synthetic importance of the preparation of β-aminoketones and their applications given that this type of core is the basic structure of many drugs. The various synthetic strategies for β-aminoketones included multicomponent synthesis, e.g., Mannich reactions under catalytic and non-catalytic conditions, addition to imines and unsaturated compounds, and condensation reactions of alcohols with amines. The multicomponent synthesis is a beneficial route for the synthesis of piperidines, while morpholinones, and piperazinones are prepared from alkenyl Grignard reagents with the corresponding ester. The spirocyclic systems are prepared from reactions of cycloalkanol-1H-indenes with 1-(arylsulfonamido)-pyridinium tetrafluoroborates. Also, imidazolines are synthesized by applying regio-, stereo-, and chemoselective deamination of enones employing electrophiles such as N-chlorosuccinimide and tosyl amine. Furthermore, indolizine-based β-aminoketones are efficiently synthesized through aza-Cope rearrangement–Mannich cyclization with high stereoselectivity. These protocols were extended to include the synthesis of pyrido-isoindoles from reactions of N-aryl-1,2,3,4-tetrahydroisoquinolines with α-diazoketones under catalytic conditions. Besides, propargylic alcohols are beneficial substrates for the synthesis of β-aminoketones and their reactions with N-Ts substituted hydrazines. Other methods are applied for the synthesis of β-aminoketones such as reductive hydroamination of ynones, alkylation of alicyclic imines with β-ketoacids, carbonylative coupling, coupling of halo ketones with amines, and acid hydrolysis of metal complexes. The synthetic applications of β-aminoketones covered the synthesis of amino alcohol and oxime derivatives, metal complexes, fluoxetine, sotolon, (S)-ketamine, indolines, and benzoazepinones. The compounds with β-aminoketone fragments underwent tertiary amino group substitutions. The reactions of substituents were extended to include the synthesis of imines and reductive cyclization.
8. Future prospective
N-Mannich bases have been extensively employed as prodrugs of amine drugs. The corresponding C-Mannich bases (β-aminoketones) have attracted much less interest perhaps because they are not appropriately liable to undergo in vivo elimination at the biological pH.167 In addition, β-aminoketones were firstly observed to be inhibitors of TR coactivator interaction through the utility of high-throughput screening employing a library of 138000 compounds in an in vitro fluorescence polarization assay.168 Although researchers are interested in preparing many compounds that contain the β-aminoketone nucleus to explore the multiple biological properties of these compounds, many researchers have focused on developing and finding different preparation approaches instead of exploiting these compounds by including them in various biological assessments.
Conflicts of interest
The authors state no conflict of interest.
Abbreviations
| Polycarbonates | (PC); |
| 2,2′-Bipyridine | (bpy); |
| Argon | (Ar); |
| Room temperature | (rt); |
| Boron hydrogen sulfate | (B(HSO4)3); |
| Iron-doped multi-walled carbon nanotubes | (Fe/MWCNTs); |
| Sulfonated-polyethylene glycol-coated Fe3O4 nanocomposite | (Fe3O4/PEG-SO3H); |
| Tri-methylammonium-butane sulfonate | (TMBSA) ionic liquid (IL); |
| 3-(N,N-Dimethyldodecylammonium) propanesulfonic acid hydrogen sulfate | ([DDPA][HSO4]); |
| Human ovarian cancer cell line | (A2780); |
| A cell line isolated from fragments of a primary uterine tissue | (SiHa); |
| An immortal cell line | (HeLa); |
| An epithelial cell line isolated from the breast tissue | (MCF7); |
| An epithelial, human breast cancer cell line | (MDA-MB-231); |
| Lithium diisopropylamide | (LDA); |
| Trifluoroacetic acid palladium(II) salt | (Pd(TFA)2); |
| 2-Nitrobenzenesulfonamide | (2-NsNH2); |
| Bismuth(III) trifluoromethanesulfonate | (Bi(OTf)3); |
| 1,1′-Ferrocenediyl-bis(diphenylphosphine) | (dppf); |
| Human ovarian cancer cell line | (A2780); |
| 1,8-Diazabicyclo[5.4.0]undec-7-ene | (DBU); and |
| N-Bromosuccinimide | (NBS) |
References
- A. Ravn, M. Vilstrup, P. Noerby, K. Daasbjerg and T. Daasbjerg, J. Am. Chem. Soc., 2019, 141, 11821–11826 CrossRef CAS PubMed.
- N. H. Nguyen, A. B. Hughes and B. E. Sleebs, Curr. Org. Chem., 2014, 18, 260–289 CrossRef CAS.
- M. Altmeyer, E. Amtmann, C. Heyl, A. J. Scheidig and C. D. Klein, Bioorg. Med. Chem. Lett., 2014, 24, 5310–5314 CrossRef CAS PubMed.
- Y. Du, Q. Li, B. Xiong, D. Zhang and M. Wang, Bioorg. Med. Chem., 2010, 18, 4255–4268 CrossRef CAS PubMed.
- S. Krechetov, G. Nifontova, O. Dolotova and M. Veselov, Pharm. Chem. J., 2018, 52, 41–45 Search PubMed.
- G. Nifontova, S. Krechetov, O. Dolotova, S. Buyukli, A. Akhmetzyanova and I. Krasnyuk, Pharm. Chem. J., 2018, 52, 48–52 Search PubMed.
- C. Chamseddin and T. Jira, Anal. Methods, 2014, 6, 6702–6710 RSC.
- D. Hofer, B. Lohberger, B. Steinecker, K. Schmidt, S. Quasthoff and W. A. Schreibmayer, Eur. J. Pharmacol., 2006, 538, 5 CrossRef CAS PubMed.
- J. C. Kaski, L. Araujo and A. Maseri, Drugs Ther., 1991, 5, 991 CAS.
- C. F. Deacon, Nat. Rev. Endocrinol., 2020, 16, 642 CrossRef CAS PubMed.
- H. Wang, X. Li, Y. Tu and J. Zhang, Science, 2020, 23, 101138 CAS.
- Y. You and S. Luo, Org. Lett., 2018, 20, 7137 CrossRef CAS PubMed.
- C. Cabrele, T. A. Martinek, O. Reiser and L. Berlicki, J. Med. Chem., 2014, 57, 9718–9739 CrossRef CAS.
- M. J. Koyack and R. P. Cheng, Methods Mol. Biol., 2006, 340, 95–109 CAS.
- C. M. Czekster, W. E. Robertson, A. S. Walker, D. Soll and A. Schepartz, J. Am. Chem. Soc., 2016, 138, 5194–5197 CrossRef PubMed.
- F. Kudo, A. Miyanaga and T. Eguchi, Nat. Prod. Rep., 2014, 31, 1056–1073 RSC.
- A. L. Simplicio, J. M. Clancy and J. F. Gilmer, Int. J. Pharm., 2007, 336, 208–214 CrossRef CAS PubMed.
- R. L. Robinson, J. Chem. Soc., 1917, 111, 762–763 RSC.
- K. Sun, Y. Lv, Z. Zhu, B. Xiao and X. Wang, RSC Adv., 2015, 5, 3094–3097 RSC.
- K. Matsumoto, S. Hashimoto, S. Otani, F. Atnita and J. Osugi, Synth. Commun., 1984, 14, 585–590 CrossRef CAS.
- S. Kobayashi and H. Ishitani, Chem. Rev., 1999, 99, 1069–1094 CrossRef CAS PubMed.
- C. Solé and E. Fernández, Angew. Chem., Int. Ed., 2013, 52, 11351–11355 CrossRef PubMed.
- X.-J. Tang, Z.-L. Yan, W.-L. Chen, Y.-L. Zhang and Y.-Q. Wang, Tetrahedron Lett., 2013, 54, 2669–2673 CrossRef CAS.
- D. Trubitsõn, J. Martõnova, M. Kudrjasova, I. Järving and T. Kanger, Org. Lett., 2021, 23, 1820–1824 CrossRef PubMed.
- S. D. Bull, S. G. Davies, S. Delgado-Ballester, G. Fenton, P. M. Kelly and A. D. Smith, Synlett, 2000, 1257–1260 CAS.
- S. G. Davies and T. D. McCarthy, Synlett, 1995, 700–702 CrossRef CAS.
- N. Srivastava and B. K. Banik, J. Org. Chem., 2003, 68, 2109–2114 CrossRef CAS PubMed.
- G. Bartoli, M. Bosco, E. Marcantoni, M. Petrini, L. Sambri and E. Torregiani, J. Org. Chem., 2001, 66, 9052–9055 CrossRef CAS PubMed.
- I. Reboule, R. Gil and J. Collin, Tetrahedron Lett., 2005, 46, 7761–7764 CrossRef CAS.
- H. Firouzabadi, N. Iranpoor and A. A. Jafari, Adv. Synth. Catal., 2005, 347, 655–661 CrossRef CAS.
- P. Schuda, C. Ebner and T. Morgan, Tetrahedron Lett., 1986, 27, 2567–2570 CrossRef CAS.
- R. SanMardín, E. Marigorta and E. Domífnguez, Tetrahedron, 1994, 50, 2255–2264 CrossRef.
- Y. Fukumoto, H. Asai, M. Shimizu and N. Chatani, J. Am. Chem. Soc., 2007, 129, 13792–13793 CrossRef CAS PubMed.
- K. Hesp and M. Stradiotto, J. Am. Chem. Soc., 2010, 132, 18026–18029 CrossRef CAS PubMed.
- D. Leitch, P. Payne, C. Dunbar and L. Schafer, J. Am. Chem. Soc., 2009, 131, 18246–18247 CrossRef CAS.
- J. Bahri, R. Blieck, B. Jamoussi, M. Taillefer and F. Monnier, Chem. Commun., 2015, 51, 11210–11212 RSC.
- Y. You and S. Ge, Angew. Chem., Int. Ed., 2021, 60, 20684–20688 CrossRef CAS PubMed.
- J. Seah, Y. Li, S. Pullarkat and P.-H. Leung, Organometallics, 2021, 40, 2118–2122 CrossRef CAS.
- W.-J. Hao, B. Jiang, S.-J. Tu, Z.-G. Han and F. Shi, Org. Biomol. Chem., 2009, 7, 1410–1414 RSC.
- J.-T. Xu, G.-Q. Xu, Z.-Y. Wang and P.-F. Xu, J. Org. Chem., 2019, 84, 14760–14769 CrossRef CAS PubMed.
- O. Demirkol, D. Akbaşlar, S. Giray and B. Anıl, Synth. Commun., 2014, 44, 1279–1285 CrossRef CAS.
- A. Kumar, M. K. Gupta and M. Kumar, Tetrahedron Lett., 2011, 52, 4521–4525 CrossRef CAS.
- B. Karmakar and J. Banerji, Tetrahedron Lett., 2011, 52, 4957–4960 CrossRef CAS.
- K. Arya, U. C. Rajesh and D. S. Rawat, Green Chem., 2012, 14, 3344–3351 RSC.
- X. L. Liu, X. M. Zhang and W. C. Yuan, Tetrahedon Lett, 2011, 52, 903–906 CrossRef CAS.
- Y. Wu, L. Wan, G. Lu and C. Cai, Eur. J. Org. Chem., 2017, 24, 3438–3441 CrossRef.
- K. M. Elattar, A. Fekri, N. M. Bayoumy and A. A. Fadda, Res. Chem. Intermed., 2017, 43(7), 4227–4264 CrossRef CAS.
- M. Monier, D. Abdel-Latif, A. El-Mekabaty, B. D. Mert and K. M. Elattar, Curr. Org. Synth., 2019, 16(6), 812–854 CrossRef CAS PubMed.
- A. A. Fadda, N. M. Bayoumy and K. M. Elattar, Synth. Commun., 2015, 45(23), 2637–2675 CrossRef CAS.
- K. M. Elattar and B. D. Mert, RSC Adv., 2016, 6, 71827–71851 RSC.
- A. A. Fadda and S. A. El-Hadidy, Synth. Commun., 2015, 45(24), 2765–2801 CrossRef CAS.
- A. A. Fadda and A. El-Mekabaty, Synth. Commun., 2013, 43(20), 2685–2719 CrossRef CAS.
- K. M. Elattar, I. Youssef and A. A. Fadda, Synth. Commun., 2016, 46, 719–744 CrossRef CAS.
- K. M. Elattar, R. Rabie and M. M. Hammouda, Synth. Commun., 2016, 46, 1477–1498 CrossRef CAS.
- K. M. Elattar and A. A. Fadda, Synth. Commun., 2016, 46(19), 1567–1594 CrossRef CAS.
- K. M. Elattar, R. Rabie and M. M. Hammouda, Monatsh. Chem., 2017, 148, 601–627 CrossRef CAS.
- M. Monier, D. Abdel-Latif, A. El-Mekabaty and K. M. Elattar, J. Heterocycl. Chem., 2019, 56(12), 3172–3196, DOI:10.1002/jhet.3727.
- M. Monier, D. Abdel-Latif, A. El-Mekabaty and K. M. Elattar, Synth. Commun., 2020, 50(1), 1–32, DOI:10.1080/00397911.2019.1686644.
- M. Monier, A. El-Mekabaty, D. Abdel-Latif and K. M. Elattar, Synth. Commun., 2019, 49(20), 2591–2629, DOI:10.1080/00397911.2019.1643889.
-
(a) A. Y. El-Khateeb, S. E. Hamed and K. M. Elattar, RSC Adv., 2022, 12(19), 11808–11842 RSC;
(b) K. M. Elattar, A. Y. El-Khateeb and S. E. Hamed, RSC Med. Chem., 2022, 13, 522–567, 10.1039/D2MD00076H.
- A. A. Fadda and K. M. Elattar, Synth. Commun., 2016, 46(1), 1–30 CrossRef CAS.
- H. P. Mettler,and A. G. Lonza, US. Pat. 20060252945A1, 2006, https://patents.google.com/patent/EP1510517A1/en?oq=beta-ketoamines.
- N. R. Easton, and R. D. Dillard, Eli Lilly and Co, US. Pat. US3359313A, 1967, https://patents.google.com/patent/US3359313A/en.
- R. G. Charles, and J. G. Cleary, Westinghouse Electric Corp, US. Pat. US3594216A, 1971.
- D. Michel and A. G. Lonza, US. Pat. US20050256318A1, 2005.
- D. Michel, R. Fuchs,and A. G. Lonza, US. Pat. US8962865B2, 2015.
- H. Jendralla, W. Schwab, and T. Stuedemann, Optically active β-aminoketones, optically active 1,3-aminoalcohols and method for the production thereof Application PCT/EP2003/004127 events 2002-05-03 Priority to DE10219987.62002-05-03 Priority to DE20021199872003-04-22, Application filed by Aventis Pharma Deutschland Gmbh, Application filed by Aventis Pharma Deutschland Gmbh, WIPO (PCT), Internationale Veröffentlichungsnummer, WO2003093259A1, 2003 Search PubMed.
- H. Jendralla, W. Schwab, and T. Stuedemann, Sanofi Aventis Deutschland GmbH, US. Pat. US20040030145A1, 2007.
- H. Jendralla, W. Schwab, and T. Stuedemann, Application filed by Sanofi Aventis Deutschland GmbH, Priority to PCT/EP2003/004127, Eur. Patent, EP1504002B1, 2003.
- E. Manghisi, G. Cascio, and L. Bastianini, Application filed by Istituto Luso Farmaco dItalia SpA, US. pat. US3933803A, 1976.
- S. Kobayashi, and M. Sugiura, Japan Science and Technology Agency, US. Pat. EP1491525B1, 2007.
- F. Dong, Priority to CN2010106242025A, Chinese Patent, CN102070472A, 2011, https://patents.google.com/patent/CN102070472A/en?oq=%CE%B2-aminoketones.
- Y. Dachengang, C. Xineiyu, L. Hongping, H. Zuweniia, and S. Xiaoyan, Priority to CN 200810237001, Chinese Patent, CN101538230B, 2008, https://patents.google.com/patent/CN101538230B/en?oq=%CE%B2-aminoketones.
- H. Jiang, X. Yu, C. G. Daniliuc and A. Studer, Angew. Chem.,
Int. Ed., 2021, 60(26), 14399–14404 CrossRef CAS PubMed.
- M. Hadizadeh, M. H. Mosslemin and B. Sadeghi, Bulg. Chem. Commun., 2018, 50, 262–269 Search PubMed.
- M. S. Menkudle, A. V. Chakrawar, P. M. Kulkarni, W. N. Jadhav and S. R. Bhusare, Asian J. Green Chem., 2020, 4(3), 249–255 CAS.
- J. Riss, J. Cloyd, J. Gates and S. Collins, Acta Neurol. Scand., 2008, 118, 69–86 CrossRef CAS PubMed.
- S. Samshuddin, B. Narayana, B. K. Sarojini, H. S. Yathirajan and R. Raghavendra, Pharma Chem., 2012, 1445–1457 CAS.
- X. Fu, J. Feng, Z. Dong, L. Lin, X. Liu and X. Feng, Eur. J. Org. Chem., 2011, 27, 5233–5236 CrossRef.
- G. Grossi, M. D. Braccio, G. Roma, V. Ballabeni, M. Tognolini, F. Calcina and E. Barocelli, Eur. J. Med. Chem., 2002, 37, 933–944 CrossRef CAS.
- Z. Karimi-Jaberi and A. Hooshmandpour, Polycyclic Aromat. Compd., 2020, 40(2), 432–436, DOI:10.1080/10406638.2018.1441876.
- H. Sharghi, J. Aboonajmi, M. Mozaffari, M. M. Doroodmand and M. Aberi, Appl. Organomet. Chem., 2018, 32(3), e4124 CrossRef.
- M. Heidarpour, H. Anaraki-Ardakani, N. Hasanzadeh and A. Rayatzadeh, Appl. Organomet. Chem., 2020, 34(10), e5834 CrossRef CAS.
- M. Kiani, H. Anaraki-Ardakani, N. Hasanzadeh and A. Rayatzadeh, J. Iran. Chem. Soc., 2020, 17(9), 2243–2256 CrossRef CAS.
- M. A. Bigdeli, M. M. Heravi, F. Nemati and G. H. Mahdavinia, General Paper, 2008, 243–248 CAS.
- N. Saadatjoo, M. Golshekan, S. Shariati, P. Azizi and F. Nemati, Arabian J. Chem., 2017, 10, S735–S741 CrossRef CAS.
- M. A. Bigdeli, F. Nemati and G. H. Mahdavinia, Tetrahedron Lett., 2007, 48, 6801–6804 CrossRef CAS.
- R. Khoshnavazi, L. Bahrami, F. Havasi and E. Naseri, RSC Adv., 2017, 7, 11510–11521 RSC.
- M. Kooti, F. Kooshki, E. Nasiri and A. Naghdi Sedeh, J. Iran. Chem. Soc., 2018, 15, 943–953 CrossRef CAS.
- W. Shen, L. M. Wang and H. Tian, J. Fluorine Chem., 2008, 129, 267–273 CrossRef CAS.
- A. Mansoori, H. Eshghi and J. Lari, J. Chin. Chem. Soc., 2018, 65, 548–553 CrossRef CAS.
- F. Dong, L. Jun, Z. Xin-Li and L. Zu-Liang, Catal. Lett., 2007, 116, 76–80 CrossRef.
- A. Maleki, P. Zand, Z. Mohseni and R. Firouzi-Haji, Nano-Struct. Nano-Objects, 2018, 16, 31–37 CrossRef CAS.
- J. Safaei-Ghomi and S. Zahedi, Polycyclic Aromat. Compd., 2018, 38(4), 338–345 CrossRef CAS.
- J. Safaei-Ghomi and S. Zahedi, Appl. Organomet. Chem., 2015, 29, 566–571 CrossRef CAS.
- J. Lei, L. Peng, R. Qiu, Y. Liu, Y. Chen, C. T. Au and S. F. Yin, Dalton Trans., 2019, 48(23), 8478–8487 RSC.
- M. Gupta, B. Chowhan, M. Gupta and S. Paul, J. Chem. Sci., 2022, 134(1), 1–16 CrossRef.
- X. He, X. Wang, Y. L. Tse, Z. Ke and Y. Y. Yeung, Angew. Chem., Int. Ed., 2018, 57(39), 12869–12873 CrossRef CAS PubMed.
-
(a) B. B. Thompson, J. Pharm. Sci., 1968, 57, 715–733 CrossRef CAS PubMed;
(b) W. Notz, F. Tanaka and C. F. Barbas III, Acc. Chem. Res., 2004, 37, 580–591 CrossRef CAS PubMed;
(c) J. M. M. Verkade, L. J. C. van Hemert, P. J. L. M. Quaedflieg and F. P. J. T. Rutjes, Chem. Soc. Rev., 2008, 37, 29–41 RSC;
(d) A. Moyano and R. Rios, Chem. Rev., 2011, 111, 4703–4832 CrossRef CAS PubMed.
- J. Iwanejko, E. Wojaczyńska and T. K. Olszewski, Curr. Opin. Green Sustainable Chem., 2018, 10, 27–34 CrossRef.
- R. Qui, S. Yin, X. Zhang, J. Xia, X. Xu and S. Luo, Chem. Commun., 2009, 4759–4761 Search PubMed.
- F. Dong, F. Zheghao and L. Zu-Liang, Catal. Commun., 2009, 10, 1267–1270 CrossRef CAS.
- T. Chang, L. He, L. Bian, H. Han, M. Yuan and X. Gao, RSC Adv., 2014, 4, 727–731 RSC.
- G. Roman, R. Oghină and L. Săcărescu, Sci. Study Res.: Chem. Chem. Eng., Biotechnol., Food Ind., 2020, 21(4), 511–521 CAS.
- R. M. Coates and H. Y. Kang, J. Org. Chem., 1987, 52, 2065–2074 CrossRef CAS.
- Z. Szakonyi, T. Gonda, S. B. Ötvös and F. Fülöp, Tetrahedron: Asymmetry, 2014, 25, 1138–1145 CrossRef CAS.
- D. Ozsvár, V. Nagy, I. Zupkó and Z. Szakonyi, Int. J. Mol. Sci., 2021, 22, 11232, DOI:10.3390/ijms222011232.
- S. M. Habibi-Khorassani, M. Shahraki, E. Aghdaei and B. Mostafa, J. Taibah Univ. Sci., 2018, 12(1), 46–55 CrossRef.
- R. Martin-Escolano, E. Moreno-Viguri, M. Santivanez-Veliz, A. Martin-Montes, E. Medina-Carmona, R. Paucar, C. Marin, A. Azqueta, N. Cirauqui, A. L. Pey and S. Perez-Silanes, J. Med. Chem., 2018, 61(13), 5643–5663 CrossRef CAS PubMed.
- E. Moreno-Viguri, C. Jimenez-Montes, R. Martin-Escolano, M. Santivaez-Veliz, A. Martin-Montes, A. Azqueta, M. Jimenez-Lopez, S. Zamora Ledesma, N. Cirauqui, A. Lopez de Cerain, C. Marin, M. Sanchez-Moreno and S. Perez-Silanes, J. Med. Chem., 2016, 59(24), 10929–10945 CrossRef CAS PubMed.
- J. Martinez-Esparza, A. M. Oficialdegui, S. Perez-Silanes, B. Heras, L. Orus, J. A. Palop, B. Lasheras, J. Roca, M. Mourelle, A. Bosch, J. C. Del Castillo, R. Tordera, J. Del Rio and A. Monge, J. Med. Chem., 2001, 44(3), 418–428 CrossRef CAS PubMed.
- G. Mazzeo, G. Longhi, S. Abbate, F. Mangiavacchi, C. Santi, J. Han, V. A. Soloshonok, L. Melensi and R. Ruzziconi, Org. Biomol. Chem., 2018, 16(45), 8742–8750 RSC.
- Y. V. Rassukana, O. V. Stanko and P. P. Onysko, J. Fluorine Chem., 2019, 219, 123–128 CrossRef CAS.
- M. Fischer, K. Schwitalla, S. Baues, M. Schmidtmann and R. Beckhaus, Dalton Trans., 2019, 48(4), 1516–1523 RSC.
- B. M. Trost, C.-I. J. Hung and E. Gnanamani, ACS Catal., 2019, 9, 1549–1557, DOI:10.1021/acscatal.8b04685.
- J. H. Kim, A. Paul, I. Ghiviriga and D. Seidel, Org. Lett., 2021, 23, 797–801, DOI:10.1021/acs.orglett.0c04024.
- S. Fukumoto, M. Shigenobu and K. Ishimaru, Int. J. Org. Chem., 2019, 9(4), 163–173, DOI:10.4236/ijoc.2019.94014.
- G. S. Kumar, D. Singh, M. Kumar and M. Kapur, J. Org. Chem., 2018, 83(7), 3941–3951 CrossRef CAS PubMed.
- A. L. Nguyen, H. R. Khatri, J. R. Woods, C. S. Baldwin, F. R. Fronczek and D. A. Colby, J. Org. Chem., 2018, 83(6), 3109–3118 CrossRef CAS PubMed.
- K. Uoto, S. Ohsuki, H. Takenoshita, T. Ishiyama, S. Iimura, Y. Hirota, I. Mitsui, H. Terasawa and T. Soga, Chem. Pharm. Bull., 1997, 45, 1793–1804 CrossRef CAS PubMed.
- K. Nakayama, H. C. Kawato, H. Inagaki, R. Nakajima, A. Kitamura, K. Someya and T. Ohta, Org. Lett., 2000, 2, 977–980 CrossRef CAS PubMed.
- A. M. Silva, R. E. Cachau, H. L. Sham and J. W. Erickson, J. Mol. Biol., 1996, 255, 321–346 CrossRef CAS PubMed.
- S. Thaisrivongs, H. J. Schostarez, D. T. Pals and S. R. Turner, J. Med. Chem., 1987, 30, 1837–1842 CrossRef CAS PubMed.
- I. Vergely, N. Boggetto, V. Okochi, S. Golpayegani, M. Reboud-Ravaux, R. Kobaiter, R. Joyeau and M. Wakselman, Eur. J. Med. Chem., 1995, 30, 199–208 CrossRef CAS.
- G. A. Molander and A. R. Brown, J. Org. Chem., 2006, 71, 9681–9686 CrossRef CAS PubMed.
- M. M. Sadhu, S. K. Ray, R. A. Unhale and V. K. Singh, Org. Biomol. Chem., 2022, 20, 410–414, 10.1039/d1ob02162a.
- C. Miao, L. Jiang, L. Ren, Q. Xue, F. Yan, W. Shi, X. Li, J. Sheng and S. Kai, Tetrahedron, 2019, 75(14), 2215–2228 CrossRef CAS.
- D. Shanthi, K. Rajeswari, C. U. Kumar, T. Vidhyasagar and M. V. Pillai, J. Mol. Struct., 2019, 1198, 126907 CrossRef.
- W. Yuan, X. Wei and J. Yang, Tetrahedron, 2021, 89, 132079 CrossRef CAS.
- W. Yuan, J. H. Xia, X. K. Zhang, P. Liang, J. C. Zhang, W. Jiao and H. W. Shao, Tetrahedron, 2016, 72, 3994–4000, DOI:10.1016/j.tet.2016.05.023.
- A. Gomtsyan, Org. Lett., 2000, 2(1), 11–13, DOI:10.1021/ol9911122.
- A. O. Farah, M. Rabah and T. K. Beng, RSC Adv., 2020, 10(38), 22454–22459 RSC.
- T. C. Ma, S. Yao, M. M. Qiao, F. Yuan, D. Q. Shi and W. J. Xiao, Org. Chem. Front., 2021, 8(15), 4224–4229 RSC.
- V. V. Astakhova, M. Y. Moskalik and B. A. Shainyan, Org. Biomol. Chem., 2019, 17(34), 7927–7937 RSC.
- I. Ojima, in The Organic Chemistry of β-Lactams ed. G. I. Georg, VCH Publishers, New York, 1992, pp. 197–255 Search PubMed.
- A. Vico and R. Fernandez de la Pradilla, Recent Res. Dev. Org. Chem., 2000, 4, 327–334 Search PubMed.
-
(a) E. J. Corey, D.-H. Lee and S. Sarshar, Tetrahedron: Asymmetry, 1995, 6, 3–6 CrossRef CAS;
(b) A. O. Chong, K. Oshima and K. B. Sharpless, J. Am. Chem. Soc., 1977, 99, 3420–3426 CrossRef CAS.
-
(a) S. E. Denmark, X. Su, Y. Nishigaichi, D. M. Coe, K.-T. Wong, S. B. D. Winter and J. Y. Choi, J. Org. Chem., 1999, 64, 1958–1967 CrossRef CAS PubMed;
(b) A. Alexakis, I. Aujard and P. Mangeney, Synlett, 1998, 873–874 CrossRef CAS;
(c) R. D. Dghaym, R. Dhawan and B. A. Arndtsen, Angew. Chem., Int. Ed., 2001, 40, 3228–3230 CrossRef CAS.
-
(a) W. Zhang, J. L. Loebach, S. R. Wilson and E. N. Jacobsen, J. Am. Chem. Soc., 1990, 112, 2801–2802 CrossRef CAS;
(b) L. Deng and E. N. Jacobsen, J. Org. Chem., 1992, 57, 4320–4523 CrossRef CAS;
(c) R. Irie, K. Noda, Y. Ito, N. Matsumoto and Y. Katsuki, Tetrahedron Lett., 1990, 31, 7345–7348 CrossRef CAS;
(d) R. Irie, Y. Ito and Y. Katsuki, Synlett, 1991, 265–266 CrossRef CAS.
- C. Timmons, D. Chen, X. Xu and G. Li, Eur. J. Org. Chem., 2003, 2003(19), 3850–3854 CrossRef.
- C. Timmons, D. Chen, C. E. Barney, S. Kirtane and G. Li, Tetrahedron, 2004, 60(52), 12095–12099 CrossRef CAS.
- K. Chinthapally, R. Karthik, S. Senthilkumar and S. Baskaran, Chem. Eur J., 2017, 23, 533–536, DOI:10.1002/chem.201604376.
- T. S. Zhang, Q. Zhao, W. J. Hao, S. J. Tu and B. Jiang, Chem. - Asian J., 2019, 14(7), 1042–1049 CrossRef CAS PubMed.
- L. Wang, H. Han, L. Gu, W. Zhang, J. Zhao and Q. Wang, Chem. Sci., 2021, 12(46), 15389–15398 RSC.
- H. Guo, Q. Zhang, W. Pan, H. Yang, K. Pei, J. Zhai, T. Li, Z. Wang, Y. Wang and Y. Yin, Asian J. Org. Chem., 2021, 10(8), 2231–2237 CrossRef CAS.
- Q. Zhang, Y. Duan, H. Guo, H. Yang, J. Zhai, T. Li, Z. Wang, X. Lu, Y. Wang and Y. Yin, Chem. - Asian J., 2021, 16(13), 1832–1838 CrossRef CAS PubMed.
- V. Laserna, M. J. Porter and T. D. Sheppard, J. Org. Chem., 2019, 84(18), 11391–11406 CrossRef CAS PubMed.
-
(a) M. N. Pennell, M. G. Unthank, P. Turner and T. D. Sheppard, J. Org. Chem., 2011, 76, 1476–1482 CrossRef PubMed;
(b) M. N. Pennell, P. G. Turner and T. D. Sheppard, Chem. - Eur. J., 2012, 18, 4748–4758 CrossRef CAS PubMed.
-
(a) G. Barker, D. G. Johnson, P. C. Young, S. A. Macgregor and A.-L. Lee, Chem. - Eur. J., 2015, 21, 13748–13757 CrossRef CAS PubMed;
(b) P. Mukherjee and R. A. Widenhoefer, Org. Lett., 2011, 13, 1334–1337 CrossRef CAS PubMed;
(c) P. Mukherjee and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2012, 51, 1405–1407 CrossRef CAS PubMed;
(d) P. C. Young, N. A. Schopf and A.-L. Lee, Chem. Commun., 2013, 49, 4262–4264 RSC;
(e) L. Herkert, S. L. J. Green, G. Barker, D. G. Johnson, P. C. Young, S. A. Macgregor and A.-L. Lee, Chem. - Eur. J., 2014, 20, 11540–11548 CrossRef CAS PubMed;
(f) T. Ghebreghiorgis, B. Biannic, B. H. Kirk, D. H. Ess and A. Aponick, J. Am. Chem. Soc., 2012, 134, 16307–16318 CrossRef CAS PubMed.
- R. Fu, Y. Liu, T. Wu, X. Zhang, Y. Zhu, J. Luo, Z. Zhang and Y. Jiang, Chem. Commun., 2022, 58(21), 3525–3528, 10.1039/D2CC00169A.
- A. Paul, J. H. Kim, S. D. Daniel and D. Seidel, Angew. Chem., Int. Ed., 2021, 60(3), 1625–1628 CrossRef CAS PubMed.
- J. B. Peng, F. P. Wu, D. Li, H. Q. Geng, X. Qi, J. Ying and X. F. Wu, ACS Catal., 2019, 9(4), 2977–2983, DOI:10.1021/acscatal.9b00774.
- J. Yoon and C. H. Cheon, Asian J. Org. Chem., 2019, 8(9), 1631–1636 CrossRef CAS.
- G. A. Gevorgyan, N. Z. Hakobyan, S. S. Hovakimyan, A. G. Melkonyan and G. A. Panosyan, Russ. J. Gen. Chem., 2019, 89(11), 2328–2332, DOI:10.1134/S0044460X19110222.
- A. U. Isakhanyan, G. A. Gevorgyan and S. G. Chshmarityan, et al., Zh. Org. Khim., 2016, 52(4), 608–611 Search PubMed.
- G. A. Gevorgyan, A. U. Isakhanyan, N. K. Gasparyan, et al., Data on the Pharmacological Properties of Tertiary Arylaliphatic Aminopropanols” in: Progress in Organic and Pharmaceutical Chemistry [in Russian], Erevan, 2012, pp. 87–99 Search PubMed.
- E. Blum, J. Zhang, J. Zaluski, D. E. Einstein, E. E. Korshin, A. Kubas, A. Gruzman, G. P. Tochtrop, P. D. Kiser and K. Palczewski, J. Med. Chem., 2021, 64(12), 8287–8302 CrossRef CAS PubMed.
- M. Liaqat, T. Mahmud, M. Imran, M. Ashraf, A. U. Haq, M. Muddassar and T. Ahmad, Bulg. Chem. Commun., 2018, 50(1), 37–43 Search PubMed.
- C. Rhein, S. Löber, P. Gmeiner, E. Gulbins, P. Tripal and J. Kornhuber, J. Neural Transm., 2018, 125(12), 1837–1845 CrossRef CAS PubMed.
- B. T. Trang, C. H. Thuong, P. X. Thao, D. H. Mac and R. Gree, Vietnam J. Chem., 2021, 59(1), 42–46 CAS.
- S. J. Gohari, A. Javidan, A. Moghimi, M. J. Taghizadeh and M. Iman, Can. J. Chem., 2019, 97(5), 331–336 CrossRef CAS.
- D. Wei, T. Liu, Y. He, B. Wei, J. Pan, J. Zhang, N. Jiao and B. Han, Angew. Chem., Int. Ed., 2021, 60(50), 26308–26313 CrossRef CAS PubMed.
-
(a) G. A. Gevorgyan, A. G. Agababyan and O. L. Mndzhoyan, Russ. Chem. Rev., 1984, 53(6), 561 CrossRef;
(b) G. A. Gevorgyan, A. G. Agababyan and O. L. Mndzhoyan, Russ. Chem. Rev., 1985, 54(5), 495 CrossRef.
- A. L. Simplício, J. M. Clancy and J. F. Gilmer, Prodrugs for amines, Molecules, 2008, 13(3), 519–547 CrossRef PubMed.
- J. M. Verkade, L. J. van Hemert, P. J. Quaedflieg and F. P. Rutjes, Chem. Soc. Rev., 2008, 37(1), 29–41 RSC.
- J. F. Allochio Filho, B. C. Lemos, A. S. de Souza, S. Pinheiro and S. J. Greco, Tetrahedron, 2017, 73(50), 6977–7004 CrossRef CAS.
- A. L. Simplício, J. M. Clancy and J. F. Gilmer, Int. J. Pharm., 2007, 336(2), 208–214, DOI:10.1016/j.ijpharm.2006.11.055.
- L. A. Arnold, E. Estebanez-Perpina, M. Togashi, N. Jouravel, A. Shelat, A. C. McReynolds, E. Mar, P. Nguyen, J. D. Baxter, R. J. Fletterick, P. Webb and R. K. Guy, J. Biol. Chem., 2005, 280, 43048–43055 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *c
*c